Further Defining the Phenotypic Spectrum of B3GAT3 Mutations and Literature Review on Linkeropathy Syndromes

 ,
,  ,
,  ,
,  and
and 
Abstract
1. Introduction
2. Patient and Methods
2.1. Ethical Compliance
2.2. Amplicon-Based Exome Sequencing
2.3. Sanger Sequencing
3. Results
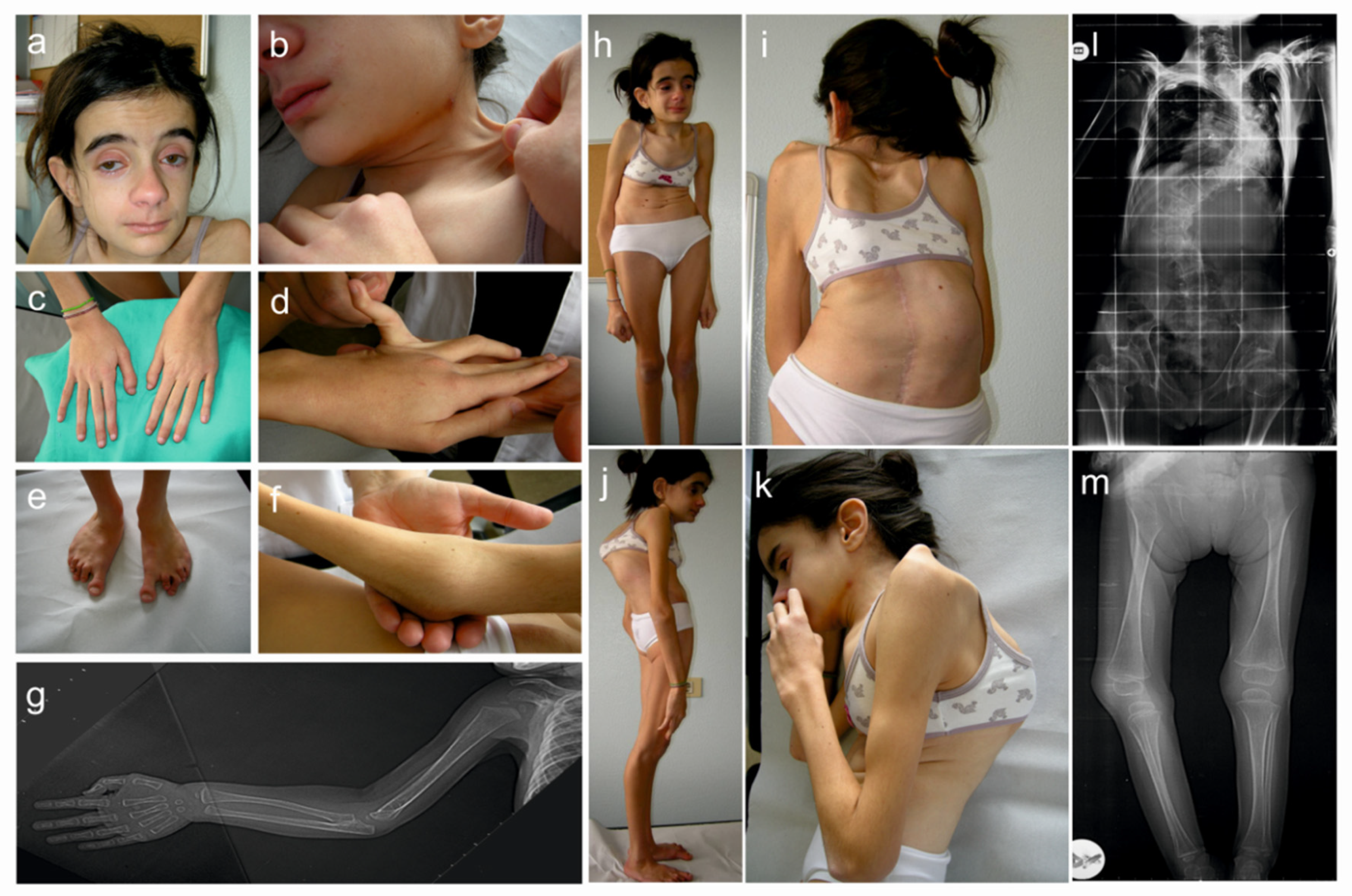
3.1. Clinical Findings
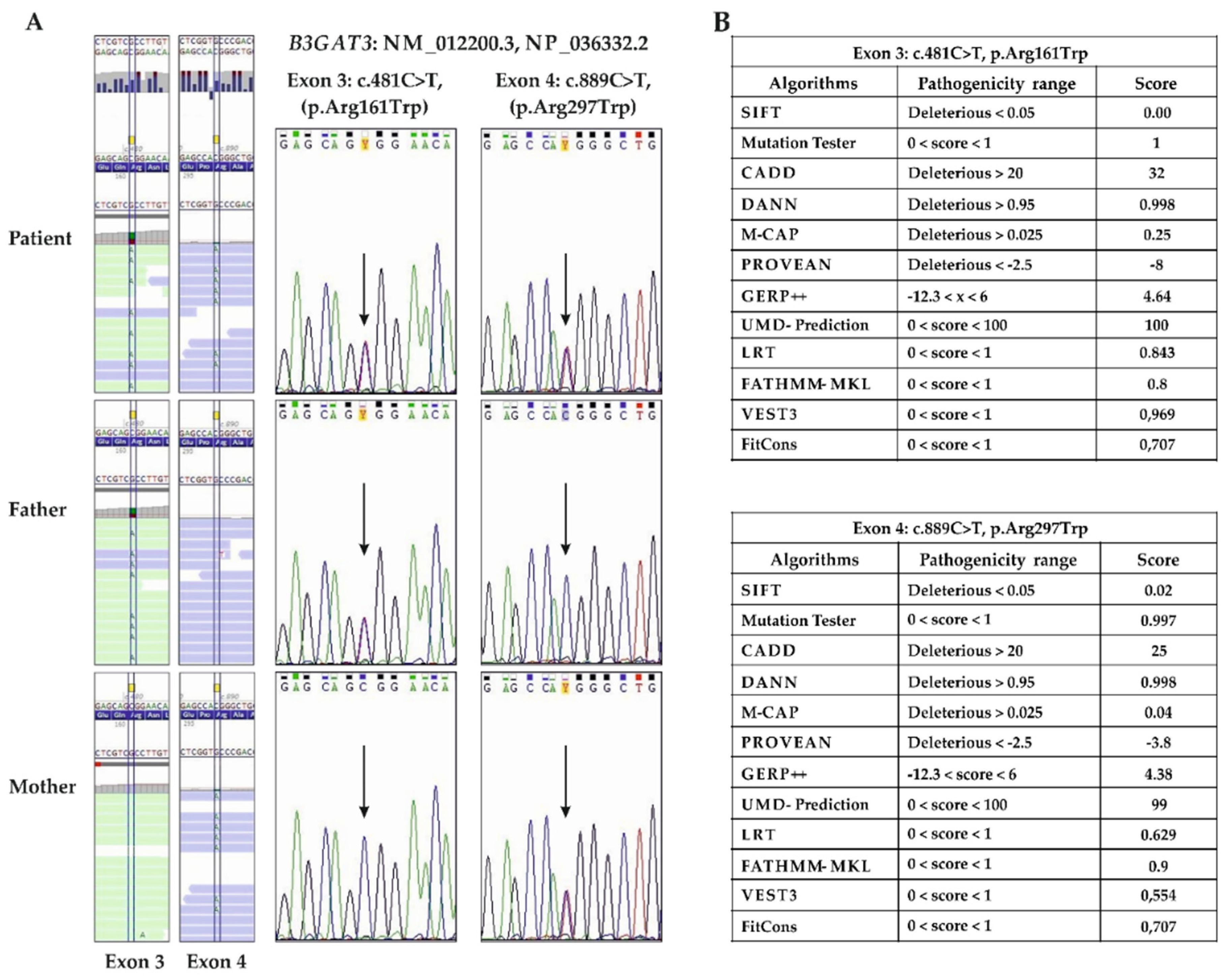
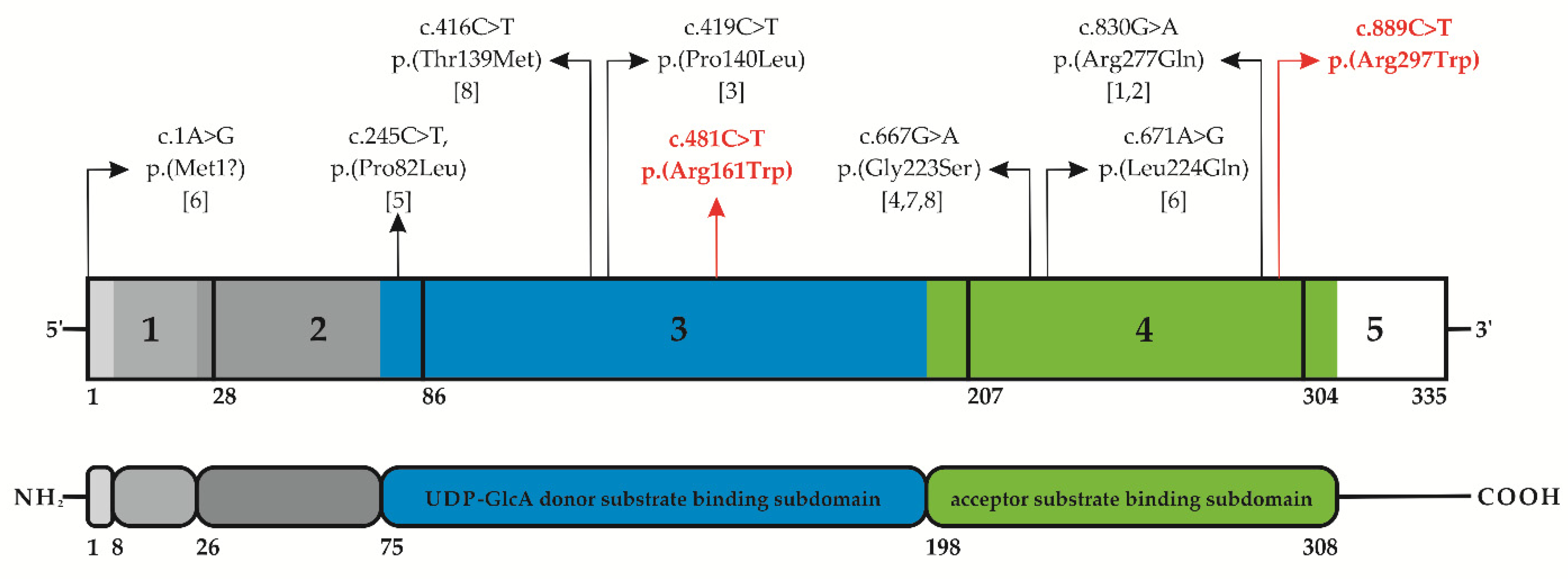
3.2. Molecular Findings
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C 2017, 175, 8–26. [Google Scholar] [CrossRef] [PubMed]
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-allelic alterations in AEBP1 lead to defective collagen assembly and connective tissue structure resulting in a variant of Ehlers-Danlos syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef] [PubMed]
- Hebebrand, M.; Vasileiou, G.; Krumbiegel, M.; Kraus, C.; Uebe, S.; Ekici, A.B.; Thiel, C.T.; Reis, A.; Popp, B. A biallelic truncating AEBP1 variant causes connective tissue disorder in two siblings. Am. J. Med. Genet. A 2019, 179, 50–56. [Google Scholar] [CrossRef] [PubMed]
- Syx, D.; De Wandele, I.; Symoens, S.; De Rycke, R.; Hougrand, O.; Voermans, N.; De Paepe, A.; Malfait, F. Bi-allelic AEBP1 mutations in two patients with Ehlers-Danlos syndrome. Hum. Mol. Genet. 2019, 28, 1853–1864. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Cinquina, V.; Venturini, M.; Pezzaioli, L.; Formenti, A.M.; Chiarelli, N.; Colombi, M. Expanding the Clinical and Mutational Spectrum of Recessive AEBP1-Related Classical-Like Ehlers-Danlos Syndrome. Genes 2019, 10, 135. [Google Scholar] [CrossRef] [PubMed]
- Castori, M.; Colombi, M. Generalized joint hypermobility, joint hypermobility syndrome and Ehlers-Danlos syndrome, hypermobility type. Am. J. Med. Genet. C Semin. Med. Genet. 2015, 169, 1–5. [Google Scholar] [CrossRef] [PubMed]
- Colombi, M.; Dordoni, C.; Chiarelli, N.; Ritelli, M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am. J. Med. Genet. Part C 2015, 169, 6–22. [Google Scholar] [CrossRef] [PubMed]
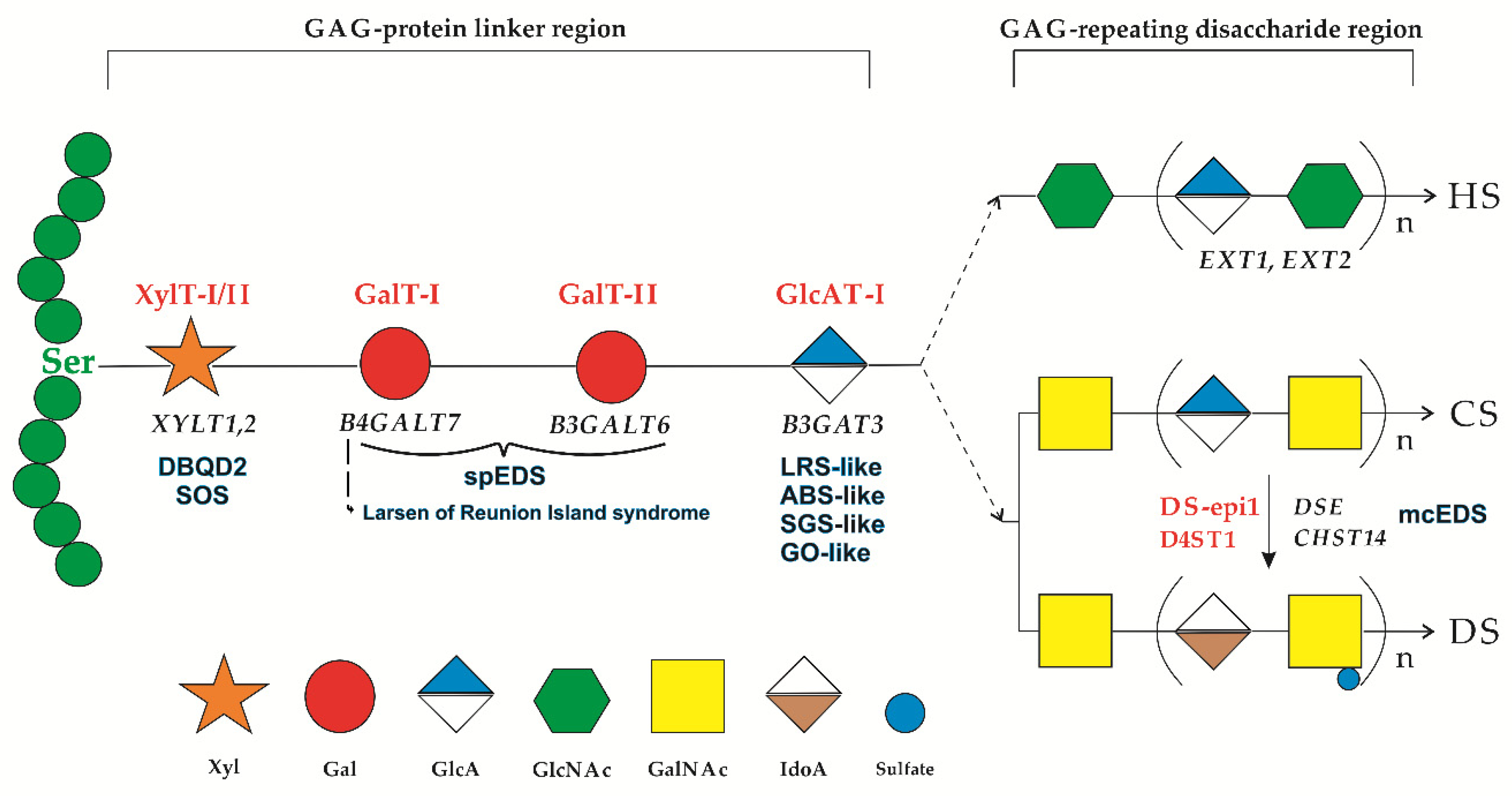
- Sugahara, K.; Kitagawa, H. Recent advances in the study of the biosynthesis and functions of sulfated glycosaminoglycans. Curr. Opin. Struct. Biol. 2000, 10, 518–527. [Google Scholar] [CrossRef]
- Bülow, H.E.; Hober, O. The molecular diversity of glycosaminoglycans shapes animal development. Annu. Rev. Cell Dev. Biol. 2006, 22, 375–407. [Google Scholar] [CrossRef]
- Esko, J.D.; Kimata, K.; Lindahl, U. Proteoglycans and Sulfated Glycosaminoglycans. In Essential of Glycobiology, 2nd ed.; Varki, A., Cummings, R.D., Esko, J.D., Freeze, H.H., Stanley, P., Bertozzi, C.R., Hart, G.W., Etzler, M.E., Eds.; Cold Spring Harbor Laboratory Press: Cold Spring Harbor, NY, USA, 2009; Chapter 16. [Google Scholar]
- Mizumoto, S.; Ikegawa, S.; Sugahara, K. Human genetic disorders caused by mutations in genes encoding biosynthetic enzymes for sulfated glycosaminoglycans. J. Biol. Chem. 2013, 19, 10953–10961. [Google Scholar] [CrossRef]
- Taylan, F.; Mäkitie, O. Abnormal Proteoglycan Synthesis Due to Gene Defects Causes Skeletal Diseases with Overlapping Phenotypes. Horm. Metab. Res. 2016, 48, 745–754. [Google Scholar] [CrossRef] [PubMed]
- Mizumoto, S.; Kosho, T.; Yamada, S.; Sugahara, K. Pathophysiological Significance of Dermatan Sulfate Proteoglycans Revealed by Human Genetic Disorders. Pharmaceuticals 2017, 10, 34. [Google Scholar] [CrossRef] [PubMed]
- Brady, A.F.; Demirdas, S.; Fournel-Gigleux, S.; Ghali, N.; Giunta, C.; Kapferer-Seebacher, I.; Kosho, T.; Mendoza-Londono, R.; Pope, M.F.; Rohrbach, M.; et al. The Ehlers-Danlos syndromes, rare types. Am. J. Med. Genet. C 2017, 175, 70–115. [Google Scholar] [CrossRef] [PubMed]
