Exploring the Genetic Landscape of Retinal Diseases in North-Western Pakistan Reveals a High Degree of Autozygosity and a Prevalent Founder Mutation in ABCA4

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
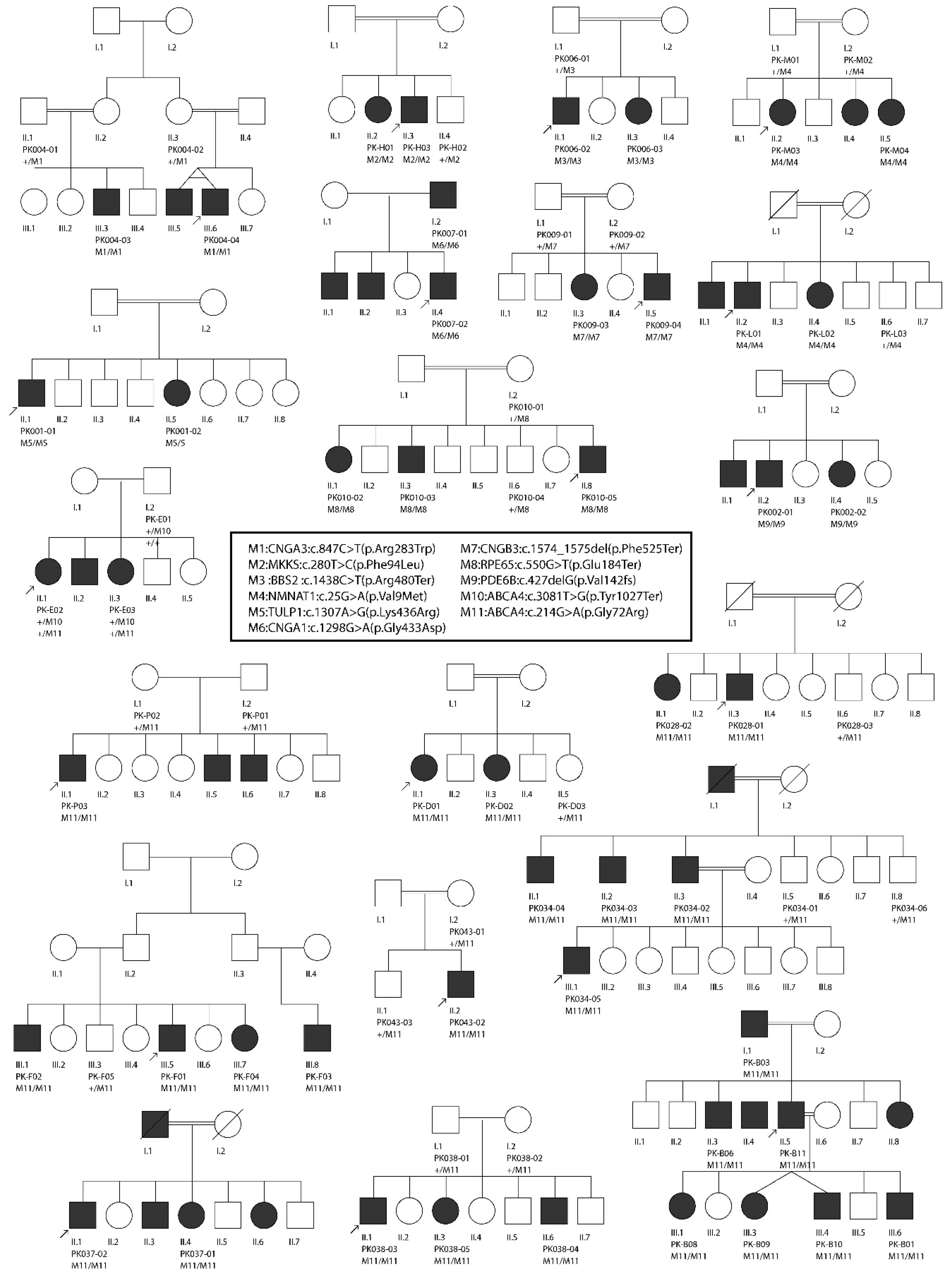
2.1. Enrollment of Families and Collection of Samples
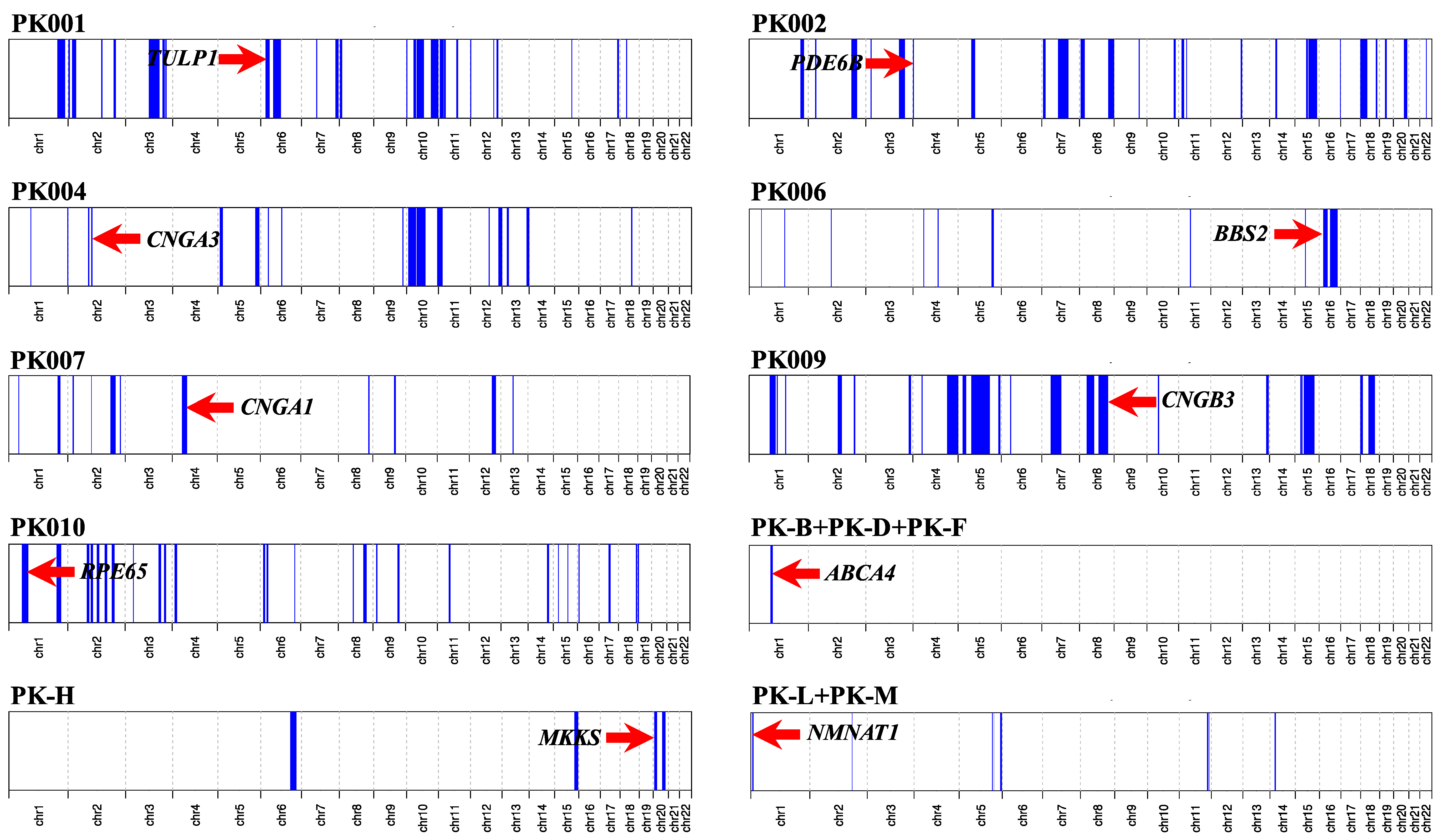
2.2. Genotyping and Homozygosity Mapping
2.3. Whole-Exome Sequencing
3. Results
3.1. Clinical Synopsis
3.2. Molecular Findings
3.3. Macular Dystrophy (Possibly Including Stargardt Disease and Cone-Rod Degeneration)
3.4. Retinitis Pigmentosa
3.5. Leber Congenital Amaurosis (Early-Onset Retinal Blindness)
3.6. Bardet–Biedl Syndrome
3.7. Achromatopsia
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Hamel, C.P. Gene discovery and prevalence in inherited retinal dystrophies. C. R. Biol. 2014, 337, 160–166. [Google Scholar] [CrossRef] [PubMed]
- Berger, W.; Kloeckener-Gruissem, B.; Neidhardt, J. The molecular basis of human retinal and vitreoretinal diseases. Prog. Retin. Res 2010, 29, 335–375. [Google Scholar] [CrossRef] [PubMed]
- Duncan, J.L.; Pierce, E.A.; Laster, A.M.; Daiger, S.P.; Birch, D.G.; Ash, J.D.; Iannaccone, A.; Flannery, J.G.; Sahel, J.A.; Zack, D.J.; et al. Inherited Retinal degenerations: Current Landscape and knowledge gaps. Transl. Vis. Sci. Technol. 2018, 7, 6. [Google Scholar] [CrossRef] [PubMed]
- Brown, M.D.; Voljavec, A.S.; Lott, M.T.; MacDonald, I.; Wallace, D.C. Leber’s hereditary optic neuropathy: A model for mitochondrial neurodegenerative diseases. Faseb. J. 1992, 6, 2791–2799. [Google Scholar] [CrossRef]
- Traboulsi, E.I. Hope and major strides for genetic diseases of the eye. J. Genet. 2009, 88, 395–397. [Google Scholar] [CrossRef] [PubMed]
- Stone, E.M.; Andorf, J.L.; Whitmore, S.S.; DeLuca, A.P.; Giacalone, J.C.; Streb, L.M.; Braun, T.A.; Mullins, R.F.; Scheetz, T.E.; Sheffield, V.C.; et al. Clinically focused molecular investigation of 1000 consecutive families with inherited retinal disease. Ophthalmology 2017, 124, 1314–1331. [Google Scholar] [CrossRef]
