groEL Gene-Based Phylogenetic Analysis of Lactobacillus Species by High-Throughput Sequencing

 ,
,
Abstract

1. Introduction
2. Materials and Methods
2.1. Bacterial Culture and Genome Extraction
2.2. Sample Collection and Genome Extraction
2.3. Selection of Lactobacillus groEL Gene
2.4. Phylogenetic Analysis
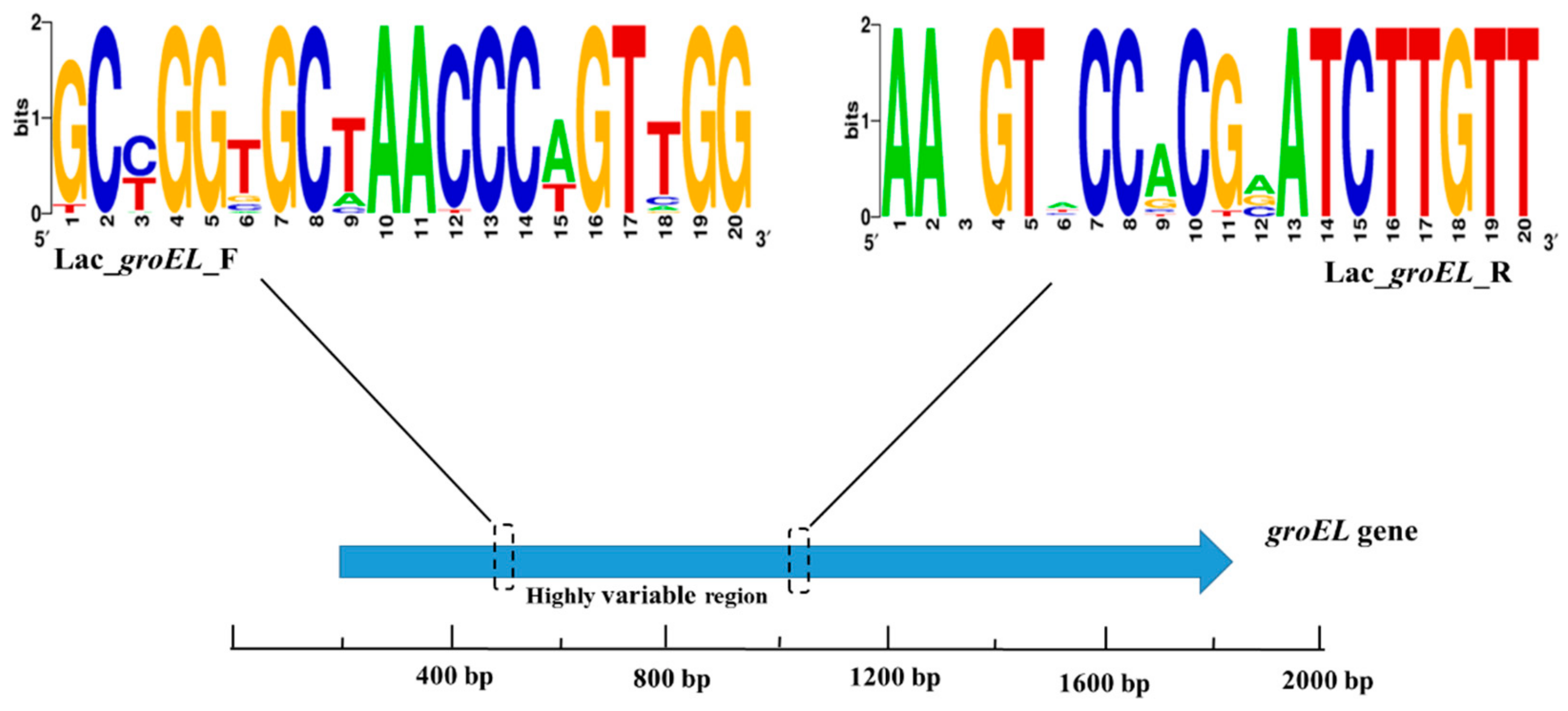
2.5. Lactobacillus groEL-Specific Primer Design
2.6. Evaluation of the Accuracy and Specificity of the Degenerate Primers
2.7. PCR Conditions, Quantifiable and Sequencing Measures
2.8. Cross-Alignment Analysis
3. Results
3.1. Selection of the Lactobacillus groEL Gene
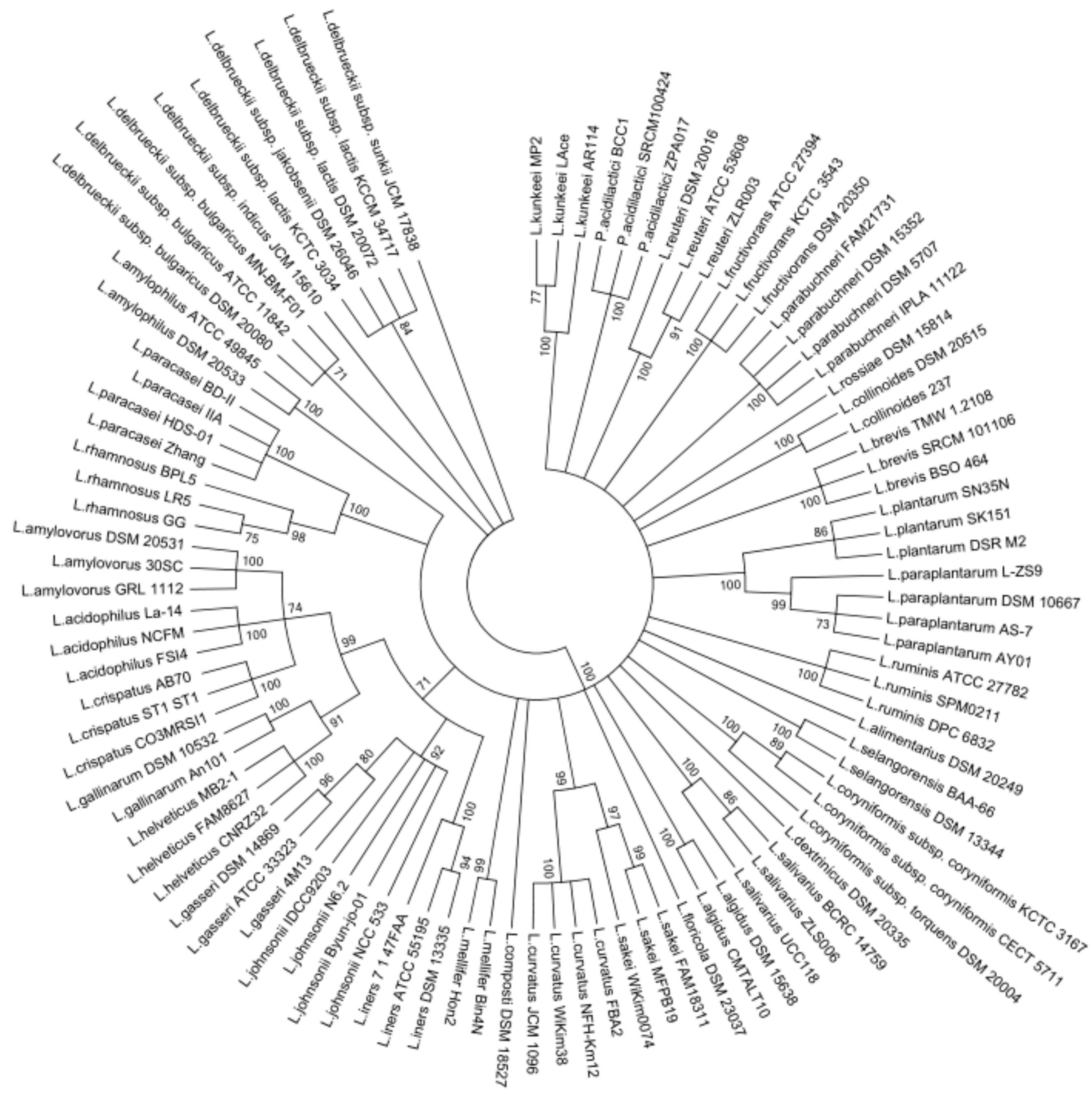
3.2. Phylogenetic Analysis of the Lactobacillus groEL Gene
3.3. Design of Novel Primer Sets for Identifying Lactobacillus
3.4. Development of Biological Information Analysis Tool for Lactobacillus
3.5. Performance Evaluation of the Degenerate Primers for Identifying Lactobacillus Species
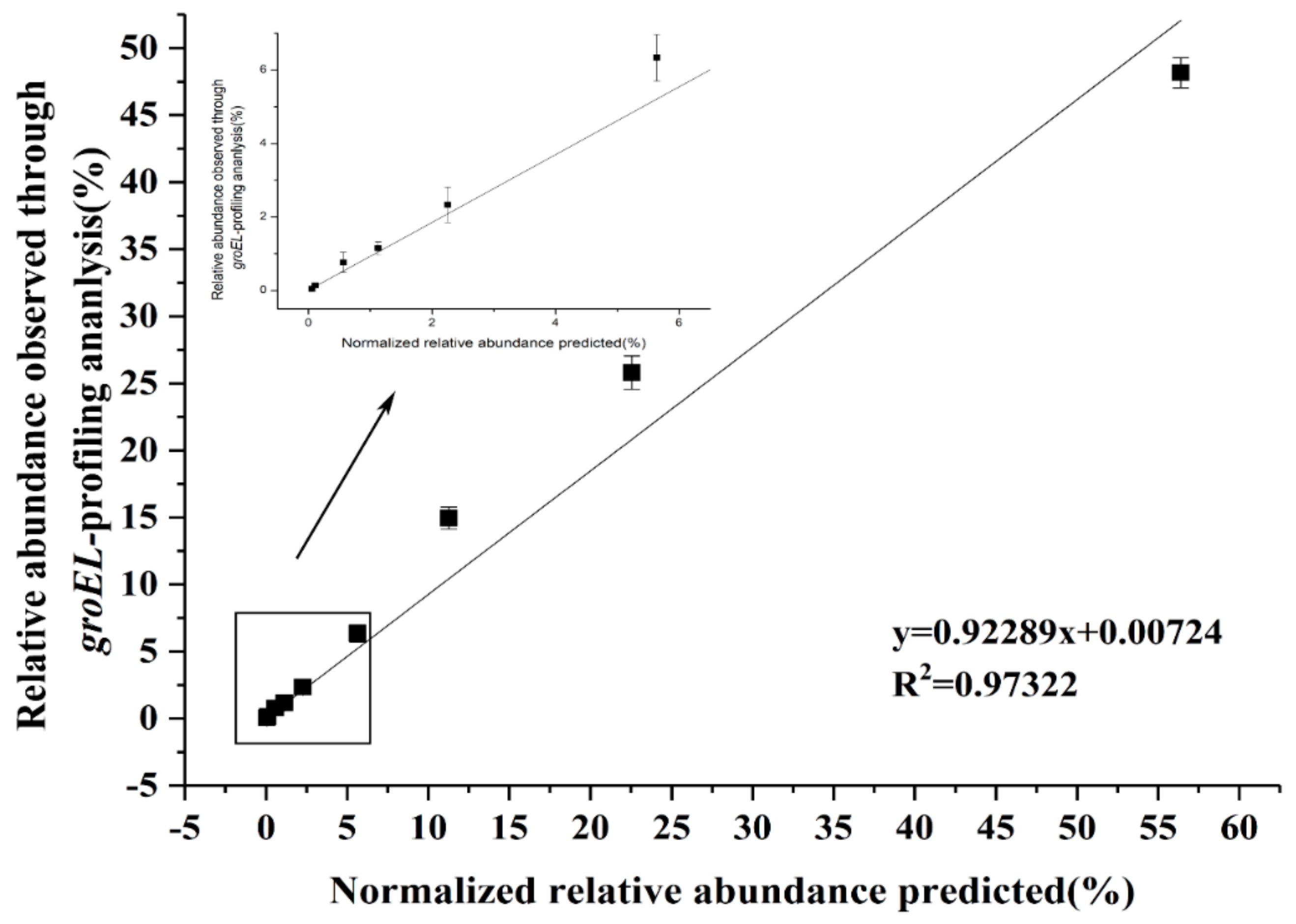
3.6. Resolution of Partial groEL Gene of Lactobacillus by Analyzing Samples from Varied Habitats
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Makarova, K.; Slesarev, A.; Wolf, Y.; Sorokin, A.; Mirkin, B.; Koonin, E.; Pavlov, A.; Pavlov, N.; Karamychev, V.; Polouchine, N.; et al. Comparative genomics of the lactic acid bacteria. Proc. Natl. Acad. Sci. USA 2006, 103, 15611–15616. [Google Scholar] [CrossRef] [PubMed]
- Duar, R.M.; Lin, X.B.; Zheng, J.; Martino, M.E.; Grenier, T.; Perez-Munoz, M.E.; Leulier, F.; Ganzle, M.; Walter, J. Lifestyles in transition: Evolution and natural history of the genus Lactobacillus. FEMS Microbiol. Rev. 2017, 41, S27–S48. [Google Scholar] [CrossRef] [PubMed]
- Walter, J. Ecological role of lactobacilli in the gastrointestinal tract: Implications for fundamental and biomedical research. Appl. Environ. Microbiol. 2008, 74, 4985–4996. [Google Scholar] [CrossRef] [PubMed]