- Kresse, H.; Rosthøj, S.; Quentin, E.; Hollmann, J.; Glössl, J.; Okada, S.; Tønnesen, T. Glycosaminoglycan-free small proteoglycan core protein is secreted by fibroblasts from a patient with a syndrome resembling progeroid. Am. J. Hum. Genet. 1987, 41, 436–453. [Google Scholar] [PubMed]
- Faiyaz-Ul-Haque, M.; Zaidi, S.H.; Al-Ali, M.; Al-Mureikhi, M.S.; Kennedy, S.; Al-Thani, G.; Tsui, L.C.; Teebi, A.S. A novel missense mutation in the galactosyltransferase-I (B4GALT7) gene in a family exhibiting facioskeletal anomalies and Ehlers-Danlos syndrome resembling the progeroid type. Am. J. Med. Genet. A 2004, 128, 39–45. [Google Scholar] [CrossRef] [PubMed]
- Guo, M.H.; Stoler, J.; Lui, J.; Nilsson, O.; Bianchi, D.W.; Hirschhorn, J.N.; Dauber, A. Redefining the progeroid form of Ehlers-Danlos syndrome: Report of the fourth patient with B4GALT7 deficiency and review of the literature. Am. J. Med. Genet. A 2013, 161, 2519–2527. [Google Scholar] [CrossRef]
- Salter, C.G.; Davies, J.H.; Moon, R.J.; Fairhurst, J.; Bunyan, D.; DDD Study; Foulds, N. Further defining the phenotypic spectrum of B4GALT7 mutations. Am. J. Med. Genet. A 2016, 170, 1556–1563. [Google Scholar] [CrossRef] [PubMed]
- Arunrut, T.; Sabbadini, M.; Jain, M.; Machol, K.; Scaglia, F.; Slavotinek, A. Corneal clouding, cataract, and colobomas with a novel missense mutation in B4GALT7-a review of eye anomalies in the linkeropathy syndromes. Am. J. Med. Genet. A 2016, 170, 2711–2718. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Dordoni, C.; Cinquina, V.; Venturini, M.; Calzavara-Pinton, P.; Colombi, M. Expanding the clinical and mutational spectrum of B4GALT7-spondylodysplastic Ehlers-Danlos syndrome. Orphanet J. Rare Dis. 2017, 7, 153. [Google Scholar] [CrossRef] [PubMed]
- Sandler-Wilson, C.; Wambach, J.A.; Marshall, B.A.; Wegner, D.J.; McAlister, W.; Cole, F.S.; Shinawi, M. Phenotype and response to growth hormone therapy in siblings with B4GALT7 deficiency. Bone 2019, 124, 14–21. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F.; Kariminejad, A.; Van Damme, T.; Gauche, C.; Syx, D.; Merhi-Soussi, F.; Gulberti, S.; Symoens, S.; Vanhauwaert, S.; Willaert, A.; et al. Defective initiation of glycosaminoglycan synthesis due to B3GALT6 mutations causes a pleiotropic Ehlers-Danlos-syndrome-like connective tissue disorder. Am. J. Hum. Genet. 2013, 92, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Nakajima, M.; Mizumoto, S.; Miyake, N.; Kogawa, R.; Iida, A.; Ito, H.; Kitoh, H.; Hirayama, A.; Mitsubuchi, H.; Miyazaki, O.; et al. Mutations in B3GALT6, which encodes a glycosaminoglycan linker region enzyme, cause a spectrum of skeletal and connective tissue disorders. Am. J. Hum. Genet. 2013, 92, 927–934. [Google Scholar] [CrossRef] [PubMed]
- Sellars, E.A.; Bosanko, K.A.; Lepard, T.; Garnica, A.; Schaefer, G.B. A newborn with complex skeletal abnormalities, joint contractures, and bilateral corneal clouding with sclerocornea. Semin. Pediatr. Neurol. 2014, 21, 84–87. [Google Scholar] [CrossRef] [PubMed]
- Ritelli, M.; Chiarelli, N.; Zoppi, N.; Dordoni, C.; Quinzani, S.; Traversa, M.; Venturini, M.; Calzavara-Pinton, P.; Colombi, M. Insights in the etiopathology of galactosyltransferase II (GalT-II) deficiency from transcriptome-wide expression profiling of skin fibroblasts of two sisters with compound heterozygosity for two novel B3GALT6 mutations. Mol. Genet. Metab. Rep. 2014, 20, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Alazami, A.M.; Al-Qattan, S.M.; Faqeih, E.; Alhashem, A.; Alshammari, M.; Alzahrani, F.; Al-Dosari, M.S.; Patel, N.; Alsagheir, A.; Binabbas, B.; et al. Expanding the clinical and genetic heterogeneity of hereditary disorders of connective tissue. Hum. Genet. 2016, 135, 525–540. [Google Scholar] [CrossRef] [PubMed]
