The Expanded SWEET Gene Family Following Whole Genome Triplication in Brassica rapa




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Identification and Characterization of SWEET Genes in B. rapa
2.2. Multiple Sequence Alignment and Phylogenetic Analysis
2.3. Expression Pattern in Different Tissues
2.4. Plant Materials, Growth Conditions, and Stress Treatment
2.5. RNA Isolation and qRT-PCR
3. Results
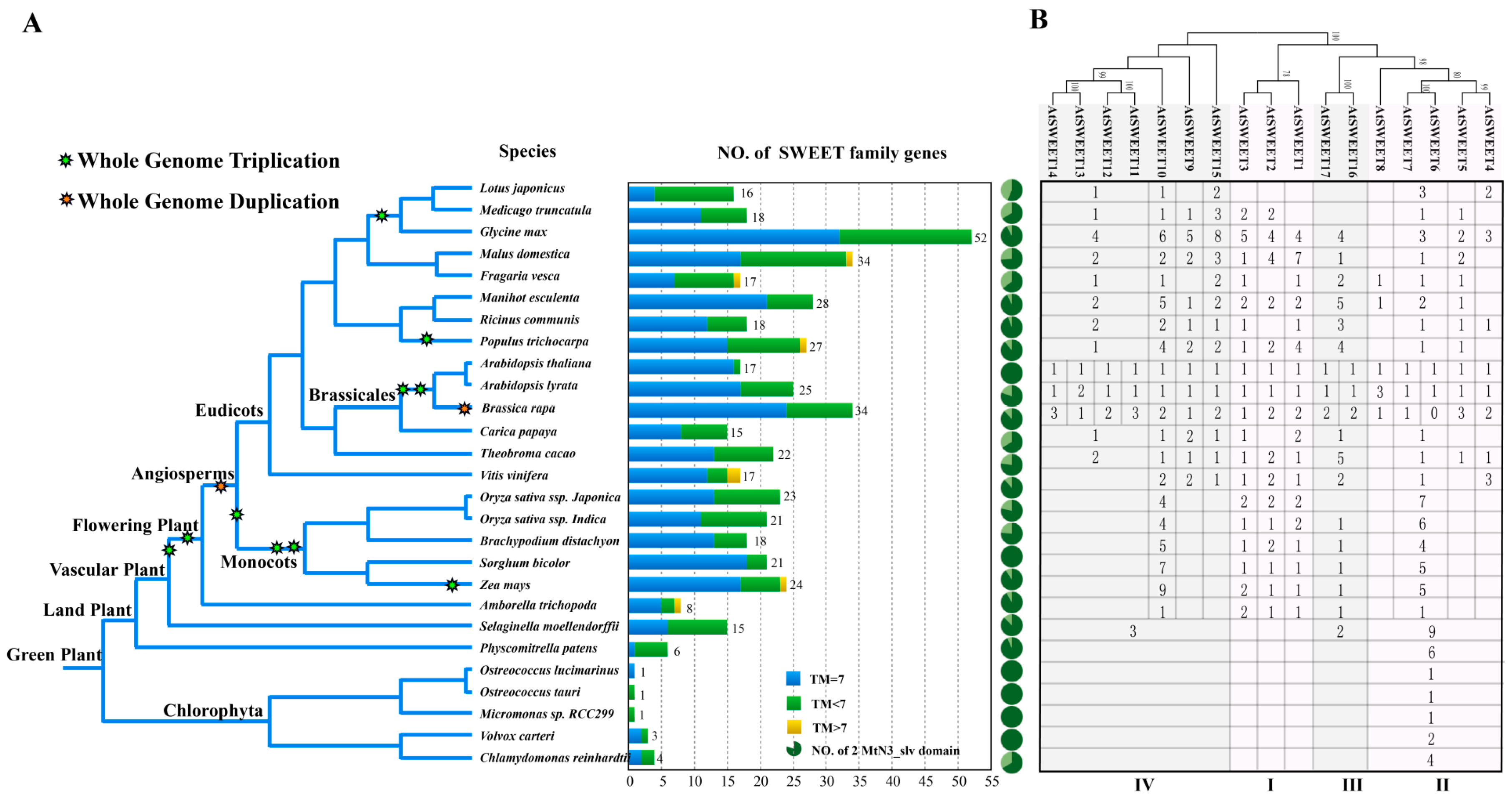
3.1. The Evolutionary Footprint of SWEETs in Plants
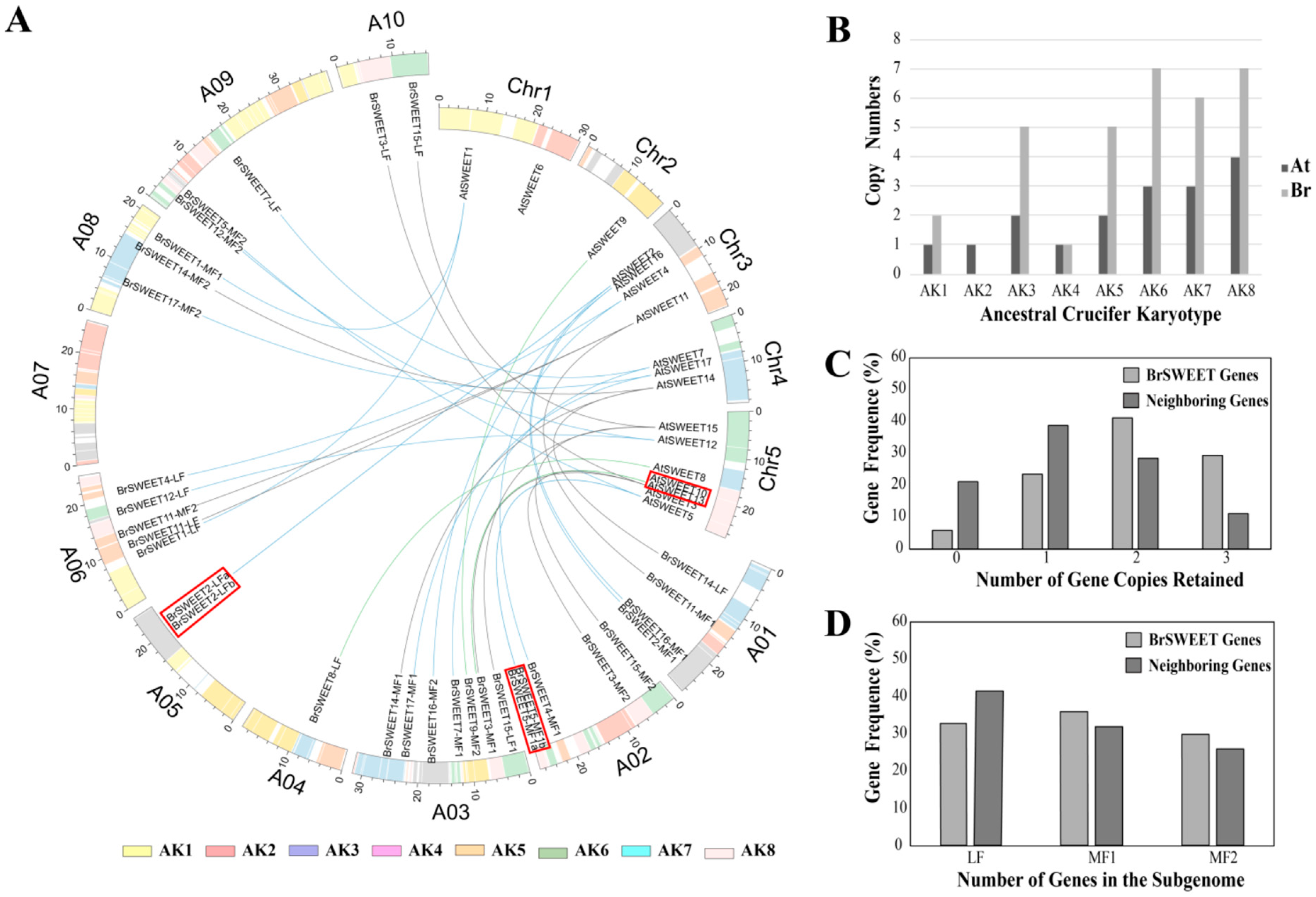
3.2. SWEETs Homologs in the B. rapa Genome
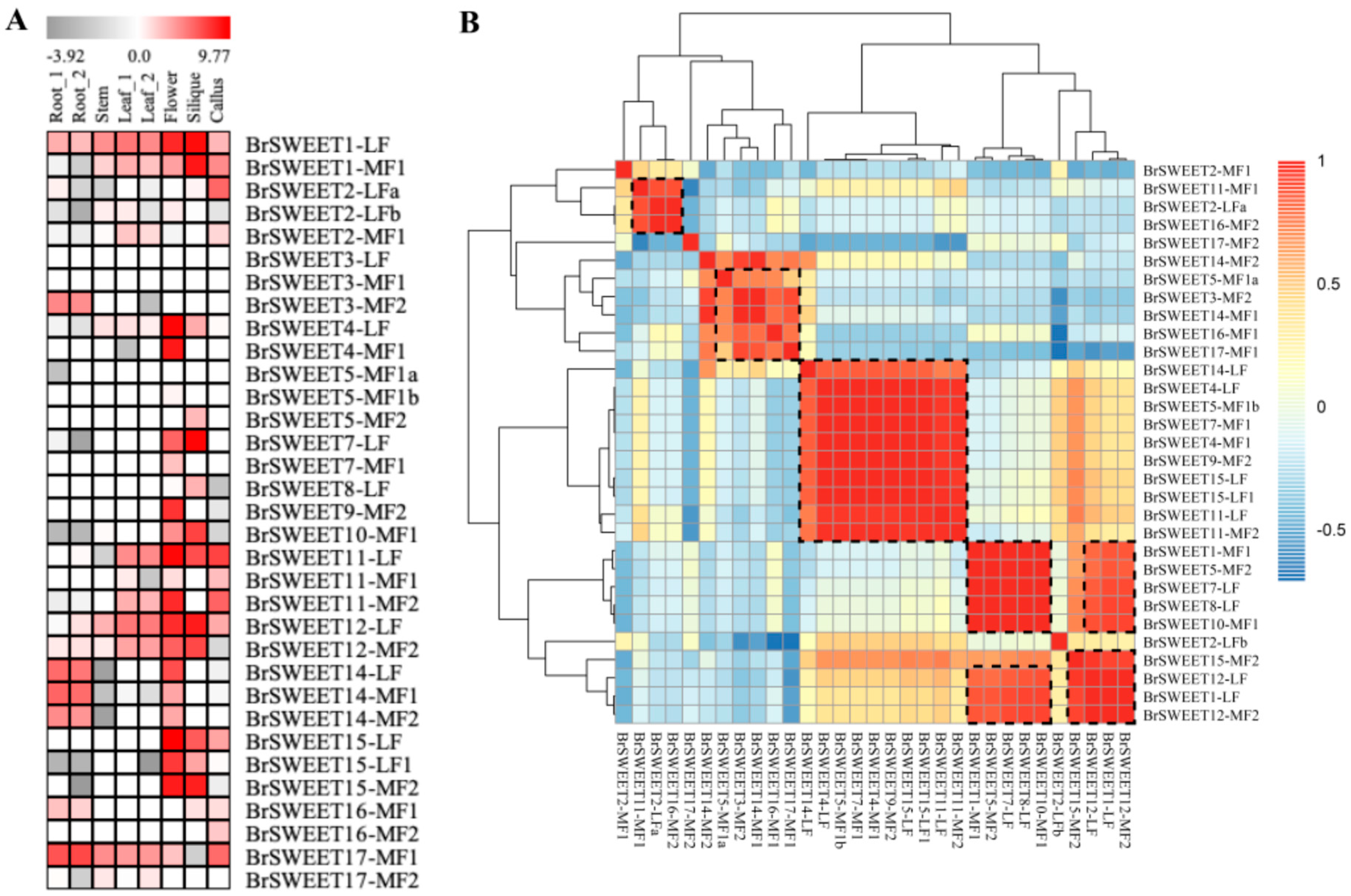
3.3. In-Silico Assessment of Transcript Impact within Co-Orthologous BrSWEETs
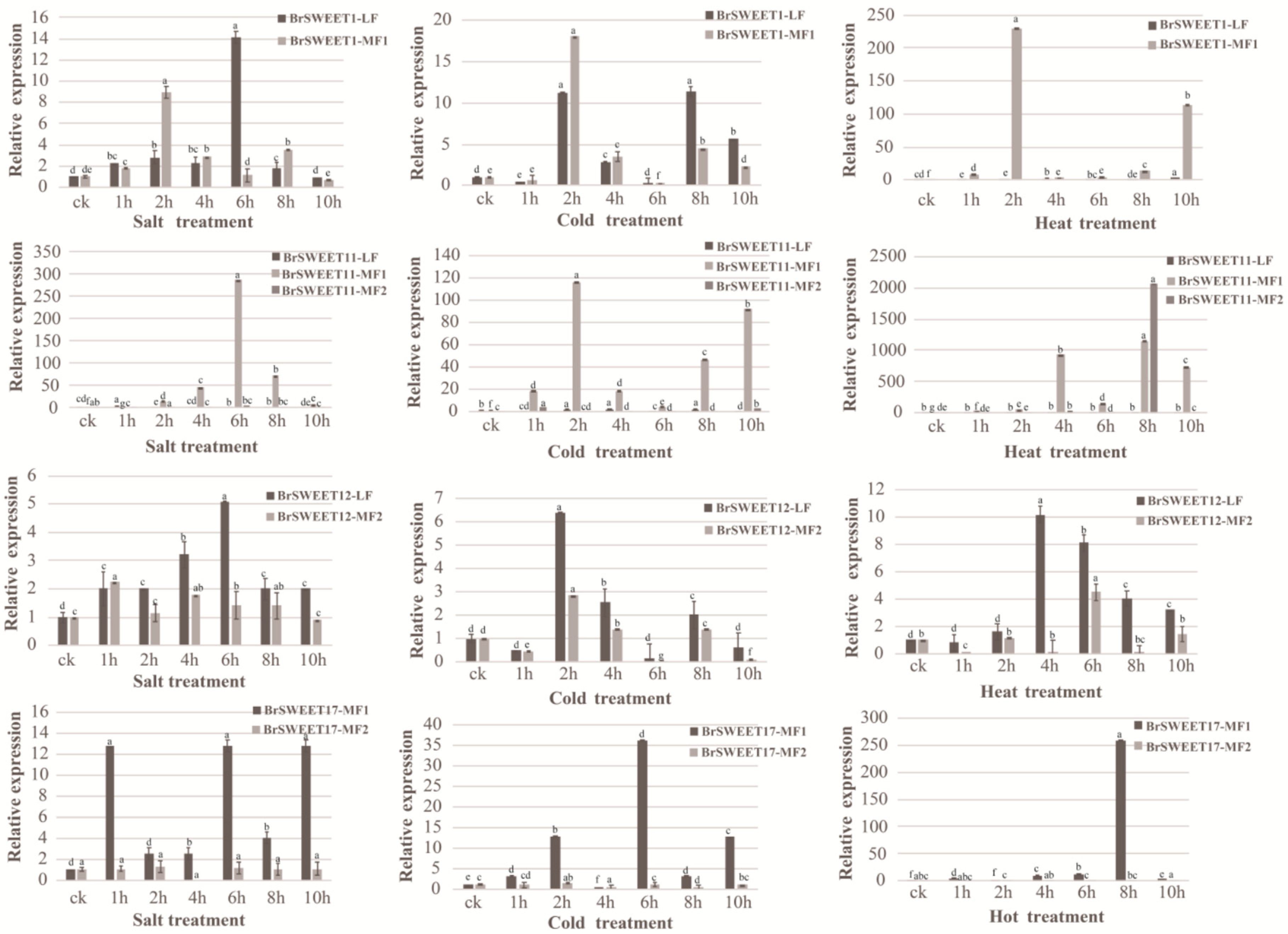
3.4. Expanded Co-Orthologous Gene Expression in Response to Abiotic Stress
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ruan, Y.L. Sucrose metabolism: Gateway to diverse carbon use and sugar signaling. Annu. Rev. Plant Biol. 2014, 65, 33–67. [Google Scholar] [CrossRef] [PubMed]
- Lemoine, R.; La Camera, S.; Atanassova, R.; Dédaldéchamp, F.; Allario, T.; Pourtau, N.; Bonnemain, J.-L.; Laloi, M.; Coutos-Thévenot, P.; Maurousset, L. Source-to-sink transport of sugar and regulation by environmental factors. Front. Plant Sci. 2013, 4, 272. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.Q.; Cheung, L.S.; Feng, L.; Tanner, W.; Frommer, W.B. Transport of sugars. Annu. Rev. Biochem. 2015, 84, 865–894. [Google Scholar] [CrossRef] [PubMed]
- Tao, Y.; Cheung, L.S.; Li, S.; Eom, J.S.; Chen, L.Q.; Xu, Y.; Perry, K.; Frommer, W.B.; Feng, L. Structure of a eukaryotic SWEET transporter in a homotrimeric complex. Nature 2015, 527, 259–263. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.-Q.; Hou, B.-H.; Lalonde, S.; Takanaga, H.; Hartung, M.L.; Qu, X.-Q.; Guo, W.-J.; Kim, J.-G.; Underwood, W.; Chaudhuri, B.; et al. Sugar transporters for intercellular exchange and nutrition of pathogens. Nature 2010, 468, 527–532. [Google Scholar] [CrossRef]