- Goldstein, E.J.; Tyrrell, K.L.; Citron, D.M. Lactobacillus species: Taxonomic complexity and controversial susceptibilities. Clin. Infect. Dis. Off. Publ. Infect. Dis. Soc. Am. 2015, 60, S98–S107. [Google Scholar] [CrossRef] [PubMed]
- Foster, L.M.; Tompkins, T.A.; Dahl, W.J. A comprehensive post-market review of studies on a probiotic product containing Lactobacillus helveticus R0052 and Lactobacillus rhamnosus R0011. Benef. Microbes 2011, 2, 319–334. [Google Scholar] [CrossRef] [PubMed]
- Salvetti, E.; Harris, H.M.B.; Felis, G.; O’Toole, P.W. Comparative genomics reveals robust phylogroups in the genus Lactobacillus as the basis for reclassification. Appl. Environ. Microbiol. 2018. [Google Scholar] [CrossRef] [PubMed]
- Bao, Q.; Song, Y.; Xu, H.; Yu, J.; Zhang, W.; Menghe, B.; Zhang, H.; Sun, Z. Multilocus sequence typing of Lactobacillus casei isolates from naturally fermented foods in China and Mongolia. J. Dairy Sci. 2016, 99, 5202–5213. [Google Scholar] [CrossRef] [PubMed]
- Zheng, J.; Ruan, L.; Sun, M.; Ganzle, M. A Genomic View of Lactobacilli and Pediococci Demonstrates that Phylogeny Matches Ecology and Physiology. Appl. Environ. Microbiol. 2015, 81, 7233–7243. [Google Scholar] [CrossRef]
- Sun, Z.; Harris, H.M.; McCann, A.; Guo, C.; Argimon, S.; Zhang, W.; Yang, X.; Jeffery, I.B.; Cooney, J.C.; Kagawa, T.F.; et al. Expanding the biotechnology potential of lactobacilli through comparative genomics of 213 strains and associated genera. Nat. Commun. 2015, 6, 8322. [Google Scholar] [CrossRef]
- Milani, C.; Duranti, S.; Mangifesta, M.; Lugli, G.A.; Turroni, F.; Mancabelli, L.; Viappiani, A.; Anzalone, R.; Alessandri, G.; Ossiprandi, M.C.; et al. Phylotype-Level Profiling of Lactobacilli in Highly Complex Environments by Means of an Internal Transcribed Spacer-Based Metagenomic Approach. Appl. Environ. Microbiol. 2018, 84, e00706–e00718. [Google Scholar] [CrossRef]
