The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye



Abstract
1. Introduction
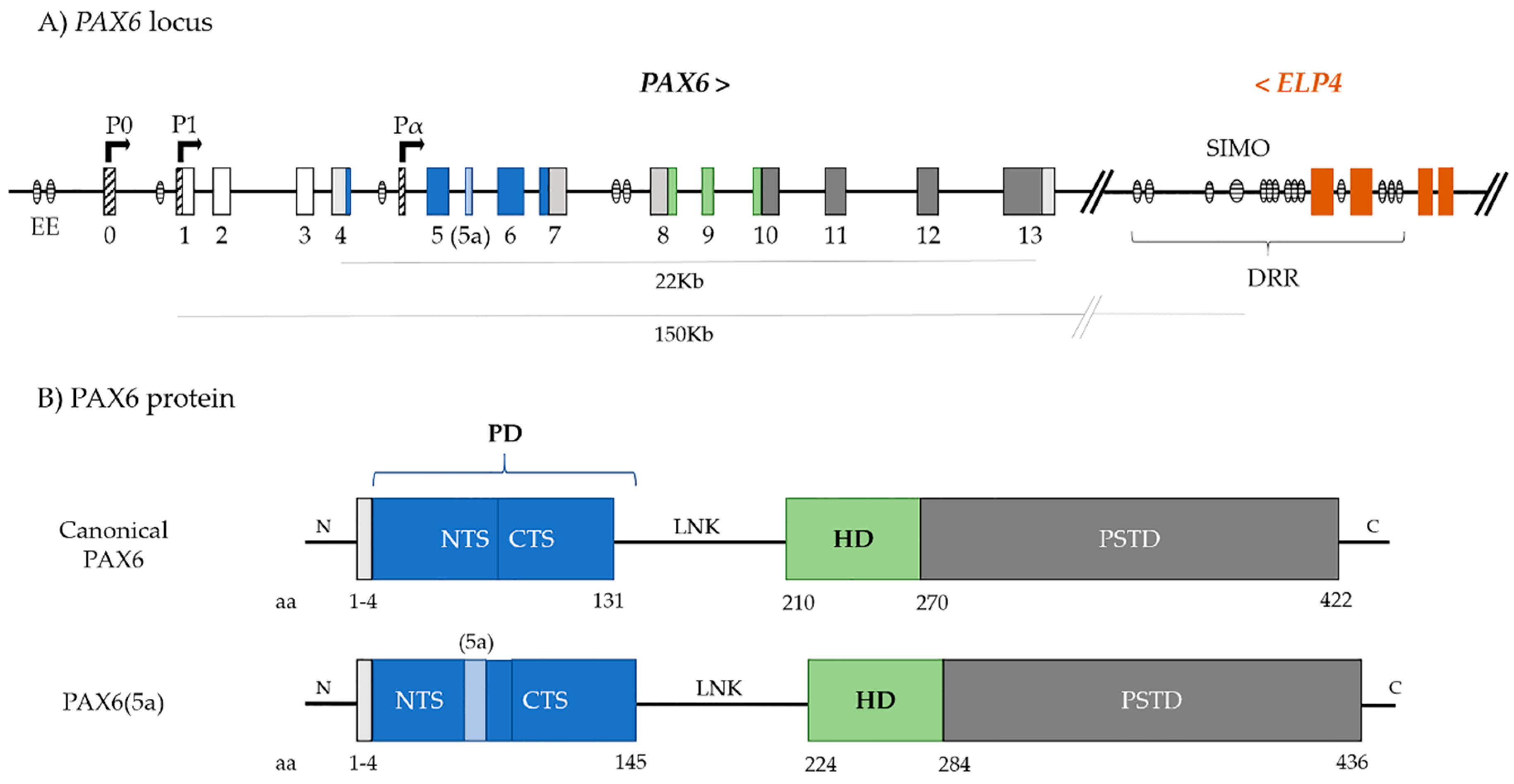
2. PAX6
3. PAX6 Regulation
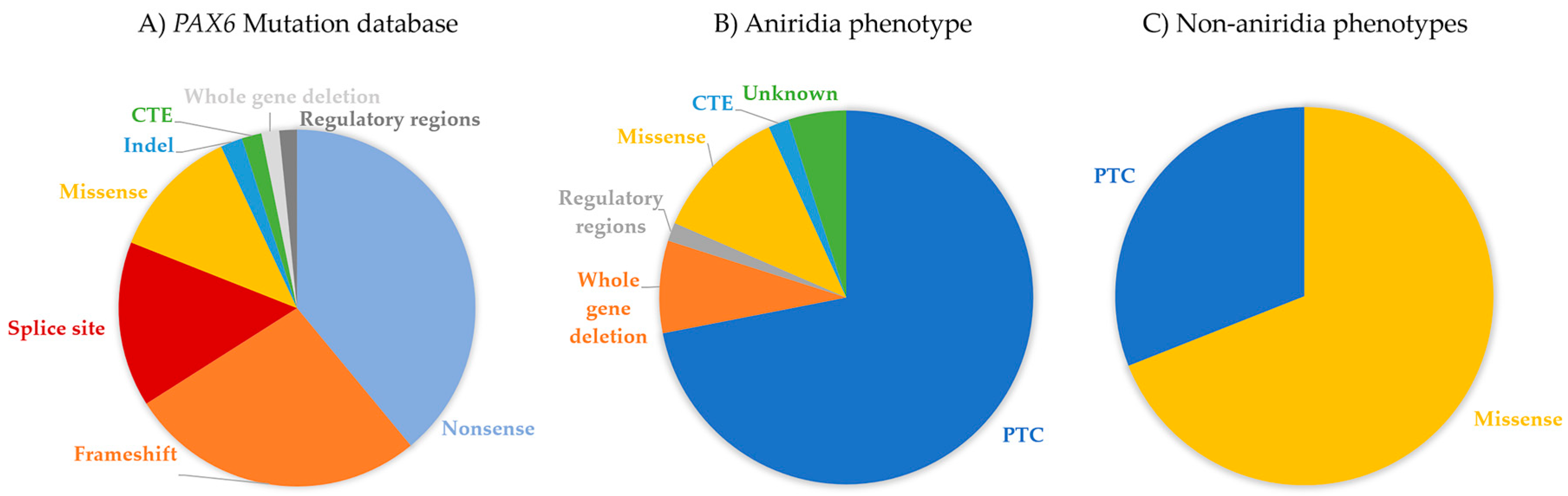
4. PAX6 Mutation Spectrum
4.1. Premature Termination Codon (PTC) Mutations—Nonsense, Frameshift, and Splice Site Variants
4.2. C-Terminal Exptension (CTE) Variants
4.3. Missense Variants
4.4. Non-Coding Variants
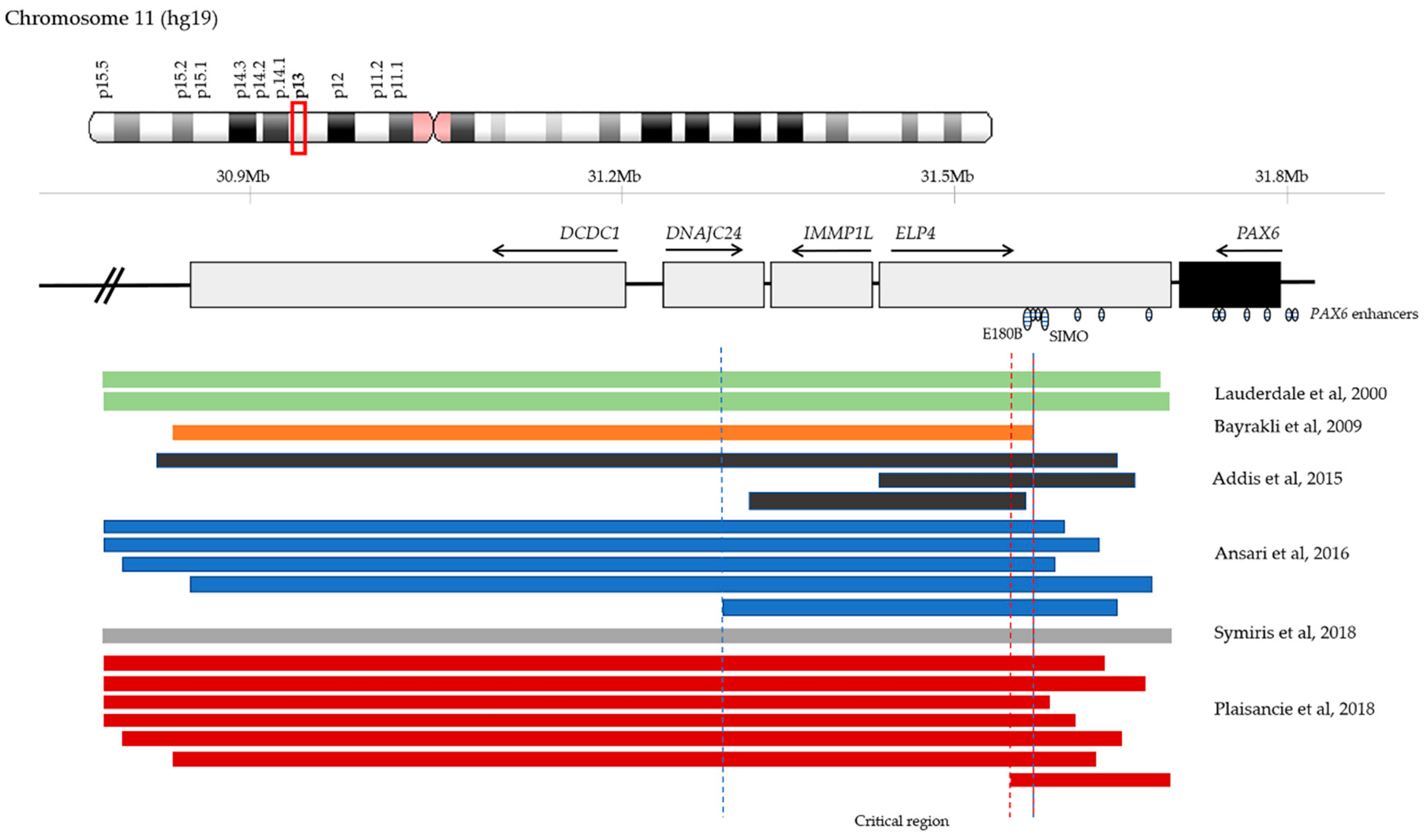
4.5. Chromosomal Rearrangements and Large Deletions
4.6. Biallelic Mutations
5. Inheritance of PAX6 Mutations
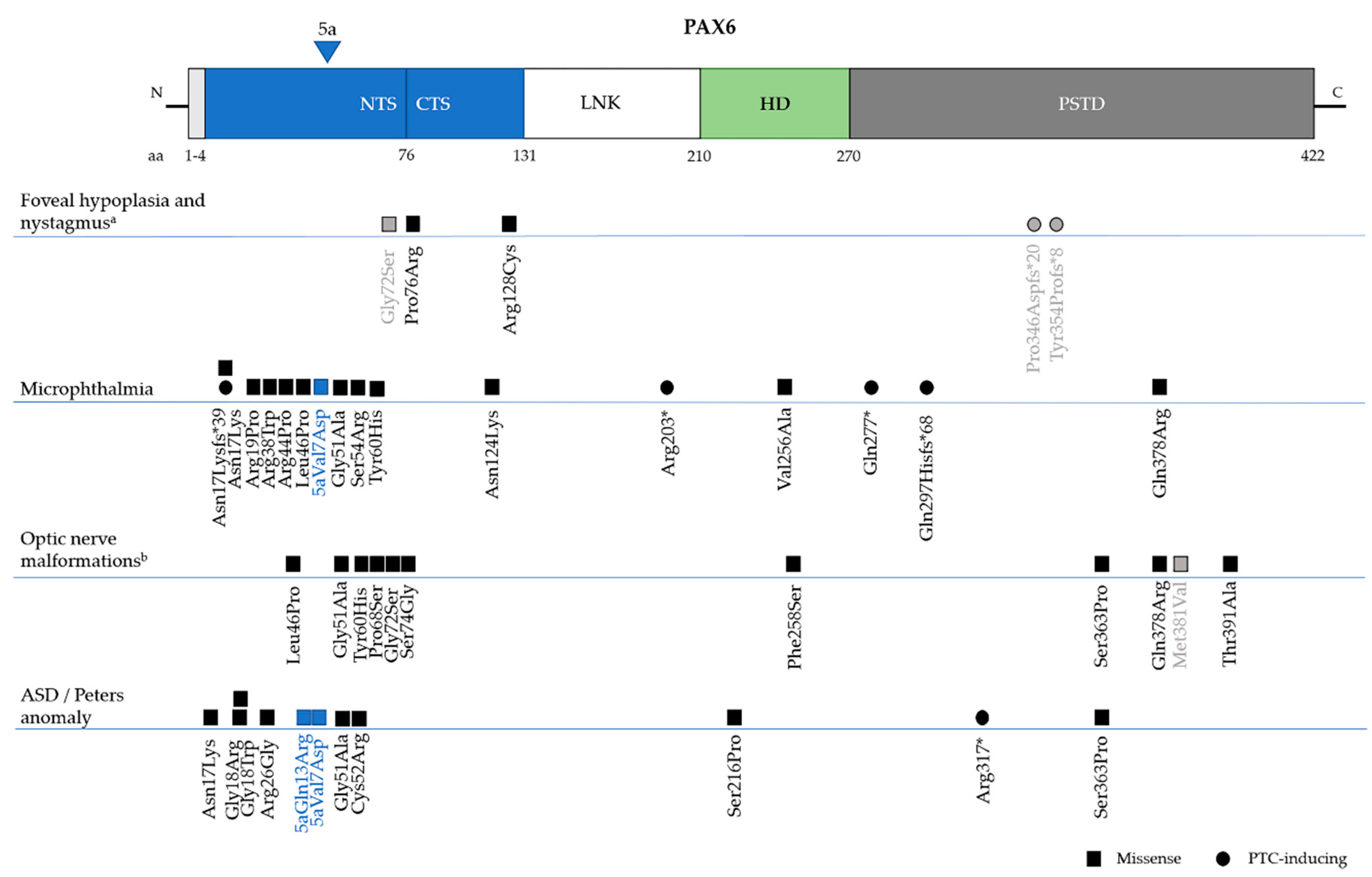
6. Genotype-Phenotype Correlations
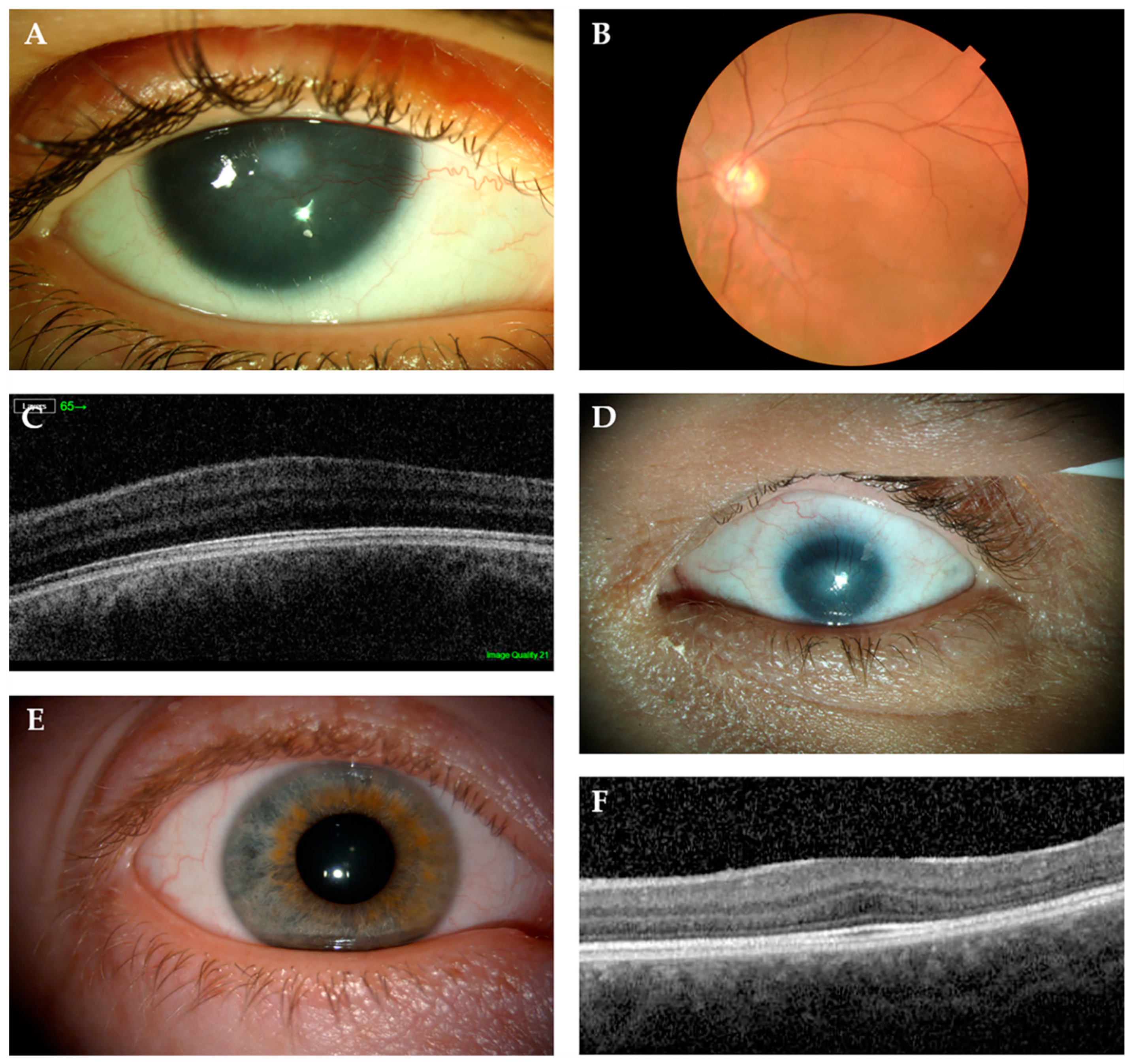
6.1. Aniridia (MIM 106210)
6.2. WAGR (MIM 194072)
6.3. Non-Aniridia Phenotypes
6.3.1. Isolated Foveal Hypoplasia
6.3.2. Microphthalmia, Anophthalmia and Coloboma (MAC)
6.3.3. Gillespie Syndrome (MIM 206700)
6.3.4. Anterior Segment Dysgenesis–Peters Anomaly (MIM 604229)
7. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Callaerts, P.; Halder, G.; Gehring, W.J. PAX-6 in development and evolution. Annu. Rev. Neurosci. 1997, 20, 483–532. [Google Scholar] [CrossRef] [PubMed]
- Ton, C.C.; Hirvonen, H.; Miwa, H.; Weil, M.M.; Monaghan, P.; Jordan, T.; van Heyningen, V.; Hastie, N.D.; Meijers-Heijboer, H.; Drechsler, M.; et al. Positional cloning and characterization of a paired box- and homeobox-containing gene from the aniridia region. Cell 1991, 67, 1059–1074. [Google Scholar] [CrossRef]
- Walther, C.; Guenet, J.L.; Simon, D.; Deutsch, U.; Jostes, B.; Goulding, M.D.; Plachov, D.; Balling, R.; Gruss, P. Pax: A murine multigene family of paired box-containing genes. Genomics 1991, 11, 424–434. [Google Scholar] [CrossRef]
- Hill, R.E.; Favor, J.; Hogan, B.L.; Ton, C.C.; Saunders, G.F.; Hanson, I.M.; Prosser, J.; Jordan, T.; Hastie, N.D.; van Heyningen, V. Mouse small eye results from mutations in a paired-like homeobox-containing gene. Nature 1991, 354, 522–525. [Google Scholar] [CrossRef] [PubMed]
- Jordan, T.; Hanson, I.; Zaletayev, D.; Hodgson, S.; Prosser, J.; Seawright, A.; Hastie, N.; van Heyningen, V. The human PAX6 gene is mutated in two patients with aniridia. Nat. Genet. 1992, 1, 328–332. [Google Scholar] [CrossRef]
- Krauss, S.; Johansen, T.; Korzh, V.; Fjose, A. Expression pattern of zebrafish pax genes suggests a role in early brain regionalization. Nature 1991, 353, 267–270. [Google Scholar] [CrossRef]
- Martin, P.; Carriere, C.; Dozier, C.; Quatannens, B.; Mirabel, M.A.; Vandenbunder, B.; Stehelin, D.; Saule, S. Characterization of a paired box- and homeobox-containing quail gene (Pax-QNR) expressed in the neuroretina. Oncogene 1992, 7, 1721–1728. [Google Scholar]
- Quiring, R.; Walldorf, U.; Kloter, U.; Gehring, W.J. Homology of the eyeless gene of Drosophila to the Small eye gene in mice and Aniridia in humans. Science 1994, 265, 785–789. [Google Scholar] [CrossRef]
- Halder, G.; Callaerts, P.; Gehring, W.J. Induction of ectopic eyes by targeted expression of the eyeless gene in Drosophila. Science 1995, 267, 1788–1792. [Google Scholar] [CrossRef]
- Chow, R.L.; Altmann, C.R.; Lang, R.A.; Hemmati-Brivanlou, A. Pax6 induces ectopic eyes in a vertebrate. Development 1999, 126, 4213–4222. [Google Scholar]
- Terzic, J.; Saraga-Babic, M. Expression pattern of PAX3 and PAX6 genes during human embryogenesis. Int. J. Dev. Biol. 1999, 43, 501–508. [Google Scholar] [PubMed]
- Shaham, O.; Menuchin, Y.; Farhy, C.; Ashery-Padan, R. Pax6: A multi-level regulator of ocular development. Prog. Retin. Eye Res. 2012, 31, 351–376. [Google Scholar] [CrossRef] [PubMed]
- Stanescu, D.; Iseli, H.P.; Schwerdtfeger, K.; Ittner, L.M.; Reme, C.E.; Hafezi, F. Continuous expression of the homeobox gene Pax6 in the ageing human retina. Eye 2007, 21, 90–93. [Google Scholar] [CrossRef] [PubMed]
- Richardson, R.; Hingorani, M.; Van Heyningen, V.; Gregory-Evans, C.; Moosajee, M. Clinical utility gene card for: Aniridia. Eur. J. Hum. Genet. 2016, 24, 1649. [Google Scholar] [CrossRef] [PubMed]
- Moosajee, M.; Hingorani, M.; Moore, A.T. PAX6-Related Aniridia. In GeneReviews((R)); Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, DC, USA, 2018. [Google Scholar]
- Maekawa, M.; Iwayama, Y.; Nakamura, K.; Sato, M.; Toyota, T.; Ohnishi, T.; Yamada, K.; Miyachi, T.; Tsujii, M.; Hattori, E.; et al. A novel missense mutation (Leu46Val) of PAX6 found in an autistic patient. Neurosci. Lett. 2009, 462, 267–271. [Google Scholar] [CrossRef]
- Kikkawa, T.; Casingal, C.R.; Chun, S.H.; Shinohara, H.; Hiraoka, K.; Osumi, N. The role of Pax6 in brain development and its impact on pathogenesis of autism spectrum disorder. Brain Res. 2019, 1705, 95–103. [Google Scholar] [CrossRef]
- Davis, L.K.; Meyer, K.J.; Rudd, D.S.; Librant, A.L.; Epping, E.A.; Sheffield, V.C.; Wassink, T.H. Pax6 3′ deletion results in aniridia, autism and mental retardation. Hum. Genet. 2008, 123, 371–378. [Google Scholar] [CrossRef]
- Hergott-Faure, L.; Borot, S.; Kleinclauss, C.; Abitbol, M.; Penfornis, A. Pituitary function and glucose tolerance in a family with a PAX6 mutation. Ann. D’Endocrinol. 2012, 73, 510–514. [Google Scholar] [CrossRef]
- Xatzipsalti, M.; Voutetakis, A.; Stamoyannou, L.; Chrousos, G.P.; Kanaka-Gantenbein, C. Congenital Hypopituitarism: Various Genes, Various Phenotypes. Horm. Metab. Res. 2019, 51, 81–90. [Google Scholar] [CrossRef]
- Hanish, A.E.; Butman, J.A.; Thomas, F.; Yao, J.; Han, J.C. Pineal hypoplasia, reduced melatonin and sleep disturbance in patients with PAX6 haploinsufficiency. J. Sleep Res. 2016, 25, 16–22. [Google Scholar] [CrossRef]
- Mitchell, T.N.; Free, S.L.; Williamson, K.A.; Stevens, J.M.; Churchill, A.J.; Hanson, I.M.; Shorvon, S.D.; Moore, A.T.; van Heyningen, V.; Sisodiya, S.M. Polymicrogyria and absence of pineal gland due to PAX6 mutation. Ann. Neurol. 2003, 53, 658–663. [Google Scholar] [CrossRef] [PubMed]
