Detection of Differentially Methylated Regions Using Bayes Factor for Ordinal Group Responses



Abstract
1. Introduction
2. Materials and Methods
2.1. Methods
2.2. Simulation Study of the Properties of BFM
3. Results
3.1. Simulation Results
3.2. Data analysis
3.3. Comparison of Bayesian Method with Scan Statistic Method for Two Groups
3.4. Bayesian Method for Ordinal Group Responses
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Yokota, J. Tumor progression and metastasis. Carcinogenesis 2000, 21, 497–503. [Google Scholar] [CrossRef] [PubMed]
- Torre, L.A.; Siegel, R.L.; Jemal, A. Lung Cancer Statistics. In Lung Cancer and Personalized Medicine: Current Knowledge and Therapies; Ahmad, A., Gadgeel, S., Eds.; Advances in Experimental Medicine and Biology; Springer International Publishing: Cham, Switzerland, 2016; pp. 1–19. ISBN 978-3-319-24223-1. [Google Scholar]
- Qureshi, S.A.; Bashir, M.U.; Yaqinuddin, A. Utility of DNA methylation markers for diagnosing cancer. Int J Surg 2010, 8, 194–198. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Varley, K.E.; Gertz, J.; Bowling, K.M.; Parker, S.L.; Reddy, T.E.; Pauli-Behn, F.; Cross, M.K.; Williams, B.A.; Stamatoyannopoulos, J.A.; Crawford, G.E.; et al. Dynamic DNA methylation across diverse human cell lines and tissues. Genome Res. 2013, 23, 555–567. [Google Scholar] [CrossRef] [PubMed]
- Klajic, J.; Fleischer, T.; Dejeux, E.; Edvardsen, H.; Warnberg, F.; Bukholm, I.; Lønning, P.E.; Solvang, H.; Børresen-Dale, A.-L.; Tost, J.; et al. Quantitative DNA methylation analyses reveal stage dependent DNA methylation and association to clinico-pathological factors in breast tumors. BMC Cancer 2013, 13, 456. [Google Scholar] [CrossRef]
- Watts, G.S.; Futscher, B.W.; Holtan, N.; Degeest, K.; Domann, F.E.; Rose, S.L. DNA methylation changes in ovarian cancer are cumulative with disease progression and identify tumor stage. BMC Med Genomics 2008, 1, 47. [Google Scholar] [CrossRef] [PubMed]
- Hoque, M.O. DNA methylation changes in prostate cancer: current developments and future clinical implementation. Expert Rev. Mol. Diagn. 2009, 9, 243–257. [Google Scholar] [CrossRef] [PubMed]
- Mitomi, H.; Fukui, N.; Tanaka, N.; Kanazawa, H.; Saito, T.; Matsuoka, T.; Yao, T. Aberrant p16 INK4a methylation is a frequent event in colorectal cancers: prognostic value and relation to mRNA expression and immunoreactivity. J. Cancer Res. Clin. Oncol. 2010, 136, 323–331. [Google Scholar] [CrossRef] [PubMed]