- Li-Qing, C.; Xiao-Qing, Q.; Bi-Huei, H.; Davide, S.; Sonia, O.; Fernie, A.R.; Frommer, W.B. Sucrose efflux mediated by SWEET proteins as a key step for phloem transport. Science 2012, 335, 207–211. [Google Scholar] [CrossRef]
- Sun, M.-X.; Huang, X.-Y.; Yang, J.; Guan, Y.-F.; Yang, Z.-N. Arabidopsis RPG1 is important for primexine deposition and functions redundantly with RPG2 for plant fertility at the late reproductive stage. Plant Reprod. 2013, 26, 83–91. [Google Scholar] [CrossRef]
- Lin, I.W.; Sosso, D.; Chen, L.Q.; Gase, K.; Kim, S.G.; Kessler, D.; Klinkenberg, P.M.; Gorder, M.K.; Hou, B.H.; Qu, X.Q.; et al. Nectar secretion requires sucrose phosphate synthases and the sugar transporter SWEET9. Nature 2014, 508, 546–549. [Google Scholar] [CrossRef]
- Seo, P.J.; Park, J.M.; Kang, S.K.; Kim, S.G.; Park, C.M. An Arabidopsis senescence-associated protein SAG29 regulates cell viability under high salinity. Planta 2011, 233, 189–200. [Google Scholar] [CrossRef]
- Lu, Y.; Gehan, J.P.; Sharkey, T.D. Daylength and circadian effects on starch degradation and maltose metabolism. Plant Physiol. 2005, 138, 2280–2291. [Google Scholar] [CrossRef]
- Sosso, D.; Luo, D.; Li, Q.-B.; Sasse, J.; Yang, J.; Gendrot, G.; Suzuki, M.; Koch, K.E.; McCarty, D.R.; Chourey, P.S.; et al. Seed filling in domesticated maize and rice depends on SWEET-mediated hexose transport. Nat. Genet. 2015, 47, 1489–1493. [Google Scholar] [CrossRef] [PubMed]
- Klemens, P.A.W.; Patzke, K.; Deitmer, J.; Spinner, L.; Hir, R.L.; Bellini, C.; Bedu, M.; Chardon, F.; Krapp, A.; Neuhaus, H.E. Overexpression of the vacuolar sugar carrier AtSWEET16 modifies germination, growth, and stress tolerance in Arabidopsis. Plant Physiol. 2013, 163, 1338–1352. [Google Scholar] [CrossRef] [PubMed]
- Zhou, J.; Peng, Z.; Long, J.; Sosso, D.; Liu, B.; Eom, J.S.; Huang, S.; Liu, S.; Vera Cruz, C.; Frommer, W.B. Gene targeting by the TAL effector PthXo2 reveals cryptic resistance gene for bacterial blight of rice. Plant J. 2015, 82, 632–643. [Google Scholar] [CrossRef] [PubMed]
