3.1. Sequencing and Data Polishing
In this study, we constructed three ISO-Seq libraries for one sample and sequenced eight cells in total using the Pacific Bioscience RS II platform. Finally, 489,270 ROIs with 1,041,604,967 bp were generated (
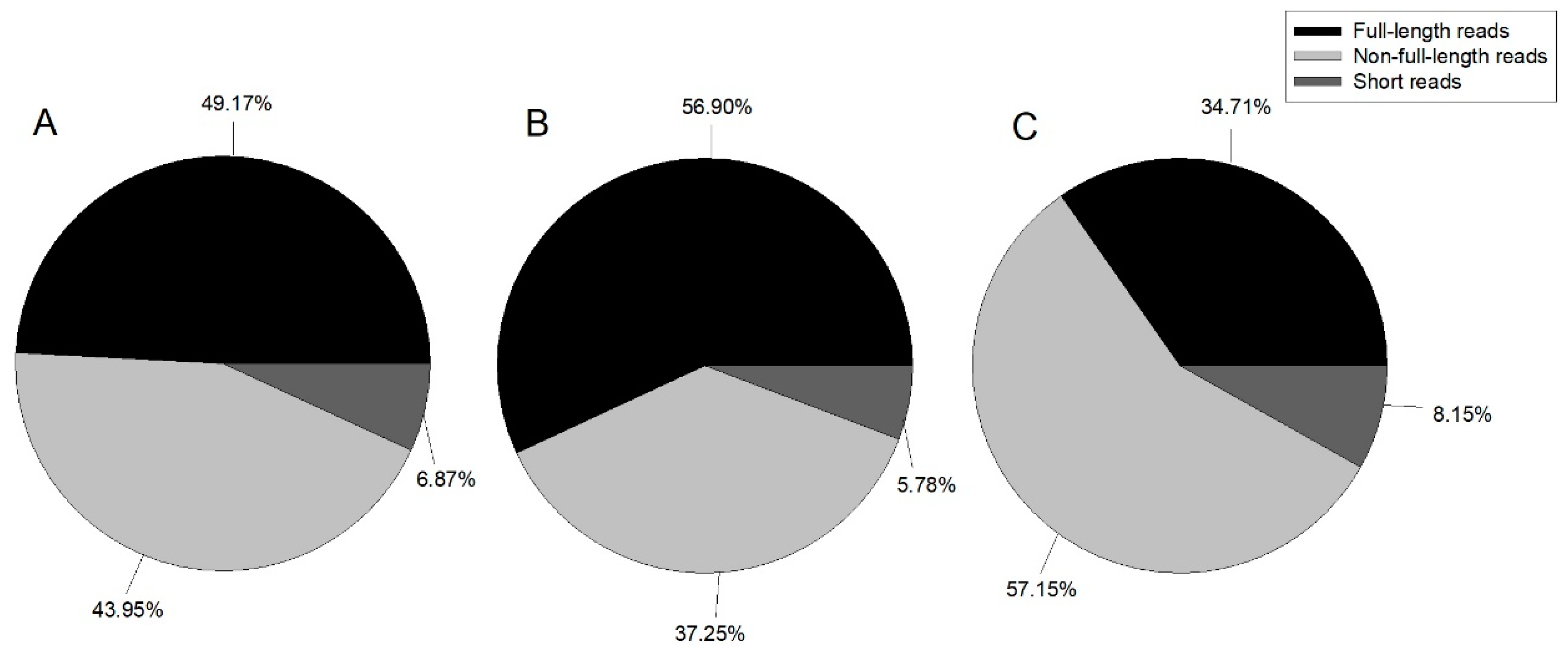
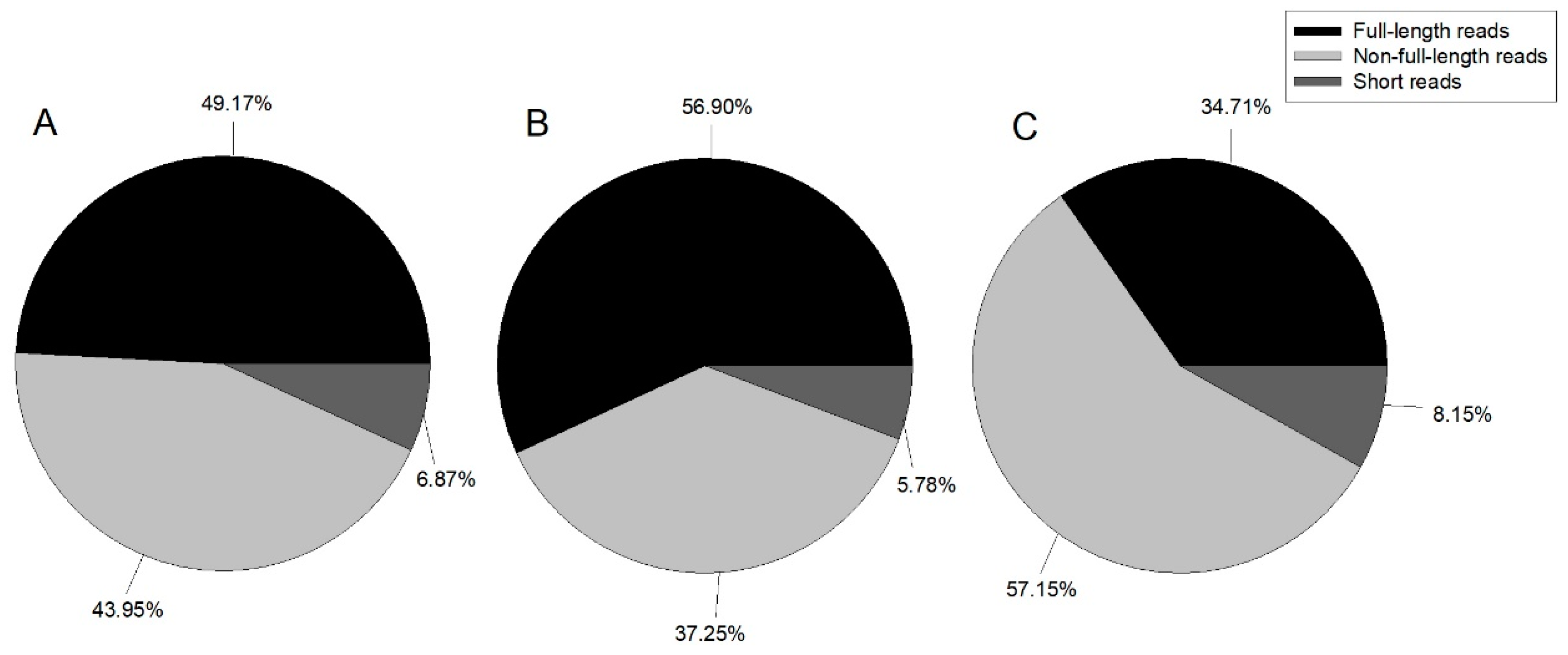
Table 1). Reads were classified by whether the 5’ primer, 3’ primer, or poly A tail were detected, with reads containing 5’ primer, 3’ primer, and poly A tail considered as full-length; those with the 5’ primer and 3’ primer, 3’ primer and poly A tail, or 5’ primer and poly A tail classified as non-full-length; and all other reads classified as short reads.
Figure 1 illustrates the proportion of each category.