- Boese, E.A.; Tollefson, M.R.; Schnieders, M.J.; Darbro, B.W.; Alward, W.L.M.; Fingert, J.H. Novel Intragenic PAX6 Deletion in a Pedigree with Aniridia, Morbid Obesity, and Diabetes. Curr. Eye Res. 2020, 45, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Panneerselvam, A.; Kannan, A.; Mariajoseph-Antony, L.F.; Prahalathan, C. PAX proteins and their role in pancreas. Diabetes Res. Clin. Pract. 2019, 155, 107792. [Google Scholar] [CrossRef] [PubMed]
- Kleinjan, D.A.; Seawright, A.; Schedl, A.; Quinlan, R.A.; Danes, S.; van Heyningen, V. Aniridia-associated translocations, DNase hypersensitivity, sequence comparison and transgenic analysis redefine the functional domain of PAX6. Hum. Mol. Genet. 2001, 10, 2049–2059. [Google Scholar] [CrossRef]
- Bhatia, S.; Bengani, H.; Fish, M.; Brown, A.; Divizia, M.T.; de Marco, R.; Damante, G.; Grainger, R.; van Heyningen, V.; Kleinjan, D.A. Disruption of autoregulatory feedback by a mutation in a remote, ultraconserved PAX6 enhancer causes aniridia. Am. J. Hum. Genet. 2013, 93, 1126–1134. [Google Scholar] [CrossRef]
- Glaser, T.; Walton, D.S.; Maas, R.L. Genomic structure, evolutionary conservation and aniridia mutations in the human PAX6 gene. Nat. Genet. 1992, 2, 232–239. [Google Scholar] [CrossRef]
- Bruun, J.A.; Thomassen, E.I.; Kristiansen, K.; Tylden, G.; Holm, T.; Mikkola, I.; Bjorkoy, G.; Johansen, T. The third helix of the homeodomain of paired class homeodomain proteins acts as a recognition helix both for DNA and protein interactions. Nucleic Acids Res. 2005, 33, 2661–2675. [Google Scholar] [CrossRef]
- Singh, S.; Chao, L.Y.; Mishra, R.; Davies, J.; Saunders, G.F. Missense mutation at the C-terminus of PAX6 negatively modulates homeodomain function. Hum. Mol. Genet. 2001, 10, 911–918. [Google Scholar] [CrossRef]
- Tang, H.K.; Singh, S.; Saunders, G.F. Dissection of the transactivation function of the transcription factor encoded by the eye developmental gene PAX6. J. Biol. Chem. 1998, 273, 7210–7221. [Google Scholar] [CrossRef]
- Epstein, J.A.; Glaser, T.; Cai, J.; Jepeal, L.; Walton, D.S.; Maas, R.L. Two independent and interactive DNA-binding subdomains of the Pax6 paired domain are regulated by alternative splicing. Genes Dev. 1994, 8, 2022–2034. [Google Scholar] [CrossRef]
- Epstein, J.; Cai, J.; Glaser, T.; Jepeal, L.; Maas, R. Identification of a Pax paired domain recognition sequence and evidence for DNA-dependent conformational changes. J. Biol. Chem. 1994, 269, 8355–8361. [Google Scholar]
- Azuma, N.; Yamaguchi, Y.; Handa, H.; Hayakawa, M.; Kanai, A.; Yamada, M. Missense mutation in the alternative splice region of the PAX6 gene in eye anomalies. Am. J. Hum. Genet. 1999, 65, 656–663. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.; Lauderdale, J.D. Analysis of Pax6 expression using a BAC transgene reveals the presence of a paired-less isoform of Pax6 in the eye and olfactory bulb. Dev. Biol. 2006, 292, 486–505. [Google Scholar] [CrossRef] [PubMed]
- Carriere, C.; Plaza, S.; Martin, P.; Quatannens, B.; Bailly, M.; Stehelin, D.; Saule, S. Characterization of quail Pax-6 (Pax-QNR) proteins expressed in the neuroretina. Mol. Cell. Biol. 1993, 13, 7257–7266. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Kim, J.; Lauderdale, J.D. Overexpression of pairedless Pax6 in the retina disrupts corneal development and affects lens cell survival. Dev. Biol. 2008, 313, 434–454. [Google Scholar] [CrossRef]
- Kleinjan, D.A.; Seawright, A.; Mella, S.; Carr, C.B.; Tyas, D.A.; Simpson, T.I.; Mason, J.O.; Price, D.J.; van Heyningen, V. Long-range downstream enhancers are essential for Pax6 expression. Dev. Biol. 2006, 299, 563–581. [Google Scholar] [CrossRef]
- Lakowski, J.; Majumder, A.; Lauderdale, J.D. Mechanisms controlling Pax6 isoform expression in the retina have been conserved between teleosts and mammals. Dev. Biol. 2007, 307, 498–520. [Google Scholar] [CrossRef]
- Azuma, N.; Tadokoro, K.; Asaka, A.; Yamada, M.; Yamaguchi, Y.; Handa, H.; Matsushima, S.; Watanabe, T.; Kohsaka, S.; Kida, Y.; et al. The Pax6 isoform bearing an alternative spliced exon promotes the development of the neural retinal structure. Hum. Mol. Genet. 2005, 14, 735–745. [Google Scholar] [CrossRef]
- Dominguez, M.; Ferres-Marco, D.; Gutierrez-Avino, F.J.; Speicher, S.A.; Beneyto, M. Growth and specification of the eye are controlled independently by Eyegone and Eyeless in Drosophila melanogaster. Nat. Genet. 2004, 36, 31–39. [Google Scholar] [CrossRef]
- Xu, P.X.; Zhang, X.; Heaney, S.; Yoon, A.; Michelson, A.M.; Maas, R.L. Regulation of Pax6 expression is conserved between mice and flies. Development 1999, 126, 383–395. [Google Scholar]
- Zhang, W.; Cveklova, K.; Oppermann, B.; Kantorow, M.; Cvekl, A. Quantitation of PAX6 and PAX6 (5a) transcript levels in adult human lens, cornea, and monkey retina. Mol. Vis. 2001, 7, 1–5. [Google Scholar] [PubMed]
- Cvekl, A.; Callaerts, P. PAX6: 25th anniversary and more to learn. Exp. Eye Res. 2017, 156, 10–21. [Google Scholar] [CrossRef] [PubMed]
- Azuma, N.; Nishina, S.; Yanagisawa, H.; Okuyama, T.; Yamada, M. PAX6 missense mutation in isolated foveal hypoplasia. Nat. Genet. 1996, 13, 141–142. [Google Scholar] [CrossRef] [PubMed]
- Van Heyningen, V.; Williamson, K.A. PAX6 in sensory development. Hum. Mol. Genet. 2002, 11, 1161–1167. [Google Scholar] [CrossRef]
- Sasamoto, Y.; Hayashi, R.; Park, S.J.; Saito-Adachi, M.; Suzuki, Y.; Kawasaki, S.; Quantock, A.J.; Nakai, K.; Tsujikawa, M.; Nishida, K. PAX6 Isoforms, along with Reprogramming Factors, Differentially Regulate the Induction of Cornea-specific Genes. Sci. Rep. 2016, 6, 20807. [Google Scholar] [CrossRef]