- Cohn, M.; Bart, R.S.; Shybut, M.; Dahlbeck, D.; Gomez, M.; Morbitzer, R.; Hou, B.H.; Frommer, W.B.; Lahaye, T.; Staskawicz, B.J. Xanthomonas axonopodis virulence is promoted by a transcription activator-like effector-mediated induction of a SWEET sugar transporter in cassava. Mol. Plant Microbe Interact. 2014, 27, 1186–1198. [Google Scholar] [CrossRef] [PubMed]
- Eom, J.-S.; Chen, L.-Q.; Sosso, D.; Julius, B.T.; Lin, I.W.; Qu, X.-Q.; Braun, D.M.; Frommer, W.B. SWEETs, transporters for intracellular and intercellular sugar translocation. Curr. Opin. Plant Biol. 2015, 25, 53–62. [Google Scholar] [CrossRef]
- Feng, L.; Frommer, W.B. Structure and function of semiSWEET and SWEET sugar transporters. Trends Biochem. Sci. 2015, 40, 480–486. [Google Scholar] [CrossRef] [PubMed]
- Xu, Y.; Tao, Y.; Cheung, L.S.; Fan, C.; Chen, L.Q.; Xu, S.; Perry, K.; Frommer, W.B.; Feng, L. Structures of bacterial homologues of SWEET transporters in two distinct conformations. Nature 2014, 515, 448–452. [Google Scholar] [CrossRef]
- Le Hir, R.; Spinner, L.; Klemens, P.A.; Chakraborti, D.; de Marco, F.; Vilaine, F.; Wolff, N.; Lemoine, R.; Porcheron, B.; Gery, C.; et al. Disruption of the sugar transporters AtSWEET11 and AtSWEET12 affects vascular development and freezing tolerance in Arabidopsis. Mol. Plant 2015, 8, 1687–1690. [Google Scholar] [CrossRef]
- Chen, L.-Q.; Lin, I.W.; Qu, X.-Q.; Sosso, D.; McFarlane, H.E.; Londoño, A.; Samuels, A.L.; Frommer, W.B. A cascade of sequentially expressed sucrose transporters in the seed coat and endosperm provides nutrition for the Arabidopsis embryo. Plant Cell 2015, 27, 607–619. [Google Scholar] [CrossRef]
- Phukan, U.J.; Jeena, G.S.; Tripathi, V.; Shukla, R.K. MaRAP 2–4, a waterlogging-responsive erf from mentha, regulates bidirectional sugar transporter at SWEET10 to modulate stress response in Arabidopsis. Plant Biotechnol. J. 2018, 16, 221–233. [Google Scholar] [CrossRef]