Although PacBio single molecule sequencing yields long reads, it has a high error rate; therefore, we used Quiver to polish the consensus isoforms. The basic steps were as follows: full-length reads were clustered into consensus reads. For each cluster, if there was sufficient full-length and non-full-length coverage, then Quiver was run to polish the consensus. Depending on the Quiver output quality value, which indicated how confident the consensus calls were, the script binned the Quiver polished output as either high or low quality. Cluster summaries of each library are shown in
Table 2. High-quality consensus sequences were subjected to further analysis. High-quality consensus isoforms of each library were merged into the final result, and redundancy was removed. The isoforms are summarized in
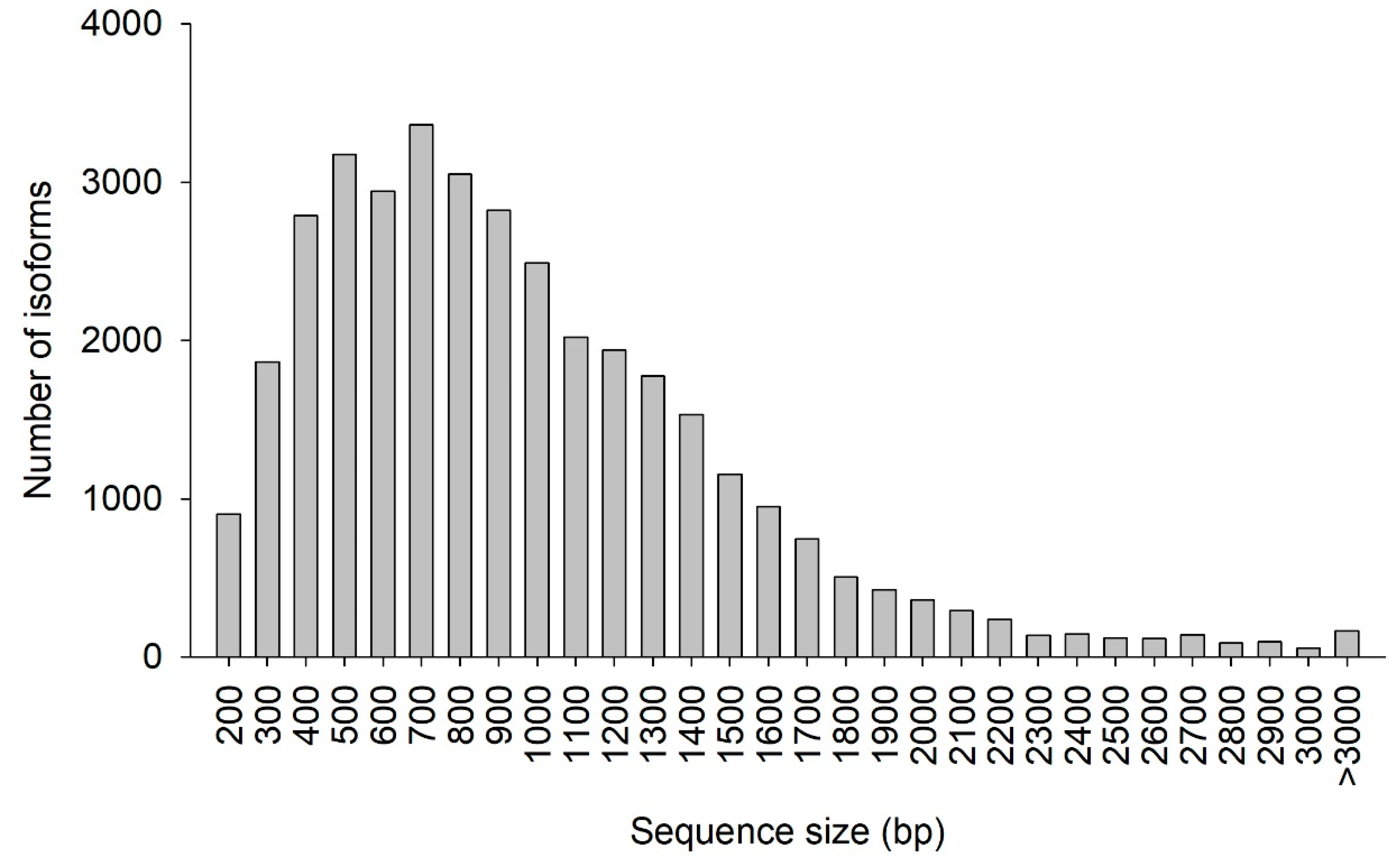
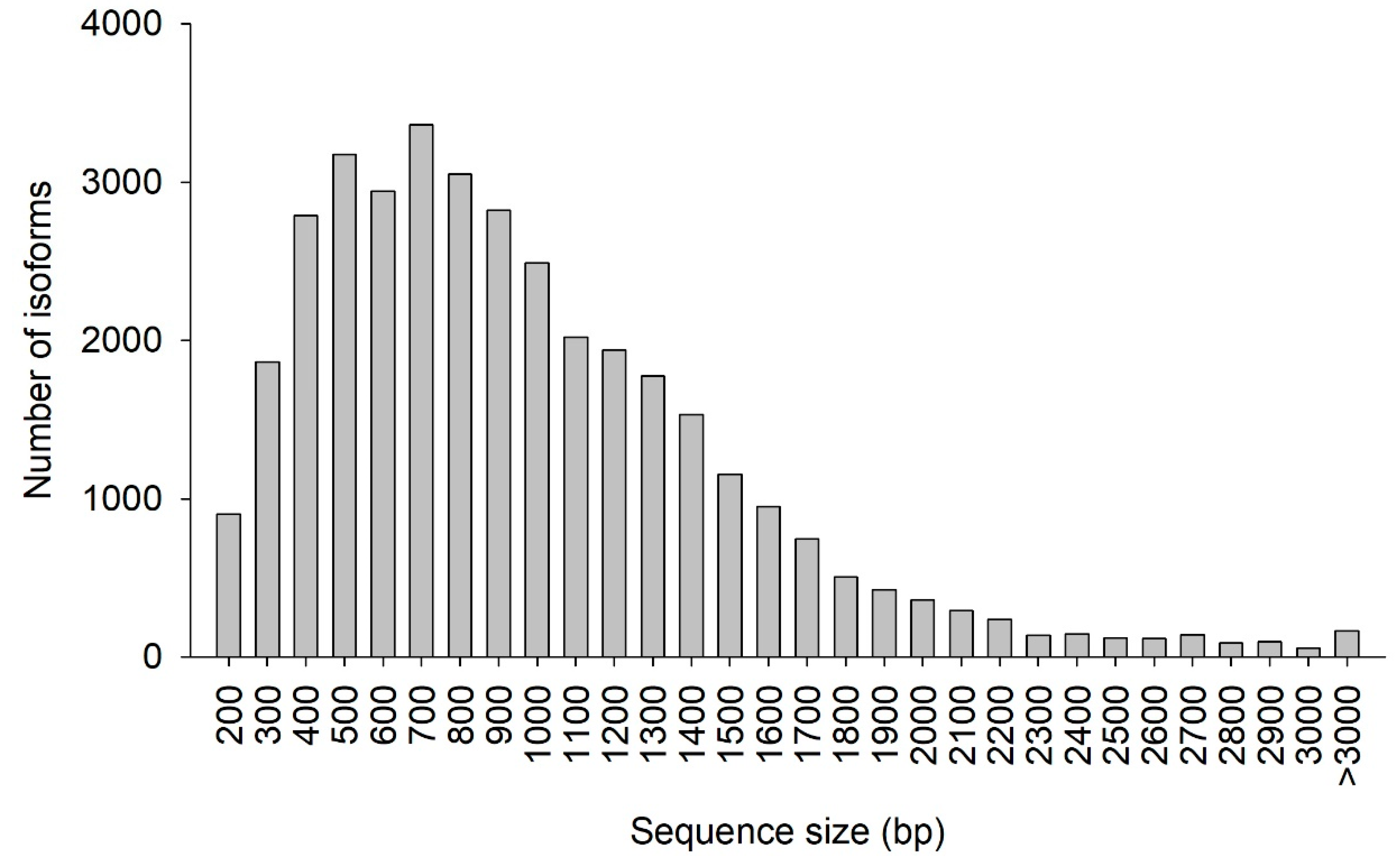
Table 3. A total of 39,129 isoforms were obtained with a total length of 76,617,709 bp, a mean length of 1958 bp, and an N50 length of 2044 bp (half of the isoforms is larger than or equal the 2044 bp). This number of isoforms is lower than that reported by Gao et al. [
7], who found a transcriptome of 47,578 unigenes, but higher than that reported by Chen et al. [
6], who assembled 16,829 unigenes. This may have been because Gao et al. [
7] used three developmental stages (third-instar, fourth-instar, and pupa) for de novo transcriptome assembly, but Chen et al. [
6] only used third-instar larvae for unigene assembly, and some genes were only expressed in the specific development stage. The number of unigenes generated from the three developmental stages in Gao’s study was greater than the number of isoforms generated from all development stages in our study. This may have been because some unigenes only have the partial sequence of the genes, and the isoforms generated in this study could produce the full length of the genes. This also explains why the mean length and N50 in this study (1958 bp and 2044 bp, respectively) are much longer than those reported by Gao et al. [
7] and Chen et al. [
6], who reported mean lengths of 860 bp and 1517 bp and N50 lengths of 576 and 762 bp, respectively.