- Pinson, J.; Simpson, T.I.; Mason, J.O.; Price, D.J. Positive autoregulation of the transcription factor Pax6 in response to increased levels of either of its major isoforms, Pax6 or Pax6 (5a), in cultured cells. BMC Dev. Biol. 2006, 6, 25. [Google Scholar] [CrossRef]
- Aota, S.; Nakajima, N.; Sakamoto, R.; Watanabe, S.; Ibaraki, N.; Okazaki, K. Pax6 autoregulation mediated by direct interaction of Pax6 protein with the head surface ectoderm-specific enhancer of the mouse Pax6 gene. Dev. Biol. 2003, 257, 1–13. [Google Scholar] [CrossRef]
- Dora, N.; Ou, J.; Kucerova, R.; Parisi, I.; West, J.D.; Collinson, J.M. PAX6 dosage effects on corneal development, growth, and wound healing. Dev. Dyn. Off. Publ. Am. Assoc. Anat. 2008, 237, 1295–1306. [Google Scholar] [CrossRef]
- Pinson, J.; Mason, J.O.; Simpson, T.I.; Price, D.J. Regulation of the Pax6: Pax6 (5a) mRNA ratio in the developing mammalian brain. BMC Dev. Biol. 2005, 5, 13. [Google Scholar] [CrossRef]
- Chauhan, B.K.; Yang, Y.; Cveklova, K.; Cvekl, A. Functional interactions between alternatively spliced forms of Pax6 in crystallin gene regulation and in haploinsufficiency. Nucleic Acids Res. 2004, 32, 1696–1709. [Google Scholar] [CrossRef][Green Version]
- Plaza, S.; Dozier, C.; Langlois, M.C.; Saule, S. Identification and characterization of a neuroretina-specific enhancer element in the quail Pax-6 (Pax-QNR) gene. Mol. Cell. Biol. 1995, 15, 892–903. [Google Scholar] [CrossRef] [PubMed]
- Plaza, S.; Dozier, C.; Turque, N.; Saule, S. Quail Pax-6 (Pax-QNR) mRNAs are expressed from two promoters used differentially during retina development and neuronal differentiation. Mol. Cell. Biol. 1995, 15, 3344–3353. [Google Scholar] [CrossRef] [PubMed]
- Kammandel, B.; Chowdhury, K.; Stoykova, A.; Aparicio, S.; Brenner, S.; Gruss, P. Distinct cis-essential modules direct the time-space pattern of the Pax6 gene activity. Dev. Biol. 1999, 205, 79–97. [Google Scholar] [CrossRef] [PubMed]
- Xu, Z.P.; Saunders, G.F. Transcriptional regulation of the human PAX6 gene promoter. J. Biol. Chem. 1997, 272, 3430–3436. [Google Scholar] [CrossRef] [PubMed]
- Bhatia, S.; Monahan, J.; Ravi, V.; Gautier, P.; Murdoch, E.; Brenner, S.; van Heyningen, V.; Venkatesh, B.; Kleinjan, D.A. A survey of ancient conserved non-coding elements in the PAX6 locus reveals a landscape of interdigitated cis-regulatory archipelagos. Dev. Biol. 2014, 387, 214–228. [Google Scholar] [CrossRef] [PubMed]
- McBride, D.J.; Buckle, A.; van Heyningen, V.; Kleinjan, D.A. DNaseI hypersensitivity and ultraconservation reveal novel, interdependent long-range enhancers at the complex Pax6 cis-regulatory region. PLoS ONE 2011, 6, e28616. [Google Scholar] [CrossRef]
- Griffin, C.; Kleinjan, D.A.; Doe, B.; van Heyningen, V. New 3′ elements control Pax6 expression in the developing pretectum, neural retina and olfactory region. Mech. Dev. 2002, 112, 89–100. [Google Scholar] [CrossRef]
- Kleinjan, D.A.; Seawright, A.; Childs, A.J.; van Heyningen, V. Conserved elements in Pax6 intron 7 involved in (auto)regulation and alternative transcription. Dev. Biol. 2004, 265, 462–477. [Google Scholar] [CrossRef]
- Williams, S.C.; Altmann, C.R.; Chow, R.L.; Hemmati-Brivanlou, A.; Lang, R.A. A highly conserved lens transcriptional control element from the Pax-6 gene. Mech. Dev. 1998, 73, 225–229. [Google Scholar] [CrossRef]
- Vance, K.W.; Sansom, S.N.; Lee, S.; Chalei, V.; Kong, L.; Cooper, S.E.; Oliver, P.L.; Ponting, C.P. The long non-coding RNA Paupar regulates the expression of both local and distal genes. EMBO J. 2014, 33, 296–311. [Google Scholar] [CrossRef]
- Buckle, A.; Nozawa, R.S.; Kleinjan, D.A.; Gilbert, N. Functional characteristics of novel pancreatic Pax6 regulatory elements. Hum. Mol. Genet. 2018, 27, 3434–3448. [Google Scholar] [CrossRef] [PubMed]
- Ravi, V.; Bhatia, S.; Gautier, P.; Loosli, F.; Tay, B.H.; Tay, A.; Murdoch, E.; Coutinho, P.; van Heyningen, V.; Brenner, S.; et al. Sequencing of Pax6 loci from the elephant shark reveals a family of Pax6 genes in vertebrate genomes, forged by ancient duplications and divergences. PLoS Genet. 2013, 9, e1003177. [Google Scholar] [CrossRef] [PubMed]
- Dimanlig, P.V.; Faber, S.C.; Auerbach, W.; Makarenkova, H.P.; Lang, R.A. The upstream ectoderm enhancer in Pax6 has an important role in lens induction. Development 2001, 128, 4415–4424. [Google Scholar] [PubMed]
- Rowan, S.; Siggers, T.; Lachke, S.A.; Yue, Y.; Bulyk, M.L.; Maas, R.L. Precise temporal control of the eye regulatory gene Pax6 via enhancer-binding site affinity. Genes Dev. 2010, 24, 980–985. [Google Scholar] [CrossRef]
- Goudreau, G.; Petrou, P.; Reneker, L.W.; Graw, J.; Loster, J.; Gruss, P. Mutually regulated expression of Pax6 and Six3 and its implications for the Pax6 haploinsufficient lens phenotype. Proc. Natl. Acad. Sci. USA 2002, 99, 8719–8724. [Google Scholar] [CrossRef]
- Inoue, M.; Kamachi, Y.; Matsunami, H.; Imada, K.; Uchikawa, M.; Kondoh, H. PAX6 and SOX2-dependent regulation of the Sox2 enhancer N-3 involved in embryonic visual system development. Genes Cells Devoted Mol. Cell. Mech. 2007, 12, 1049–1061. [Google Scholar] [CrossRef]
- Lauderdale, J.D.; Wilensky, J.S.; Oliver, E.R.; Walton, D.S.; Glaser, T. 3′ deletions cause aniridia by preventing PAX6 gene expression. Proc. Natl. Acad. Sci. USA 2000, 97, 13755–13759. [Google Scholar] [CrossRef]
- Zhang, X.; Friedman, A.; Heaney, S.; Purcell, P.; Maas, R.L. Meis homeoproteins directly regulate Pax6 during vertebrate lens morphogenesis. Genes Dev. 2002, 16, 2097–2107. [Google Scholar] [CrossRef]