- Honey, E.M. Spondyloepimetaphyseal dysplasia with joint laxity (Beighton type): A unique South African disorder. S. Afr. Med. J. 2016, 106, S54–S56. [Google Scholar] [CrossRef] [PubMed]
- Van Damme, T.; Pang, X.; Guillemyn, B.; Gulberti, S.; Syx, D.; De Rycke, R.; Kaye, O.; De Die-Smulders, C.E.M.; Pfundt, R.; Kariminejad, A.; et al. Biallelic B3GALT6 mutations cause spondylodysplastic Ehlers-Danlos syndrome. Hum. Mol. Genet. 2018, 27, 3475–3487. [Google Scholar] [CrossRef] [PubMed]
- Giunta, C.; Elçioglu, N.H.; Albrecht, B.; Eich, G.; Chambaz, C.; Janecke, A.R.; Yeowell, H.; Weis, M.; Eyre, D.R.; Kraenzlin, M.; et al. Spondylocheiro dysplastic form of the Ehlers-Danlos syndrome—An autosomal-recessive entity caused by mutations in the zinc transporter gene SLC39A13. Am. J. Hum. Genet. 2008, 82, 1290–1305. [Google Scholar] [CrossRef]
- Fukada, T.; Civic, N.; Furuichi, T.; Shimoda, S.; Mishima, K.; Higashiyama, H.; Idaira, Y.; Asada, Y.; Kitamura, H.; Yamasaki, S.T.; et al. The zinc transporter SLC39A13/ZIP13 is required for connective tissue development; its involvement in BMP/TGF-β signaling pathways. PLoS ONE 2008, 3, e3642. [Google Scholar] [CrossRef]
- Dusanic, M.; Dekomien, G.; Lücke, T.; Vorgerd, M.; Weis, J.; Epplen, J.T.; Köhler, C.; Hoffjan, S. Novel Nonsense Mutation in SLC39A13 Initially Presenting as Myopathy: Case Report and Review of the Literature. Mol. Syndromol. 2018, 9, 100–109. [Google Scholar] [CrossRef]
- Cartault, F.; Munier, P.; Jacquemont, M.L.; Vellayoudom, J.; Doray, B.; Payet, C.; Randrianaivo, H.; Laville, J.M.; Munnich, A.; Cormier-Daire, V. Expanding the clinical spectrum of B4GALT7 deficiency: Homozygous p.R270C mutation with founder effect causes Larsen of Reunion Island syndrome. Eur. J. Hum. Genet. 2015, 23, 49–53. [Google Scholar] [CrossRef]
- Schreml, J.; Durmaz, B.; Cogulu, O.; Keupp, K.; Beleggia, F.; Pohl, E.; Milz, E.; Coker, M.; Ucar, S.K.; Nürnberg, G.; et al. The missing “link”: An autosomal recessive short stature syndrome caused by a hypofunctional XYLT1 mutation. Hum. Genet. 2014, 133, 29–39. [Google Scholar] [CrossRef]
- Bui, C.; Huber, C.; Tuysuz, B.; Alanay, Y.; Bole-Feysot, C.; Leroy, J.G.; Mortier, G.; Nitschke, P.; Munnich, A.; Cormier-Daire, V. XYLT1 mutations in Desbuquois dysplasia type 2. Am. J. Hum. Genet. 2014, 94, 405–414. [Google Scholar] [CrossRef]
- Van Koningsbruggen, S.; Knoester, H.; Bakx, R.; Mook, O.; Knegt, L.; Cobben, J.M. Complete and partial XYLT1 deletion in a patient with neonatal short limb skeletal dysplasia. Am. J. Med. Genet. A 2016, 170, 510–514. [Google Scholar] [CrossRef]
- Jamsheer, A.; Olech, E.M.; Kozłowski, K.; Niedziela, M.; Sowińska-Seidler, A.; Obara-Moszyńska, M.; Latos-Bieleńska, A.; Karczewski, M.; Zemojtel, T. Exome sequencing reveals two novel compound heterozygous XYLT1 mutations in a Polish patient with Desbuquois dysplasia type 2 and growth hormone deficiency. J. Hum. Genet. 2016, 61, 577–583. [Google Scholar] [CrossRef]
- Silveira, C.; Leal, G.F.; Cavalcanti, D.P. Desbuquois dysplasia type II in a patient with a homozygous mutation in XYLT1 and new unusual findings. Am. J. Med. Genet. A 2016, 170, 3043–3047. [Google Scholar] [CrossRef]
- Guo, L.; Elcioglu, N.H.; Iida, A.; Demirkol, Y.K.; Aras, S.; Matsumoto, N.; Nishimura, G.; Miyake, N.; Ikegawa, S. Novel and recurrent XYLT1 mutations in two Turkish families with Desbuquois dysplasia, type 2. J. Hum. Genet. 2017, 62, 447–451. [Google Scholar] [CrossRef]