- Shevtsov, A.; Kushugulova, A.; Tynybayeva, I.K.; Kozhakhmetov, S.; Abzhalelov, A.B.; Momynaliev, K.T.; Stoyanova, L.G. Identification of phenotypically and genotypically related Lactobacillus strains based on nucleotide sequence analysis of the groEL, rpoB, rplB, and 16S rRNA genes. Microbiology 2011, 80, 672–681. [Google Scholar] [CrossRef]
- Viale, A.M.; Arakaki, A.K.; Soncini, F.C.; Ferreyra, R.G. Evolutionary relationships among eubacterial groups as inferred from GroEL (chaperonin) sequence comparisons. Int. J. Syst. Bacteriol. 1994, 44, 527–533. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bergonzelli, G.E.; Granato, D.; Pridmore, R.D.; Marvin-Guy, L.F.; Donnicola, D.; Corthesy-Theulaz, I.E. GroEL of Lactobacillus johnsonii La1 (NCC 533) is cell surface associated: Potential role in interactions with the host and the gastric pathogen Helicobacter pylori. Infect. Immun. 2006, 74, 425–434. [Google Scholar] [CrossRef] [PubMed]
- Blaiotta, G.; Fusco, V.; Ercolini, D.; Aponte, M.; Pepe, O.; Villani, F. Lactobacillus strain diversity based on partial hsp60 gene sequences and design of PCR-restriction fragment length polymorphism assays for species identification and differentiation. Appl. Environ. Microbiol. 2008, 74, 208–215. [Google Scholar] [CrossRef] [PubMed]
- Hu, L.; Lu, W.; Wang, L.; Pan, M.; Zhang, H.; Zhao, J.; Chen, W. Assessment of Bifidobacterium Species Using groEL Gene on the Basis of Illumina MiSeq High-Throughput Sequencing. Genes 2017, 8, 336. [Google Scholar] [CrossRef] [PubMed]
- Dobson, C.M.; Chaban, B.; Deneer, H.; Ziola, B. Lactobacillus casei, Lactobacillus rhamnosus, and Lactobacillus zeae isolates identified by sequence signature and immunoblot phenotype. Can. J. Microbiol. 2004, 50, 482–488. [Google Scholar] [CrossRef]
