A Homeostasis Hypothesis of Avian Influenza Resistance in Chickens

 ,
,  ,
, {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Samples and Experimental Design
2.2. RNA-seq Data Analysis
2.3. Whole Genome Bisulfite Sequencing Data Analysis
2.4. Resequencing Data Analysis
2.5. dn/ds Ratio Calculation
2.6. Data Availability
3. Results
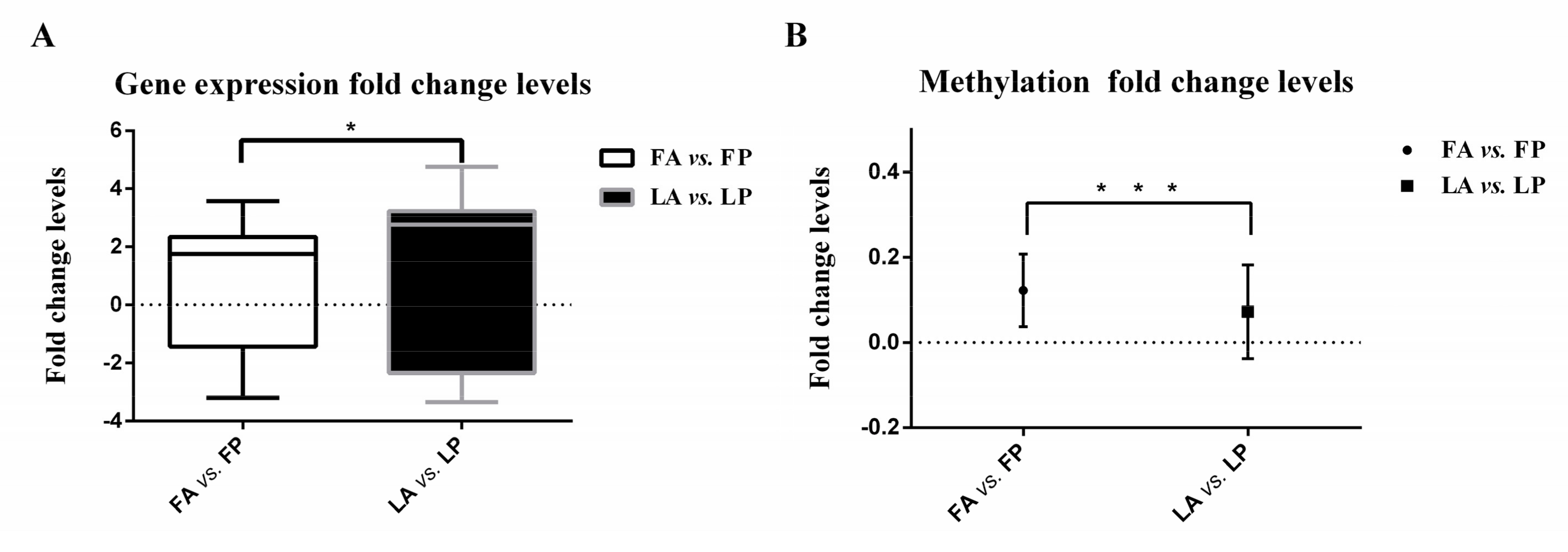
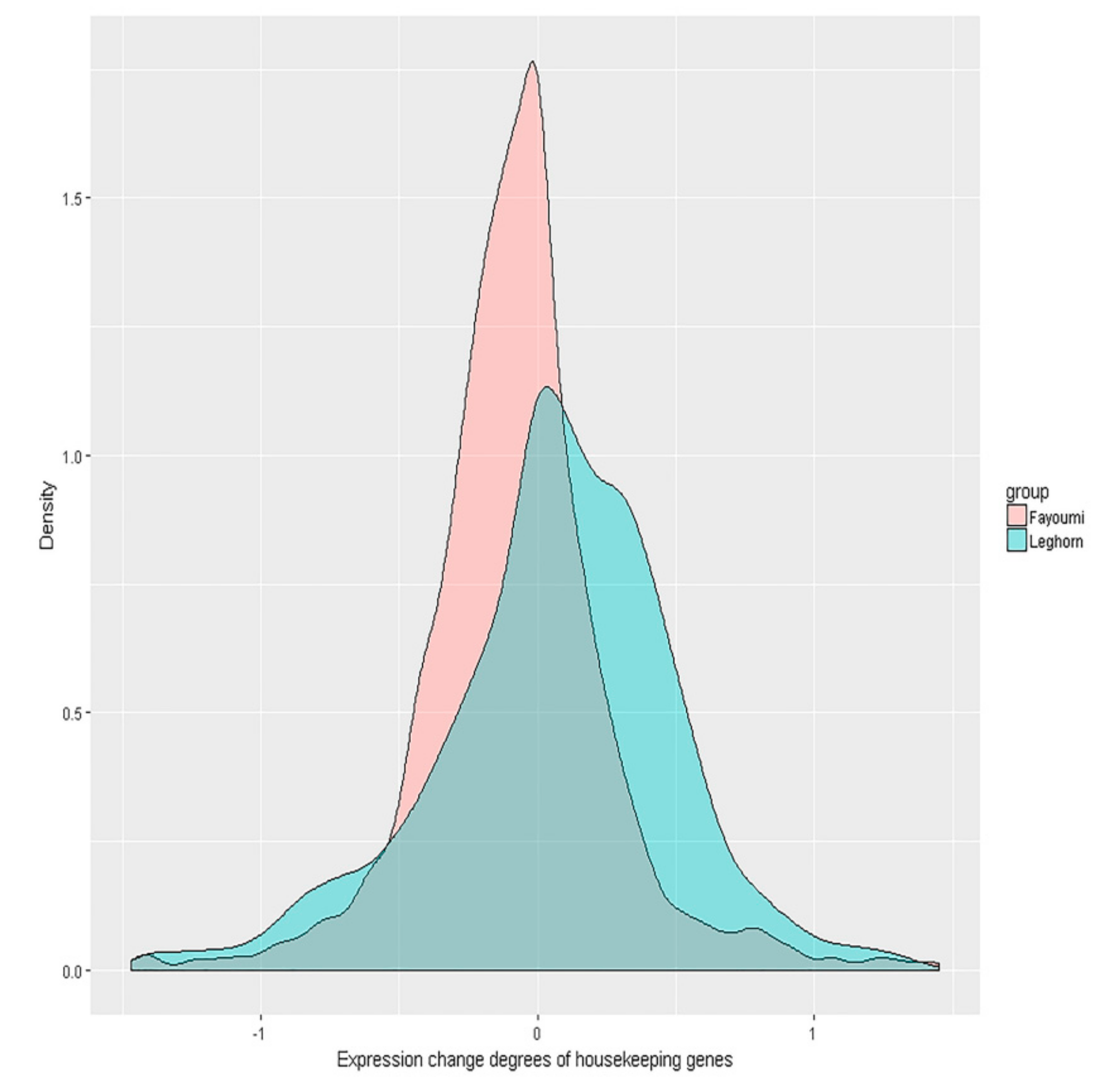
3.1. Less Gene Expression Fold Change Levels in Response to Avian Influenza Virus in Fayoumi
3.2. Avian Influenza Virus Inoculation Introduced Changes in DNA Methylation in Both Breeds
3.3. Less DNA Methylation Fold Change Levels in Response to Avian Influenza Virus in Fayoumi
3.4. Further Tests for Homeostasis Hypothesis in Fayoumi and Leghorn Breeds
3.5. Genetic Differentiation Between Fayoumi and Leghorn Chickens
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dhama, K.; Chakraborty, S.; Tiwari, R.; Kumar, A.; Rahal, A.; Latheef, K.S.; Wani, M.Y.; Kapoor, S. Avian/Bird Flu Virus: Poultry Pathogen Having Zoonotic and Pandemic Threats: A Review. J. Med. Sci. 2013, 13, 301–315. [Google Scholar] [CrossRef]
- Francisco, N.; Donadel, M.; Jit, M.; Hutubessy, R. A systematic review of the social and economic burden of influenza in low- and middle-income countries. Vaccine 2015, 33, 6537–6544. [Google Scholar] [CrossRef]
- Suarez, D.L.; Schultz-Cherry, S. Immunology of avian influenza virus: A review. Dev. Comp. Immunol. 2000, 24, 269–283. [Google Scholar] [CrossRef]
- Ewald, S.J.; Kapczynski, D.R.; Livant, E.J.; Suarez, D.L.; Ralph, J.; McLeod, S.; Miller, C. Association of Mx1 Asn631 variant alleles with reductions in morbidity, early mortality, viral shedding, and cytokine responses in chickens infected with a highly pathogenic avian influenza virus. Immunogenetics 2011, 63, 363–375. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Brahmakshatriya, V.; Lupiani, B.; Reddy, S.; Okimoto, R.; Li, X.; Chiang, H.; Zhou, H. Associations of chicken Mx1 polymorphism with antiviral responses in avian influenza virus infected embryos and broilers. Poult. Sci. 2012, 91, 3019–3024. [Google Scholar] [CrossRef] [PubMed]
- Lyall, J.; Irvine, R.M.; Sherman, A.; McKinley, T.J.; Núñez, A.; Purdie, A.; Outtrim, L.; Brown, I.H.; Rolleston, S.G.; Sang, H.; et al. Suppression of avian influenza transmission in genetically modified chickens. Science 2011, 331, 223–226. [Google Scholar] [CrossRef] [PubMed]