- Kanno, Y.; Oikawa, T.; Chiba, Y.; Ishimaru, Y.; Shimizu, T.; Sano, N.; Koshiba, T.; Kamiya, Y.; Ueda, M.; Seo, M. AtSWEET13 and AtSWEET14 regulate gibberellin-mediated physiological processes. Nat. Commun. 2016, 7, 13245. [Google Scholar] [CrossRef] [PubMed]
- Liu, X.; Zhang, Y.; Yang, C.; Tian, Z.; Li, J. AtSWEET4, a hexose facilitator, mediates sugar transport to axial sinks and affects plant development. Sci. Rep. 2016, 6, 24563. [Google Scholar] [CrossRef] [PubMed]
- Guan, Y.-F.; Huang, X.-Y.; Zhu, J.; Gao, J.-F.; Zhang, H.-X.; Yang, Z.-N. Ruptured pollen grain1, a member of the MtN3/Saliva gene family, is crucial for exine pattern formation and cell integrity of microspores in Arabidopsis. Plant Physiol. 2008, 147, 852–863. [Google Scholar] [CrossRef] [PubMed]
- Yang, B.; Sugio, A.; White, F.F. Os8N3 is a host disease-susceptibility gene for bacterial blight of rice. Proc. Natl. Acad. Sci. USA 2006, 103, 10503–10508. [Google Scholar] [CrossRef] [PubMed]
- Chardon, F.; Bedu, M.; Calenge, F.; Klemens, P.A.; Spinner, L.; Clement, G.; Chietera, G.; Léran, S.; Ferrand, M.; Lacombe, B. Leaf fructose content is controlled by the vacuolar transporter SWEET17 in Arabidopsis. Curr. Biol. 2013, 23, 697–702. [Google Scholar] [CrossRef] [PubMed]
- Yuan, M.; Zhao, J.; Huang, R.; Li, X.; Xiao, J.; Wang, S. Rice MtN3/Saliva/SWEET gene family: Evolution, expression profiling, and sugar transport. J. Integr. Plant Biol. 2014, 56, 559–570. [Google Scholar] [CrossRef]
- Zhou, Y.; Liu, L.; Huang, W.; Yuan, M.; Zhou, F.; Li, X.; Lin, Y. Overexpression of OsSWEET5 in rice causes growth retardation and precocious senescence. PLoS ONE 2014, 9, e94210. [Google Scholar] [CrossRef]
- Feng, C.-Y.; Han, J.-X.; Han, X.-X.; Jiang, J. Genome-wide identification, phylogeny, and expression analysis of the SWEET gene family in tomato. Gene 2015, 573, 261–272. [Google Scholar] [CrossRef]
- Ho, L.-H.; Klemens, P.A.; Neuhaus, H.E.; Ko, H.-Y.; Hsieh, S.-Y.; Guo, W.-J. SlSWEET1a is involved in glucose import to young leaves in tomato plants. J. Exp. Bot. 2019. [Google Scholar] [CrossRef]