- Dunson, D.B.; Neelon, B. Bayesian Inference on Order-Constrained Parameters in Generalized Linear Models. Biometrics 2003, 59, 286–295. [Google Scholar] [CrossRef]
- Leek, J.T.; Scharpf, R.B.; Bravo, H.C.; Simcha, D.; Langmead, B.; Johnson, W.E.; Geman, D.; Baggerly, K.; Irizarry, R.A. Tackling the widespread and critical impact of batch effects in high-throughput data. Nat. Rev. Genet. 2010, 11, 733–739. [Google Scholar] [CrossRef]
- Bartholomew, D.J. A test of homogeneity for ordered alternatives. Biometrika 1959, 46, 36–48. [Google Scholar] [CrossRef]
- Robertson, T.; Wegman, E.J. Likelihood ratio tests for order restrictions in exponential families. The Annals of Statistics 1978, 485–505. [Google Scholar] [CrossRef]
- Ayer, M.; Brunk, H.D.; Ewing, G.M.; Reid, W.T.; Silverman, E. An empirical distribution function for sampling with incomplete information. The annals of mathematical statistics 1955, 641–647. [Google Scholar] [CrossRef]
- Taylor, J.M.G.; Wang, L.; Li, Z. Analysis on binary responses with ordered covariates and missing data. Stat Med 2007, 26, 3443–3458. [Google Scholar] [CrossRef] [PubMed]
- Teschendorff, A.E.; Menon, U.; Gentry-Maharaj, A.; Ramus, S.J.; Weisenberger, D.J.; Shen, H.; Campan, M.; Noushmehr, H.; Bell, C.G.; Maxwell, A.P.; et al. Age-dependent DNA methylation of genes that are suppressed in stem cells is a hallmark of cancer. Genome Res. 2010, 20, 440–446. [Google Scholar] [CrossRef] [PubMed]
- Kibriya, M.G.; Raza, M.; Jasmine, F.; Roy, S.; Paul-Brutus, R.; Rahaman, R.; Dodsworth, C.; Rakibuz-Zaman, M.; Kamal, M.; Ahsan, H. A genome-wide DNA methylation study in colorectal carcinoma. BMC Med Genomics 2011, 4, 50. [Google Scholar] [CrossRef] [PubMed]
- Jeffreys, H. Theory of probability, Clarendon; Oxford University Press: Oxford, UK, 1961. [Google Scholar]
- Kass, R.E.; Raftery, A.E. Bayes factors. Journal of the american statistical association 1995, 90, 773–795. [Google Scholar] [CrossRef]
- Dai, B.; Ding, S.; Wahba, G. Multivariate bernoulli distribution. Bernoulli 2013, 19, 1465–1483. [Google Scholar] [CrossRef]
- Chiorazzi, N.; Rai, K.R.; Ferrarini, M. Chronic lymphocytic leukemia. N. Engl. J. Med. 2005, 352, 804–815. [Google Scholar] [CrossRef]
- Keating, M.J.; Chiorazzi, N.; Messmer, B.; Damle, R.N.; Allen, S.L.; Rai, K.R.; Ferrarini, M.; Kipps, T.J. Biology and treatment of chronic lymphocytic leukemia. Hematology Am Soc Hematol Educ Program 2003, 153–175. [Google Scholar] [CrossRef]