- June, B.S.; Yuk, S.; Jang, Y.; Choi, H.; Jeon, M.; Erdene-Ochir, T.O.; Kwon, J.; Noh, J.; Sun, K.J.; Gyu, Y.J.; et al. Transgenic Chickens Expressing the 3D8 Single Chain Variable Fragment Protein Suppress Avian Influenza Transmission. Sci. Rep. 2017, 7, 1–11. [Google Scholar] [CrossRef]
- Ledur, M.C.; Fairfull, R.W.; McMillan, I.; Asseltine, L. Genetic effects of aging on egg production traits in the first laying cycle of wWhite Leghorn strains and strain crosses. Poult. Sci. 2000, 3, 296–304. [Google Scholar] [CrossRef]
- Abernathy, J.; Li, X.; Jia, X.; Chou, W.; Lamont, S.J.; Crooijmans, R.; Zhou, H. Copy number variation in Fayoumi and Leghorn chickens analyzed using array comparative genomic hybridization. Anim. Genet. 2014, 45, 400–411. [Google Scholar] [CrossRef]
- Marie-Hélène, P.L.; Bertrand, B.; Jean-Luc, C.; Frédérique, P.; Katia, F.; Leroux, S.; Hélène, L.; Aurélie, T.; David, G.; Jean-Michel, R.; et al. Microsatellite mapping of QTLs affecting resistance to coccidiosis (Eimeria tenella) in a Fayoumi × White Leghorn cross. BMC Genom. 2009, 10, 31–44. [Google Scholar] [CrossRef]
- Li, J.; Li, R.; Wang, Y.; Hu, X.; Zhao, Y.; Li, L.; Feng, C.; Gu, X.; Liang, F.; Lamont, S.J.; et al. Genome-wide DNA methylome variation in two genetically distinct chicken lines using MethylC-seq. BMC Genom. 2015, 16, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Lupiani, B.; Reddy, S.M.; Lamont, S.J.; Zhou, H. RNA-seq analysis revealed novel genes and signaling pathway associated with disease resistance to avian influenza virus infection in chickens. Poult. Sci. 2014, 93, 485–493. [Google Scholar] [CrossRef] [PubMed]
- Fleming, D.S.; Koltes, J.E.; Fritz-Waters, E.R.; Rothschild, M.F.; Schmidt, C.J.; Ashwell, C.M.; Persia, M.E.; Reecy, J.M.; Lamont, S.J. Single nucleotide variant discovery of highly inbred Leghorn and Fayoumi chicken breeds using pooled whole genome resequencing data reveals insights into phenotype differences. BMC Genom. 2016, 17, 812. [Google Scholar] [CrossRef] [PubMed]
- Ramsay, D.S.; Woods, S.C. Clarifying the roles of homeostasis and allostasis in physiological regulation. Psychol. Rev. 2014, 121, 25–247. [Google Scholar] [CrossRef] [PubMed]
- Pacis, A.; Tailleux, L.; Morin, A.M.; Lambourne, J.; MacIsaac, J.L.; Yotova, V.; Dumaine, A.; Danckaert, A.; Luca, F.; Grenier, J.; et al. Bacterial infection remodels the DNA methylation landscape of human dendritic cells. Genome Res. 2015, 25, 1801–1811. [Google Scholar] [CrossRef] [PubMed]
- Shang, X.; Su, J.; Wan, Q.; Su, J.; Feng, X. CpG methylation in the 5′-flanking region of LGP2 gene lacks association with resistance/susceptibility to GCRV but contributes to the differential expression between muscle and spleen tissues in grass carp, Ctenopharyngodon idella. Fish Shellfish Immunol. 2014, 40, 154–163. [Google Scholar] [CrossRef] [PubMed]