- Antosova, B.; Smolikova, J.; Klimova, L.; Lachova, J.; Bendova, M.; Kozmikova, I.; Machon, O.; Kozmik, Z. The Gene Regulatory Network of Lens Induction Is Wired through Meis-Dependent Shadow Enhancers of Pax6. PLoS Genet. 2016, 12, e1006441. [Google Scholar] [CrossRef]
- Blanco-Kelly, F.; Palomares, M.; Vallespin, E.; Villaverde, C.; Martin-Arenas, R.; Velez-Monsalve, C.; Lorda-Sanchez, I.; Nevado, J.; Trujillo-Tiebas, M.J.; Lapunzina, P.; et al. Improving molecular diagnosis of aniridia and WAGR syndrome using customized targeted array-based CGH. PLoS ONE 2017, 12, e0172363. [Google Scholar] [CrossRef]
- Ansari, M.; Rainger, J.; Hanson, I.M.; Williamson, K.A.; Sharkey, F.; Harewood, L.; Sandilands, A.; Clayton-Smith, J.; Dollfus, H.; Bitoun, P.; et al. Genetic Analysis of ‘PAX6-Negative’ Individuals with Aniridia or Gillespie Syndrome. PLoS ONE 2016, 11, e0153757. [Google Scholar] [CrossRef] [PubMed]
- Syrimis, A.; Nicolaou, N.; Alexandrou, A.; Papaevripidou, I.; Nicolaou, M.; Loukianou, E.; Christophidou-Anastasiadou, V.; Malas, S.; Sismani, C.; Tanteles, G.A. Aniridia due to a novel microdeletion affecting PAX6 regulatory enhancers: Case report and review of the literature. J. Genet. 2018, 97, 555–562. [Google Scholar] [CrossRef] [PubMed]
- Macdonald, G.C.; Hesselson, S.E.; Chan, J.Y.; Jenkins, A.B.; Laybutt, D.R.; Hesselson, D.; Campbell, L.V. Deletion distal to the PAX6 coding region reveals a novel basis for familial cosegregation of aniridia and diabetes mellitus. Diabetes Res. Clin. Pract. 2019, 148, 64–71. [Google Scholar] [CrossRef] [PubMed]
- Plaisancie, J.; Tarilonte, M.; Ramos, P.; Jeanton-Scaramouche, C.; Gaston, V.; Dollfus, H.; Aguilera, D.; Kaplan, J.; Fares-Taie, L.; Blanco-Kelly, F.; et al. Implication of non-coding PAX6 mutations in aniridia. Hum. Genet. 2018, 137, 831–846. [Google Scholar] [CrossRef] [PubMed]
- Bayrakli, F.; Guney, I.; Bayri, Y.; Ercan-Sencicek, A.G.; Ceyhan, D.; Cankaya, T.; Mason, C.; Bilguvar, K.; Bayrakli, S.; Mane, S.M.; et al. A novel heterozygous deletion within the 3’ region of the PAX6 gene causing isolated aniridia in a large family group. J. Clin. Neurosci. 2009, 16, 1610–1614. [Google Scholar] [CrossRef] [PubMed]
- Addis, L.; Ahn, J.W.; Dobson, R.; Dixit, A.; Ogilvie, C.M.; Pinto, D.; Vaags, A.K.; Coon, H.; Chaste, P.; Wilson, S.; et al. Microdeletions of ELP4 Are Associated with Language Impairment, Autism Spectrum Disorder, and Mental Retardation. Hum. Mutat. 2015, 36, 842–850. [Google Scholar] [CrossRef] [PubMed]
- Tzoulaki, I.; White, I.M.; Hanson, I.M. PAX6 mutations: Genotype-phenotype correlations. BMC Genet. 2005, 6, 27. [Google Scholar] [CrossRef]
- Celik, A.; Kervestin, S.; Jacobson, A. NMD: At the crossroads between translation termination and ribosome recycling. Biochimie 2015, 114, 2–9. [Google Scholar] [CrossRef]
- Vincent, M.C.; Pujo, A.L.; Olivier, D.; Calvas, P. Screening for PAX6 gene mutations is consistent with haploinsufficiency as the main mechanism leading to various ocular defects. Eur. J. Hum. Genet. 2003, 11, 163–169. [Google Scholar] [CrossRef]
- Singh, S.; Tang, H.K.; Lee, J.Y.; Saunders, G.F. Truncation mutations in the transactivation region of PAX6 result in dominant-negative mutants. J. Biol. Chem. 1998, 273, 21531–21541. [Google Scholar] [CrossRef]
- Inoue, K.; Khajavi, M.; Ohyama, T.; Hirabayashi, S.; Wilson, J.; Reggin, J.D.; Mancias, P.; Butler, I.J.; Wilkinson, M.F.; Wegner, M.; et al. Molecular mechanism for distinct neurological phenotypes conveyed by allelic truncating mutations. Nat. Genet. 2004, 36, 361–369. [Google Scholar] [CrossRef] [PubMed]
- Vasilyeva, T.A.; Voskresenskaya, A.A.; Kasmann-Kellner, B.; Khlebnikova, O.V.; Pozdeyeva, N.A.; Bayazutdinova, G.M.; Kutsev, S.I.; Ginter, E.K.; Semina, E.V.; Marakhonov, A.V.; et al. Molecular analysis of patients with aniridia in Russian Federation broadens the spectrum of PAX6 mutations. Clin. Genet. 2017, 92, 639–644. [Google Scholar] [CrossRef] [PubMed]
- Robinson, D.O.; Howarth, R.J.; Williamson, K.A.; van Heyningen, V.; Beal, S.J.; Crolla, J.A. Genetic analysis of chromosome 11p13 and the PAX6 gene in a series of 125 cases referred with aniridia. Am. J. Med. Genet. Part A 2008, 146, 558–569. [Google Scholar] [CrossRef] [PubMed]
- Bobilev, A.M.; McDougal, M.E.; Taylor, W.L.; Geisert, E.E.; Netland, P.A.; Lauderdale, J.D. Assessment of PAX6 alleles in 66 families with aniridia. Clin. Genet. 2016, 89, 669–677. [Google Scholar] [CrossRef] [PubMed]
- Hingorani, M.; Williamson, K.A.; Moore, A.T.; van Heyningen, V. Detailed ophthalmologic evaluation of 43 individuals with PAX6 mutations. Investig. Ophthalmol. Vis. Sci. 2009, 50, 2581–2590. [Google Scholar] [CrossRef] [PubMed]
- Chao, L.Y.; Mishra, R.; Strong, L.C.; Saunders, G.F. Missense mutations in the DNA-binding region and termination codon in PAX6. Hum. Mutat. 2003, 21, 138–145. [Google Scholar] [CrossRef]
- Tang, H.K.; Chao, L.Y.; Saunders, G.F. Functional analysis of paired box missense mutations in the PAX6 gene. Hum. Mol. Genet. 1997, 6, 381–386. [Google Scholar] [CrossRef]
- Hanson, I.M. PAX6 and congenital eye malformations. Pediatr. Res. 2003, 54, 791–796. [Google Scholar] [CrossRef]
- Azuma, N.; Yamaguchi, Y.; Handa, H.; Tadokoro, K.; Asaka, A.; Kawase, E.; Yamada, M. Mutations of the PAX6 gene detected in patients with a variety of optic-nerve malformations. Am. J. Hum. Genet. 2003, 72, 1565–1570. [Google Scholar] [CrossRef]
- Williamson, K.A.; FitzPatrick, D.R. The genetic architecture of microphthalmia, anophthalmia and coloboma. Eur. J. Med. Genet. 2014, 57, 369–380. [Google Scholar] [CrossRef]
- Harding, P.; Moosajee, M. The Molecular Basis of Human Anophthalmia and Microphthalmia. J. Dev. Biol. 2019, 7, 16. [Google Scholar] [CrossRef] [PubMed]