- Al-Jezawi, N.K.; Ali, B.R.; Al-Gazali, L. Endoplasmic reticulum retention of xylosyltransferase 1 (XYLT1) mutants underlying Desbuquois dysplasia type II. Am. J. Med. Genet. A 2017, 173, 1773–1781. [Google Scholar] [CrossRef]
- LaCroix, A.J.; Stabley, D.; Sahraoui, R.; Adam, M.P.; Mehaffey, M.; Kernan, K.; Myers, C.T.; Fagerstrom, C.; Anadiotis, G.; Akkari, Y.M.; et al. GGC Repeat Expansion and Exon 1 Methylation of XYLT1 Is a Common Pathogenic Variant in Baratela-Scott Syndrome. Am. J. Hum. Genet. 2019, 104, 35–44. [Google Scholar] [CrossRef]
- Munns, C.F.; Fahiminiya, S.; Poudel, N.; Munteanu, M.C.; Majewski, J.; Sillence, D.O.; Metcalf, J.P.; Biggin, A.; Glorieux, F.; Fassier, F.; et al. Homozygosity for frameshift mutations in XYLT2 result in a spondylo-ocular syndrome with bone fragility, cataracts, and hearing defects. Am. J. Hum. Genet. 2015, 96, 971–978. [Google Scholar] [CrossRef]
- Taylan, F.; Costantini, A.; Coles, N.; Pekkinen, M.; Héon, E.; Şıklar, Z.; Berberoğlu, M.; Kämpe, A.; Kıykım, E.; Grigelioniene, G.; et al. Spondyloocular Syndrome: Novel Mutations in XYLT2 Gene and Expansion of the Phenotypic Spectrum. J. Bone Miner. Res. 2016, 31, 1577–1585. [Google Scholar] [CrossRef]
- Taylan, F.; Yavaş Abalı, Z.; Jäntti, N.; Güneş, N.; Darendeliler, F.; Baş, F.; Poyrazoğlu, Ş.; Tamçelik, N.; Tüysüz, B.; Mäkitie, O. Two novel mutations in XYLT2 cause spondyloocular syndrome. Am. J. Med. Genet. A 2017, 173, 3195–3200. [Google Scholar] [CrossRef]
- Umair, M.; Eckstein, G.; Rudolph, G.; Strom, T.; Graf, E.; Hendig, D.; Hoover, J.; Alanay, J.; Meitinger, T.; Schmidt, H.; et al. Homozygous XYLT2 variants as a cause of spondyloocular syndrome. Clin. Genet. 2018, 93, 913–918. [Google Scholar] [CrossRef]
- Guleray, N.; Simsek Kiper, P.O.; Utine, G.E.; Boduroglu, K.; Alikasifoglu, M. Intrafamilial variability of XYLT2-related spondyloocular syndrome. Eur. J. Med. Genet. 2018, 1769, 30611–30616. [Google Scholar] [CrossRef]
- Kausar, M.; Chew, E.G.Y.; Ullah, H.; Anees, M.; Khor, C.C.; Foo, J.N.; Makitie, O.; Siddiqi, S. A Novel Homozygous Frameshift Variant in XYLT2 Causes Spondyloocular Syndrome in a Consanguineous Pakistani Family. Front. Genet. 2019, 10, 144. [Google Scholar] [CrossRef]
- Baasanjav, S.; Al-Gazali, L.; Hashiguchi, T.; Mizumoto, S.; Fischer, B.; Horn, D.; Seelow, D.; Ali, B.R.; Aziz, S.A.; Langer, R.; et al. Faulty initiation of proteoglycan synthesis causes cardiac and joint defects. Am. J. Hum. Genet. 2011, 15, 15–27. [Google Scholar] [CrossRef]
- Von Oettingen, J.E.; Tan, W.H.; Dauber, A. Skeletal dysplasia, global developmental delay, and multiple congenital anomalies in a 5-year-old boy-report of the second family with B3GAT3 mutation and expansion of the phenotype. Am. J. Med. Genet. A 2014, 164, 1580–1586. [Google Scholar] [CrossRef]
- Budde, B.S.; Mizumoto, S.; Kogawa, R.; Becker, C.; Altmüller, J.; Thiele, H.; Rüschendorf, F.; Toliat, M.R.; Kaleschke, G.; Hämmerle, J.M.; et al. Skeletal dysplasia in a consanguineous clan from the island of Nias/Indonesia is caused by a novel mutation in B3GAT3. Hum. Genet. 2015, 134, 691–704. [Google Scholar] [CrossRef]
- Jones, K.L.; Schwarze, U.; Adam, M.P.; Byers, P.H.; Mefford, H.C. A homozygous B3GAT3 mutation causes a severe syndrome with multiple fractures, expanding the phenotype of linkeropathy syndromes. Am. J. Med. Genet. A 2015, 167, 2691–2696. [Google Scholar] [CrossRef]
- Job, F.; Mizumoto, S.; Smith, L.; Couser, N.; Brazil, A.; Saal, H.; Patterson, M.; Gibson, M.I.; Soden, S.; Miller, N.; et al. Functional validation of novel compound heterozygous variants in B3GAT3 resulting in severe osteopenia and fractures: Expanding the disease phenotype. BMC Med. Genet. 2016, 17, 86. [Google Scholar] [CrossRef]