3.2. Functional Annotation
After obtaining the isoforms, we used BLAST, Blast2GO, and InterProScan5 to perform functional annotation based on the NR, NT, GO, COG, KEGG, Swiss-Prot, and InterPro databases (
Table 4). Overall, 91.10% of the isoforms had annotation results with different databases. In the previous
B. odoriphaga short-read sequence transcriptome study, 12,480 (74.16%) out of 16,829 and 21,985 (46.21%) out of 47,578 unigenes had annotation results against different databases in Chen’s and Gao’s studies, respectively [
6,
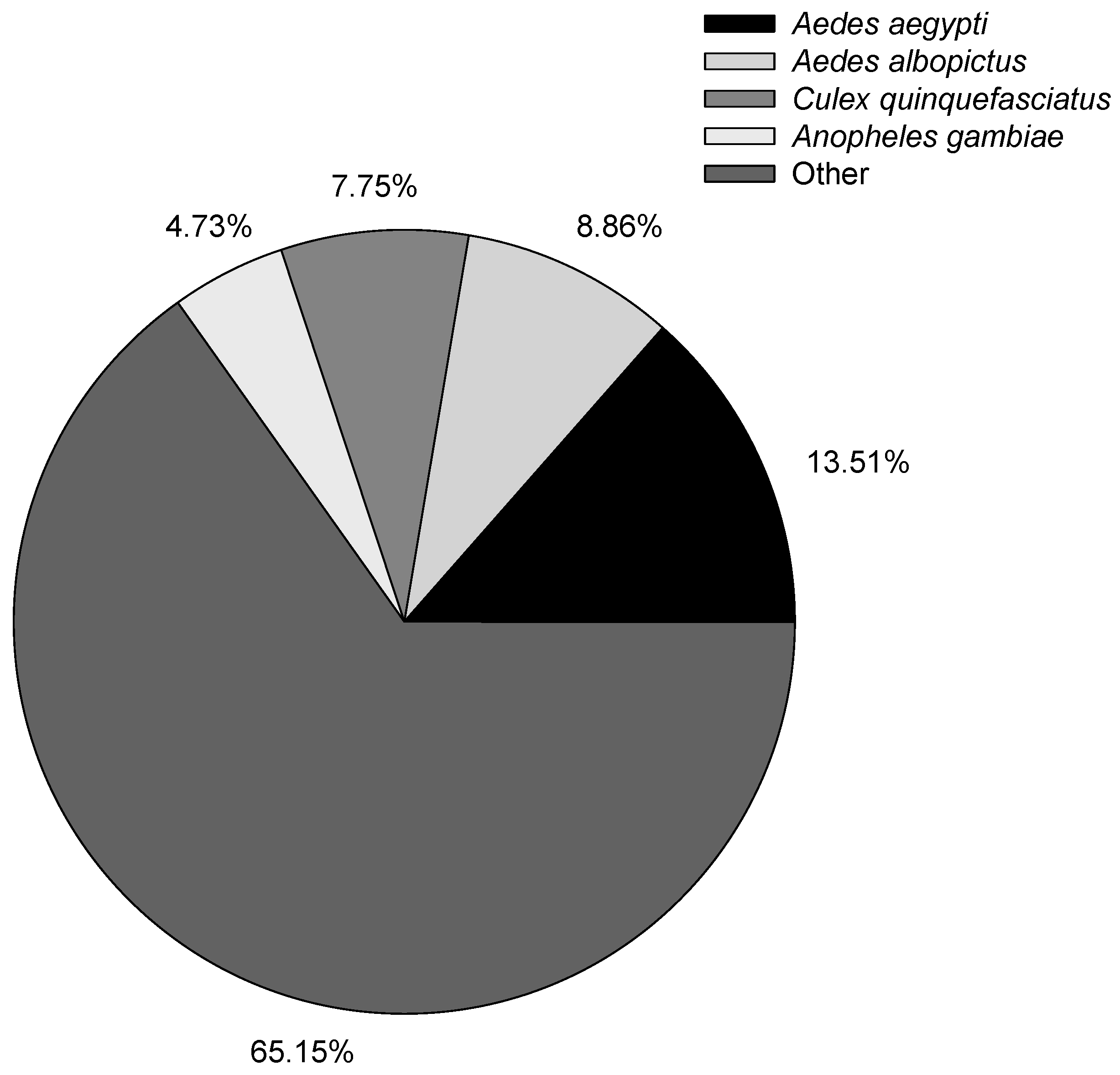
7]. The higher annotated percentage of the isoform shows the good quality of long-read sequence results obtained using the Pacific Bioscience RS II platform. The species distribution with NR annotation is shown in
Figure 2, and the most annotated species were mosquitoes, with 13.51% of the isoforms closely matching
Aedes aegypti, followed by
Aedes albopictus (8.86%),
Culex quinquefasciatus (7.75%), and
Anopheles gambiae (4.73%). Evaluation of the species distribution confirmed that
B. odoriphaga belonged to the suborder of Nematocera rather than Brachycera. The larvae of
B. odoriphaga look like fly larvae, but the adults look like mosquitos, which has led to confusion regarding which sub-order they belong to. However, the species distribution showed that the top four species were midges, indicating that