- Döhner, H.; Stilgenbauer, S.; Benner, A.; Leupolt, E.; Kröber, A.; Bullinger, L.; Döhner, K.; Bentz, M.; Lichter, P. Genomic aberrations and survival in chronic lymphocytic leukemia. N. Engl. J. Med. 2000, 343, 1910–1916. [Google Scholar] [CrossRef]
- Hamblin, T.J.; Davis, Z.; Gardiner, A.; Oscier, D.G.; Stevenson, F.K. Unmutated Ig V(H) genes are associated with a more aggressive form of chronic lymphocytic leukemia. Blood 1999, 94, 1848–1854. [Google Scholar] [PubMed]
- Hamblin, T.J.; Orchard, J.A.; Gardiner, A.; Oscier, D.G.; Davis, Z.; Stevenson, F.K. Immunoglobulin V genes and CD38 expression in CLL. Blood 2000, 95, 2455–2457. [Google Scholar] [PubMed]
- Damle, R.N.; Wasil, T.; Fais, F.; Ghiotto, F.; Valetto, A.; Allen, S.L.; Buchbinder, A.; Budman, D.; Dittmar, K.; Kolitz, J.; et al. Ig V gene mutation status and CD38 expression as novel prognostic indicators in chronic lymphocytic leukemia. Blood 1999, 94, 1840–1847. [Google Scholar] [PubMed]
- Kanduri, M.; Cahill, N.; Göransson, H.; Enström, C.; Ryan, F.; Isaksson, A.; Rosenquist, R. Differential genome-wide array-based methylation profiles in prognostic subsets of chronic lymphocytic leukemia. Blood 2010, 115, 296–305. [Google Scholar] [CrossRef] [PubMed]
- Rahmatpanah, F.B.; Carstens, S.; Guo, J.; Sjahputera, O.; Taylor, K.H.; Duff, D.; Shi, H.; Davis, J.W.; Hooshmand, S.I.; Chitma-Matsiga, R.; et al. Differential DNA methylation patterns of small B-cell lymphoma subclasses with different clinical behavior. Leukemia 2006, 20, 1855–1862. [Google Scholar] [CrossRef]
- Pei, L.; Choi, J.-H.; Liu, J.; Lee, E.-J.; McCarthy, B.; Wilson, J.M.; Speir, E.; Awan, F.; Tae, H.; Arthur, G.; et al. Genome-wide DNA methylation analysis reveals novel epigenetic changes in chronic lymphocytic leukemia. Epigenetics 2012, 7, 567–578. [Google Scholar] [CrossRef][Green Version]
- Meissner, A.; Gnirke, A.; Bell, G.W.; Ramsahoye, B.; Lander, E.S.; Jaenisch, R. Reduced representation bisulfite sequencing for comparative high-resolution DNA methylation analysis. Nucleic Acids Res. 2005, 33, 5868–5877. [Google Scholar] [CrossRef]
- Tong, W.-G.; Wierda, W.G.; Lin, E.; Kuang, S.-Q.; Bekele, B.N.; Estrov, Z.; Wei, Y.; Yang, H.; Keating, M.J.; Garcia-Manero, G. Genome-wide DNA methylation profiling of chronic lymphocytic leukemia allows identification of epigenetically repressed molecular pathways with clinical impact. Epigenetics 2010, 5, 499–508. [Google Scholar] [CrossRef]