- Deml, B.; Reis, L.M.; Lemyre, E.; Clark, R.D.; Kariminejad, A.; Semina, E.V. Novel mutations in PAX6, OTX2 and NDP in anophthalmia, microphthalmia and coloboma. Eur. J. Hum. Genet. 2016, 24, 535–541. [Google Scholar] [CrossRef] [PubMed]
- Perenthaler, E.; Yousefi, S.; Niggl, E.; Barakat, T.S. Beyond the Exome: The Non-coding Genome and Enhancers in Neurodevelopmental Disorders and Malformations of Cortical Development. Front. Cell. Neurosci. 2019, 13, 352. [Google Scholar] [CrossRef] [PubMed]
- Protas, M.E.; Weh, E.; Footz, T.; Kasberger, J.; Baraban, S.C.; Levin, A.V.; Katz, L.J.; Ritch, R.; Walter, M.A.; Semina, E.V.; et al. Mutations of conserved non-coding elements of PITX2 in patients with ocular dysgenesis and developmental glaucoma. Hum. Mol. Genet. 2017, 26, 3630–3638. [Google Scholar] [CrossRef] [PubMed]
- Eisenberger, T.; Neuhaus, C.; Khan, A.O.; Decker, C.; Preising, M.N.; Friedburg, C.; Bieg, A.; Gliem, M.; Charbel Issa, P.; Holz, F.G.; et al. Increasing the yield in targeted next-generation sequencing by implicating CNV analysis, non-coding exons and the overall variant load: The example of retinal dystrophies. PLoS ONE 2013, 8, e78496. [Google Scholar] [CrossRef] [PubMed]
- Gronskov, K.; Rosenberg, T.; Sand, A.; Brondum-Nielsen, K. Mutational analysis of PAX6: 16 novel mutations including 5 missense mutations with a mild aniridia phenotype. Eur. J. Hum. Genet. 1999, 7, 274–286. [Google Scholar] [CrossRef]
- Axton, R.A.; Hanson, I.M.; Love, J.; Seawright, A.; Prosser, J.; van Heyningen, V. Combined SSCP/heteroduplex analysis in the screening for PAX6 mutations. Mol. Cell. Probes 1997, 11, 287–292. [Google Scholar] [CrossRef]
- Filatova, A.Y.; Vasilyeva, T.A.; Marakhonov, A.V.; Voskresenskaya, A.A.; Zinchenko, R.A.; Skoblov, M.Y. Functional reassessment of PAX6 single nucleotide variants by in vitro splicing assay. Eur. J. Hum. Genet. 2019, 27, 488–493. [Google Scholar] [CrossRef]
- Yokoi, T.; Nishina, S.; Fukami, M.; Ogata, T.; Hosono, K.; Hotta, Y.; Azuma, N. Genotype-phenotype correlation of PAX6 gene mutations in aniridia. Hum. Genome Var. 2016, 3, 15052. [Google Scholar] [CrossRef]
- Crolla, J.A.; van Heyningen, V. Frequent chromosome aberrations revealed by molecular cytogenetic studies in patients with aniridia. Am. J. Hum. Genet. 2002, 71, 1138–1149. [Google Scholar] [CrossRef]
- Hall, H.N.; Williamson, K.A.; FitzPatrick, D.R. The genetic architecture of aniridia and Gillespie syndrome. Hum. Genet. 2019, 138, 881–898. [Google Scholar] [CrossRef] [PubMed]
- Glaser, T.; Jepeal, L.; Edwards, J.G.; Young, S.R.; Favor, J.; Maas, R.L. PAX6 gene dosage effect in a family with congenital cataracts, aniridia, anophthalmia and central nervous system defects. Nat. Genet. 1994, 7, 463–471. [Google Scholar] [CrossRef] [PubMed]
- Schmidt-Sidor, B.; Szymanska, K.; Williamson, K.; van Heyningen, V.; Roszkowski, T.; Wierzba-Bobrowicz, T.; Zaremba, J. Malformations of the brain in two fetuses with a compound heterozygosity for two PAX6 mutations. Folia Neuropathol. 2009, 47, 372–382. [Google Scholar] [PubMed]
- Solomon, B.D.; Pineda-Alvarez, D.E.; Balog, J.Z.; Hadley, D.; Gropman, A.L.; Nandagopal, R.; Han, J.C.; Hahn, J.S.; Blain, D.; Brooks, B.; et al. Compound heterozygosity for mutations in PAX6 in a patient with complex brain anomaly, neonatal diabetes mellitus, and microophthalmia. Am. J. Med. Genet. Part A 2009, 149, 2543–2546. [Google Scholar] [CrossRef]
- Lee, H.J.; Colby, K.A. A review of the clinical and genetic aspects of aniridia. Semin. Ophthalmol. 2013, 28, 306–312. [Google Scholar] [CrossRef]
- Qin, L.; Wang, J.; Tian, X.; Yu, H.; Truong, C.; Mitchell, J.J.; Wierenga, K.J.; Craigen, W.J.; Zhang, V.W.; Wong, L.C. Detection and Quantification of Mosaic Mutations in Disease Genes by Next-Generation Sequencing. J. Mol. Diagn. 2016, 18, 446–453. [Google Scholar] [CrossRef]
- Riera, M.; Wert, A.; Nieto, I.; Pomares, E. Panel-based whole exome sequencing identifies novel mutations in microphthalmia and anophthalmia patients showing complex Mendelian inheritance patterns. Mol. Genet. Genom. Med. 2017, 5, 709–719. [Google Scholar] [CrossRef]
- Bai, Z.; Kong, X. Extension of the mutation spectrum of PAX6 from three Chinese congenital aniridia families and identification of male gonadal mosaicism. Mol. Genet. Genom. Med. 2018, 6, 1053–1067. [Google Scholar] [CrossRef]
- Tarilonte, M.; Morin, M.; Ramos, P.; Galdos, M.; Blanco-Kelly, F.; Villaverde, C.; Rey-Zamora, D.; Rebolleda, G.; Munoz-Negrete, F.J.; Tahsin-Swafiri, S.; et al. Parental Mosaicism in PAX6 Causes Intra-Familial Variability: Implications for Genetic Counseling of Congenital Aniridia and Microphthalmia. Front. Genet. 2018, 9, 479. [Google Scholar] [CrossRef]
- Dubey, S.K.; Mahalaxmi, N.; Vijayalakshmi, P.; Sundaresan, P. Mutational analysis and genotype-phenotype correlations in southern Indian patients with sporadic and familial aniridia. Mol. Vis. 2015, 21, 88–97. [Google Scholar]
- Gramer, E.; Reiter, C.; Gramer, G. Glaucoma and frequency of ocular and general diseases in 30 patients with aniridia: A clinical study. Eur. J. Ophthalmol. 2012, 22, 104–110. [Google Scholar] [CrossRef] [PubMed]
- Lim, H.T.; Kim, D.H.; Kim, H. PAX6 aniridia syndrome: Clinics, genetics, and therapeutics. Curr. Opin. Ophthalmol. 2017, 28, 436–447. [Google Scholar] [CrossRef] [PubMed]
- McCulley, T.J.; Mayer, K.; Dahr, S.S.; Simpson, J.; Holland, E.J. Aniridia and optic nerve hypoplasia. Eye 2005, 19, 762–764. [Google Scholar] [CrossRef] [PubMed]
- Pedersen, H.R.; Hagen, L.A.; Landsend, E.C.S.; Gilson, S.J.; Utheim, O.A.; Utheim, T.P.; Neitz, M.; Baraas, R.C. Color Vision in Aniridia. Investig. Ophthalmol. Vis. Sci. 2018, 59, 2142–2152. [Google Scholar] [CrossRef]