- Lai, A.Y.; Mav, D.; Shah, R.; Grimm, S.A.; Phadke, D.; Hatzi, K.; Melnick, A.; Geigerman, C.; Sobol, S.E.; Jaye, D.L.; et al. DNA methylation profiling in human B cells reveals immune regulatory elements and epigenetic plasticity at Alu elements during B-cell activation. Genome Res. 2013, 23, 2030–2041. [Google Scholar] [CrossRef]
- Xu, H.; Zhu, X.; Hu, Y.; Li, Z.; Zhang, X.; Nie, Q.; Nolan, L.K.; Lamont, S.J. DNA methylome in spleen of avian pathogenic escherichia coli-challenged broilers and integration with mRNA expression. Sci. Rep. 2015, 4, 8303. [Google Scholar] [CrossRef]
- Yu, A.; Lepere, G.; Jay, F.; Wang, J.; Bapaume, L.; Wang, Y.; Abraham, A.L.; Penterman, J.; Fischer, R.L.; Voinnet, O.; et al. Dynamics and biological relevance of DNA demethylation in Arabidopsis antibacterial defense. Proc. Natl. Acad. Sci. USA 2013, 110, 2389–2394. [Google Scholar] [CrossRef]
- Xu, S.; Yao, H.; Zhang, J.; Zhang, Z.; Wang, J.; Zhang, J.; Jiang, Z. The oxidative damage and disbalance of calcium homeostasis in brain of chicken induced by selenium deficiency. Biol. Trace Elem. Res. 2013, 151, 225–233. [Google Scholar] [CrossRef]
- Waddington, C.H. Canalization of development and the inheritance of acquired characters. Nature 1942, 150, 563–565. [Google Scholar] [CrossRef]
- Takahashi, K.H. Multiple modes of canalization: Links between genetic, environmental canalizations and developmental stability, and their trait-specificity. Semin. Cell Dev. Biol. 2019, 88, 14–20. [Google Scholar] [CrossRef] [PubMed]
- Trapnell, C.; Roberts, A.; Goff, L.; Pertea, G.; Kim, D. Differential gene and transcript expression analysis of RNA-seq experiments with TopHat and Cufflinks. Nat. Protoc. 2012, 7, 562–578. [Google Scholar] [CrossRef] [PubMed]
- Krueger, F.; Andrews, S.R. Bismark: A flexible aligner and methylation caller for Bisulfite-Seq applications. Bioinformatics 2011, 27, 1571–1572. [Google Scholar] [CrossRef] [PubMed]
- Dolzhenko, E.; Smith, A.D. Using beta-binomial regression for high-precision differential methylation analysis in multifactor whole-genome bisulfite sequencing experiments. BMC Bioinform. 2014, 15, 210–215. [Google Scholar] [CrossRef] [PubMed]
- Li, H.; Durbin, R. Fast and accurate short read alignment with Burrows-Wheeler transform. Bioinformatics 2009, 25, 1754–1760. [Google Scholar] [CrossRef]
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef]