- Dunbar, F.; Xu, H.; Ryu, D.; Ghosh, S.; Shi, H.; George, V. Computational Methods for Detection of Differentially Methylated Regions Using Kernel Distance and Scan Statistics. Genes (Basel) 2019, 10. [Google Scholar] [CrossRef]
- French, D.; Hamilton, L.H.; Mattano, L.A.; Sather, H.N.; Devidas, M.; Nachman, J.B.; Relling, M.V. Children’s Oncology Group A PAI-1 (SERPINE1) polymorphism predicts osteonecrosis in children with acute lymphoblastic leukemia: a report from the Children’s Oncology Group. Blood 2008, 111, 4496–4499. [Google Scholar] [CrossRef]
- Wang, T.; Qian, D.; Hu, M.; Li, L.; Zhang, L.; Chen, H.; Yang, R.; Wang, B. Human cytomegalovirus inhibits apoptosis by regulating the activating transcription factor 5 signaling pathway in human malignant glioma cells. Oncol Lett 2014, 8, 1051–1057. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Glodkowska-Mrowka, E.; Solarska, I.; Mrowka, P.; Bajorek, K.; Niesiobedzka-Krezel, J.; Seferynska, I.; Borg, K.; Stoklosa, T. Differential expression of BIRC family genes in chronic myeloid leukaemia--BIRC3 and BIRC8 as potential new candidates to identify disease progression. Br. J. Haematol. 2014, 164, 740–742. [Google Scholar] [CrossRef] [PubMed]
- Chae, H.-D.; Mitton, B.; Lacayo, N.J.; Sakamoto, K.M. Replication factor C3 is a CREB target gene that regulates cell cycle progression through the modulation of chromatin loading of PCNA. Leukemia 2015, 29, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- Xu, W.; Zhou, L.; Chen, Q.; Chen, C.; Fang, L.; Fang, X.; Shen, H. [Effect of YB-1 gene knockdown on human leukemia cell line K562/A02]. Zhonghua Yi Xue Yi Chuan Xue Za Zhi 2009, 26, 400–405. [Google Scholar] [PubMed]
- Lasa, A.; Serrano, E.; Carricondo, M.; Carnicer, M.J.; Brunet, S.; Badell, I.; Sierra, J.; Aventín, A.; Nomdedéu, J.F. High expression of CEACAM6 and CEACAM8 mRNA in acute lymphoblastic leukemias. Ann. Hematol. 2008, 87, 205–211. [Google Scholar] [CrossRef] [PubMed]
- Camgoz, A.; Gencer, E.B.; Ural, A.U.; Baran, Y. Mechanisms responsible for nilotinib resistance in human chronic myeloid leukemia cells and reversal of resistance. Leuk. Lymphoma 2013, 54, 1279–1287. [Google Scholar] [CrossRef] [PubMed]
- Caldow, M.K.; Digby, M.R.; Cameron-Smith, D. Short communication: Bovine-derived proteins activate STAT3 in human skeletal muscle in vitro. J. Dairy Sci. 2015, 98, 3016–3019. [Google Scholar] [CrossRef]