- Pedersen, H.R.; Neitz, M.; Gilson, S.J.; Landsend, E.C.S.; Utheim, O.A.; Utheim, T.P.; Baraas, R.C. The Cone Photoreceptor Mosaic in Aniridia: Within-Family Phenotype-Genotype Discordance. Ophthalmol. Retin. 2019, 3, 523–534. [Google Scholar] [CrossRef]
- Lagali, N.; Wowra, B.; Fries, F.N.; Latta, L.; Moslemani, K.; Utheim, T.P.; Wylegala, E.; Seitz, B.; Kasmann-Kellner, B. Early phenotypic features of aniridia-associated keratopathy and association with PAX6 coding mutations. Ocul. Surf. 2019. [Google Scholar] [CrossRef]
- Lagali, N.; Wowra, B.; Fries, F.N.; Latta, L.; Moslemani, K.; Utheim, T.P.; Wylegala, E.; Seitz, B.; Kasmann-Kellner, B. PAX6 Mutational Status Determines Aniridia-Associated Keratopathy Phenotype. Ophthalmology 2019. [Google Scholar] [CrossRef]
- Aggarwal, S.; Jinda, W.; Limwongse, C.; Atchaneeyasakul, L.O.; Phadke, S.R. Run-on mutation in the PAX6 gene and chorioretinal degeneration in autosomal dominant aniridia. Mol. Vis. 2011, 17, 1305–1309. [Google Scholar]
- Ito, Y.A.; Footz, T.K.; Berry, F.B.; Mirzayans, F.; Yu, M.; Khan, A.O.; Walter, M.A. Severe molecular defects of a novel FOXC1 W152G mutation result in aniridia. Investig. Ophthalmol. Vis. Sci. 2009, 50, 3573–3579. [Google Scholar] [CrossRef]
- Law, S.K.; Sami, M.; Piri, N.; Coleman, A.L.; Caprioli, J. Asymmetric phenotype of Axenfeld-Rieger anomaly and aniridia associated with a novel PITX2 mutation. Mol. Vis. 2011, 17, 1231–1238. [Google Scholar]
- Ito, Y.A.; Walter, M.A. Genomics and anterior segment dysgenesis: A review. Clin. Exp. Ophthalmol. 2014, 42, 13–24. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Shan, X.; Gregory-Evans, C.Y. A mouse model of aniridia reveals the in vivo downstream targets of Pax6 driving iris and ciliary body development in the eye. Biochim. Biophys. Acta Mol. Basis Dis. 2017, 1863, 60–67. [Google Scholar] [CrossRef] [PubMed]
- Seo, S.; Singh, H.P.; Lacal, P.M.; Sasman, A.; Fatima, A.; Liu, T.; Schultz, K.M.; Losordo, D.W.; Lehmann, O.J.; Kume, T. Forkhead box transcription factor FoxC1 preserves corneal transparency by regulating vascular growth. Proc. Natl. Acad. Sci. USA 2012, 109, 2015–2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Qin, G.; Chen, G.; Li, T.; Gao, L.; Huang, L.; Zhang, Y.; Ouyang, K.; Wang, Y.; Pang, Y.; et al. Variants in TRIM44 Cause Aniridia by Impairing PAX6 Expression. Hum. Mutat. 2015, 36, 1164–1167. [Google Scholar] [CrossRef] [PubMed]
- Han, J.C.; Liu, Q.R.; Jones, M.; Levinn, R.L.; Menzie, C.M.; Jefferson-George, K.S.; Adler-Wailes, D.C.; Sanford, E.L.; Lacbawan, F.L.; Uhl, G.R.; et al. Brain-derived neurotrophic factor and obesity in the WAGR syndrome. N. Engl. J. Med. 2008, 359, 918–927. [Google Scholar] [CrossRef] [PubMed]
- Fischbach, B.V.; Trout, K.L.; Lewis, J.; Luis, C.A.; Sika, M. WAGR syndrome: A clinical review of 54 cases. Pediatrics 2005, 116, 984–988. [Google Scholar] [CrossRef]
- Shinawi, M.; Sahoo, T.; Maranda, B.; Skinner, S.A.; Skinner, C.; Chinault, C.; Zascavage, R.; Peters, S.U.; Patel, A.; Stevenson, R.E.; et al. 11p14.1 microdeletions associated with ADHD, autism, developmental delay, and obesity. Am. J. Med. Genet. Part A 2011, 155, 1272–1280. [Google Scholar] [CrossRef]
- Harcourt, B.E.; Bullen, D.V.R.; Kao, K.T.; Tassoni, D.; Alexander, E.J.; Burgess, T.; White, S.M.; Sabin, M.A. Maternal inheritance of BDNF deletion, with phenotype of obesity and developmental delay in mother and child. Am. J. Med. Genet. Part A 2018, 176, 194–200. [Google Scholar] [CrossRef]
- Szatmari, P.; Paterson, A.D.; Zwaigenbaum, L.; Roberts, W.; Brian, J.; Liu, X.Q.; Vincent, J.B.; Skaug, J.L.; Thompson, A.P.; Senman, L.; et al. Mapping autism risk loci using genetic linkage and chromosomal rearrangements. Nat. Genet. 2007, 39, 319–328. [Google Scholar] [CrossRef]
- Sisodiya, S.M.; Free, S.L.; Williamson, K.A.; Mitchell, T.N.; Willis, C.; Stevens, J.M.; Kendall, B.E.; Shorvon, S.D.; Hanson, I.M.; Moore, A.T.; et al. PAX6 haploinsufficiency causes cerebral malformation and olfactory dysfunction in humans. Nat. Genet. 2001, 28, 214–216. [Google Scholar] [CrossRef]
- Marlin, S.; Couet, D.; Lacombe, D.; Cessans, C.; Bonneau, D. Obesity: A new feature of WAGR (del 11p) syndrome. Clin. Dysmorphol. 1994, 3, 255–257. [Google Scholar] [CrossRef] [PubMed]
- Gul, D.; Ogur, G.; Tunca, Y.; Ozcan, O. Third case of WAGR syndrome with severe obesity and constitutional deletion of chromosome (11)(p12p14). Am. J. Med. Genet. 2002, 107, 70–71. [Google Scholar] [CrossRef] [PubMed]
- Thomas, S.; Thomas, M.G.; Andrews, C.; Chan, W.M.; Proudlock, F.A.; McLean, R.J.; Pradeep, A.; Engle, E.C.; Gottlob, I. Autosomal-dominant nystagmus, foveal hypoplasia and presenile cataract associated with a novel PAX6 mutation. Eur. J. Hum. Genet. 2014, 22, 344–349. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.; Li, S.; Zhang, Q. Microphthalmia, late onset keratitis, and iris coloboma/aniridia in a family with a novel PAX6 mutation. Ophthalmic Genet. 2012, 33, 119–121. [Google Scholar] [CrossRef]
- Dansault, A.; David, G.; Schwartz, C.; Jaliffa, C.; Vieira, V.; de la Houssaye, G.; Bigot, K.; Catin, F.; Tattu, L.; Chopin, C.; et al. Three new PAX6 mutations including one causing an unusual ophthalmic phenotype associated with neurodevelopmental abnormalities. Mol. Vis. 2007, 13, 511–523. [Google Scholar]
- Kawase, E.; Tanaka, K.; Honna, T.; Azuma, N. A case of atypical WAGR syndrome with anterior segment anomaly and microphthalmos. Arch. Ophthalmol. 2001, 119, 1855–1856. [Google Scholar]
- Kamachi, Y.; Uchikawa, M.; Tanouchi, A.; Sekido, R.; Kondoh, H. Pax6 and SOX2 form a co-DNA-binding partner complex that regulates initiation of lens development. Genes Dev. 2001, 15, 1272–1286. [Google Scholar] [CrossRef]