- Caporaso, J.G.; Lauber, C.L.; Walters, W.A.; Berg-Lyons, D.; Huntley, J.; Fierer, N.; Owens, S.M.; Betley, J.; Fraser, L.; Bauer, M.; et al. Ultra-high-throughput microbial community analysis on the Illumina HiSeq and MiSeq platforms. ISME J. 2012, 6, 1621–1624. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y.; Kellingray, L.; Le Gall, G.; Zhao, J.; Zhang, H.; Narbad, A.; Zhai, Q.; Chen, W. The divergent restoration effects of Lactobacillus strains in antibiotic-induced dysbiosis. J. Funct. Foods 2018, 51, 142–152. [Google Scholar] [CrossRef]
- Castro, V.S.; Teixeira, L.A.C.; Rodrigues, D.D.P.; Dos Santos, L.F.; Conte-Junior, C.A.; Figueiredo, E.E.S. Occurrence and antimicrobial resistance of E. coli non-O157 isolated from beef in Mato Grosso, Brazil. Trop. Anim. Health Prod. 2019, 51, 1117–1123. [Google Scholar] [CrossRef]
- Tan, H.; Zhao, J.; Zhang, H.; Zhai, Q.; Chen, W. Novel strains of Bacteroides fragilis and Bacteroides ovatus alleviate the LPS-induced inflammation in mice. Appl. Microbiol. Biotechnol. 2019, 103, 2353–2365. [Google Scholar] [CrossRef]
- Sun, Z.; Liu, W.; Gao, W.; Yang, M.; Zhang, J.; Wu, L.; Wang, J.; Menghe, B.; Sun, T.; Zhang, H. Identification and characterization of the dominant lactic acid bacteria from kurut: The naturally fermented yak milk in Qinghai, China. J. Gen. Appl. Microbiol. 2010, 56, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.; Xu, J.; Wang, J.; Menghebilige; Sun, T.; Li, H.; Guo, M. A survey on chemical and microbiological composition of kurut, naturally fermented yak milk from Qinghai in China. Food Control 2008, 19, 578–586. [Google Scholar] [CrossRef]
- Ferrario, C.; Milani, C.; Mancabelli, L.; Lugli, G.A.; Turroni, F.; Duranti, S.; Mangifesta, M.; Viappiani, A.; Sinderen, D.; Ventura, M. A genome-based identification approach for members of the genus Bifidobacterium. FEMS Microbiol. Ecol. 2015, 91, fiv009. [Google Scholar] [CrossRef]