- Tang, H.-M.V.; Gao, W.-W.; Chan, C.-P.; Cheng, Y.; Deng, J.-J.; Yuen, K.-S.; Iha, H.; Jin, D.-Y. SIRT1 Suppresses Human T-Cell Leukemia Virus Type 1 Transcription. J. Virol. 2015, 89, 8623–8631. [Google Scholar] [CrossRef] [PubMed]
- Carbonnelle-Puscian, A.; Copie-Bergman, C.; Baia, M.; Martin-Garcia, N.; Allory, Y.; Haioun, C.; Crémades, A.; Abd-Alsamad, I.; Farcet, J.-P.; Gaulard, P.; et al. The novel immunosuppressive enzyme IL4I1 is expressed by neoplastic cells of several B-cell lymphomas and by tumor-associated macrophages. Leukemia 2009, 23, 952–960. [Google Scholar] [CrossRef]
- Kang, X.; Lu, Z.; Cui, C.; Deng, M.; Fan, Y.; Dong, B.; Han, X.; Xie, F.; Tyner, J.W.; Coligan, J.E.; et al. The ITIM-containing receptor LAIR1 is essential for acute myeloid leukaemia development. Nat. Cell Biol. 2015, 17, 665–677. [Google Scholar] [CrossRef]
- Haimovici, A.; Brigger, D.; Torbett, B.E.; Fey, M.F.; Tschan, M.P. Induction of the autophagy-associated gene MAP1S via PU.1 supports APL differentiation. Leuk. Res. 2014, 38, 1041–1047. [Google Scholar] [CrossRef] [PubMed]
- O’Connor, C.; Campos, J.; Osinski, J.M.; Gronostajski, R.M.; Michie, A.M.; Keeshan, K. Nfix expression critically modulates early B lymphopoiesis and myelopoiesis. PLoS ONE 2015, 10, e0120102. [Google Scholar] [CrossRef] [PubMed]
- Moon, E.; Lee, R.; Near, R.; Weintraub, L.; Wolda, S.; Lerner, A. Inhibition of PDE3B augments PDE4 inhibitor-induced apoptosis in a subset of patients with chronic lymphocytic leukemia. Clin. Cancer Res. 2002, 8, 589–595. [Google Scholar] [PubMed]
- Runne, C.; Chen, S. PLEKHG2 promotes heterotrimeric G protein βγ-stimulated lymphocyte migration via Rac and Cdc42 activation and actin polymerization. Mol. Cell. Biol. 2013, 33, 4294–4307. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Rantakari, P.; Auvinen, K.; Jäppinen, N.; Kapraali, M.; Valtonen, J.; Karikoski, M.; Gerke, H.; Iftakhar-E-Khuda, I.; Keuschnigg, J.; Umemoto, E.; et al. The endothelial protein PLVAP in lymphatics controls the entry of lymphocytes and antigens into lymph nodes. Nat. Immunol. 2015, 16, 386–396. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Smith, L.; Johnson, M.R.; Wang, K.; Diasio, R.B.; Smith, J.B. Activation of protein kinase C induces nuclear translocation of RFX1 and down-regulates c-myc via an intron 1 X box in undifferentiated leukemia HL-60 cells. J. Biol. Chem. 2000, 275, 32227–32233. [Google Scholar] [CrossRef] [PubMed]