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [CrossRef]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Pond, S.L.; Frost, S.D.; Muse, S.V. HyPhy: Hypothesis testing using phylogenies. Binformatics 2005, 21, 676–679. [Google Scholar] [CrossRef]
- Weaver, S.; Shank, S.D.; Spielman, S.J.; Li, M.; Muse, S.V.; Kosakovsky Pond, S.L. Datamonkey 2.0: A modern web application for characterizing selective and other evolutionary processes. Mol. Biol. Evol. 2018, 35, 773–777. [Google Scholar] [CrossRef] [PubMed]
- Murrell, B.; Wertheim, J.O.; Moola, S.; Weighill, T.; Scheffler, K.; Kosakovsky, P.S.L. Detecting individual sites subject to episodic diversifying selection. PLoS Genet. 2012, 8, e1002764. [Google Scholar] [CrossRef] [PubMed]
- Mikko, T.; Daniel, F.J.; Lindquist, S. HSP90 at the hub of protein homeostasis: Emerging mechanistic insights. Nat. Rev. Mol. Cell Biol. 2010, 11, 515–528. [Google Scholar] [CrossRef]
- Suzuki, M.M.; Bird, A. DNA methylation landscapes: Provocative insights from epigenomics. Nat. Rev. Genet. 2008, 9, 465–476. [Google Scholar] [CrossRef] [PubMed]
- Jones, P.A. Functions of DNA methylation: Islands, start sites, gene bodies and beyond. Nat. Rev. Genet. 2012, 13, 484–492. [Google Scholar] [CrossRef]
- Maunakea, K.A.; Chepelev, I.; Cui, K.; Zhao, K. Intragenic DNA methylation modulates alternative splicing by recruiting MeCP2 to promote exon recognition. Cell Res. 2013, 23, 1256–1269. [Google Scholar] [CrossRef] [PubMed]
- Costa, T.; Chaves, A.J.; Valle, R.; Darji, A.; van Riel, D.; Kuiken, T.; Majo, N.; Ramis, A. Distribution patterns of influenza virus receptors and viral attachment patterns in the respiratory and intestinal tracts of seven avian species. Vet. Res. 2012, 43, 1–13. [Google Scholar] [CrossRef]
- Jiang, M.; Zhang, S.; Yang, Z.; Lin, H.; Zhu, J.; Liu, L.; Wang, W.; Liu, S.; Liu, W.; Ma, Y.; et al. Self-recognition of an inducible host lncRNA by RIG-I feedback restricts innate immune response. Cell 2018, 173, 906–919. [Google Scholar] [CrossRef]
- Félix, M.; Barkoulas, M. Pervasive robustness in biological systems. Nat. Rev. Genet. 2015, 16, 483–496. [Google Scholar] [CrossRef]
- Phillips, A.M.; Ponomarenko, A.I.; Chen, K.; Ashenberg, O.; Miao, J.; McHugh, S.M.; Butty, V.L.; Whittaker, C.A.; Moore, C.L.; Bloom, J.D.; et al. Destabilized adaptive influenza variants critical for innate immune system escape are potentiated by host chaperones. PLoS Biol. 2018, 16, e3000008. [Google Scholar] [CrossRef]
