Cellular and Molecular Mechanisms in the Pathogenesis of Classical, Vascular, and Hypermobile Ehlers‒Danlos Syndromes





Abstract
1. The Extracellular Matrix: An Overview
2. Pathological ECM Remodeling and Perturbation of Cellular Homeostasis
3. Ehlers‒Danlos Syndromes
4. Classical Ehlers‒Danlos Syndrome
5. Altered ECM Turnover, Wound Healing, and Inflammation in cEDS Fibroblasts
6. Perturbation of ER Homeostasis and Autophagy in cEDS Fibroblasts
7. Vascular Ehlers‒Danlos Syndrome
8. Disturbance of ECM Organization, Collagens Processing, and ER Homeostasis in vEDS Fibroblasts
9. Hypermobile Ehlers‒Danlos Syndrome
10. Pathological ECM Remodeling and Defective Cell-Cell Interactions in hEDS/HSD Cells
11. Differential Expression of Genes Involved in Inflammatory, Immune, and Pain Response in hEDS/HSD Cells
12. Emerging Aspects of hEDS/HSD Pathophysiology by microRNAs Profiling
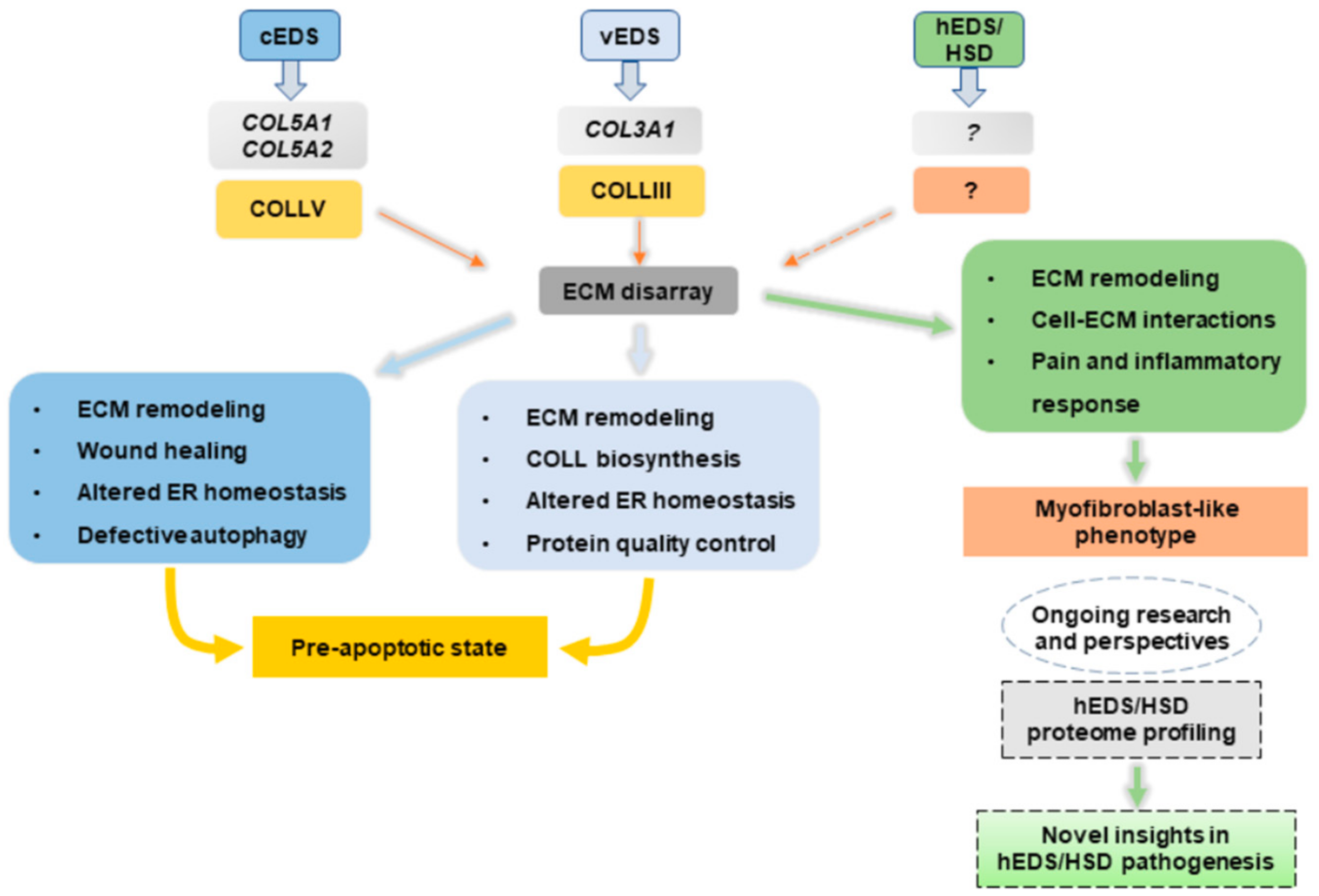
13. Conclusions and Perspectives
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Theocharis, A.D.; Manou, D.; Karamanos, N.K. The extracellular matrix as a multitasking player in disease. FEBS J. 2019. [Google Scholar] [CrossRef] [PubMed]
- Arseni, L.; Lombardi, A.; Orioli, D. From Structure to Phenotype: Impact of Collagen Alterations on Human Health. Int. J. Mol. Sci. 2018, 19, 5. [Google Scholar] [CrossRef] [PubMed]
- Murphy-Ullrich, J.E.; Sage, E.H. Revisiting the matricellular concept. Matrix Biol. 2014, 37, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Hynes, R.O.; Naba, A. Overview of the matrisome—An inventory of extracellular matrix constituents and functions. Cold Spring Harb. Perspect. Biol. 2012, 4, a004903. [Google Scholar] [CrossRef] [PubMed]
- Naba, A.; Clauser, K.R.; Ding, H.; Whittaker, C.A.; Carr, S.A.; Hynes, R.O. The extracellular matrix: Tools and insights for the “omics” era. Matrix Biol. 2016, 49, 10–24. [Google Scholar] [CrossRef] [PubMed]
- Myllyharju, J.; Kivirikko, K.I. Collagens, modifying enzymes and their mutations in humans, flies and worms. Trends Genet. 2004, 20, 33–43. [Google Scholar] [CrossRef] [PubMed]
- Nyström, A.; Bruckner-Tuderman, L. Matrix molecules and skin biology. Semin. Cell Dev. Biol. 2019, 89, 136–146. [Google Scholar] [CrossRef] [PubMed]
- Gjaltema, R.A.; Bank, R.A. Molecular insights into prolyl and lysyl hydroxylation of fibrillar collagens in health and disease. Crit. Rev. Biochem. Mol. Biol. 2017, 52, 74–95. [Google Scholar] [CrossRef] [PubMed]
- Sorushanova, A.; Delgado, L.M.; Wu, Z.; Shologu, N.; Kshirsagar, A.; Raghunath, R.; Mullen, A.M.; Bayon, Y.; Pandit, A.; Raghunath, M.; et al. The Collagen Suprafamily: From Biosynthesis to Advanced Biomaterial Development. Adv. Mater. 2019, 31, e1801651. [Google Scholar] [CrossRef] [PubMed]
- Malfait, F. Vascular aspects of the Ehlers-Danlos Syndromes. Matrix Biol. 2018, 71–72, 380–395. [Google Scholar] [CrossRef] [PubMed]
- Lenselink, E.A. Role of fibronectin in normal wound healing. Int. Wound J. 2015, 12, 313–316. [Google Scholar] [CrossRef] [PubMed]
- Iozzo, R.V.; Gubbiotti, M.A. Extracellular matrix: The driving force of mammalian diseases. Matrix Biol. 2018, 71–72, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Karamanos, N.K.; Theocharis, A.D.; Neill, T.; Iozzo, R.V. Matrix modeling and remodeling: A biological interplay regulating tissue homeostasis and diseases. Matrix Biol. 2019, 75–76, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Schwartz, M.A. Integrins and extracellular matrix in mechanotransduction. Cold Spring Harb. Perspect. Biol. 2010, 2, a005066. [Google Scholar] [CrossRef] [PubMed]
- Campbell, I.D.; Humphries, M.J. Integrin structure, activation, and interactions. Cold Spring Harb. Perspect. Biol. 2011, 3, a004994. [Google Scholar] [CrossRef] [PubMed]
- Frisch, S.M.; Francis, H. Disruption of epithelial cell matrix interactions induces apoptosis. J. Cell Biol. 1994, 124, 619–626. [Google Scholar] [CrossRef] [PubMed]
- Vlahakis, A.; Debnath, J. The Interconnections between Autophagy and Integrin-Mediated Cell Adhesion. J. Mol. Biol. 2017, 429, 515–530. [Google Scholar] [CrossRef]
- Lock, R.; Debnath, J. Extracellular matrix regulation of autophagy. Curr. Opin. Cell Biol. 2008, 20, 583–588. [Google Scholar] [CrossRef]
- Neill, T.; Schaefer, L.; Iozzo, R.V. Instructive roles of extracellular matrix on autophagy. Am. J. Pathol. 2014, 184, 2146–2153. [Google Scholar] [CrossRef]
- Settembre, C.; Cinque, L.; Bartolomeo, R.; Di Malta, C.; De Leonibus, C.; Forrester, A. Defective collagen proteostasis and matrix formation in the pathogenesis of lysosomal storage disorders. Matrix Biol. 2018, 71–72, 283–293. [Google Scholar] [CrossRef]
- Lamandé, S.R.; Bateman, J.F. Collagen VI disorders: Insights on form and function in the extracellular matrix and beyond. Matrix Biol. 2018, 71–72, 348–367. [Google Scholar] [CrossRef]
- Wilkinson, S. Emerging Principles of Selective ER Autophagy. J. Mol. Biol. 2019. [Google Scholar] [CrossRef]
- Bonnans, C.; Chou, J.; Werb, Z. Remodelling the extracellular matrix in development and disease. Nat. Rev. Mol. Cell Biol. 2014, 15, 786–801. [Google Scholar] [CrossRef]
- Frevert, C.W.; Felgenhauer, J.; Wygrecka, M.; Nastase, M.V.; Schaefer, L. Danger-Associated Molecular Patterns Derived From the Extracellular Matrix Provide Temporal Control of Innate Immunity. J. Histochem. Cytochem. 2018, 66, 213–227. [Google Scholar] [CrossRef]
- Genovese, F.; Karsdal, M.A. Protein degradation fragments as diagnostic and prognostic biomarkers of connective tissue diseases: Understanding the extracellular matrix message and implication for current and future serological biomarkers. Expert Rev. Proteom. 2016, 13, 213–225. [Google Scholar] [CrossRef]