- Yauy, K.; Tran Mau-Them, F.; Willems, M.; Coubes, C.; Blanchet, P.; Herlin, C.; Taleb Arrada, I.; Sanchez, E.; Faure, J.M.; Le Gac, M.P.; et al. B3GAT3-related disorder with craniosynostosis and bone fragility due to a unique mutation. Genet. Med. 2018, 20, 269–274. [Google Scholar] [CrossRef]
- Colman, M.; Van Damme, T.; Steichen-Gersdorf, E.; Laccone, F.; Nampoothiri, S.; Syx, D.; Guillemyn, B.; Symoens, S.; Malfait, F. The clinical and mutational spectrum of B3GAT3 linkeropathy: Two case reports and literature review. Orphanet. J. Rare Dis. 2019, 14, 138. [Google Scholar] [CrossRef]
- San Lucas, F.A.; Wang, G.; Scheet, P.; Peng, B. Integrated annotation and analysis of genetic variants from next-generation sequencing studies with variant tools. Bioinformatics 2012, 1, 421–422. [Google Scholar] [CrossRef]
- Ravasio, V.; Ritelli, M.; Legati, A.; Giacopuzzi, E. GARFIELD-NGS: Genomic vARiants FIltering by dEep Learning moDels in NGS. Bioinformatics 2018, 1, 3038–3040. [Google Scholar] [CrossRef]
- Wang, K.; Li, M.; Hakonarson, H. Annovar: Functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids. Res. 2010, 38, 164. [Google Scholar] [CrossRef]
- Quang, D.; Chen, Y.; Xie, X. Dann: A deep learning approach for annotating the pathogenicity of genetic variants. Bioinformatics 2015, 31, 761–763. [Google Scholar] [CrossRef]
- Jagadeesh, K.A.; Wenger, A.M.; Berger, M.J.; Guturu, H.; Stenson, P.D.; Cooper, D.N.; Bernstein, J.A.; Bejerano, G. M-CAP eliminates a majority of variants of uncertain significance in clinical exomes at high sensitivity. Nat. Genet. 2016, 48, 1581–1586. [Google Scholar] [CrossRef]
- Petrovski, S.; Wang, Q.; Heinzen, E.L.; Allen, A.S.; Goldstein, D.B. Genic intolerance to functional variation and the interpretation of personal genomes. PLoS Genet. 2013, 9, e1003709. [Google Scholar] [CrossRef]
- Itan, Y.; Shang, L.; Boisson, B.; Patin, E.; Bolze, A.; Moncada-Vélez, M.; Scott, E.; Ciancanelli, M.J.; Lafaille, F.G.; Markle, J.G.; et al. The human gene damage index as a gene-level approach to prioritizing exome variants. Proc. Natl. Acad. Sci. USA 2015, 112, 13615–13620. [Google Scholar] [CrossRef]
- Sim, N.L.; Kumar, P.; Hu, J.; Henikoff, S.; Schneider, G.; Ng, P.C. SIFT web server: Predicting effects of amino acid substitutions on proteins. Nucleic Acids Res. 2012, 40, W452–W457. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef]
- Rentzsch, P.; Witten, D.; Cooper, G.M.; Shendure, J.; Kircher, M. CADD: Predicting the deleteriousness of variants throughout the human genome. Nucleic Acids Res. 2018. [Google Scholar] [CrossRef]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Eugene, V.; Davydov, E.V.; Goode, D.L.; Sirota, M.; Cooper, G.M.; Sidow, A.; Batzoglou, S. Identifying a High Fraction of the Human Genome to be under Selective Constraint Using GERP++. PLoS Comput. Biol. 2010, 6, e1001025. [Google Scholar] [CrossRef]
- Salgado, D.; Desvignes, J.P.; Rai, G.; Blanchard, A.; Miltgen, M.; Pinard, A.; Lévy, N.; Collod-Béroud, G.; Béroud, C. UMD-Predictor: A High-Throughput Sequencing Compliant System for Pathogenicity Prediction of any Human cDNA Substitution. Hum. Mutat. 2016, 35, 439–446. [Google Scholar] [CrossRef]
- Chun, S.; Fay, J.C. Identification of deleterious mutations within three human genomes. Genom. Res. 2009, 19, 1553–1561. [Google Scholar] [CrossRef]
- Shihab, H.A.; Rogers, M.F.; Gough, J.; Mort, M.; Cooper, D.N.; Day, I.N.; Gaunt, T.R.; Campbell, C. An integrative approach to predicting the functional effects of non-coding and coding sequence variation. Bioinformatics 2015, 31, 1536–1543. [Google Scholar] [CrossRef]
- Carter, H.; Douville, C.; Stenson, P.D.; Cooper, D.N.; Karchin, R. Identifying Mendelian disease genes with the Variant Effect Scoring Tool BMC. Genomics 2013, 14, S3. [Google Scholar] [CrossRef]