- Zhang, Z.; Zou, L.; Ren, C.; Ren, F.; Wang, Y.; Fan, P.; Li, S.; Liang, Z. VvSWEET10 mediates sugar accumulation in grapes. Genes 2019, 10, 255. [Google Scholar] [CrossRef]
- Chong, J.; Piron, M.-C.; Meyer, S.; Merdinoglu, D.; Bertsch, C.; Mestre, P. The SWEET family of sugar transporters in grapevine: VvSWEET4 is involved in the interaction with botrytis cinerea. J. Exp. Bot. 2014, 65, 6589–6601. [Google Scholar] [CrossRef] [PubMed]
- Kryvoruchko, I.S.; Sinharoy, S.; Torres-Jerez, I.; Sosso, D.; Pislariu, C.I.; Guan, D.; Murray, J.; Benedito, V.A.; Frommer, W.B.; Udvardi, M.K. MtSWEET11, a nodule-specific sucrose transporter of Medicago truncatula. Plant Physiol. 2016, 171, 554–565. [Google Scholar] [CrossRef] [PubMed]
- Artemieva, A.M. Genetic diversity and intraspecific phylogenetic relationships of Brassica rapa l. Species crops based on microsatellite analysis. Вестник ВОГиС (Рoссийская Федерация) 2008. [Google Scholar] [CrossRef]
- Wang, X.; Wang, H.; Wang, J.; Sun, R.; Wu, J.; Liu, S.; Bai, Y.; Mun, J.H.; Bancroft, I.; Cheng, F. The genome of the mesopolyploid crop species Brassica rapa. Nat. Genet. 2011, 43, 1035–1039. [Google Scholar] [CrossRef]
- Ramchiary, N.; Yong, P.L. Genetics of brassica rapa L. In Genetics and Genomics of the Brassicaceae; Springer: New York, NY, USA, 2010; pp. 215–260. [Google Scholar]
- Beilstein, M.A.; Nagalingum, N.S.; Clements, M.D.; Manchester, S.R.; Sarah, M. Dated molecular phylogenies indicate a miocene origin for Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2010, 107, 18724–18728. [Google Scholar] [CrossRef]
- Lukens, L.N.; Quijada, P.A.; Udall, J.; Pires, J.C.; Schranz, M.E.; Osborn, T.C. Genome redundancy and plasticity within ancient and recent Brassica crop species. Biol. J. Linn. Soc. 2004, 82, 665–674. [Google Scholar] [CrossRef]
- Lysak, M.A.; Koch, M.A.; Pecinka, A.; Schubert, I. Chromosome triplication found across the Brassiceae. Genome Res. 2005, 15, 516–525. [Google Scholar] [CrossRef]