- Bhattacharyya, S.; Midwood, K.S.; Yin, H.; Varga, J. Toll-Like Receptor-4 Signaling Drives Persistent Fibroblast Activation and Prevents Fibrosis Resolution in Scleroderma. Adv. Wound Care 2017, 6, 356–369. [Google Scholar] [CrossRef]
- Lamandé, S.R.; Bateman, J.F. Genetic Disorders of the Extracellular Matrix. Anat. Rec. 2019. [Google Scholar] [CrossRef]
- Colombi, M.; Dordoni, C.; Chiarelli, N.; Ritelli, M. Differential diagnosis and diagnostic flow chart of joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type compared to other heritable connective tissue disorders. Am. J. Med. Genet. Part C 2015, 169, 6–22. [Google Scholar] [CrossRef]
- Bateman, J.F.; Boot-Handford, R.P.; Lamande, S.R. Genetic diseases of connective tissues: Cellular and extra cellular effects of ECM mutations. Nat. Rev. Genet. 2009, 10, 173–183. [Google Scholar] [CrossRef]
- Lu, P.; Takai, K.; Weaver, V.M.; Werb, Z. Extracellular matrix degradation and remodeling in development and disease. Cold Spring Harb. Perspect. Biol. 2011, 3, a005058. [Google Scholar] [CrossRef]
- Karsdal, M.A.; Nielsen, M.J.; Sand, J.M.; Henriksen, K.; Genovese, F.; Bay-Jensen, A.C.; Smith, V.; Adamkewicz, J.I.; Christiansen, C.; Leeming, D.J. Extracellular matrix remodeling: The common denominator in connective tissue diseases. Possibilities for evaluation and current understanding of the matrix as more than a passive architecture, but a key player in tissue failure. ASSAY Drug Dev. Technol. 2013, 11, 70–92. [Google Scholar] [CrossRef]
- Malfait, F.; Francomano, C.; Byers, P.; Belmont, J.; Berglund, B.; Black, J.; Bloom, L.; Bowen, J.M.; Brady, A.F.; Burrows, N.P.; et al. The 2017 international classification of the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 8–26. [Google Scholar] [CrossRef]
- Blackburn, P.R.; Xu, Z.; Tumelty, K.E.; Zhao, R.W.; Monis, W.J.; Harris, K.G.; Gass, J.M.; Cousin, M.A.; Boczek, N.J.; Mitkov, M.V.; et al. Bi-allelic alterations in AEBP1 lead to defective collagen assembly and connective tissue structure resulting in a variant of Ehlers-Danlos syndrome. Am. J. Hum. Genet. 2018, 102, 696–705. [Google Scholar] [CrossRef]
- Hebebrand, M.; Vasileiou, G.; Krumbiegel, M.; Kraus, C.; Uebe, S.; Ekici, A.B.; Thiel, C.T.; Reis, A.; Popp, B. A biallelic truncating AEBP1 variant causes connective tissue disorder in two siblings. Am. J. Med. Genet. A 2019, 179, 50–56. [Google Scholar] [CrossRef]
- Syx, D.; De Wandele, I.; Symoens, S.; De Rycke, R.; Hougrand, O.; Voermans, N.; De Paepe, A.; Malfait, F. Bi-allelic AEBP1 mutations in two patients with Ehlers-Danlos syndrome. Hum. Mol. Genet. 2019, 28, 1853–1864. [Google Scholar] [CrossRef]
- Ritelli, M.; Cinquina, V.; Venturini, M.; Pezzaioli, L.; Formenti, A.M.; Chiarelli, N.; Colombi, M. Expanding the Clinical and Mutational Spectrum of Recessive AEBP1-Related Classical-Like Ehlers-Danlos Syndrome. Genes. 2019, 10, 135. [Google Scholar] [CrossRef]
- Maugeri, A.; De Paepe, A.; Malfait, F. Genetics and testing of Ehlers-Danlos syndrome and of differential diagnostic diseases. In Ehlers-Danlos Syndrome: A Multidisciplinary Approach, 1st ed.; Jacobs, J.W.G., Cornelissens, L.J.M., Eds.; IOS Press BV: Amsterdam, The Netherlands, 2018; Chapter 3; pp. 33–46. [Google Scholar]
- Damme, T.M.; Syx, D.; Coucke, P.; Symoens, S.; De Paepe, A.; Malfait, F. Genetics of the Ehlers–Danlos syndrome: More than collagen disorders. Expert Opin. Orphan Drugs 2015, 3, 379–392. [Google Scholar] [CrossRef]
- Bowen, J.M.; Sobey, G.J.; Burrows, N.P.; Colombi, M.; Lavallee, M.E.; Malfait, F.; Francomano, C.A. Ehlers-Danlos syndrome, classical type. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 75, 27–39. [Google Scholar] [CrossRef]
- Symoens, S.; Syx, D.; Malfait, F.; Callewaert, B.; De Backer, J.; Vanakker, O.; Coucke, P.; De Paepe, A. Comprehensive molecular analysis demonstrates type V collagen mutations in over 90% of patients with classic EDS and allows to refine diagnostic criteria. Hum. Mutat. 2012, 33, 1485–1493. [Google Scholar] [CrossRef]
- Ritelli, M.; Dordoni, C.; Venturini, M.; Chiarelli, N.; Quinzani, S.; Traversa, M.; Zoppi, N.; Vascellaro, A.; Wischmeijer, A.; Manfredini, E.; et al. Clinical and molecular characterization of 40 patients with classic Ehlers-Danlos syndrome: Identification of 18 COL5A1 and 2 COL5A2 novel mutations. Orphanet J. Rare Dis. 2013, 8, 58. [Google Scholar] [CrossRef]
- Wenstrup, R.J.; Florer, J.B.; Brunskill, E.W.; Bell, S.M.; Chervoneva, I.; Birk, D.E. Type V collagen controls the initiation of collagen fibril assembly. J. Biol. Chem. 2004, 279, 53331–53337. [Google Scholar] [CrossRef]
- Mak, K.M.; Png, C.Y.; Lee, D.J. Type V Collagen in Health, Disease, and Fibrosis. Anat. Rec. 2016, 299, 613–629. [Google Scholar] [CrossRef]
- Zoppi, N.; Gardella, R.; De Paepe, A.; Barlati, S.; Colombi, M. Human fibroblasts with mutations in COL5A1 and COL3A1 genes do not organize collagens and fibronectin in the extracellular matrix, down-regulate α2β1 integrin, and recruit αvβ3 instead of α5β1 integrin. J. Biol. Chem. 2004, 30, 18157–18168. [Google Scholar] [CrossRef]
- Zoppi, N.; Barlati, S.; Colombi, M. FAK-independent αvβ3 integrin-EGFR complexes rescue from anoikis matrix-defective fibroblasts. Biochim. Biophys. Acta 2008, 1783, 1177–1188. [Google Scholar] [CrossRef]