- Aberdam, E.; Petit, I.; Sangari, L.; Aberdam, D. Induced pluripotent stem cell-derived limbal epithelial cells (LiPSC) as a cellular alternative for in vitro ocular toxicity testing. PLoS ONE 2017, 12, e0179913. [Google Scholar] [CrossRef]
- Williamson, K.A.; Hall, H.N.; Owen, L.J.; Livesey, B.J.; Hanson, I.M.; Adams, G.G.W.; Bodek, S.; Calvas, P.; Castle, B.; Clarke, M.; et al. Recurrent heterozygous PAX6 missense variants cause severe bilateral microphthalmia via predictable effects on DNA-protein interaction. Genet. Med. Off. J. Am. Coll. Med. Genet. 2019. [Google Scholar] [CrossRef]
- Guo, H.; Dai, L.; Huang, Y.; Liao, Q.; Bai, Y. A large novel deletion downstream of PAX6 gene in a Chinese family with ocular coloboma. PLoS ONE 2013, 8, e83073. [Google Scholar] [CrossRef]
- Goolam, S.; Carstens, N.; Ross, M.; Bentley, D.; Lopes, M.; Peden, J.; Kingsbury, Z.; Tsogka, E.; Barlow, R.; Carmichael, T.R.; et al. Familial congenital cataract, coloboma, and nystagmus phenotype with variable expression caused by mutation in PAX6 in a South African family. Mol. Vis. 2018, 24, 407–413. [Google Scholar] [PubMed]
- Gregory-Evans, C.Y.; Williams, M.J.; Halford, S.; Gregory-Evans, K. Ocular coloboma: A reassessment in the age of molecular neuroscience. J. Med. Genet. 2004, 41, 881–891. [Google Scholar] [CrossRef] [PubMed]
- Lee, B.; Choi, D.G.; Chun, B.Y.; Oh, E.H.; Lee, Y.J.; Kim, U.K.; Park, J.S. A family with a mild form of congenital nystagmus and optic disc coloboma caused by a novel PAX6 mutation. Gene 2019, 705, 177–180. [Google Scholar] [CrossRef] [PubMed]
- Schwarz, M.; Cecconi, F.; Bernier, G.; Andrejewski, N.; Kammandel, B.; Wagner, M.; Gruss, P. Spatial specification of mammalian eye territories by reciprocal transcriptional repression of Pax2 and Pax6. Development 2000, 127, 4325–4334. [Google Scholar]
- Gerber, S.; Alzayady, K.J.; Burglen, L.; Bremond-Gignac, D.; Marchesin, V.; Roche, O.; Rio, M.; Funalot, B.; Calmon, R.; Durr, A.; et al. Recessive and Dominant De Novo ITPR1 Mutations Cause Gillespie Syndrome. Am. J. Hum. Genet. 2016, 98, 971–980. [Google Scholar] [CrossRef]
- McEntagart, M.; Williamson, K.A.; Rainger, J.K.; Wheeler, A.; Seawright, A.; De Baere, E.; Verdin, H.; Bergendahl, L.T.; Quigley, A.; Rainger, J.; et al. A Restricted Repertoire of De Novo Mutations in ITPR1 Cause Gillespie Syndrome with Evidence for Dominant-Negative Effect. Am. J. Hum. Genet. 2016, 98, 981–992. [Google Scholar] [CrossRef]
- Ticho, B.H.; Hilchie-Schmidt, C.; Egel, R.T.; Traboulsi, E.I.; Howarth, R.J.; Robinson, D. Ocular findings in Gillespie-like syndrome: Association with a new PAX6 mutation. Ophthalmic Genet. 2006, 27, 145–149. [Google Scholar] [CrossRef]
- Hanson, I.M.; Fletcher, J.M.; Jordan, T.; Brown, A.; Taylor, D.; Adams, R.J.; Punnett, H.H.; van Heyningen, V. Mutations at the PAX6 locus are found in heterogeneous anterior segment malformations including Peters’ anomaly. Nat. Genet. 1994, 6, 168–173. [Google Scholar] [CrossRef]
- Wolf, M.T.; Lorenz, B.; Winterpacht, A.; Drechsler, M.; Schumacher, V.; Royer-Pokora, B.; Blankenagel, A.; Zabel, B.; Wildhardt, G. Ten novel mutations found in Aniridia. Hum. Mutat. 1998, 12, 304–313. [Google Scholar] [CrossRef]
- Sun, J.; Zhao, Y.; McGreal, R.; Cohen-Tayar, Y.; Rockowitz, S.; Wilczek, C.; Ashery-Padan, R.; Shechter, D.; Zheng, D.; Cvekl, A. Pax6 associates with H3K4-specific histone methyltransferases Mll1, Mll2, and Set1a and regulates H3K4 methylation at promoters and enhancers. Epigenet. Chromatin 2016, 9, 37. [Google Scholar] [CrossRef]
- Sun, J.; Rockowitz, S.; Xie, Q.; Ashery-Padan, R.; Zheng, D.; Cvekl, A. Identification of in vivo DNA-binding mechanisms of Pax6 and reconstruction of Pax6-dependent gene regulatory networks during forebrain and lens development. Nucleic Acids Res. 2015, 43, 6827–6846. [Google Scholar] [CrossRef] [PubMed]
- Tsui, S.; Wang, J.; Wang, L.; Dai, W.; Lu, L. CTCF-Mediated and Pax6-Associated Gene Expression in Corneal Epithelial Cell-Specific Differentiation. PLoS ONE 2016, 11, e0162071. [Google Scholar] [CrossRef] [PubMed]
- Ryan, B.C.; Lowe, K.; Hanson, L.; Gil, T.; Braun, L.; Howard, P.L.; Chow, R.L. Mapping the Pax6 3′ untranslated region microRNA regulatory landscape. BMC Genom. 2018, 19, 820. [Google Scholar] [CrossRef] [PubMed]
- Bhinge, A.; Poschmann, J.; Namboori, S.C.; Tian, X.; Jia Hui Loh, S.; Traczyk, A.; Prabhakar, S.; Stanton, L.W. MiR-135b is a direct PAX6 target and specifies human neuroectoderm by inhibiting TGF-beta/BMP signaling. EMBO J. 2014, 33, 1271–1283. [Google Scholar] [CrossRef]
- Shaham, O.; Gueta, K.; Mor, E.; Oren-Giladi, P.; Grinberg, D.; Xie, Q.; Cvekl, A.; Shomron, N.; Davis, N.; Keydar-Prizant, M.; et al. Pax6 regulates gene expression in the vertebrate lens through miR-204. PLoS Genet. 2013, 9, e1003357. [Google Scholar] [CrossRef]
- Shalom-Feuerstein, R.; Serror, L.; De La Forest Divonne, S.; Petit, I.; Aberdam, E.; Camargo, L.; Damour, O.; Vigouroux, C.; Solomon, A.; Gaggioli, C.; et al. Pluripotent stem cell model reveals essential roles for miR-450b-5p and miR-184 in embryonic corneal lineage specification. Stem Cells 2012, 30, 898–909. [Google Scholar] [CrossRef]
- Berdasco, M.; Gomez, A.; Rubio, M.J.; Catala-Mora, J.; Zanon-Moreno, V.; Lopez, M.; Hernandez, C.; Yoshida, S.; Nakama, T.; Ishikawa, K.; et al. DNA Methylomes Reveal Biological Networks Involved in Human Eye Development, Functions and Associated Disorders. Sci. Rep. 2017, 7, 11762. [Google Scholar] [CrossRef]
- Singer, R.A.; Arnes, L.; Cui, Y.; Wang, J.; Gao, Y.; Guney, M.A.; Burnum-Johnson, K.E.; Rabadan, R.; Ansong, C.; Orr, G.; et al. The Long Noncoding RNA Paupar Modulates PAX6 Regulatory Activities to Promote Alpha Cell Development and Function. Cell Metab. 2019, 30, 1091–1106.e8. [Google Scholar] [CrossRef]