- Arias, T.; Pires, J.C. Diversification times among Brassica (Brassicaceae) crops suggest hybrid formation after 20 million years of divergence. Am. J. Bot. 2014, 101, 86–91. [Google Scholar] [CrossRef]
- Haibao, T.; Woodhouse, M.R.; Feng, C.; Schnable, J.C.; Pedersen, B.S.; Gavin, C.; Xiaowu, W.; Michael, F.; Chris, P. Altered patterns of fractionation and exon deletions in brassica rapa support a two-step model of paleohexaploidy. Genetics 2012, 190, 1563–1574. [Google Scholar] [CrossRef]
- Peer, Y.V.D. A mystery unveiled. Genome Biol. 2011, 12, 113. [Google Scholar] [CrossRef]
- Wang, Y.; Tang, H.; Debarry, J.D.; Tan, X.; Li, J.; Wang, X.; Lee, T.H.; Jin, H.; Marler, B.; Guo, H. MCScanX: A toolkit for detection and evolutionary analysis of gene synteny and collinearity. Nucleic Acids Res. 2012, 40, e49. [Google Scholar] [CrossRef] [PubMed]
- Feng, C.; Jian, W.; Lu, F.; Wang, X. Syntenic gene analysis between Brassica rapa and other brassicaceae species. Front. Plant Sci. 2012, 3, 198. [Google Scholar] [CrossRef]
- Mount, D.W. Using the Basic Local Alignment Search Tool (BLAST). Cold Spring Harb. Protoc. 2007, 2007, 17. [Google Scholar] [CrossRef] [PubMed]
- Kazutaka, K.; Standley, D.M. Mafft multiple sequence alignment software version 7: Improvements in performance and usability. Mol. Biol. Evol. 2013, 30, 102–343. [Google Scholar] [CrossRef]
- Beitz, E. Texshade: Shading and labeling of multiple sequence alignments using LATEX2 epsilon. Bioinformatics 2000, 16, 135–139. [Google Scholar] [CrossRef] [PubMed]
- Saitou, N.; Nei, M. The neighbor-joining method: A new method for reconstructing phylogenetic trees. Mol. Biol. Evol. 1987, 4, 406–425. [Google Scholar] [CrossRef]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870. [Google Scholar] [CrossRef]
- Stéphane, G.; Olivier, G. Phyml-a simple, fast, and accurate algorithm to estimate large phylogenies by maximum likelihood. Syst. Biol. 2003, 52, 696–704. [Google Scholar] [CrossRef]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. ProtTest 3: Fast selection of best-fit models of protein evolution. Bioinformatics 2011, 27, 1164–1165. [Google Scholar] [CrossRef]