- Zoppi, N.; Ritelli, M.; Colombi, M. Type III and V collagens modulate the expression and assembly of EDA+fibronectin in the extracellular matrix of defective Ehlers-Danlos syndrome fibroblasts. Biochim. Biophys. Acta 2012, 1820, 1576–1587. [Google Scholar] [CrossRef]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Ritelli, M.; Colombi, M. Molecular insights in the pathogenesis of classical Ehlers-Danlos syndrome from transcriptome-wide expression profiling of patients’ skin fibroblasts. PLoS ONE 2019, 14, e0211647. [Google Scholar] [CrossRef]
- Viglio, S.; Zoppi, N.; Sangalli, A.; Gallanti, A.; Barlati, S.; Mottes, M.; Colombi, M.; Valli, M. Rescue of migratory defects of Ehlers-Danlos syndrome fibroblasts in vitro by type V collagen but not insulin-like binding protein-1. J. Investig. Dermatol. 2008, 128, 1915–1919. [Google Scholar] [CrossRef]
- Zoppi, N.; Chiarelli, N.; Binetti, S.; Ritelli, M.; Colombi, M. Dermal fibroblast-to-myofibroblast transition sustained by αvβ3 integrin-ILK-Snail1/Slug signaling is a common feature for hypermobile Ehlers-Danlos syndrome and hypermobility spectrum disorders. Biochim. Biophys. Acta 2018, 1864, 1010–1023. [Google Scholar] [CrossRef]
- Zoppi, N.; Chiarelli, N.; Ritelli, M.; Colombi, M. Multifaced Roles of the αvβ3 Integrin in Ehlers-Danlos and Arterial Tortuosity Syndromes’ Dermal Fibroblasts. Int. J. Mol. Sci. 2018, 19, 982. [Google Scholar] [CrossRef]
- DeNigris, J.; Yao, Q.; Birk, E.K.; Birk, D.E. Altered dermal fibroblast behavior in a collagen V haploinsufficient murine model of classic Ehlers-Danlos syndrome. Connect. Tissue Res. 2016, 57, 1–9. [Google Scholar] [CrossRef]
- Park, A.C.; Phan, N.; Massoudi, D.; Liu, Z.; Kernien, J.F.; Adams, S.M.; Davidson, J.M.; Birk, D.E.; Liu, B.; Greenspan, D.S. Deficits in Col5a2 Expression Result in Novel Skin and Adipose Abnormalities and Predisposition to Aortic Aneurysms and Dissections. Am. J. Pathol. 2017, 187, 2300–2311. [Google Scholar] [CrossRef]
- Rousselle, P.; Montmasson, M.; Garnier, C. Extracellular matrix contribution to skin wound re-epithelialization. Matrix Biol. 2019, 75–76, 12–26. [Google Scholar] [CrossRef]
- Tracy, L.E.; Minasian, R.A.; Caterson, E.J. Extracellular Matrix and Dermal Fibroblast Function in the Healing Wound. Adv. Wound Care 2016, 5, 119–136. [Google Scholar] [CrossRef]
- Chester, D.; Brown, A.C. The role of biophysical properties of provisional matrix proteins in wound repair. Matrix Biol. 2017, 60–61, 124–140. [Google Scholar] [CrossRef]
- Wang, W.; Li, P.; Li, W.; Jiang, J.; Cui, Y.; Li, S.; Wang, Z. Osteopontin activates mesenchymal stem cells to repair skin wound. PLoS ONE 2017, 12, e0185346. [Google Scholar] [CrossRef]
- Icer, M.A.; Gezmen-Karadag, M. The multiple functions and mechanisms of osteopontin. Clin. Biochem. 2018, 59, 17–24. [Google Scholar] [CrossRef]
- Ross, F.P.; Chappel, J.; Alvarez, J.; Sander, D.; Butler, W.; Farach-Carson, M.; Mintz, K.; Robey, P.G.; Teitelbaum, S.; Cheresh, D. Interactions between the bone matrix proteins osteopontin and bone sialoprotein and the osteoclast integrin alpha v beta 3 potentiate bone resorption. J. Biol. Chem. 1993, 268, 9901–9907. [Google Scholar]
- Giachelli, C.M.; Steitz, S. Osteopontin: A versatile regulator of inflammation and biomineralization. Matrix Biol. 2000, 19, 615–622. [Google Scholar] [CrossRef]
- Liaw, L.; Birk, D.E.; Ballas, C.B.; Whitsitt, J.S.; Davidson, J.M.; Hogan, B.L. Altered wound healing in mice lacking a functional osteopontin gene (spp1). J. Clin. Investig. 1998, 101, 1468–1478. [Google Scholar] [CrossRef]
- Maruhashi, T. Interaction between periostin and BMP-1 promotes proteolytic activation of lysyl oxidase. J. Biol. Chem. 2010, 285, 13294–13303. [Google Scholar] [CrossRef]
- Egbert, M.; Ruetze, M.; Sattler, M.; Wenck, H.; Gallinat, S.; Lucius, R.; Weise, J.M. The matricellular protein periostin contributes to proper collagen function and is downregulated during skin aging. J. Dermatol. Sci. 2014, 73, 40–48. [Google Scholar] [CrossRef]
- González-González, L.; Alonso, J. Periostin: A Matricellular Protein with Multiple Functions in Cancer Development and Progression. Front. Oncol. 2018, 8, 225. [Google Scholar] [CrossRef]
- Li, G.; Jin, R.; Norris, R.A.; Zhang, L.; Yu, S.; Wu, F.; Markwald, R.R.; Nanda, A.; Conway, S.J.; Smyth, S.S.; et al. Periostin mediates vascular smooth muscle cell migration through the integrins alphavbeta3 and alphavbeta5 and focal adhesion kinase (FAK) pathway. Atherosclerosis 2010, 208, 358–365. [Google Scholar] [CrossRef]
- Walker, J.T.; McLeod, K.; Kim, S.; Conway, S.J.; Hamilton, D.W. Periostin as a multifunctional modulator of the wound healing response. Cell Tissue Res. 2016, 365, 453–465. [Google Scholar] [CrossRef]
- Nunomura, S.; Nanri, Y.; Ogawa, M.; Arima, K.; Mitamura, Y.; Yoshihara, T.; Hasuwa, H.; Conway, S.J.; Izuhara, K. Constitutive overexpression of periostin delays wound healing in mouse skin. Wound Repair Regen. 2018, 26, 6–15. [Google Scholar] [CrossRef]
- Lim, P.J.; Lindert, U.; Opitz, L.; Hausser, I.; Rohrbach, M.; Giunta, C. Transcriptome Profiling of Primary Skin Fibroblasts Reveal Distinct Molecular Features Between PLOD1-and FKBP14-Kyphoscoliotic Ehlers-Danlos Syndrome. Genes 2019, 10, 517. [Google Scholar] [CrossRef]
- Penta, K.; Varner, J.A.; Liaw, L.; Hidai, C.; Schatzman, R.; Quertermous, T. Del1 induces integrin signaling and angiogenesis by ligation of alphaVbeta3. J. Biol. Chem. 1999, 274, 11101–11109. [Google Scholar] [CrossRef]
- Shen, W.; Zhu, S.; Qin, H.; Zhong, M.; Wu, J.; Zhang, R.; Song, H. EDIL3 knockdown inhibits retinal angiogenesis through the induction of cell cycle arrest in vitro. Mol. Med. Rep. 2017, 16, 4054–4060. [Google Scholar] [CrossRef]