- Tumpey, T.M.; Szretter, K.J.; Van Hoeven, N.; Katz, J.M.; Kochs, G.; Haller, O.; Garcia-Sastre, A.; Staeheli, P. The Mx1 gene protects mice against the pandemic 1918 and highly lethal human H5N1 influenza viruses. J. Virol. 2007, 81, 10818–10821. [Google Scholar] [CrossRef] [PubMed]
- Pavlovic, J.; Haller, O.; Staeheli, P. Human and mouse Mx proteins inhibit different steps of the influenza virus multiplication cycle. J. Virol. 1992, 66, 2564–2569. [Google Scholar] [PubMed]
- Pillai, P.S.; Molony, R.D.; Martinod, K.; Dong, H.; Pang, I.K.; Tal, M.C.; Solis, A.G.; Bielecki, P.; Mohanty, S.; Trentalange, M.; et al. Mx1 reveals innate pathways to antiviral resistance and lethal influenza disease. Science 2016, 352, 463–466. [Google Scholar] [CrossRef] [PubMed]
- Kaminski, M.M.; Ohnemus, A.; Cornitescu, M.; Staeheli, P. Plasmacytoid dendritic cells and Toll-like receptor 7-dependent signalling promote efficient protection of mice against highly virulent influenza A virus. J. Gen. Virol. 2012, 93, 555–559. [Google Scholar] [CrossRef] [PubMed]
- Grimm, D.; Staeheli, P.; Hufbauer, M.; Koerner, I.; Martinez-Sobrido, L.; Solorzano, A.; Garcia-Sastre, A.; Haller, O.; Kochs, G. Replication fitness determines high virulence of influenza A virus in mice carrying functional Mx1 resistance gene. Proc. Natl. Acad. Sci. USA 2007, 104, 6806–6811. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.H. Novel genetic capacitors and potentiators for the natural genetic variation of sensory bristles and their trait specificity inDrosophila melanogaster. Mol. Ecol. 2015, 24, 5561–5572. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Piel, W.H.; Gui, L.; Bruford, E.; Monteiro, A. The HSP90 family of genes in the human genome: Insights into their divergence and evolution. Genomics 2005, 86, 627–637. [Google Scholar] [CrossRef] [PubMed]
- Makhnevych, T.; Houry, W.A. The role of Hsp90 in protein complex assembly. Biochim. Biophys. Acta 2012, 1823, 674–682. [Google Scholar] [CrossRef] [PubMed]
- Wahl, A.; Schafer, F.; Bardet, W.; Hildebrand, W.H. HLA class I molecules reflect an altered host proteome after influenza virus infection. Hum. Immunol. 2010, 71, 14–22. [Google Scholar] [CrossRef] [PubMed]
- Grabek, K.R.; Karimpour-Fard, A.; Epperson, L.E.; Hindle, A.; Hunter, L.E.; Martin, S.L. Multistate proteomics analysis reveals novel strategies used by a hibernator to precondition the heart and conserve ATP for winter heterothermy. Physiol. Genom. 2011, 43, 1263–1275. [Google Scholar] [CrossRef] [PubMed]
- Wong, S.L.; To, J.; Santos, J.; Allam, V.S.R.R.; Dalton, J.P.; Djordjevic, S.P.; Donnelly, S.; Padula, M.P.; Sukkar, M.B. Proteomic analysis of extracellular HMGB1 identifies binding partners and exposes its potential role in airway epithelial cell homeostasis. J. Proteome Res. 2018, 17, 33–45. [Google Scholar] [CrossRef] [PubMed]