- Guindon, S.; Dufayard, J.F.; Lefort, V.; Anisimova, M.; Hordijk, W.; Gascuel, O. New algorithms and methods to estimate maximum-likelihood phylogenies: Assessing the performance of phyml 3.0. Syst. Biol. 2010, 59, 307–321. [Google Scholar] [CrossRef]
- Devisetty, U.K.; Covington, M.F.; An, V.T.; Lekkala, S.; Maloof, J.N. Polymorphism identification and improved genome annotation of Brassica rapa through deep RNA sequencing. G3 Genes Genet. 2014, 4, 2065–2078. [Google Scholar] [CrossRef]
- Xiao, D.; Zhang, N.-W.; Zhao, J.-J.; Bonnema, G.; Hou, X.-L. Validation of reference genes for real-time quantitative PCR normalisation in non-heading Chinese cabbage. Funct. Plant Biol. 2012, 39, 342–350. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The evolutionary fate and consequences of duplicate genes. Science 2000, 290, 1151–1155. [Google Scholar] [CrossRef] [PubMed]
- Chandran, D. Co-option of developmentally regulated plant SWEET transporters for pathogen nutrition and abiotic stress tolerance. IUBMB Life 2015, 67, 461–471. [Google Scholar] [CrossRef] [PubMed]
- Gamalei, Y. Structure and function of leaf minor veins in trees and herbs. Trees 1989, 3, 96–110. [Google Scholar] [CrossRef]
- Gamalei, Y. Phloem loading and its development related to plant evolution from trees to herbs. Trees 1991, 5, 50–64. [Google Scholar] [CrossRef]
- Xuan, Y.H.; Hu, Y.B.; Chen, L.Q.; Sosso, D.; Ducat, D.C.; Hou, B.H.; Frommer, W.B. Functional role of oligomerization for bacterial and plant SWEET sugar transporter family. Proc. Natl. Acad. Sci. USA 2013, 110, E3685–E3694. [Google Scholar] [CrossRef] [PubMed]
- Proels, R.K.; Huckelhoven, R. Cell-wall invertases, key enzymes in the modulation of plant metabolism during defence responses. Mol. Plant Pathol. 2014, 15, 858–864. [Google Scholar] [CrossRef]