- Wang, Z.; Boyko, T.; Tran, M.C.; LaRussa, M.; Bhatia, N.; Rashidi, V.; Longaker, M.T.; Yang, G.P. DEL1 protects against chondrocyte apoptosis through integrin binding. J. Surg. Res. 2018, 231, 1–9. [Google Scholar] [CrossRef]
- Hajishengallis, G.; Chavakis, T. DEL-1-Regulated Immune Plasticity and Inflammatory Disorders. Trends Mol. Med. 2019, 25, 444–459. [Google Scholar] [CrossRef]
- Brandt, K.; Grunler, J.; Brismar, K.; Wang, J. Effects of IGFBP-1 and IGFBP-2 and their fragments on migration and IGF-induced proliferation of human dermal fibroblasts. Growth Horm. IGF Res. 2015, 25, 34–40. [Google Scholar] [CrossRef]
- Katayama, H.; Tamai, K.; Shibuya, R.; Nakamura, M.; Mochizuki, M.; Yamaguchi, K.; Kawamura, S.; Tochigi, T.; Sato, I.; Okanishi, T.; et al. Long non-coding RNA HOTAIR promotes cell migration by upregulating insulin growth factor-binding protein 2 in renal cell carcinoma. Sci. Rep. 2017, 7, 12016. [Google Scholar] [CrossRef]
- Ahmad, S.; Bhatia, K.; Kindelin, A.; Ducruet, A.F. The Role of Complement C3a Receptor in Stroke. Neuromol. Med. 2019. [Google Scholar] [CrossRef]
- Korkmaz, H.I.; Krijnen, P.A.J.; Ulrich, M.M.W.; de Jong, E.; van Zuijlen, P.P.M.; Niessen, H.W.M. The role of complement in the acute phase response after burns. Burns 2017, 43, 1390–1399. [Google Scholar] [CrossRef]
- Rafail, S.; Kourtzelis, I.; Foukas, P.G.; Markiewski, M.M.; DeAngelis, R.A.; Guariento, M.; Ricklin, D.; Grice, E.A.; Lambris, J.D. Complement deficiency promotes cutaneous wound healing in mice. J. Immunol. 2015, 194, 1285–1291. [Google Scholar] [CrossRef]
- Colombi, M.; Dordoni, C.; Venturini, M.; Ciaccio, C.; Morlino, S.; Chiarelli, N.; Zanca, A.; Calzavara-Pinton, P.; Zoppi, N.; Castori, M.; et al. Spectrum of mucocutaneous, ocular and facial features and delineation of novel presentations in 62 classical Ehlers-Danlos syndrome patients. Clin. Genet. 2017, 92, 624–631. [Google Scholar] [CrossRef]
- Smith, M.; Wilkinson, S. ER homeostasis and autophagy. Essays Biochem. 2017, 61, 625–635. [Google Scholar] [CrossRef]
- Wong, M.Y.; Shoulders, M.D. Targeting defective proteostasis in the collagenopathies. Curr. Opin. Chem. Biol. 2019, 50, 80–88. [Google Scholar] [CrossRef]
- Moon, H.W.; Han, H.G.; Jeon, Y.J. Protein Quality Control in the Endoplasmic Reticulum and Cancer. Int. J. Mol. Sci. 2018, 19, 3020. [Google Scholar] [CrossRef]
- Mogk, A.; Bukau, B.; Kampinga, H.H. Cellular Handling of Protein Aggregates by Disaggregation Machines. Mol. Cell 2018, 69, 214–226. [Google Scholar] [CrossRef]
- Wilkinson, S. ER-phagy: Shaping up and destressing the endoplasmic reticulum. FEBS J. 2019. [Google Scholar] [CrossRef]
- Smith, M.D.; Harley, M.E.; Kemp, A.J.; Wills, J.; Lee, M.; Arends, M.; von Kriegsheim, A.; Behrends, C.; Wilkinson, S. CCPG1 is a non-canonical autophagy cargo receptor essential for ER-phagy and pancreatic ER proteostasis. Dev. Cell 2018, 44, 217–232. [Google Scholar] [CrossRef]
- Forrester, A.; De Leonibus, C.; Grumati, P.; Fasana, E.; Piemontese, M.; Staiano, L.; Fregno, I.; Raimondi, A.; Marazza, A.; Bruno, G.; et al. A selective ER-phagy exerts procollagen quality control via a Calnexin-FAM134B complex. EMBO J. 2019, 38, e99847. [Google Scholar] [CrossRef]
- Ishida, Y.; Yamamoto, A.; Kitamura, A.; Lamandé, S.R.; Yoshimori, T.; Bateman, J.F.; Kubota, H.; Nagata, K. Autophagic elimination of misfolded procollagen aggregates in the endoplasmic reticulum as a means of cell protection. Mol. Biol. Cell 2009, 20, 2744–2754. [Google Scholar] [CrossRef]
- Nagahama, M.; Suzuki, M.; Hamada, Y.; Hatsuzawa, K.; Tani, K.; Yamamoto, A.; Tagaya, M. SVIP is a novel VCP/p97-interacting protein whose expression causes cell vacuolation. Mol. Biol. Cell 2003, 14, 262–273. [Google Scholar] [CrossRef]
- Ballar, P.; Zhong, Y.; Nagahama, M.; Tagaya, M.; Shen, Y.; Fang, S. Identification of SVIP as an endogenous inhibitor of endoplasmic reticulum-associated degradation. J. Biol. Chem. 2007, 282, 33908–33914. [Google Scholar] [CrossRef]
- Wang, Y.; Ballar, P.; Zhong, Y.; Zhang, X.; Liu, C.; Zhang, Y.J.; Monteiro, M.J.; Li, J.; Fang, S. SVIP induces localization of p97/VCP to the plasma and lysosomal membranes and regulates autophagy. PLoS ONE 2011, 6, e24478. [Google Scholar] [CrossRef]
- Jia, D.; Wang, Y.Y.; Wang, P.; Huang, Y.; Liang, D.Y.; Wang, D.; Cheng, C.; Zhang, C.; Guo, L.; Liang, P.; et al. SVIP alleviates CCl4-induced liver fibrosis via activating autophagy and protecting hepatocytes. Cell Death Dis. 2019, 10, 71. [Google Scholar] [CrossRef]
- Fung, C.; Lock, R.; Gao, S.; Salas, E.; Debnath, J. Induction of autophagy during extracellular matrix detachment promotes cell survival. Mol. Biol. Cell 2008, 19, 797–806. [Google Scholar] [CrossRef]
- Kawano, S.; Torisu, T.; Esaki, M.; Torisu, K.; Matsuno, Y.; Kitazono, T. Autophagy promotes degradation of internalized collagen and regulates distribution of focal adhesions to suppress cell adhesion. Biol. Open 2017, 6, 1644–1653. [Google Scholar] [CrossRef]
- Byers, P.H.; Belmont, J.; Black, J.; De Backer, J.; Frank, M.; Jeunemaitre, X.; Johnson, D.; Pepin, M.; Robert, L.; Sanders, L.; et al. Diagnosis, natural history, and management in vascular Ehlers-Danlos syndrome. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 40–47. [Google Scholar] [CrossRef]
- Ritelli, M.; Rovati, C.; Venturini, M.; Chiarelli, N.; Cinquina, V.; Castori, M.; Colombi, M. Application of the 2017 criteria for vascular Ehlers-Danlos syndrome in 50 patients ascertained according to the Villefranche nosology. Clin. Genet. under review.