- Arbiza, L.; Gronau, I.; Aksoy, B.A.; Hubisz, M.J.; Gulko, B.; Keinan, A.; Siepel, A. Genome-wide inference of natural selection on human transcription factor binding sites. Nat. Genet. 2013, 45, 723–729. [Google Scholar] [CrossRef]
- Pedersen, L.C.; Tsuchida, K.; Kitagawa, H.; Sugahara, K.; Darden, T.A.; Negishi, M. Heparan/chondroitin sulfate biosynthesis. Structure and mechanism of human glucuronyltransferase I. J. Biol. Chem. 2000, 277, 34580–34585. [Google Scholar] [CrossRef]
- Pedersen, L.C.; Darden, T.A.; Negishi, M. Crystal structure of β1,3-glucuronyltransferase I in complex with active donor substrate UDP-GlcUA. J. Biol. Chem. 2002, 277, 21869–21873. [Google Scholar] [CrossRef]
- Li, Q.; Wang, K. InterVar: Clinical Interpretation of Genetic Variants by the 2015 ACMG-AMP Guidelines. Am. J. Hum. Genet. 2017, 100, 267–280. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. ACMG Laboratory Quality Assurance Committee. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Oldani, E.; Garel, C.; Bucourt, M.; Carbillon, L. Prenatal diagnosis of Antley-Bixler syndrome and POR deficiency. Am. J. Case. 2015, 16, 882–885. [Google Scholar] [CrossRef]
- Greally, M.T. Shprintzen-Goldberg Syndrome. In GeneReviews®; Adam, M.P., Ardinger, H.H., Pagon, R.A., Wallace, S.E., Bean, L.J.H., Stephens, K., Amemiya, A., Eds.; University of Washington: Seattle, WA, USA, 2006; pp. 1993–2019. [Google Scholar]
- Ouzzine, M.; Gulberti, S.; Levoin, N.; Netter, P.; Magdalou, J.; Fournel-Gigleux, S. The donor substrate specificity of the human β 1,3-glucuronosyltransferase I toward UDP-glucuronic acid is determined by two crucial histidine and arginine residues. J. Biol. Chem. 2002, 227, 25439–25445. [Google Scholar] [CrossRef]
- Fondeur-Gelinotte, M.; Lattard, V.; Oriol, R.; Mollicone, R.; Jacquinet, J.C.; Mulliert, G.; Gulberti, S.; Netter, P.; Magdalou, J.; Ouzzine, M.; et al. Phylogenetic and mutational analyses reveal key residues for UDP-glucuronic acid binding and activity of β 1,3-glucuronosyltransferase I (GlcAT-I). Protein Sci. 2006, 15, 1667–1678. [Google Scholar] [CrossRef]
- Venselaar, H.; Te Beek, T.A.; Kuipers, R.K.; Hekkelman, M.L.; Vriend, G. Protein structure analysis of mutations causing inheritable diseases. An e-Science approach with life scientist friendly interfaces. BMC Bioinform. 2010, 11, 548. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Variant | Effect | Inheritance Model | Genotype | Clinvar Phenotype |
|---|---|---|---|---|---|
| B3GAT3 (NM_00122200) | c.481C>T (father) c.889C>T (mother) | p.(Arg161Trp) p.(Arg297Trp) | Recessive (comp het) | comp het | Multiple joint dislocations, short stature, craniofacial dysmorphism, and congenital heart defects |
| BMP8A (NM_181809) | c.333G>T | p.(Met111Ile) | Dominant (de novo) | het | |
| FES (NM_0002005.3) | c.1778G>A | p.(Arg593Gln) | Dominant (de novo) | het | |
| NR2F6 (NM_005234) | c.806C>T | p.(Pro269Leu) | Dominant (de novo) | het | |
| PAK2 (NM_002577) | c.303G>C | p.(Gln101His) | Dominant (de novo) | het | |
| TRAK1 (NM_001042646) | c.1327G>A | p.(Ser443Gly) | Dominant (de novo) | het |
| References | Present Patient | [1,2] | [3] | [6] | [8] | [5] | [4,7,8] |
|---|---|---|---|---|---|---|---|
| Number of patients | n = 1 | n = 6 | n = 8 | n = 1 | n=1 | n = 1 | n = 8 |
| Phenotype | LRS-like | LRS-like | LRS-like | LRS-like | LRS-like | GO-like | ABS/SGS-like |
| Consanguinity | - | + | + | - | + | + | + |
| B3GAT3 variant(s) (NM_012200.3) | c.481C>T c.889C>T | c.830G>A homozygous | c.419C>T homozygous | c.1A>G c.671T>A | c.416C>T homozygous | c.245C>T homozygous | c.667G>A homozygous |