- Kumar, S.; Nei, M.; Dudley, J.; Tamura, K. MEGA: A biologist-centric software for evolutionary analysis of DNA and protein sequences. Brief. Bioinform. 2008, 9, 299–306. [Google Scholar] [CrossRef] [PubMed]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Chenna, R.; Sugawara, H.; Koike, T.; Lopez, R.; Gibson, T.J.; Higgins, D.G.; Thompson, J.D. Multiple sequence alignment with the Clustal series of programs. Nucleic Acids Res. 2003, 31, 3497–3500. [Google Scholar] [CrossRef] [PubMed]
- Untergasser, A.; Cutcutache, I.; Koressaar, T.; Ye, J.; Faircloth, B.C.; Remm, M.; Rozen, S.G. Primer3—new capabilities and interfaces. Nucleic Acids Res. 2012, 40, e115. [Google Scholar] [CrossRef]
- Wojciech, R. OLIGO 7 Primer Analysis Software. Methods Mol. Biol. 2007, 402, 35–59. [Google Scholar]
- Ye, J.; Coulouris, G.; Zaretskaya, I.; Cutcutache, I.; Rozen, S.; Madden, T.L. Primer-BLAST: A tool to design target-specific primers for polymerase chain reaction. BMC Bioinform. 2012, 13, 134. [Google Scholar] [CrossRef]
- Jia, H.R.; Geng, L.L.; Li, Y.H.; Wang, Q.; Diao, Q.Y.; Zhou, T.; Dai, P.L. The effects of Bt Cry1Ie toxin on bacterial diversity in the midgut of Apis mellifera ligustica (Hymenoptera: Apidae). Sci. Rep. 2016, 6, 24664. [Google Scholar] [CrossRef]
- Dieffenbach, C.W.; Lowe, T.M.; Dveksler, G.S. General concepts for PCR primer design. PCR Methods Appl. 1993, 3, S30–S37. [Google Scholar] [CrossRef] [PubMed]
- Stsepetova, J.; Sepp, E.; Kolk, H.; Loivukene, K.; Songisepp, E.; Mikelsaar, M. Diversity and metabolic impact of intestinal Lactobacillus species in healthy adults and the elderly. Br. J. Nutr. 2011, 105, 1235–1244. [Google Scholar] [CrossRef] [PubMed]