- Pepin, M.; Schwarze, U.; Superti-Furga, A.; Byers, P.H. Clinical and genetic features of Ehlers-Danlos syndrome type IV, the vascular type. N. Engl. J. Med. 2000, 342, 673–680. [Google Scholar] [CrossRef]
- Smith, L.T.; Schwarze, U.; Goldstein, J.; Byers, P.H. Mutations in the COL3A1 gene result in the Ehlers-Danlos syndrome type IV and alterations in the size and distribution of the major collagen fibrils of the dermis. J. Investig. Dermatol. 1997, 108, 241–247. [Google Scholar] [CrossRef]
- Liu, X.; Wu, H.; Byrne, M.; Krane, S.; Jaenisch, R. Type III collagen is crucial for collagen I fibrillogenesis and for normal cardiovascular development. Proc. Natl. Acad. Sci. USA 1997, 94, 1852–1856. [Google Scholar] [CrossRef]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Ritelli, M.; Colombi, M. Transcriptome analysis of skin fibroblasts with dominant negative COL3A1 mutations provides molecular insights into the etiopathology of vascular Ehlers-Danlos syndrome. PLoS ONE 2018, 13, e0191220. [Google Scholar] [CrossRef]
- Sengle, G.; Sakai, L.Y. The fibrillin microfibril scaffold: A niche for growth factors and mechanosensation? Matrix Biol. 2015, 47, 3–12. [Google Scholar] [CrossRef]
- Schiavinato, A.; Keene, D.R.; Wohl, A.P.; Corallo, D.; Colombatti, A.; Wagener, R.; Paulsson, M.; Bonaldo, P.; Sengle, G. Targeting of EMILIN-1 and EMILIN-2 to fibrillin microfibrils facilitates their incorporation into the extracellular matrix. J. Investig. Dermatol. 2016, 136, 1150–1160. [Google Scholar] [CrossRef]
- Boregowda, R.; Paul, E.; White, J.; Ritty, T.M. Bone and soft connective tissue alterations result from loss of fibrillin-2 expression. Matrix Biol. 2008, 27, 661–666. [Google Scholar] [CrossRef]
- Brinckmann, J.; Hunzelmann, N.; Kahle, B.; Rohwedel, J.; Kramer, J.; Gibson, M.A.; Hubmacher, D.; Reinhardt, D.P. Enhanced fibrillin-2 expression is a general feature of wound healing and sclerosis: Potential alteration of cell attachment and storage of TGF-beta. Lab. Investig. 2010, 90, 739–752. [Google Scholar] [CrossRef]
- D’Hondt, S.; Guillemyn, B.; Syx, D.; Symoens, S.; De Rycke, R.; Vanhoutte, L.; Toussaint, W.; Lambrecht, B.N.; De Paepe, A.; Keene, D.R.; et al. Type III collagen affects dermal and vascular collagen fibrillogenesis and tissue integrity in a mutant Col3a1 transgenic mouse model. Matrix Biol. 2018, 70, 72–83. [Google Scholar] [CrossRef]
- Ishikawa, Y.; Bächinger, H.P. A substrate preference for the rough endoplasmic reticulum resident protein FKBP22 during collagen biosynthesis. J. Biol. Chem. 2014, 289, 18189–18201. [Google Scholar] [CrossRef]
- Baumann, M.; Giunta, C.; Krabichler, B.; Rüschendorf, F.; Zoppi, N.; Colombi, M.; Bittner, R.E.; Quijano-Roy, S.; Muntoni, F.; Cirak, S.; et al. Mutations in FKBP14 cause a variant of Ehlers-Danlos syndrome with progressive kyphoscoliosis, myopathy, and hearing loss. Am. J. Hum. Genet. 2012, 90, 201–216. [Google Scholar] [CrossRef]
- Soares Moretti, A.; Martins Laurindo, F.R. Protein disulfide isomerases: Redox connections in and out of the endoplasmic reticulum. Arch. Biochem. Biophys. 2017, 617, 106–119. [Google Scholar] [CrossRef]
- Szegezdi, E.; Macdonald, D.C.; Ni Chonghaile, T.; Gupta, S.; Samali, A. Bcl-2 family on guard at the ER. Am. J. Physiol.-Cell Physiol. 2009, 296, C941–C953. [Google Scholar] [CrossRef]
- Sisinni, L.; Pietrafesa, M.; Lepore, S.; Maddalena, F.; Condelli, V.; Esposito, F.; Landriscina, M. Endoplasmic Reticulum Stress and Unfolded Protein Response in Breast Cancer: The Balance between Apoptosis and Autophagy and Its Role in Drug Resistance. Int. J. Mol. Sci. 2019, 20, 857. [Google Scholar] [CrossRef]
- Boot-Handford, R.P.; Briggs, M.D. The unfolded protein response and its relevance to connective tissue diseases. Cell Tissue Res. 2010, 339, 197–211. [Google Scholar] [CrossRef]
- Hughes, A.; Oxford, A.E.; Tawara, K.; Jorcyk, C.L.; Oxford, J.T. Endoplasmic Reticulum Stress and Unfolded Protein Response in Cartilage Pathophysiology; Contributing Factors to Apoptosis and Osteoarthritis. Int. J. Mol. Sci. 2017, 18, 665. [Google Scholar] [CrossRef]
- Tinkle, B.; Castori, M.; Berglund, B.; Cohen, H.; Grahame, R.; Kazkaz, H.; Levy, H. Hypermobile Ehlers-Danlos syndrome (a.k.a. Ehlers-Danlos syndrome type III and Ehlers-Danlos syndrome hypermobility type): Clinical description and natural history. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 48–69. [Google Scholar] [CrossRef]
- Castori, M.; Tinkle, B.; Levy, H.; Grahame, R.; Malfait, F.; Hakim, A. A framework for the classification of joint hypermobility and related conditions. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 148–157. [Google Scholar] [CrossRef]
- Schalkwijk, J.; Zweers, M.C.; Steijlen, P.M.; Dean, W.B.; Taylor, G.; van Vlijmen, I.M.; van Haren, B.; Miller, W.L.; Bristow, J. A recessive form of the Ehlers-Danlos syndrome caused by tenascin-X deficiency. N. Engl. J. Med. 2001, 345, 1167–1175. [Google Scholar] [CrossRef]
- Merke, D.P.; Chen, W.; Morissette, R.; Xu, Z.; Van Ryzin, C.; Sachdev, V.; Hannoush, H.; Shanbhag, S.M.; Acevedo, A.T.; Nishitani, M.; et al. Tenascin-X haploinsufficiency associated with Ehlers-Danlos syndrome in patients with congenital adrenal hyperplasia. J. Clin. Endocrinol. Metab. 2013, 98, 379–387. [Google Scholar] [CrossRef]
- Morissette, R.; Chen, W.; Perritt, A.F.; Dreiling, J.L.; Arai, A.E.; Sachdev, V.; Hannoush, H.; Mallappa, A.; Xu, Z.; McDonnell, N.B.; et al. Broadening the Spectrum of Ehlers Danlos Syndrome in Patients with Congenital Adrenal Hyperplasia. J. Clin. Endocrinol. Metab. 2015, 100, 1143–1152. [Google Scholar] [CrossRef]
- Syx, D.; Symoens, S.; Steyaert, W.; De Paepe, A.; Coucke, P.J.; Malfait, F. Ehlers-Danlos Syndrome, Hypermobility Type, Is Linked to Chromosome 8p22-8p21.1 in an Extended Belgian Family. Dis. Markers 2015, 2015, 828970. [Google Scholar] [CrossRef]
- Copetti, M.; Morlino, S.; Colombi, M.; Grammatico, P.; Fontana, A.; Castori, M. Severity classes in adults with hypermobile Ehlers-Danlos syndrome/hypermobility spectrum disorders: A pilot study of 105 Italian patients. Rheumatology 2019, kez029. [Google Scholar] [CrossRef]