- Abu-Safieh, L.; Alrashed, M.; Anazi, S.; Alkuraya, H.; Khan, A.O.; Al-Owain, M.; Al-Zahrani, J.; Al-Abdi, L.; Hashem, M.; Al-Tarimi, S.; et al. Autozygome-guided exome sequencing in retinal dystrophy patients reveals pathogenetic mutations and novel candidate disease genes. Genome Res. 2013, 23, 236–247. [Google Scholar] [CrossRef]
- Khalak, H.G.; Wakil, S.M.; Imtiaz, F.; Ramzan, K.; Baz, B.; Almostafa, A.; Hagos, S.; Alzahrani, F.; Abu-Dhaim, N.; Abu Safieh, L.; et al. Autozygome maps dispensable DNA and reveals potential selective bias against nullizygosity. Genet. Med. 2012, 14, 515–519. [Google Scholar] [CrossRef]
- Alkuraya, F.S. Autozygome decoded. Genet. Med. 2010, 12, 765–771. [Google Scholar] [CrossRef]
- Alkuraya, F.S. The application of next-generation sequencing in the autozygosity mapping of human recessive diseases. Hum. Genet. 2013, 132, 1197–1211. [Google Scholar] [CrossRef]
- Chinthapalli, K. First cousin marriage can double risk of birth defects, finds study. Br. Med. J. 2013, 347, f4374. [Google Scholar] [CrossRef] [PubMed]
- Hamamy, H.; Antonarakis, S.E.; Cavalli-Sforza, L.L.; Temtamy, S.; Romeo, G.; Kate, L.P.; Bennett, R.L.; Shaw, A.; Megarbane, A.; van Duijn, C.; et al. Consanguineous marriages, pearls and perils: Geneva international consanguinity workshop report. Genet. Med. 2011, 13, 841–847. [Google Scholar] [CrossRef] [PubMed]
- Riaz, M.; Tiller, J.; Ajmal, M.; Azam, M.; Qamar, R.; Lacaze, P. Implementation of public health genomics in Pakistan. Eur. J. Hum. Genet. 2019, 27, 1485–1492. [Google Scholar] [CrossRef] [PubMed]
- National Institute of Population Studies. Pakistan Demographic and Health Survey; Pakistan, I., Ed.; NIPS: Maryland, MD, USA, 2013. [Google Scholar]
- Hassan, B.; Ahmed, R.; Li, B.; Noor, A.; Hassan, Z.U. A comprehensive study capturing vision loss burden in Pakistan (1990–2025): Findings from the global burden of disease (GBD) 2017 study. PLoS ONE 2019, 14, e0216492. [Google Scholar] [CrossRef] [PubMed]
- Adhi, M.I.; Ahmed, J. Frequency and clinical presentation of retinaly dystrophies—A Hospital based study. Pak. J. Ophthalmol. 2002, 18, 106–110. [Google Scholar]