- McHale, C.M.; Zhang, L.; Lan, Q.; Li, G.; Hubbard, A.E.; Forrest, M.S.; Vermeulen, R.; Chen, J.; Shen, M.; Rappaport, S.M.; et al. Changes in the peripheral blood transcriptome associated with occupational benzene exposure identified by cross-comparison on two microarray platforms. Genomics 2009, 93, 343–349. [Google Scholar] [CrossRef]
- Crans-Vargas, H.N.; Landaw, E.M.; Bhatia, S.; Sandusky, G.; Moore, T.B.; Sakamoto, K.M. Expression of cyclic adenosine monophosphate response-element binding protein in acute leukemia. Blood 2002, 99, 2617–2619. [Google Scholar] [CrossRef]
- Mayr, B.; Montminy, M. Transcriptional regulation by the phosphorylation-dependent factor CREB. Nat. Rev. Mol. Cell Biol. 2001, 2, 599–609. [Google Scholar] [CrossRef]
- Stewart, H.J.S.; Horne, G.A.; Bastow, S.; Chevassut, T.J.T. BRD4 associates with p53 in DNMT3A-mutated leukemia cells and is implicated in apoptosis by the bromodomain inhibitor JQ1. Cancer Med 2013, 2, 826–835. [Google Scholar] [CrossRef]
- Muto, T.; Takeuchi, M.; Yamazaki, A.; Sugita, Y.; Tsukamoto, S.; Sakai, S.; Takeda, Y.; Mimura, N.; Ohwada, C.; Sakaida, E.; et al. Efficacy of myeloablative allogeneic hematopoietic stem cell transplantation in adult patients with MLL-ELL-positive acute myeloid leukemia. Int. J. Hematol. 2015, 102, 86–92. [Google Scholar] [CrossRef] [PubMed]
- Kong, J.H.; Mun, Y.-C.; Kim, S.; Choi, H.S.; Kim, Y.-K.; Kim, H.-J.; Moon, J.H.; Sohn, S.K.; Kim, S.-H.; Jung, C.W.; et al. Polymorphisms of ERCC1 genotype associated with response to imatinib therapy in chronic phase chronic myeloid leukemia. Int. J. Hematol. 2012, 96, 327–333. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Wu, D.; Li, H.; Dong, M. The effect of XPD/ERCC2 Lys751Gln polymorphism on acute leukemia risk: a systematic review and meta-analysis. Gene 2014, 538, 209–216. [Google Scholar] [CrossRef] [PubMed]
- Secchiero, P.; Barbarotto, E.; Tiribelli, M.; Zerbinati, C.; di Iasio, M.G.; Gonelli, A.; Cavazzini, F.; Campioni, D.; Fanin, R.; Cuneo, A.; et al. Functional integrity of the p53-mediated apoptotic pathway induced by the nongenotoxic agent nutlin-3 in B-cell chronic lymphocytic leukemia (B-CLL). Blood 2006, 107, 4122–4129. [Google Scholar] [CrossRef] [PubMed]