- Castori, M.; Dordoni, C.; Valiante, M.; Sperduti, I.; Ritelli, M.; Morlino, S.; Chiarelli, N.; Celletti, C.; Venturini, M.; Camerota, F.; et al. Nosology and inheritance pattern(s) of joint hypermobility syndrome and Ehlers-Danlos syndrome, hypermobility type: A study of intrafamilial and interfamilial variability in 23 Italian pedigrees. Am. J. Med. Genet. A 2014, 164, 3010–3020. [Google Scholar] [CrossRef]
- Ritelli, M.; Chiarelli, N.; Zoppi, N.; Dordoni, C.; Quinzani, S.; Traversa, M.; Venturini, M.; Calzavara-Pinton, P.; Colombi, M. Insights in the etiopathology of galactosyltransferase II (GalT-II) deficiency from transcriptome-wide expression profiling of skin fibroblasts of two sisters with compound heterozygosity for two novel B3GALT6 mutations. Mol. Genet. Metab. Rep. 2014, 20, 1–15. [Google Scholar] [CrossRef]
- Zoppi, N.; Chiarelli, N.; Cinquina, V.; Ritelli, M.; Colombi, M. GLUT10 deficiency leads to oxidative stress and non-canonical αvβ3 integrin-mediated TGFβ signaling associated with extracellular matrix disarray in arterial tortuosity syndrome skin fibroblasts. Hum. Mol. Genet. 2015, 24, 6769–6787. [Google Scholar] [CrossRef]
- Manzoni, C.; Kia, D.A.; Vandrovcova, J.; Hardy, J.; Wood, N.W.; Lewis, P.A.; Ferrari, R. Genome, transcriptome and proteome: The rise of omics data and their integration in biomedical sciences. Brief. Bioinform. 2018, 19, 286–302. [Google Scholar] [CrossRef]
- Casamassimi, A.; Federico, A.; Rienzo, M.; Esposito, S.; Ciccodicola, A. Transcriptome Profiling in Human Diseases: New Advances and Perspectives. Int. J. Mol. Sci. 2017, 18, 1652. [Google Scholar] [CrossRef]
- Ideozu, J.E.; Zhang, X.; McColley, S.; Levy, H. Transcriptome Profiling and Molecular Therapeutic Advances in Cystic Fibrosis: Recent Insights. Genes 2019, 10, 180. [Google Scholar] [CrossRef]
- Chiarelli, N.; Carini, G.; Zoppi, N.; Dordoni, C.; Ritelli, M.; Venturini, M.; Castori, M.; Colombi, M. Transcriptome-wide expression profiling in skin fibroblasts of patients with joint hypermobility syndrome/Ehlers-Danlos syndrome hypermobility type. PLoS ONE 2016, 11, e0161347. [Google Scholar] [CrossRef]
- Jansen, K.A.; Atherton, P.; Ballestrem, C. Mechanotransduction at the cell-matrix interface. Semin. Cell Dev. Biol. 2017, 71, 75–83. [Google Scholar] [CrossRef]
- Freedman, B.R.; Bade, N.D.; Riggin, C.N.; Zhang, S.; Haines, P.G.; Ong, K.L.; Janmey, P.A. The (dys) functional extracellular matrix. Biochim. Biophys. Acta 2015, 1853, 3153–3164. [Google Scholar] [CrossRef]
- Kalluri, R.; Neilson, E.R. Epithelial-mesenchymal transition and its implications for fibrosis. J. Clin. Investig. 2003, 112, 1776–1784. [Google Scholar] [CrossRef]
- Lamouille, S.; Xu, J.; Derynck, R. Molecular mechanisms of epithelial-mesenchymal transition. Nat. Rev. Mol. Cell Biol. 2014, 15, 178–196. [Google Scholar] [CrossRef]
- McCrea, P.D.; Maher, M.T.; Gottardi, C.J. Nuclear signaling from cadherin adhesion complexes. Curr. Top. Dev. Biol. 2015, 112, 129–196. [Google Scholar]
- Hinz, B.; Gabbiani, G. Cell-matrix and cell-cell contacts of myofibroblasts: Role in connective tissue remodeling. Thromb. Haemost. 2003, 90, 993–1002. [Google Scholar]
- Michalik, M.; Wójcik-Pszczoła, K.; Paw, M.; Wnuk, D.; Koczurkiewicz, P.; Sanak, M.; Pękala, E.; Madeja, Z. Fibroblast-to-myofibroblast transition in bronchial asthma. Cell. Mol. Life Sci. 2018, 75, 3943–3961. [Google Scholar] [CrossRef]
- Hinz, B.; McCulloch, C.A.; Coelho, N.M. Mechanical regulation of myofibroblast phenoconversion and collagen contraction. Exp. Cell Res. 2019, 379, 119–128. [Google Scholar] [CrossRef]
- Pakshir, P.; Hinz, B. The big five in fibrosis: Macrophages, myofibroblasts, matrix, mechanics, and miscommunication. Matrix Biol. 2018, 68–69, 81–93. [Google Scholar] [CrossRef]
- Mullenbrock, S.; Liu, F.; Szak, S.; Hronowski, X.; Gao, B.; Juhasz, P.; Sun, C.; Liu, M.; McLaughlin, H.; Xiao, Q.; et al. Systems Analysis of Transcriptomic and Proteomic Profiles Identifies Novel Regulation of Fibrotic Programs by miRNAs in Pulmonary Fibrosis Fibroblasts. Genes 2018, 9, 588. [Google Scholar] [CrossRef]
- Van Caam, A.; Vonk, M.; van den Hoogen, F.; van Lent, P.; van der Kraan, P. Unraveling SSc Pathophysiology; The Myofibroblast. Front. Immunol. 2018, 9, 2452. [Google Scholar] [CrossRef]
- Watanabe, T.; Baker Frost, D.A.; Mlakar, L.; Heywood, J.; da Silveira, W.A.; Hardiman, G.; Feghali-Bostwick, C. A Human Skin Model Recapitulates Systemic Sclerosis Dermal Fibrosis and Identifies COL22A1 as a TGFβ Early Response Gene that Mediates Fibroblast to Myofibroblast Transition. Genes 2019, 10, 75. [Google Scholar] [CrossRef]
- Działo, E.; Tkacz, K.; Błyszczuk, P. Crosstalk between the TGF-β and WNT signalling pathways during cardiac fibrogenesis. Acta Biochim. Pol. 2018, 65, 341–349. [Google Scholar] [CrossRef]
- Cruciat, C.M.; Niehrs, C. Secreted and transmembrane wnt inhibitors and activators. Cold Spring Harb. Perspect. Biol. 2013, 5, a015081. [Google Scholar] [CrossRef]
- Carthy, J.M.; Garmaroudi, F.S.; Luo, Z.; McManus, B.M. Wnt3a induces myofibroblast differentiation by upregulating TGF-β signaling through SMAD2 in a β-catenin-dependent manner. PLoS ONE 2011, 6, e19809. [Google Scholar] [CrossRef]
- Blyszczuk, P.; Müller-Edenborn, B.; Valenta, T.; Osto, E.; Stellato, M.; Behnke, S.; Glatz, K.; Basler, K.; Lüscher, T.F.; Distler, O.; et al. Transforming growth factor-β-dependent Wnt secretion controls myofibroblast formation and myocardial fibrosis progression in experimental autoimmune myocarditis. Eur. Heart J. 2017, 38, 1413–1425. [Google Scholar] [CrossRef]
- Abonia, J.P.; Wen, T.; Stucke, E.M.; Grotjan, T.; Griffith, M.S.; Kemme, K.A.; Collins, M.H.; Putnam, P.E.; Franciosi, J.P.; Von Tiehl, K.F.; et al. High prevalence of eosinophilic esophagitis in patients with inherited connective tissue disorders. J. Allergy Clin. Immunol. 2013, 132, 378–386. [Google Scholar] [CrossRef]