- Li, L.; Chen, Y.; Jiao, X.; Jin, C.; Jiang, D.; Tanwar, M.; Ma, Z.; Huang, L.; Ma, X.; Sun, W.; et al. Homozygosity mapping and genetic analysis of autosomal recessive retinal dystrophies in 144 consanguineous pakistani families. Invest. Ophthalmol. Vis. Sci. 2017, 58, 2218–2238. [Google Scholar] [CrossRef] [PubMed]
- Maria, M.; Ajmal, M.; Azam, M.; Waheed, N.K.; Siddiqui, S.N.; Mustafa, B.; Ayub, H.; Ali, L.; Ahmad, S.; Micheal, S.; et al. Homozygosity mapping and targeted sanger sequencing reveal genetic defects underlying inherited retinal disease in families from pakistan. PLoS ONE 2015, 10, e0119806. [Google Scholar] [CrossRef]
- Maranhao, B.; Biswas, P.; Gottsch, A.D.; Navani, M.; Naeem, M.A.; Suk, J.; Chu, J.; Khan, S.N.; Poleman, R.; Akram, J.; et al. Investigating the molecular basis of retinal degeneration in a familial cohort of pakistani decent by exome sequencing. PLoS ONE 2015, 10, e0136561. [Google Scholar] [CrossRef]
- Khan, M.I.; Azam, M.; Ajmal, M.; Collin, R.W.; den Hollander, A.I.; Cremers, F.P.; Qamar, R. The molecular basis of retinal dystrophies in pakistan. Genes 2014, 5, 176–195. [Google Scholar] [CrossRef]
- Pervaiz, R.; Faisal, F.; Serakinci, N. Practice of consanguinity and attitudes towards risk in the pashtun population of khyber pakhtunkhwa, pakistan. J. Biosoc. Sci. 2018, 50, 414–420. [Google Scholar] [CrossRef]
- Ahmad, B.; Rehman, A.U.; Malik, S. Consanguinity and inbreeding coefficient in tribal pashtuns inhabiting the turbulent and war-affected territory of bajaur agency, north-west pakistan. J. Biosoc. Sci. 2016, 48, 113–128. [Google Scholar] [CrossRef] [PubMed]
- Rehman, A.U.; Ahmad, I.; Zaman, M.; Malik, S. Transition in consanguinity in dir lower district, a victim of war, natural disaster and population displacement, in north-west pakistan—A response to sthanadar et al. (2015). J. Biosoc. Sci. 2016, 48, 421–426. [Google Scholar] [CrossRef] [PubMed]
- Sthanadar, A.A.; Bittles, A.H.; Zahid, M. Civil unrest and the current profile of consanguineous marriage in Khyber Pakhtunkhwa province, Pakistan. J. Biosoc. Sci. 2014, 46, 698–701. [Google Scholar] [CrossRef]