| Protein Change (NP_036332.2) | p.(Arg161Trp) p.(Arg297Trp) | p.(Arg277Gln) | p.(Pro140Leu) | p.(Met1?) p.(Leu224Gln) | p.(Thr139Met) | p.(Pro82Leu) | p.(Gly223Ser) |
| Skeletal | |||||||
| Short stature | + | + | + | - | + | + | 2/5 |
| Joint hypermobility | + | + | - | + | + | na | 1/2 |
| Joint dislocations | + | + | + | + | + | + | 3/7 |
| Elbow joint abnormalities | + | + | 4/8 | na | - | na | + |
| Multiple fractures | - | - | na | + | - | + | 6/8 |
| Kyphoscoliosis | + | - | 4/8 | + | - | na | 1/7 |
| Scoliosis/kyphosis | + | 0/1 | 4/8 | + | - | + | 2/7 |
| Platyspondyly | - | - | 0/2 | - | - | + | - |
| Peculiar fingers (long, slender, tapered, broad, thin, arachnodactyly) | + | + | + | + | + | na | + |
| Pectus abnormality | + | 1/6 | - | - | - | na | 1/2 |
| Radioulnar synostosis | + | 1/1 | 2/2 | - | - | na | 7/7 |
| Bowing of limbs | - | na | 4/8 | + | - | na | 0/8 |
| Metaphyseal flaring | + | 1/1 | na | na | - | na | 1/2 |
| Iliac abnormalities | - | 1/1 | na | na | na | na | 1/2 |
| Radial head subluxation or dislocation | - | na | 2/2 | na | + | na | 0/1 |
| Foot deformity | + | + | 6/8 | - | + | na | + |
| pes planus | + | 1/1 | na | - | + | na | na |
| hallux valgus | + | 1/1 | 6/8 | - | + | na | na |
| club feet | - | 0/1 | na | - | - | na | + |
| sandal gap between toes | + | 1/1 | 3/8 | - | - | na | 1/1 |
| Osteopenia | + | 5/6 | na | + | + | + | 2/2 |
| Cervical spine instability | + | 1/1 | na | na | na | na | na |
| Craniofacial | |||||||
| Midface hypoplasia | + | + | + | + | + | na | 7/7 |
| Flat face | - | 1/1 | na | - | - | na | na |
| Craniosynostosis | + | + | na | + | na | na | 4/7 |
| Frontal bossing | + | 1/6 | na | + | - | na | 3/7 |
| Wide forehead | - | 1/1 | na | na | - | na | 2/2 |
| Blue sclerae | + | 0/1 | - | + | + | na | 5/5 |
| Proptosis or prominent eyes | - | + | na | - | + | na | 5/5 |
| Downslanting palpebral fissures | + | 3/5 | na | + | + | na | 1/2 |
| Low-set ears | + | 2/5 | na | na | na | na | 1/1 |
| Depressed nasal bridge | - | 5/6 | 4/8 | - | - | na | 4/7 |
| Small mouth/microstomia | - | 4/6 | 3/8 | - | + | na | 2/2 |
| Long upper lip/long philtrum | -/+ | na | na | na | + | na | 2/2 |
| Cleft palate/bifid uvula | +/- | na | na | + | + | na | 1/1 |
| Micrognathia | + | 4/6 | 4/8 | na | - | na | 0/1 |
| Short and/or webbed neck | + | + | 2/8 | + | + | na | 2/2 |
| Cutaneous | |||||||
| Skin (hyperextensibility; soft, doughy, thin, translucent skin) | + (mild) | 1/1 skin wrinkling | - | + | - | Cutis laxa | 1/2 Cutis laxa |
| Easy bruising | + | 0/1 | na | na | na | na | na |
| Atrophic scarring | + (mild) | 0/1 | na | na | na | na | 0/1 |
| Other | |||||||
| Cardiovascular abnormalities | + | 6/6 | 0/3 | + | na | + | 4/8 |
| Muscle hypotonia | + | 0/1 | na | + | na | na | 4/4 |
| Refractive errors/hypermetropia | + | 1/1 | na | + | - | na | 0/1 |
| Delayed motor development | + | 1/1 | na | + | + | na | 1/1 |
| Delayed cognitive development | - | - | - | - | - | na | 1/1 |
| Bone chondromas | - | - | - | - | - | + | - |
| Anterior ectopic anus | + | - | - | - | - | - | - |
| Genes | B3GAT3 | B4GALT7 | B3GALT6 | SLC39A13 | XYLT1 | XYLT2 |
|---|---|---|---|---|---|---|
| Number of patients | n = 26 | n = 32 | n = 46 | n = 9 | n = 28 | n = 20 |
| Skeletal | ||||||
| Short stature | 18/23 | 29/29 | 36/46 | + | + | 7/17 |
| Joint hypermobility | 10/19 | + | 37/46 | + | 13/14 | 2/5 |
| Joint dislocation | 9/25 | + | 37/46 | + | 14/14 | na |
| Joint contractures (hands, elbow) | 19/24 | 4/9 | 30/46 | 3/9 | 4/5 | na |
| Low bone density/osteopenia | 10/11 | 6/32 | 20/46 | 7/9 | 2/2 | 15/15 |
| Multiple fractures | 8/18 | 1/10 | 21/46 | na | na | 19/19 |