- Fikree, A.; Grahame, R.; Aktar, R.; Farmer, A.D.; Hakim, A.J.; Morris, J.K.; Knowles, C.H.; Aziz, Q. A prospective evaluation of undiagnosed joint hypermobility syndrome in patients with gastrointestinal symptoms. Clin. Gastroenterol. Hepatol. 2013, 27, 569–579. [Google Scholar] [CrossRef]
- Morgan, A.W.; Pearson, S.B.; Davies, S.; Gooi, H.C.; Bird, H.A. Asthma and airway collapse in two heritable disorders of connective tissue. Ann. Rheum. Dis. 2007, 66, 1369–1373. [Google Scholar] [CrossRef]
- Rodgers, K.R.; Gui, J.; Dinulos, M.B.; Chou, R.C. Ehlers-Danlos syndrome hypermobility type is associated with rheumatic diseases. Sci. Rep. 2017, 7, 39636. [Google Scholar] [CrossRef]
- Grether-Beck, S.; Felsner, I.; Brenden, H.; Kohne, Z.; Majora, M.; Marini, A.; Jaenicke, T.; Rodriguez-Martin, M.; Trullas, C.; Hupe, M.; et al. Urea uptake enhances barrier function and antimicrobial defense in humans by regulating epidermal gene expression. J. Investig. Dermatol. 2012, 132, 1561–1572. [Google Scholar] [CrossRef]
- Nagahara, M.; Waguri-Nagaya, Y.; Yamagami, T.; Aoyama, M.; Tada, T.; Inoue, K.; Asai, K.; Otsuka, T. TNF-alpha-induced aquaporin 9 in synoviocytes from patients with OA and RA. Rheumatology 2010, 49, 898–906. [Google Scholar] [CrossRef]
- Takeuchi, K.; Hayashi, S.; Matumoto, T.; Hashimoto, S.; Takayama, K.; Chinzei, N.; Kihara, S.; Haneda, M.; Kirizuki, S.; Kuroda, Y.; et al. Downregulation of aquaporin 9 decreases catabolic factor expression through nuclear factor-κB signaling in chondrocytes. Int. J. Mol. Med. 2018, 42, 1548–1558. [Google Scholar] [CrossRef]
- Xu, Y.; Ma, M.; Ippolito, G.C.; Schroeder, H.W.; Carroll, M.C.; Volanakis, J.E. Complement activation in factor D-deficient mice. Proc. Natl. Acad. Sci. USA 2001, 98, 14577–14582. [Google Scholar] [CrossRef]
- Kluzek, S.; Arden, N.K.; Newton, J. Adipokines as potential prognostic biomarkers in patients with acute knee injury. Biomarkers 2015, 20, 519–525. [Google Scholar] [CrossRef]
- Martel-Pelletier, J.; Raynauld, J.P.; Dorais, M.; Abram, F.; Pelletier, J.P. The levels of the adipokines adipsin and leptin are associated with knee osteoarthritis progression as assessed by MRI and incidence of total knee replacement in symptomatic osteoarthritis patients: A post hoc analysis. Rheumatology 2016, 55, 680–688. [Google Scholar] [CrossRef]
- Chandran, V.; Abji, F.; Perruccio, A.V.; Gandhi, R.; Li, S.; Cook, R.J.; Gladman, D.D. Serum-based soluble markers differentiate psoriatic arthritis from osteoarthritis. Ann. Rheum. Dis. 2019, 78, 796–801. [Google Scholar] [CrossRef]
- Lyons, J.J.; Yu, X.; Hughes, J.D.; Le, Q.T.; Jamil, A.; Bai, Y.; Ho, N.; Zhao, M.; Liu, Y.; O’Connell, M.P.; et al. Elevated basal serum tryptase identifies a multisystem disorder associated with increased TPSAB1 copy number. Nat. Genet. 2016, 48, 1564–1569. [Google Scholar] [CrossRef]
- He, Y.W.; Li, H.; Zhang, J.; Hsu, C.L.; Lin, E.; Zhang, N.; Guo, J.; Forbush, K.A.; Bevan, M.J. The extracellular matrix protein mindin is a pattern-recognition molecule for microbial pathogens. Nat. Immunol. 2004, 5, 88–97. [Google Scholar] [CrossRef]
- Jia, W.; Li, H.; He, Y.W. The extracellular matrix protein mindin serves as an integrin ligand and is critical for inflammatory cell recruitment. Blood 2005, 106, 3854–3859. [Google Scholar] [CrossRef]
- Liu, Y.S.; Wang, L.F.; Cheng, X.S.; Huo, Y.N.; Ouyang, X.M.; Liang, L.Y.; Lin, Y.; Wu, J.F.; Ren, J.L.; Guleng, B. The pattern-recognition molecule mindin binds integrin Mac-1 to promote macrophage phagocytosis via Syk activation and NF-κB p65 translocation. J. Cell. Mol. Med. 2019, 23, 3402–3416. [Google Scholar] [CrossRef]
- Clapp, C.; Adán, N.; Ledesma-Colunga, M.G.; Solís-Gutiérrez, M.; Triebel, J.; Martínez de la Escalera, G. The role of the prolactin/vasoinhibin axis in rheumatoid arthritis: An integrative overview. Cell. Mol. Life Sci. 2016, 73, 2929–2948. [Google Scholar] [CrossRef]
- Ledesma-Colunga, M.G.; Adán, N.; Ortiz, G.; Solís-Gutiérrez, M.; López-Barrera, F.; Martínez de la Escalera, G.; Clapp, C. Prolactin blocks the expression of receptor activator of nuclear factor κB ligand and reduces osteoclastogenesis and bone loss in murine inflammatory arthritis. Arthritis Res. Ther. 2017, 19, 93. [Google Scholar] [CrossRef]
- Patil, M.J.; Ruparel, S.B.; Henry, M.A.; Akopian, A.N. Prolactin regulates TRPV1, TRPA1, and TRPM8 in sensory neurons in a sex-dependent manner: Contribution of prolactin receptor to inflammatory pain. Am. J. Physiol. Endocrinol. Metab. 2013, 305, E1154–E1164. [Google Scholar] [CrossRef]
- Basso, L.; Altier, C. Transient Receptor Potential Channels in neuropathic pain. Curr. Opin. Pharmacol. 2017, 32, 9–15. [Google Scholar] [CrossRef]
- Rodríguez-Calvo, R.; Tajes, M.; Vázquez-Carrera, M. The NR4A subfamily of nuclear receptors: Potential new therapeutic targets for the treatment of inflammatory diseases. Expert Opin. Ther. Targets 2017, 21, 291–304. [Google Scholar] [CrossRef]
- Murphy, E.P.; Crean, D. Molecular Interactions between NR4A Orphan Nuclear Receptors and NF-Kb Are Required for Appropriate Inflammatory Responses and Immune Cell Homeostasis. Biomolecules 2015, 5, 1302–1318. [Google Scholar] [CrossRef]
- Banno, A.; Lakshmi, S.P.; Reddy, A.T.; Kim, S.C.; Reddy, R.C. Key Functions and Therapeutic Prospects of Nur77 in Inflammation Related Lung Diseases. Am. J. Pathol. 2019, 189, 482–491. [Google Scholar] [CrossRef]
- Fernandes, J.C.R.; Acuña, S.M.; Aoki, J.I.; Floeter-Winter, L.M.; Muxel, S.M. Long Non-Coding RNAs in the Regulation of Gene Expression: Physiology and Disease. Noncoding RNA 2019, 5, 17. [Google Scholar] [CrossRef]
- Filipowicz, W.; Bhattacharyya, S.N.; Sonenberg, N. Mechanisms of post-transcriptional regulation by microRNAs: Are the answers insight? Nat. Rev. Genet 2008, 9, 102–114. [Google Scholar] [CrossRef]
- Leinders, M.; Üçeyler, N.; Thomann, A.; Sommer, C. Aberrant microRNA expression in patients with painful peripheral neuropathies. J. Neurol. Sci. 2017, 380, 242–249. [Google Scholar] [CrossRef]