- Sthanadar, A.A.; Bittles, A.H.; Zahid, M. Increasing prevalence of consanguineous marriage confirmed in Khyber Pakhtunkhwa province, Pakistan. J. Biosoc. Sci. 2016, 48, 418–420. [Google Scholar] [CrossRef] [PubMed]
- Purcell, S.; Neale, B.; Todd-Brown, K.; Thomas, L.; Ferreira, M.A.; Bender, D.; Maller, J.; Sklar, P.; de Bakker, P.I.; Daly, M.J.; et al. PLINK: A tool set for whole-genome association and population-based linkage analyses. Am. J. Hum. Genet. 2007, 81, 559–575. [Google Scholar] [CrossRef] [PubMed]
- Royer-Bertrand, B.; Castillo-Taucher, S.; Moreno-Salinas, R.; Cho, T.J.; Chae, J.H.; Choi, M.; Kim, O.H.; Dikoglu, E.; Campos-Xavier, B.; Girardi, E.; et al. Mutations in the heat-shock protein A9 (HSPA9) gene cause the EVEN-PLUS syndrome of congenital malformations and skeletal dysplasia. Sci. Rep. 2015, 5, 17154. [Google Scholar] [CrossRef] [PubMed]
- Kohl, S.; Marx, T.; Giddings, I.; Jagle, H.; Jacobson, S.G.; Apfelstedt-Sylla, E.; Zrenner, E.; Sharpe, L.T.; Wissinger, B. Total colourblindness is caused by mutations in the gene encoding the alpha-subunit of the cone photoreceptor cGMP-gated cation channel. Nat. Genet. 1998, 19, 257–259. [Google Scholar] [CrossRef] [PubMed]
- Xing, D.J.; Zhang, H.X.; Huang, N.; Wu, K.C.; Huang, X.F.; Huang, F.; Tong, Y.; Pang, C.P.; Qu, J.; Jin, Z.B. Comprehensive molecular diagnosis of Bardet-Biedl syndrome by high-throughput targeted exome sequencing. PLoS ONE 2014, 9, e90599. [Google Scholar] [CrossRef]
- Falk, M.J.; Zhang, Q.; Nakamaru-Ogiso, E.; Kannabiran, C.; Fonseca-Kelly, Z.; Chakarova, C.; Audo, I.; Mackay, D.S.; Zeitz, C.; Borman, A.D.; et al. NMNAT1 mutations cause Leber congenital amaurosis. Nat. Genet. 2012, 44, 1040–1045. [Google Scholar] [CrossRef]
- Gu, S.; Lennon, A.; Li, Y.; Lorenz, B.; Fossarello, M.; North, M.; Gal, A.; Wright, A. Tubby-like protein-1 mutations in autosomal recessive retinitis pigmentosa. Lancet 1998, 351, 1103–1104. [Google Scholar] [CrossRef]
- Fujinami, K.; Zernant, J.; Chana, R.K.; Wright, G.A.; Tsunoda, K.; Ozawa, Y.; Tsubota, K.; Robson, A.G.; Holder, G.E.; Allikmets, R.; et al. Clinical and molecular characteristics of childhood-onset Stargardt disease. Ophthalmology 2015, 122, 326–334. [Google Scholar] [CrossRef] [PubMed]