- Couvreur, T.L.; Franzke, A.; Al-Shehbaz, I.A.; Bakker, F.T.; Koch, M.A.; Mummenhoff, K. Molecular phylogenetics, temporal diversification, and principles of evolution in the mustard family (Brassicaceae). Mol. Biol. Evol. 2010, 27, 55–71. [Google Scholar] [CrossRef]
- Franzke, A.; Lysak, M.A.; Alshehbaz, I.A.; Koch, M.A.; Mummenhoff, K. Cabbage family affairs: The evolutionary history of Brassicaceae. Trends Plant Sci. 2011, 16, 108–116. [Google Scholar] [CrossRef]
- Chen, H.Y.; Huh, J.H.; Yu, Y.C.; Ho, L.H.; Chen, L.Q.; Tholl, D.; Frommer, W.B.; Guo, W.J. The Arabidopsis vacuolar sugar transporter SWEET2 limits carbon sequestration from roots and restricts Pythium infection. Plant J. Cell Mol. Biol. 2015, 83, 1046–1058. [Google Scholar] [CrossRef] [PubMed]
- Van Bel, A.J.E. Different phloem-loading machineries correlated with the climate. Acta Bot. Neerl. 1992, 41, 121–141. [Google Scholar] [CrossRef]
- Miao, L.; Lv, Y.; Kong, L.; Chen, Q.; Chen, C.; Li, J.; Zeng, F.; Wang, S.; Li, J.; Huang, L. Genome-wide identification, phylogeny, evolution, and expression patterns of MtN3/Saliva/SWEET genes and functional analysis of BcNS in Brassica rapa. BMC Genom. 2018, 19, 174. [Google Scholar] [CrossRef] [PubMed]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Wei, Y.; Xiao, D.; Zhang, C.; Hou, X. The Expanded SWEET Gene Family Following Whole Genome Triplication in Brassica rapa. Genes 2019, 10, 722. https://doi.org/10.3390/genes10090722
Wei Y, Xiao D, Zhang C, Hou X. The Expanded SWEET Gene Family Following Whole Genome Triplication in Brassica rapa. Genes. 2019; 10(9):722. https://doi.org/10.3390/genes10090722
Chicago/Turabian StyleWei, Yanping, Dong Xiao, Changwei Zhang, and Xilin Hou. 2019. "The Expanded SWEET Gene Family Following Whole Genome Triplication in Brassica rapa" Genes 10, no. 9: 722. https://doi.org/10.3390/genes10090722
APA StyleWei, Y., Xiao, D., Zhang, C., & Hou, X. (2019). The Expanded SWEET Gene Family Following Whole Genome Triplication in Brassica rapa. Genes, 10(9), 722. https://doi.org/10.3390/genes10090722