- Gazon, H.; Lemasson, I.; Polakowski, N.; Césaire, R.; Matsuoka, M.; Barbeau, B.; Mesnard, J.-M.; Peloponese, J.-M. Human T-cell leukemia virus type 1 (HTLV-1) bZIP factor requires cellular transcription factor JunD to upregulate HTLV-1 antisense transcription from the 3’ long terminal repeat. J. Virol. 2012, 86, 9070–9078. [Google Scholar] [CrossRef] [PubMed]
- Sincennes, M.-C.; Humbert, M.; Grondin, B.; Lisi, V.; Veiga, D.F.T.; Haman, A.; Cazaux, C.; Mashtalir, N.; Affar, E.B.; Verreault, A.; et al. The LMO2 oncogene regulates DNA replication in hematopoietic cells. Proc. Natl. Acad. Sci. U.S.A. 2016, 113, 1393–1398. [Google Scholar] [CrossRef]
- Agrawal-Singh, S.; Isken, F.; Agelopoulos, K.; Klein, H.-U.; Thoennissen, N.H.; Koehler, G.; Hascher, A.; Bäumer, N.; Berdel, W.E.; Thiede, C.; et al. Genome-wide analysis of histone H3 acetylation patterns in AML identifies PRDX2 as an epigenetically silenced tumor suppressor gene. Blood 2012, 119, 2346–2357. [Google Scholar] [CrossRef]
- Hakata, Y.; Yamada, M.; Shida, H. A multifunctional domain in human CRM1 (exportin 1) mediates RanBP3 binding and multimerization of human T-cell leukemia virus type 1 Rex protein. Mol. Cell. Biol. 2003, 23, 8751–8761. [Google Scholar] [CrossRef]
- Talby, L.; Chambost, H.; Roubaud, M.-C.; N’Guyen, C.; Milili, M.; Loriod, B.; Fossat, C.; Picard, C.; Gabert, J.; Chiappetta, P.; et al. The chemosensitivity to therapy of childhood early B acute lymphoblastic leukemia could be determined by the combined expression of CD34, SPI-B and BCR genes. Leuk. Res. 2006, 30, 665–676. [Google Scholar] [CrossRef]
- Juric, D.; Lacayo, N.J.; Ramsey, M.C.; Racevskis, J.; Wiernik, P.H.; Rowe, J.M.; Goldstone, A.H.; O’Dwyer, P.J.; Paietta, E.; Sikic, B.I. Differential gene expression patterns and interaction networks in BCR-ABL-positive and -negative adult acute lymphoblastic leukemias. J. Clin. Oncol. 2007, 25, 1341–1349. [Google Scholar] [CrossRef]
- Schaid, D.J.; Sinnwell, J.P.; McDonnell, S.K.; Thibodeau, S.N. Detecting genomic clustering of risk variants from sequence data: cases versus controls. Hum. Genet. 2013, 132, 1301–1309. [Google Scholar] [CrossRef] [PubMed][Green Version]
- George, V.; Laud, P.W. A Bayesian approach to the transmission/disequilibrium test for binary traits. Genetic Epidemiol. 2002, 22, 41–51. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Site | 1 | 2 | … | 9 | 10 | 11 | 12 | 13 | 14 | 15 | 16 | 17 | … | 24 |
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| group 1 | 0.44 | 0.46 | … | 0.6 | 0.62 | 0.64 | 0.66 | 0.66 | 0.64 | 0.62 | 0.6 | 0.58 | … | 0.44 |