- Rivera, A.; White, K.; Stohr, H.; Steiner, K.; Hemmrich, N.; Grimm, T.; Jurklies, B.; Lorenz, B.; Scholl, H.P.; Apfelstedt-Sylla, E.; et al. A comprehensive survey of sequence variation in the ABCA4 (ABCR) gene in Stargardt disease and age-related macular degeneration. Am. J. Hum. Genet. 2000, 67, 800–813. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.; Jordan, D.M.; Sunyaev, S.R. Predicting functional effect of human missense mutations using PolyPhen-2. Curr. Protoc. Hum. Genet. 2013, 7, 7–20. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Rodelsperger, C.; Schuelke, M.; Seelow, D. MutationTaster evaluates disease-causing potential of sequence alterations. Nat. Methods 2010, 7, 575–576. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genome Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef]
- Stenson, P.D.; Ball, E.V.; Mort, M.; Phillips, A.D.; Shiel, J.A.; Thomas, N.S.; Abeysinghe, S.; Krawczak, M.; Cooper, D.N. Human gene mutation database (HGMD): 2003 update. Hum. Mutat. 2003, 21, 577–581. [Google Scholar] [CrossRef]
- Lek, M.; Karczewski, K.J.; Minikel, E.V.; Samocha, K.E.; Banks, E.; Fennell, T.; O’Donnell-Luria, A.H.; Ware, J.S.; Hill, A.J.; Cummings, B.B.; et al. Analysis of protein-coding genetic variation in 60,706 humans. Nature 2016, 536, 285–291. [Google Scholar] [CrossRef]
- Karczewski, K.J.; Francioli, L.C.; Tiao, G.; Cummings, B.B.; Alföldi, J.; Wang, Q.; Collins, R.L.; Laricchia, K.M.; Ganna, A.; Birnbaum, D.P.; et al. Variation across 141, 456 human exomes and genomes reveals the spectrum of loss-of-function intolerance across human protein-coding genes. BioRxiv 2019, 531210. [Google Scholar]
- Saleheen, D.; Natarajan, P.; Armean, I.M.; Zhao, W.; Rasheed, A.; Khetarpal, S.A.; Won, H.H.; Karczewski, K.J.; O’Donnell-Luria, A.H.; Samocha, K.E.; et al. Human knockouts and phenotypic analysis in a cohort with a high rate of consanguinity. Nature 2017, 544, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Wakeling, M.N.; Laver, T.W.; Wright, C.F.; De Franco, E.; Stals, K.L.; Patch, A.M.; Hattersley, A.T.; Flanagan, S.E.; Ellard, S.; Study, D.D.D. Homozygosity mapping provides supporting evidence of pathogenicity in recessive Mendelian disease. Genet. Med. 2019, 21, 982–986. [Google Scholar] [CrossRef]