- Hayashi, R.; Ishikawa, Y.; Sasamoto, Y.; Katori, R.; Nomura, N.; Ichikawa, T.; Araki, S.; Soma, T.; Kawasaki, S.; Sekiguchi, K.; et al. Co-ordinated ocular development from human iPS cells and recovery of corneal function. Nature 2016, 531, 376–380. [Google Scholar] [CrossRef]
- Qiu, X.; Yang, J.; Liu, T.; Jiang, Y.; Le, Q.; Lu, Y. Efficient generation of lens progenitor cells from cataract patient-specific induced pluripotent stem cells. PLoS ONE 2012, 7, e32612. [Google Scholar] [CrossRef]
- Foster, J.W.; Wahlin, K.; Adams, S.M.; Birk, D.E.; Zack, D.J.; Chakravarti, S. Cornea organoids from human induced pluripotent stem cells. Sci. Rep. 2017, 7, 41286. [Google Scholar] [CrossRef] [PubMed]
- Nakano, T.; Ando, S.; Takata, N.; Kawada, M.; Muguruma, K.; Sekiguchi, K.; Saito, K.; Yonemura, S.; Eiraku, M.; Sasai, Y. Self-formation of optic cups and storable stratified neural retina from human ESCs. Cell Stem Cell 2012, 10, 771–785. [Google Scholar] [CrossRef] [PubMed]
- Gamm, D.M.; Clark, E.; Capowski, E.E.; Singh, R. The Role of FGF9 In The Production of Neural Retina And RPE In A Pluripotent Stem Cell Model of Early Human Retinal Development. Am. J. Ophthalmol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Parfitt, D.A.; Lane, A.; Ramsden, C.M.; Carr, A.J.; Munro, P.M.; Jovanovic, K.; Schwarz, N.; Kanuga, N.; Muthiah, M.N.; Hull, S.; et al. Identification and Correction of Mechanisms Underlying Inherited Blindness in Human iPSC-Derived Optic Cups. Cell Stem Cell 2016, 18, 769–781. [Google Scholar] [CrossRef]
- Blau, H.M.; Daley, G.Q. Stem Cells in the Treatment of Disease. N. Engl. J. Med. 2019, 380, 1748–1760. [Google Scholar] [CrossRef]
- Deng, W.L.; Gao, M.L.; Lei, X.L.; Lv, J.N.; Zhao, H.; He, K.W.; Xia, X.X.; Li, L.Y.; Chen, Y.C.; Li, Y.P.; et al. Gene Correction Reverses Ciliopathy and Photoreceptor Loss in iPSC-Derived Retinal Organoids from Retinitis Pigmentosa Patients. Stem Cell Rep. 2018, 10, 1267–1281. [Google Scholar] [CrossRef]
- Richardson, R.; Smart, M.; Tracey-White, D.; Webster, A.R.; Moosajee, M. Mechanism and evidence of nonsense suppression therapy for genetic eye disorders. Exp. Eye Res. 2017, 155, 24–37. [Google Scholar] [CrossRef]
- Wang, X.; Gregory-Evans, K.; Wasan, K.M.; Sivak, O.; Shan, X.; Gregory-Evans, C.Y. Efficacy of Postnatal In Vivo Nonsense Suppression Therapy in a Pax6 Mouse Model of Aniridia. Mol. Ther. Nucleic Acids 2017, 7, 417–428. [Google Scholar] [CrossRef]
- Gregory-Evans, C.Y.; Wang, X.; Wasan, K.M.; Zhao, J.; Metcalfe, A.L.; Gregory-Evans, K. Postnatal manipulation of Pax6 dosage reverses congenital tissue malformation defects. J. Clin. Investig. 2014, 124, 111–116. [Google Scholar] [CrossRef]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Regulatory Element | Distance to PAX6 (Kb) | Location | Eye Expression | Extra-Ocular Expression | Reference |
|---|---|---|---|---|---|
| RB | −215 | Intron ELP4 | Diencephalon, telencephalon, pineal gland | [37] | |
| E180B | −176 | Intron ELP4 | RGCs and optic nerve | Trigeminal ganglia, dorsal spinal cord neurons | [56] |
| HS8B | −177 | Intron ELP4 | [25] | ||
| HS8A | −176 | Intron ELP4 | [25] | ||
| HS6 | −172 | Intron ELP4 | Neural retina | Neural structures | [57] |
| HS5 | −168 | Intron ELP4 | Optic cup and neural retina | Diencephalon | [57] |
| HS3 | −162 | Intron ELP4 | Neural retina | [25] | |
| HS2 | −161 | Intron ELP4 | Neural retina | [25] | |
| SIMO | −153 | Intron ELP4 | Lens and neural retina | Diencephalon, hindbrain, neural tube | [25] |
| E120 | −126 | Intron ELP4 | Olfactory bulbs, brain | [56] | |
| E100 | −104 | Intron ELP4 | Neural retina | Diencephalon, olfactory region | [58] |
| E60A | −54 | Intron ELP4 | Optic cup and neural retina | Neural structures | [57] |
| 7CE1 | −18 | Intron PAX6 | Late eye development | [59] | |
| NRE | −13 | Intron PAX6 | Neural retina, ciliary body, iris | [54] | |
| 0CE1 | −1 | 5’UTR PAX6 | [26] | ||
| agCNE14 (P2) | 4 | Pancreas | [56] | ||
| EE | 5 | Cornea, lens, conjunctiva and lacrimal gland | [54,60] | ||
| agCNE12 (P) | 7 | Pancreatic islets | |||
| agCNE11 | 8 | Pineal gland | [56] | ||
| agCNE10 (Up-9) | 9 | No specific pattern | [56] | ||
| agCNE9 (Up-10) | 12 | Pineal gland | [56] | ||
| Mouse-like Paupar | 54 | [61] | |||
| PE3 (E-52) | 57 | Developing pancreas and brain | [62] | ||
| E-55/C | 59 | No specific pattern | [56] | ||
| E-55/B | 60 | No specific pattern | [56] | ||
| E-55/A | 70 | Hindbrain | [56] | ||
| E-72 | 71 | No specific pattern | [56] | ||
| PE4 (E120) | 110 | Developing pancreas, olfactory tract, cerebellum, hindbrain | [62] | ||
| agCNE5 (Id855) | 178 | Forebrain | [56] | ||
| agCNE4 | 186 | Hindbrain | [56] | ||
| agCNE3 | 214 | Olfactory bulbs | [56] | ||
| E200 | 214 | Olfactory bulbs, cerebellum | [63] | ||
| agCNE1 | 224 | Trigeminal ganglia, dorsal spinal cord neurons | [56] | ||
| E250 | 247 | No specific pattern | [56] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lima Cunha, D.; Arno, G.; Corton, M.; Moosajee, M. The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes 2019, 10, 1050. https://doi.org/10.3390/genes10121050
Lima Cunha D, Arno G, Corton M, Moosajee M. The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes. 2019; 10(12):1050. https://doi.org/10.3390/genes10121050
Chicago/Turabian StyleLima Cunha, Dulce, Gavin Arno, Marta Corton, and Mariya Moosajee. 2019. "The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye" Genes 10, no. 12: 1050. https://doi.org/10.3390/genes10121050
APA StyleLima Cunha, D., Arno, G., Corton, M., & Moosajee, M. (2019). The Spectrum of PAX6 Mutations and Genotype-Phenotype Correlations in the Eye. Genes, 10(12), 1050. https://doi.org/10.3390/genes10121050