- Baker, M.L.; Schountz, T.; Wang, L.F. Antiviral immune responses of bats: A review. Zoonoses Public Health 2013, 60, 104–116. [Google Scholar] [CrossRef] [PubMed]
- Ahn, M.; Cui, J.; Irving, A.T.; Wang, L. Unique loss of the PYHIN gene family in bats amongst mammals: Implications for inflammasome sensing. Sci. Rep. 2016, 6, 21722. [Google Scholar] [CrossRef] [PubMed]
- Li, N.; Parrish, M.; Chan, T.K.; Yin, L.; Rai, P.; Yoshiyuki, Y.; Abolhassani, N.; Tan, K.B.; Kiraly, O.; Chow, V.T.K.; et al. Influenza infection induces host DNA damage and dynamic DNA damage responses during tissue regeneration. Cell Mol. Life Sci. 2015, 72, 2973–2988. [Google Scholar] [CrossRef] [PubMed]
- Xie, J.; Li, Y.; Shen, X.; Goh, G.; Zhu, Y.; Cui, J.; Wang, L.; Shi, Z.; Zhou, P. Dampened STING-dependent interferon activation in bats. Cell Host Microbe 2018, 23, 297–301. [Google Scholar] [CrossRef] [PubMed]
- Joanna, K.; Hughes, G.M.; Palsson-McDermott, E.M.; Quinn, S.R.; Puechmaille, J.S.; O’Neill, L.A.J.; Teeling, E.C. A potent anti-inflammatory response in bat macrophages may be linked to extended longevity and viral tolerance. Acta Chiropterol. 2017, 2, 219–228. [Google Scholar] [CrossRef]
- Fuchs, J.; Hölzer, M.; Schilling, M.; Patzina, C.; Andreas, S.; Hoenen, T.; Zimmer, G.; Marz, M.; Weber, F.; Müller, M.A. Evolution and antiviral specificity of interferon-induced Mx proteins of bats against Ebola-, Influenza-, and other RNA viruses. J. Virol. 2017, 91, e317–e361. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
An, J.; Li, J.; Wang, Y.; Wang, J.; Li, Q.; Zhou, H.; Hu, X.; Zhao, Y.; Li, N. A Homeostasis Hypothesis of Avian Influenza Resistance in Chickens. Genes 2019, 10, 543. https://doi.org/10.3390/genes10070543
An J, Li J, Wang Y, Wang J, Li Q, Zhou H, Hu X, Zhao Y, Li N. A Homeostasis Hypothesis of Avian Influenza Resistance in Chickens. Genes. 2019; 10(7):543. https://doi.org/10.3390/genes10070543
Chicago/Turabian StyleAn, Jing, Jinxiu Li, Ying Wang, Jing Wang, Qinghe Li, Huaijun Zhou, Xiaoxiang Hu, Yiqiang Zhao, and Ning Li. 2019. "A Homeostasis Hypothesis of Avian Influenza Resistance in Chickens" Genes 10, no. 7: 543. https://doi.org/10.3390/genes10070543
APA StyleAn, J., Li, J., Wang, Y., Wang, J., Li, Q., Zhou, H., Hu, X., Zhao, Y., & Li, N. (2019). A Homeostasis Hypothesis of Avian Influenza Resistance in Chickens. Genes, 10(7), 543. https://doi.org/10.3390/genes10070543