- Szpiech, Z.A.; Xu, J.; Pemberton, T.J.; Peng, W.; Zollner, S.; Rosenberg, N.A.; Li, J.Z. Long runs of homozygosity are enriched for deleterious variation. Am. J. Hum. Genet. 2013, 93, 90–102. [Google Scholar] [CrossRef] [PubMed]
- Alkuraya, F.S. Discovery of rare homozygous mutations from studies of consanguineous pedigrees. Curr. Protoc. Hum. Genet. 2012, 6, 6–12. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Family ID | Tentative Diagnosis | Gene Name | Transcript ID | cDNA Change | Protein Change | GnomAD MAF | Mutation Number | Zyg-Osity | ROH Size | Method | References |
|---|---|---|---|---|---|---|---|---|---|---|---|
| PK004 | ACHM | CNGA3 | NM_001298.2 | c.847C>T | p.Arg283Trp | 0.0001402 | M1 | Hom | 5-Mb | WES | [28] |
| PK-H | BBS | MKKS | NM_170784.2 | c.280T>C | p.Phe94Leu | NA | M2 | Hom | 10-Mb | SNP array | This study |
| PK006 | BBS | BBS2 | NM_031885.3 | c.1438C>T | p.Arg480Ter | 0.00001647 | M3 | Hom | 31-Mb | SNP array | [29] |
| PK-L | LCA | NMNAT1 | NM_022787.3 | c.25G>A | p.Val9Met | NA | M4 | Hom | 2-Mb | SNP array | [30] |
| PK-M | LCA | NMNAT1 | NM_022787.3 | c.25G>A | p.Val9Met | NA | M4 | Hom | 2-Mb | SNP array | [30] |
| PK001 | RP | TULP1 | NM_001289395.1 | c.1307A>G | p.Lys436Arg | 0.00002472 | M5 | Hom | 17-Mb | WES | [31] |
| PK007 | RP | CNGA1 | NM_001142564.1 | c.1298G>A | p.Gly433Asp | NA | M6 | Hom | 21-Mb | WES | [18] |
| PK009 | MD | CNGB3 | NM_019098.4 | c.1574_1575del | p.Phe525Ter | NA | M7 | Hom | 39-Mb | WES | This study |
| PK010 | RP | RPE65 | NM_000329.2 | c.550G>T | p.Glu184Ter | NA | M8 | Hom | 21-Mb | WES | This study |
| PK002 | RP | PDE6B | NM_001145291.1 | c.427del | p.Ala143LeufsTer7 | NA | M9 | Hom | 4-Mb | WES | This study |
| PK-E | MD | ABCA4 | NM_000350.2 | c.3081T>G | p.Tyr1027Ter | NA | M10 | Het | NA | SNP-WES | [32] |
| PK-E | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Het | NA | SNP-WES | [33] |