- Greco, S.; Cardinali, B.; Falcone, G.; Martelli, F. Circular RNAs in Muscle Function and Disease. Int. J. Mol. Sci. 2018, 19, 3454. [Google Scholar] [CrossRef]
- D’Agnelli, S.; Arendt-Nielsen, L.; Gerra, M.C.; Zatorri, K.; Boggiani, L.; Baciarello, M.; Bignami, E. Fibromyalgia: Genetics and epigenetics insights may provide the basis for the development of diagnostic biomarkers. Mol. Pain 2019, 15, 1744806918819944. [Google Scholar] [CrossRef]
- Kim, J.; Hyun, J.; Wang, S.; Lee, C.; Jung, Y. MicroRNA-378 is involved in hedgehog-driven epithelial-to-mesenchymal transition in hepatocytes of regenerating liver. Cell Death Dis. 2018, 9, 721. [Google Scholar] [CrossRef]
- Zhang, T.; Hu, J.; Wang, X.; Zhao, X.; Li, Z.; Niu, J.; Steer, C.J.; Zheng, G.; Song, G. MicroRNA-378 promotes hepatic inflammation and fibrosis via modulation of the NF-κB-TNFα pathway. J. Hepatol. 2019, 70, 87–96. [Google Scholar] [CrossRef]
- Xiao, Z.; Deng, D.; He, L.; Jiao, H.; Ye, Y.; Liang, L.; Ding, Y.; Liao, W. MicroRNA-224 sustains Wnt/β-catenin signaling and promotes aggressive phenotype of colorectal cancer. J. Exp. Clin. Cancer Res. 2016, 35, 21. [Google Scholar]
- Peng, Y.; Zhang, X.; Feng, X.; Fan, X.; Jin, Z. The crosstalk between microRNAs and the Wnt/β-catenin signaling pathway in cancer. Oncotarget 2017, 8, 14089–14106. [Google Scholar] [CrossRef]
- Bjersing, J.L.; Lundborg, C.; Bokarewa, M.I.; Mannerkorpi, K. Profile of cerebrospinal microRNAs in fibromyalgia. PLoS ONE 2013, 8, e78762. [Google Scholar] [CrossRef]
- Chopra, P.; Tinkle, B.; Hamonet, C.; Brock, I.; Gompel, A.; Bulbena, A.; Francomano, C. Pain management in the Ehlers-Danlos syndromes. Am. J. Med. Genet. C Semin. Med. Genet. 2017, 175, 212–219. [Google Scholar] [CrossRef]
- Wade, S.M.; Trenkmann, M.; McGarry, T.; Canavan, M.; Marzaioli, V.; Wade, S.C.; Veale, D.J.; Fearon, U. Altered expression of microRNA-23a in psoriatic arthritis modulates synovial fibroblast pro-inflammatory mechanisms via phosphodiesterase 4B. J. Autoimmun. 2019, 96, 86–93. [Google Scholar] [CrossRef]
- Tajerian, M.; Clark, J.D. The role of the extracellular matrix in chronic pain following injury. Pain 2015, 156, 366–370. [Google Scholar] [CrossRef]
- Parisien, M.; Samoshkin, A.; Tansley, S.N.; Piltonen, M.H.; Martin, L.J.; El-Hachem, N.; Dagostino, C.; Allegri, M.; Mogil, J.S.; Khoutorsky, A.; et al. Genetic pathway analysis reveals a major role for extracellular matrix organization in inflammatory and neuropathic pain. Pain 2019, 60, 932–944. [Google Scholar] [CrossRef]
- Andersen, H.H.; Duroux, M.; Gazerani, P. MicroRNA as modulators and biomarkers of inflammatory and neuropathic pain conditions. Neurobiol. Dis. 2014, 71, 159–168. [Google Scholar] [CrossRef]


{kind=link}
| EDS Type | IP | Gene | Protein |
|---|---|---|---|
| Group A: disorders of collagen primary structure and collagen processing | |||
| Classical EDS (cEDS) | AD | Major: COL5A1, COL5A2 Rare: COL1A1 | COLLV COLLI |
| Vascular EDS (vEDS) | AD | COL3A1 | COLLIII |
| Arthrochalasia EDS (aEDS) | AD | COL1A1, COL1A2 | COLLI |
| Dermatosparaxis EDS (dEDS) | AR | ADAMTS2 | ADAMTS-2 |
| Cardiac-valvular EDS (cvEDS) | AR | COL1A2 | COLLI |
| Classical-like 2 EDS A (cl2EDS) | AR | AEBP1 | ACLP |
| Group B: disorders of collagen folding and collagen cross-linking | |||
| Kyphoscoliotic EDS (kEDS) | AR | PLOD1 FKBP14 | LH1 FKBP22 |
| Group C: disorders of structure and function of myomatrix | |||
| Classical-like EDS (clEDS) Myopathic EDS (mEDS) | AR AD/AR | TNXB COL12A1 | Tenascin X COLLXII |
| Group D: disorders of glycosaminoglycan biosynthesis | |||
| Spondylodysplastic EDS (spEDS) | AR | B4GALT7 B3GALT6 | β4GalT7 β3GalT6 |
| Musculocontractural EDS (mcEDS) | AR | CHST14 DSE | D4ST1 DSE |
| Group E: disorders of complement pathway | |||
| Periodontal EDS (pEDS) | AD | C1R C1S | C1r C1s |
| Group F: disorders of intracellular processes | |||
| Spondylodysplastic EDS (spEDS) | AR | SLC39A13 | ZIP13 |
| Brittle Cornea Syndrome (BCS) | AR | ZNF469 PRDM5 | ZNF469 PRDM5 |
| EDS type molecularly unsolved | |||
| Hypermobile EDS (hEDS) | AD | Unknown | Unknown |
| Insights in the Pathogenesis of cEDS, vEDS, and hEDS | Transcriptome Findings on Patients’ Fibroblasts | In Vitro Studies on Patients’ Fibroblasts | ||||
|---|---|---|---|---|---|---|
| Perturbed Biological Processes | cEDS | vEDS | hEDS | cEDS | vEDS | hEDS |
| ECM disorganization | + | + | + | + | + | + |
| Altered cell-matrix interactions | − | − | + | + | + | + |
| Disturbed cell-cell contacts | − | − | + | − | − | + |
| Fibroblast-to-myofibroblast transition | − | − | + | − | − | + |
| Altered inflammatory responses | + | − | + | − | na | + |
| Perturbed cell migration | + | + | − | + | + | + |
| Defective wound healing | + | + | − | + | na | na |
| Survival from anoikis | − | − | − | + | + | na |
| Collagens biosynthesis/processing | + | + | − | na | + | na |
| ER homeostasis/protein folding | + | + | − | na | + | na |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Chiarelli, N.; Ritelli, M.; Zoppi, N.; Colombi, M. Cellular and Molecular Mechanisms in the Pathogenesis of Classical, Vascular, and Hypermobile Ehlers‒Danlos Syndromes. Genes 2019, 10, 609. https://doi.org/10.3390/genes10080609
Chiarelli N, Ritelli M, Zoppi N, Colombi M. Cellular and Molecular Mechanisms in the Pathogenesis of Classical, Vascular, and Hypermobile Ehlers‒Danlos Syndromes. Genes. 2019; 10(8):609. https://doi.org/10.3390/genes10080609
Chicago/Turabian StyleChiarelli, Nicola, Marco Ritelli, Nicoletta Zoppi, and Marina Colombi. 2019. "Cellular and Molecular Mechanisms in the Pathogenesis of Classical, Vascular, and Hypermobile Ehlers‒Danlos Syndromes" Genes 10, no. 8: 609. https://doi.org/10.3390/genes10080609
APA StyleChiarelli, N., Ritelli, M., Zoppi, N., & Colombi, M. (2019). Cellular and Molecular Mechanisms in the Pathogenesis of Classical, Vascular, and Hypermobile Ehlers‒Danlos Syndromes. Genes, 10(8), 609. https://doi.org/10.3390/genes10080609