- De Filippo, C.; Cavalieri, D.; Di Paola, M.; Ramazzotti, M.; Poullet, J.B.; Massart, S.; Collini, S.; Pieraccini, G.; Lionetti, P. Impact of diet in shaping gut microbiota revealed by a comparative study in children from Europe and rural Africa. Proc. Natl. Acad. Sci. USA 2010, 107, 14691–14696. [Google Scholar] [CrossRef]
- Armougom, F.; Henry, M.; Vialettes, B.; Raccah, D.; Raoult, D. Monitoring bacterial community of human gut microbiota reveals an increase in Lactobacillus in obese patients and Methanogens in anorexic patients. PLoS ONE 2009, 4, e7125. [Google Scholar] [CrossRef] [PubMed]
- Zmora, N.; Suez, J.; Elinav, E. You are what you eat: Diet, health and the gut microbiota. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 35–56. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Samples | Human | Rat | Mice | Kurut | |
|---|---|---|---|---|---|
| Genus | |||||
| Enterobacteriaceae | 0.28(0.12) | 0(0) | 0(0) | 0(0) | |
| Streptococcus | 0.11(0.09) | 0(0) | 0.02(0.02) | 0.15(0.03) | |
| Bacteroides | 0.1(0.05) | 0.03(0) | 0(0) | 0(0) | |
| Bifidobacterium | 0.09(0.03) | 0(0) | 0.07(0.05) | 0(0) | |
| other genera <1% | 0.09(0.02) | 0.07(0) | 0.05(0.01) | 0.01(0) | |
| Blautia | 0.06(0.03) | 0.01(0) | 0(0) | 0(0) | |
| Ruminococcaceae | 0.05(0.02) | 0.06(0) | 0(0) | 0(0) | |
| Clostridiales | 0.04(0.03) | 0.16(0.01) | 0.12(0.03) | 0(0) | |
| Lachnospiraceae | 0.04(0.01) | 0.03(0.01) | 0.04(0.02) | 0(0) | |
| Clostridiaceae | 0.03(0.01) | 0.01(0) | 0(0) | 0(0) | |
| Collinsella | 0.02(0.01) | 0(0) | 0(0) | 0(0) | |
| Enterococcus | 0.02(0.01) | 0(0) | 0.01(0) | 0(0) | |
| Erysipelotrichaceae | 0.01(0) | 0(0) | 0(0) | 0(0) | |
| Eubacterium | 0.01(0) | 0(0) | 0(0) | 0(0) | |
| Lactobacillus | 0.01(0) | 0.13(0.02) | 0.14(0.02) | 0.82(0.03) | |
| Mitsuokella | 0.01(0.01) | 0(0) | 0(0) | 0(0) | |
| Parabacteroides | 0.01(0) | 0(0) | 0(0) | 0(0) | |
| Prevotella | 0.01(0.01) | 0.08(0.01) | 0(0) | 0(0) | |