| PK-B | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Hom | 7-Mb | SNP-WES | [33] |
| PK-P | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Hom | NA | TSS | [33] |
| PK-F | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Hom | 24-Mb | SNP-WES | [33] |
| PK-D | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Hom | 24-Mb | SNP-WES | [33] |
| PK028 | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Hom | NA | TSS | [33] |
| PK034 | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Hom | NA | TSS | [33] |
| PK037 | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Hom | NA | TSS | [33] |
| PK038 | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Hom | NA | TSS | [33] |
| PK043 | MD | ABCA4 | NM_000350.2 | c.214G>A | p.Gly72Arg | 0.00002784 | M11 | Hom | NA | TSS | [33] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ur Rehman, A.; Peter, V.G.; Quinodoz, M.; Rashid, A.; Khan, S.A.; Superti-Furga, A.; Rivolta, C. Exploring the Genetic Landscape of Retinal Diseases in North-Western Pakistan Reveals a High Degree of Autozygosity and a Prevalent Founder Mutation in ABCA4. Genes 2020, 11, 12. https://doi.org/10.3390/genes11010012
Ur Rehman A, Peter VG, Quinodoz M, Rashid A, Khan SA, Superti-Furga A, Rivolta C. Exploring the Genetic Landscape of Retinal Diseases in North-Western Pakistan Reveals a High Degree of Autozygosity and a Prevalent Founder Mutation in ABCA4. Genes. 2020; 11(1):12. https://doi.org/10.3390/genes11010012
Chicago/Turabian StyleUr Rehman, Atta, Virginie G. Peter, Mathieu Quinodoz, Abdur Rashid, Syed Akhtar Khan, Andrea Superti-Furga, and Carlo Rivolta. 2020. "Exploring the Genetic Landscape of Retinal Diseases in North-Western Pakistan Reveals a High Degree of Autozygosity and a Prevalent Founder Mutation in ABCA4" Genes 11, no. 1: 12. https://doi.org/10.3390/genes11010012
APA StyleUr Rehman, A., Peter, V. G., Quinodoz, M., Rashid, A., Khan, S. A., Superti-Furga, A., & Rivolta, C. (2020). Exploring the Genetic Landscape of Retinal Diseases in North-Western Pakistan Reveals a High Degree of Autozygosity and a Prevalent Founder Mutation in ABCA4. Genes, 11(1), 12. https://doi.org/10.3390/genes11010012