| group 2 | 0.44 | 0.46 | … | 0.6 | 0.72 | 0.74 | 0.76 | 0.76 | 0.74 | 0.72 | 0.6 | 0.58 | … | 0.44 |
| group 3 | 0.44 | 0.46 | … | 0.6 | 0.82 | 0.84 | 0.86 | 0.86 | 0.84 | 0.82 | 0.6 | 0.58 | … | 0.44 |
| group 4 | 0.44 | 0.46 | … | 0.6 | 0.92 | 0.94 | 0.96 | 0.96 | 0.94 | 0.92 | 0.6 | 0.58 | … | 0.44 |
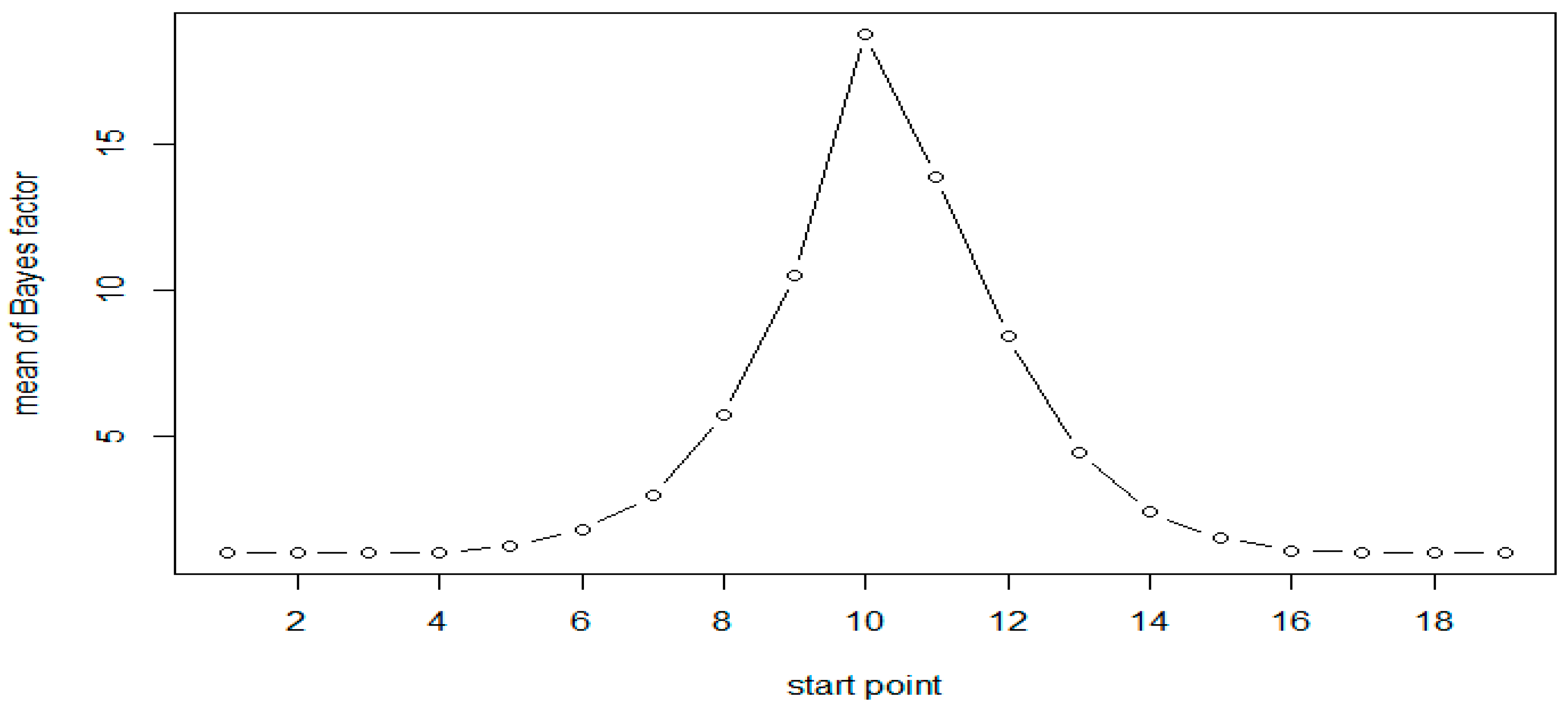
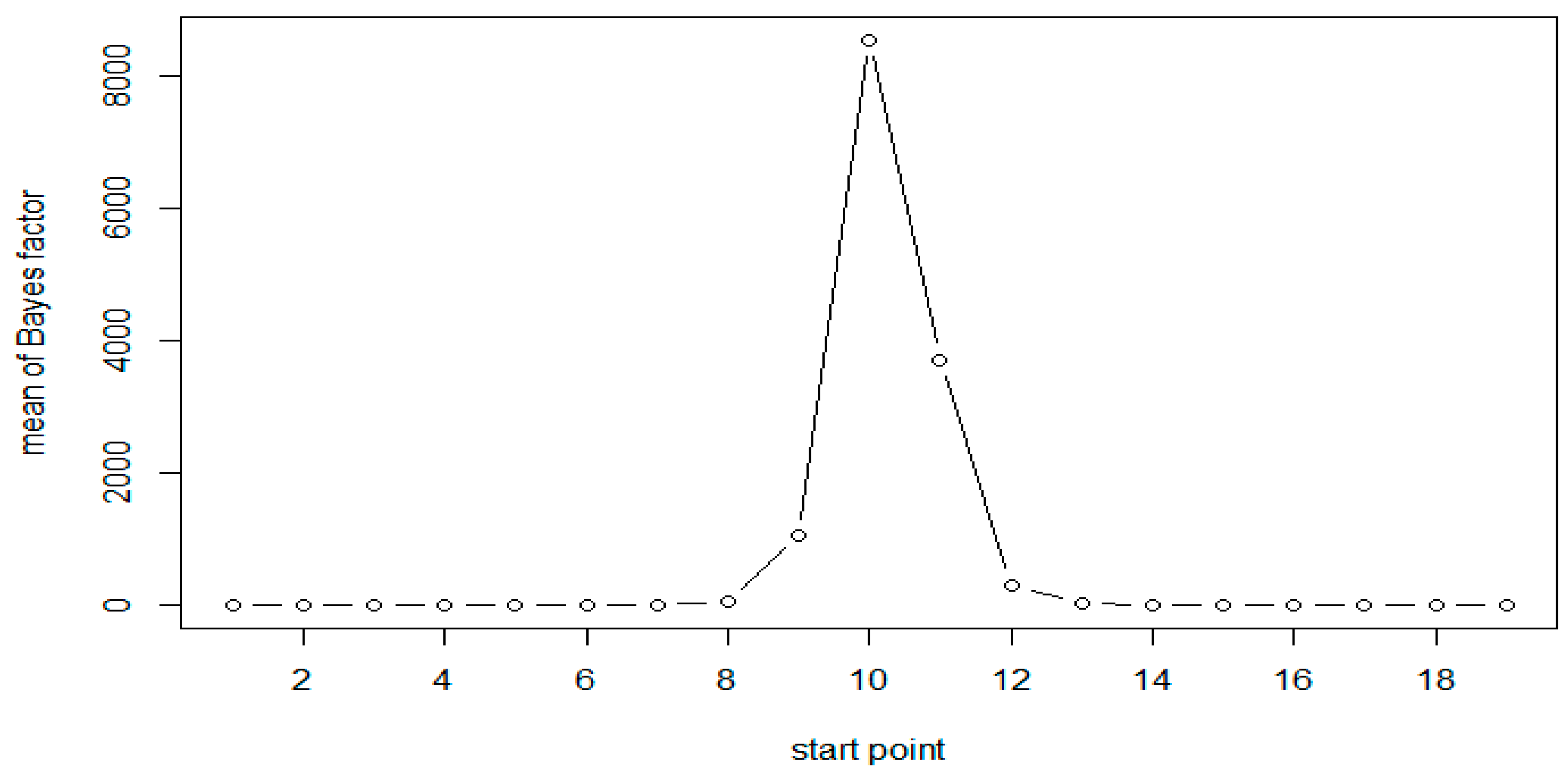
| Start | End | N = 50 (Scenario 1) | N = 100 (Scenario 1) | N = 50 (Scenario 2) |
|---|---|---|---|---|
| 1 | 6 | 1.02 | 1.02 | 1.03 |
| 2 | 7 | 1.01 | 1.02 | 1.01 |
| 3 | 8 | 1.01 | 1.02 | 1.02 |
| 4 | 9 | 1.02 | 1.01 | 1.01 |
| 5 | 10 | 1.24 | 1.53 | 1.26 |
| 6 | 11 | 1.78 | 3.12 | 1.78 |
| 7 | 12 | 2.95 | 9.16 | 2.85 |
| 8 | 13 | 5.74 | 41.42 | 4.95 |
| 9 | 14 | 10.53 | 1052.07 | 9.31 |
| 10 | 15 | 18.79 | 8554.12 | 18.31 |
| 11 | 16 | 13.9 | 3718.77 | 13.79 |
| 12 | 17 | 8.44 | 306.07 | 8.12 |
| 13 | 18 | 4.43 | 21.91 | 4.5 |
| 14 | 19 | 2.4 | 5.66 | 2.6 |
| 15 | 20 | 1.52 | 2.22 | 1.6 |
| 16 | 21 | 1.07 | 1.11 | 1.07 |
| 17 | 22 | 1.03 | 1.04 | 1.02 |
| 18 | 23 | 1.01 | 1.03 | 1.02 |
| 19 | 24 | 1.03 | 1.03 | 1.01 |
| Cut-off Point | Number of DMCs in the Windows | ||||||
|---|---|---|---|---|---|---|---|
| 0 | 1 | 2 | 3 | 4 | 5 | 6 | |
| 1.34 | 0.050 | 0.56 | 0.97 | 1 | 1 | 1 | 1 |
| 1.5 | 0.010 | 0.35 | 0.91 | 1 | 1 | 1 | 1 |
| BFM > 2 | SSM (p < 0.05) | Common | |
|---|---|---|---|
| Total | 183 | 181 | 67 |
| PubMed | 42 | 41 | 18 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Dunbar, F.; Xu, H.; Ryu, D.; Ghosh, S.; Shi, H.; George, V. Detection of Differentially Methylated Regions Using Bayes Factor for Ordinal Group Responses. Genes 2019, 10, 721. https://doi.org/10.3390/genes10090721
Dunbar F, Xu H, Ryu D, Ghosh S, Shi H, George V. Detection of Differentially Methylated Regions Using Bayes Factor for Ordinal Group Responses. Genes. 2019; 10(9):721. https://doi.org/10.3390/genes10090721
Chicago/Turabian StyleDunbar, Fengjiao, Hongyan Xu, Duchwan Ryu, Santu Ghosh, Huidong Shi, and Varghese George. 2019. "Detection of Differentially Methylated Regions Using Bayes Factor for Ordinal Group Responses" Genes 10, no. 9: 721. https://doi.org/10.3390/genes10090721
APA StyleDunbar, F., Xu, H., Ryu, D., Ghosh, S., Shi, H., & George, V. (2019). Detection of Differentially Methylated Regions Using Bayes Factor for Ordinal Group Responses. Genes, 10(9), 721. https://doi.org/10.3390/genes10090721