| Kypho/scoliosis | 8/24 | 7/32 | 32/46 | 1/7 | 10/12 | 12/13 |
| Platyspondyly | 1/20 | - | 13/36 | + | 5/9 | 18/18 |
| Peculiar fingers a | 25/25 | 1/30 | 13/36 | 7/7 | 19/20 | 9/12 |
| Foot deformity b | 22/25 | 9/10 | 24/46 | 8/8 | 9/13 | 12/12 |
| Pectus excavatum/carinatum | 3/19 | 1/1 | 2/10 | na | 10/12 | 4/4 |
| Radioulnar synostosis | 11/13 | 18/31 | 1/36 | - | na | na |
| Metaphyseal flaring | 3/5 | 4/8 | 23/46 | 4/6 | 4/4 | 0/1 |
| Monkey-wrench femora | 0/4 | 1/2 | na | na | 11/12 | na |
| Iliac abnormalities | 2/4 | - | 27/46 | - | 4/4 | 2/2 |
| Radial head subluxation or dislocation | 3/5 | 17/31 | 15/36 | 3/6 | 1/1 | na |
| Bowing of limbs | 5/19 | 7/32 | 13/46 | 8/8 | 5/5 | na |
| Advance bone age/carpal ossification | 0/10 | 1/32 | 5/16 | na | 13/14 | - |
| Craniofacial | ||||||
| Midface hypoplasia | 24/25 | na | 8/10 | 0/6 | 1/1 | na |
| Flat face | 1/4 | 29/32 | 22/36 | 1/1 | 18/18 | na |
| Craniosynostosis | 12/15 | 6/8 | 1/36 | - | na | na |
| Frontal bossing | 4/12 | - | 29/46 | + | na | na |
| Wide forehead | 2/5 | 29/32 | na | na | 1/1 | 1/1 |
| Blue sclerae | 8/17 | 6/10 | 30/46 | + | 5/6 | 2/6 |
| Proptosis or prominent eyes | 12/14 | 28/30 | 20/46 | + | 6/7 | na |
| Wide-spaced eyes | 3/7 | 28/32 | - | 1/1 | 1/1 | 4/4 |
| Low-set ears | 2/8 | 7/10 | 20/46 | na | na | 4/4 |
| Depressed nasal bridge | 13/20 | - | 10/46 | 1/1 | 21/21 | 4/4 |
| Small mouth/microstomia | 10/19 | 28/30 | - | - | 2/2 | na |
| Long upper lip/long philtrum | 4/4 | - | 15/36 | na | 6/6 | 1/1 |
| Cleft palate/bifid uvula | 4/4 | 4/31 | 6/46 | 3/8 | 7/16 | na |
| Micrognathia | 9/17 | 3/32 | 14/46 | - | 3/3 | na |
| Short and/or webbed neck | 13/19 | - | - | 8/8 | 5/5 | 4/4 |
| Abnormal dentition | 1/2 | 6/10 | 17/46 | 8/9 | 3/5 | 2/14 |
| Ocular | ||||||
| Refractive errors/hypermetropia | 3/5 | 12/29 | 1/10 | 1/9 | 1/5 | 14/14 |
| Clouded cornea | 2/4 | 1/30 | 1/46 | - | 0/5 | na |
| Cataract | 0/26 | 0/30 | na | na | 0/5 | 18/20 |
| Retinal detachment | 0/26 | Na | na | na | 0/5 | 9/16 |
| Cutaneous | ||||||
| Hyperextensible, soft, doughy, thin, translucent skin; cutis laxa | 4/15 | 30/32 | 29/46 | + | 2/2 | 1/5 |
| Atrophic scarring | 1/5 | 4/32 | 10/46 | 5/7 | na | na |
| Other | ||||||
| Cardiovascular abnormalities | 13/20 | - | 2/36 | - | 1/9 | 7/19 |
| Muscle hypotonia | 6/7 | 10/32 | 21/46 | 3/3 | 2/2 | 10/10 |
| Delayed motor development | 5/5 | 8/10 | 12/46 | 3/6 | 10/13 | 6/13 |
| Delayed cognitive development | 1/18 | 19/32 | 14/46 | 0/6 | 17/19 | 9/17 |
| Deafness | 1/3 | 2/32 | 2/46 | - | 2/8 | 12/20 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ritelli, M.; Cinquina, V.; Giacopuzzi, E.; Venturini, M.; Chiarelli, N.; Colombi, M. Further Defining the Phenotypic Spectrum of B3GAT3 Mutations and Literature Review on Linkeropathy Syndromes. Genes 2019, 10, 631. https://doi.org/10.3390/genes10090631
Ritelli M, Cinquina V, Giacopuzzi E, Venturini M, Chiarelli N, Colombi M. Further Defining the Phenotypic Spectrum of B3GAT3 Mutations and Literature Review on Linkeropathy Syndromes. Genes. 2019; 10(9):631. https://doi.org/10.3390/genes10090631
Chicago/Turabian StyleRitelli, Marco, Valeria Cinquina, Edoardo Giacopuzzi, Marina Venturini, Nicola Chiarelli, and Marina Colombi. 2019. "Further Defining the Phenotypic Spectrum of B3GAT3 Mutations and Literature Review on Linkeropathy Syndromes" Genes 10, no. 9: 631. https://doi.org/10.3390/genes10090631
APA StyleRitelli, M., Cinquina, V., Giacopuzzi, E., Venturini, M., Chiarelli, N., & Colombi, M. (2019). Further Defining the Phenotypic Spectrum of B3GAT3 Mutations and Literature Review on Linkeropathy Syndromes. Genes, 10(9), 631. https://doi.org/10.3390/genes10090631