| Ruminococcus | 0.01(0) | 0.03(0) | 0.05(0.01) | 0(0) | |
| Adlercreutzia | 0(0) | 0(0) | 0.03(0.01) | 0(0) | |
| Aerococcaceae | 0(0) | 0(0) | 0.01(0) | 0(0) | |
| Aerococcus | 0(0) | 0(0) | 0.06(0.04) | 0(0) | |
| Pseudomonas | 0(0) | 0(0) | 0(0) | 0.01(0) | |
| Bacteroidales | 0(0) | 0.17(0.01) | 0.13(0.06) | 0(0) | |
| Coprococcus | 0(0) | 0.01(0) | 0(0) | 0(0) | |
| Desulfovibrionaceae | 0(0) | 0.01(0) | 0(0) | 0(0) | |
| Helicobacteraceae | 0(0) | 0.01(0) | 0(0) | 0(0) | |
| Planococcaceae | 0(0) | 0(0) | 0.03(0.01) | 0(0) | |
| Pseudomonas | 0(0) | 0(0) | 0.01(0) | 0(0) | |
| Oscillospira | 0(0) | 0.04(0) | 0.01(0) | 0(0) | |
| Peptostreptococcaceae | 0(0) | 0.05(0.01) | 0(0) | 0(0) | |
| Staphylococcus | 0(0) | 0(0) | 0.21(0.07) | 0(0) | |
| Rikenellaceae | 0(0) | 0.04(0.01) | 0(0) | 0(0) | |
| Turicibacter | 0(0) | 0.02(0) | 0.01(0) | 0(0) | |
| unassigned genera | 0(0) | 0.04(0.01) | 0.01(0) | 0(0) | |
| Samples | Human | Rat | Mice | Kurut | |
|---|---|---|---|---|---|
| Species | |||||
| L. mucosae | 0.67(0.04) | 0(0) | 0.01(0.01) | 0(0) | |
| L. gasseri | 0.06(0.02) | 0(0) | 0(0) | 0(0) | |
| L. salivarius | 0.06(0.01) | 0(0) | 0(0) | 0(0) | |
| other Lactobacillus <1% | 0.06(0.01) | 0(0) | 0(0) | 0(0) | |
| L. oris | 0.05(0.03) | 0.01(0) | 0(0) | 0(0) | |
| L. rhamnosus | 0.03(0.01) | 0(0) | 0(0) | 0(0) | |
| L. amylovorus | 0.02(0.01) | 0(0) | 0(0) | 0(0) | |
| L. casei | 0.01(0) | 0(0) | 0(0) | 0(0) | |
| L. crispatus | 0.01(0) | 0(0) | 0(0) | 0(0) | |
| L. plantarum | 0.01(0.01) | 0.01(0) | 0.01(0) | 0(0) | |
| L. vaginalis | 0.01(0.01) | 0(0) | 0(0) | 0(0) | |
| L. acidophilus | 0(0) | 0(0) | 0.1(0.03) | 0(0) | |
| L. fermentum | 0(0) | 0.05(0.05) | 0(0) | 0(0) | |
| L. reuteri | 0(0) | 0.1(0.04) | 0.11(0.02) | 0(0) | |
| unassigned Lactobacillus | 0(0) | 0.07(0.02) | 0.1(0.02) | 0(0) | |
| L. delbrueckii subsp. bulgaricus | 0(0) | 0(0) | 0(0) | 0.97(0.03) | |
| L. helveticus | 0(0) | 0(0) | 0(0) | 0.03(0.03) | |
| L. intestinalis | 0(0) | 0.33(0.1) | 0.01(0) | 0(0) | |
| L. johnsonii | 0(0) | 0.4(0.11) | 0.65(0.04) | 0(0) | |
| L. sp. L6 | 0(0) | 0.01(0) | 0.01(0) | 0(0) | |
| L. animalis | 0(0) | 0.01(0) | 0(0) | 0(0) | |
| unassigned species | 0(0) | 0.01(0) | 0(0) | 0(0) | |
| Species | groEL identity % with the Closet Species 2 | Closest Species |
|---|---|---|
| L. acidophilus | 100 | L. amylovorus |
| L. acidophilus | 99.8 | L. kitasatonis |
| L. amylolyticus | 99.6 | L. helveticus |
| L. buchneri | 99.8 | L. hilgardii |
| L. farciminis | 98.4 | L. formosensis |
| L. gasseri | 100 | L. paragasseri |
| L. johnsonii | 98.9 | L. taiwanensis |
| L. plantarum | 100 | L. pentosus |
| L. sakei | 99.3 | L. curvatus |
| L. zeae | 100 | L. paracasei |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Xie, M.; Pan, M.; Jiang, Y.; Liu, X.; Lu, W.; Zhao, J.; Zhang, H.; Chen, W. groEL Gene-Based Phylogenetic Analysis of Lactobacillus Species by High-Throughput Sequencing. Genes 2019, 10, 530. https://doi.org/10.3390/genes10070530
Xie M, Pan M, Jiang Y, Liu X, Lu W, Zhao J, Zhang H, Chen W. groEL Gene-Based Phylogenetic Analysis of Lactobacillus Species by High-Throughput Sequencing. Genes. 2019; 10(7):530. https://doi.org/10.3390/genes10070530
Chicago/Turabian StyleXie, Miaoqi, Mingluo Pan, Yang Jiang, Xiaoming Liu, Wenwei Lu, Jianxin Zhao, Hao Zhang, and Wei Chen. 2019. "groEL Gene-Based Phylogenetic Analysis of Lactobacillus Species by High-Throughput Sequencing" Genes 10, no. 7: 530. https://doi.org/10.3390/genes10070530
APA StyleXie, M., Pan, M., Jiang, Y., Liu, X., Lu, W., Zhao, J., Zhang, H., & Chen, W. (2019). groEL Gene-Based Phylogenetic Analysis of Lactobacillus Species by High-Throughput Sequencing. Genes, 10(7), 530. https://doi.org/10.3390/genes10070530