B. odoriphaga are more closely related to midges than flies.
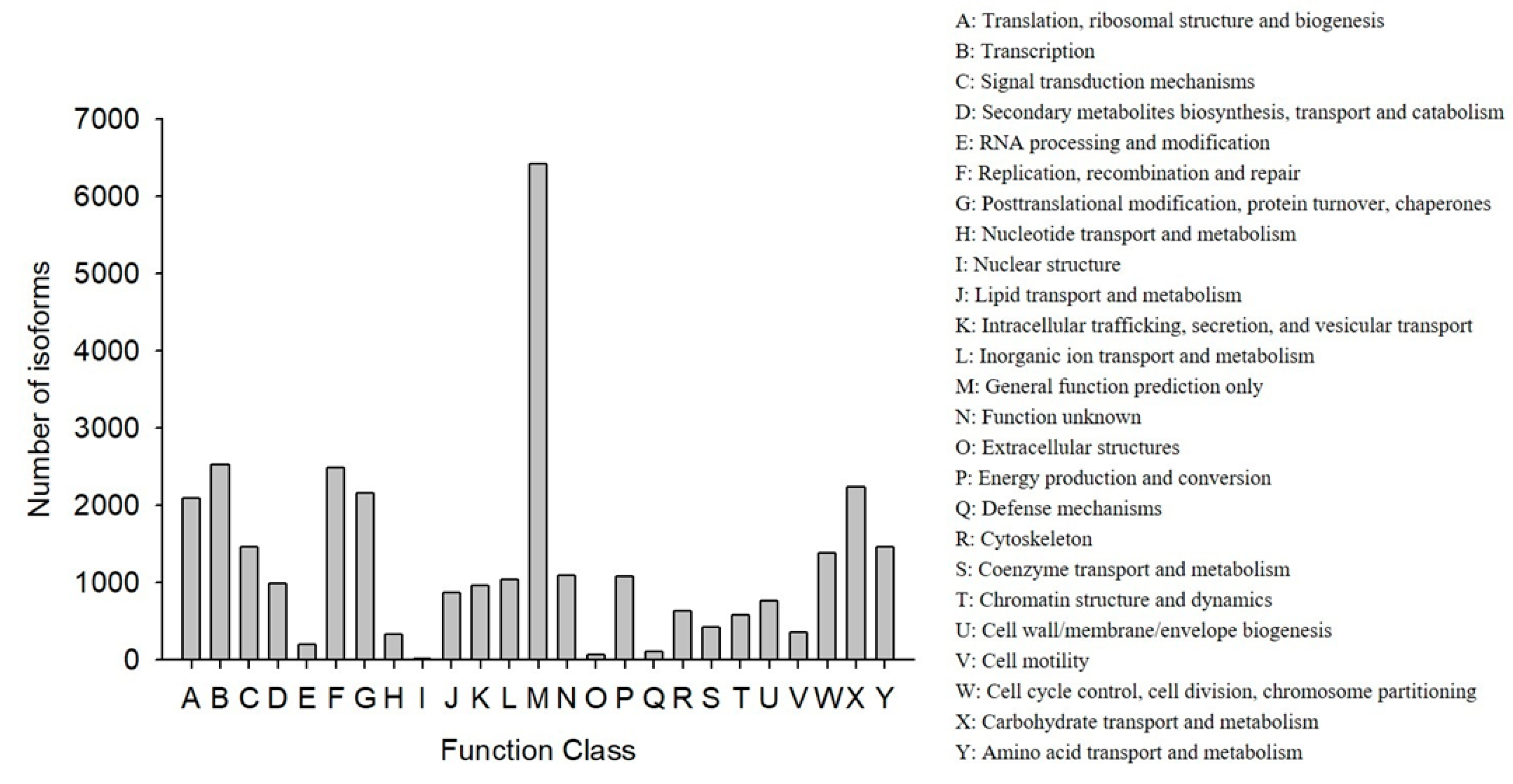
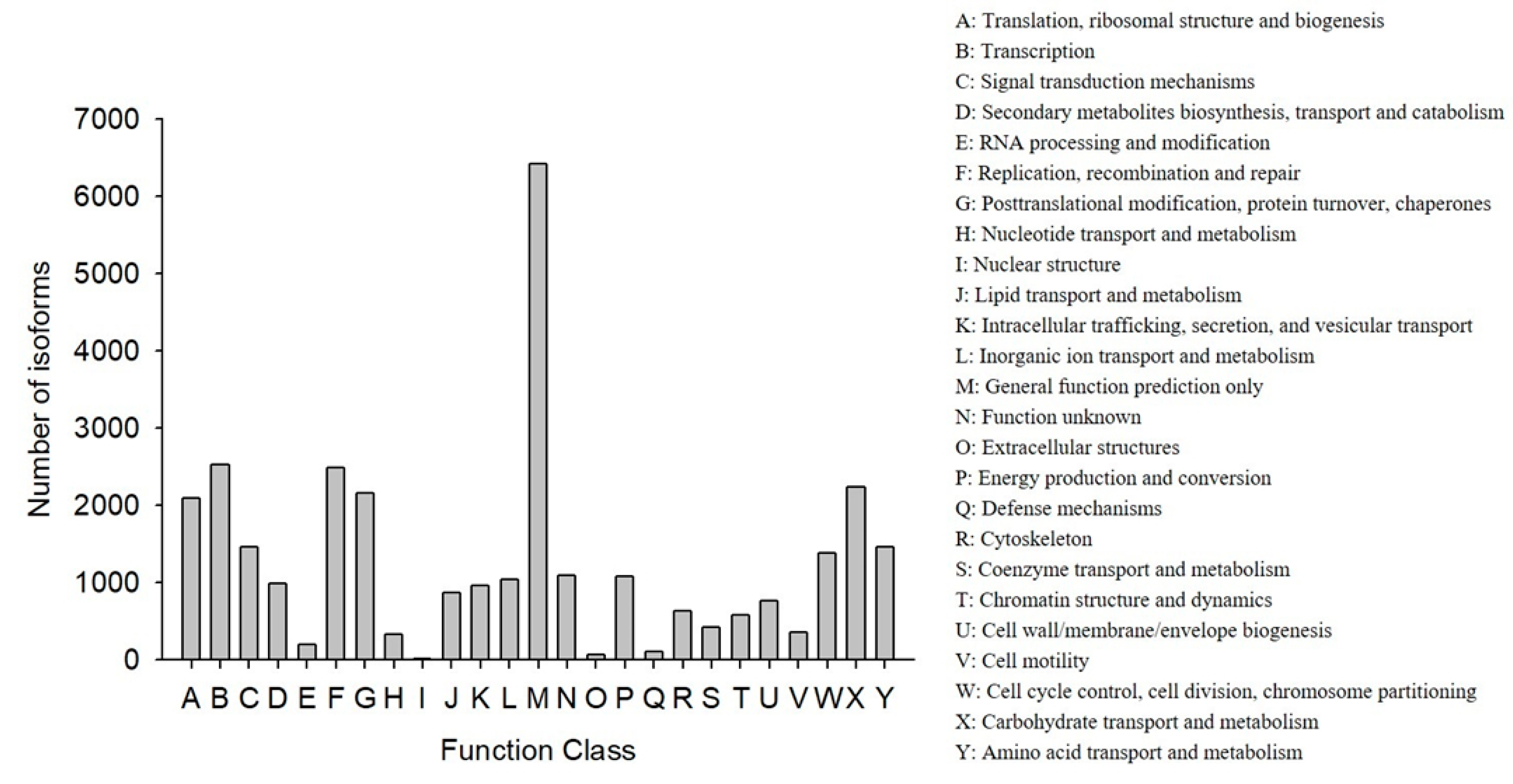
COG classification showed that 18,473 out of the 33,238 NR hit isoforms could be categorized (
Table 4). Among the 25 COG categories, the largest group was the cluster for “general function prediction only” (6417, or 20.27%), followed by “transcription” (2523, or 7.97%), and then “replication, recombination, and repair” (2483, or 7.84%), while the categories of “nuclear structure” (16, or 0.05%), “extracellular structures” (65, or 0.21%), and “defense mechanisms” (110, or 0.35%) were the smallest clusters (
Figure 3).
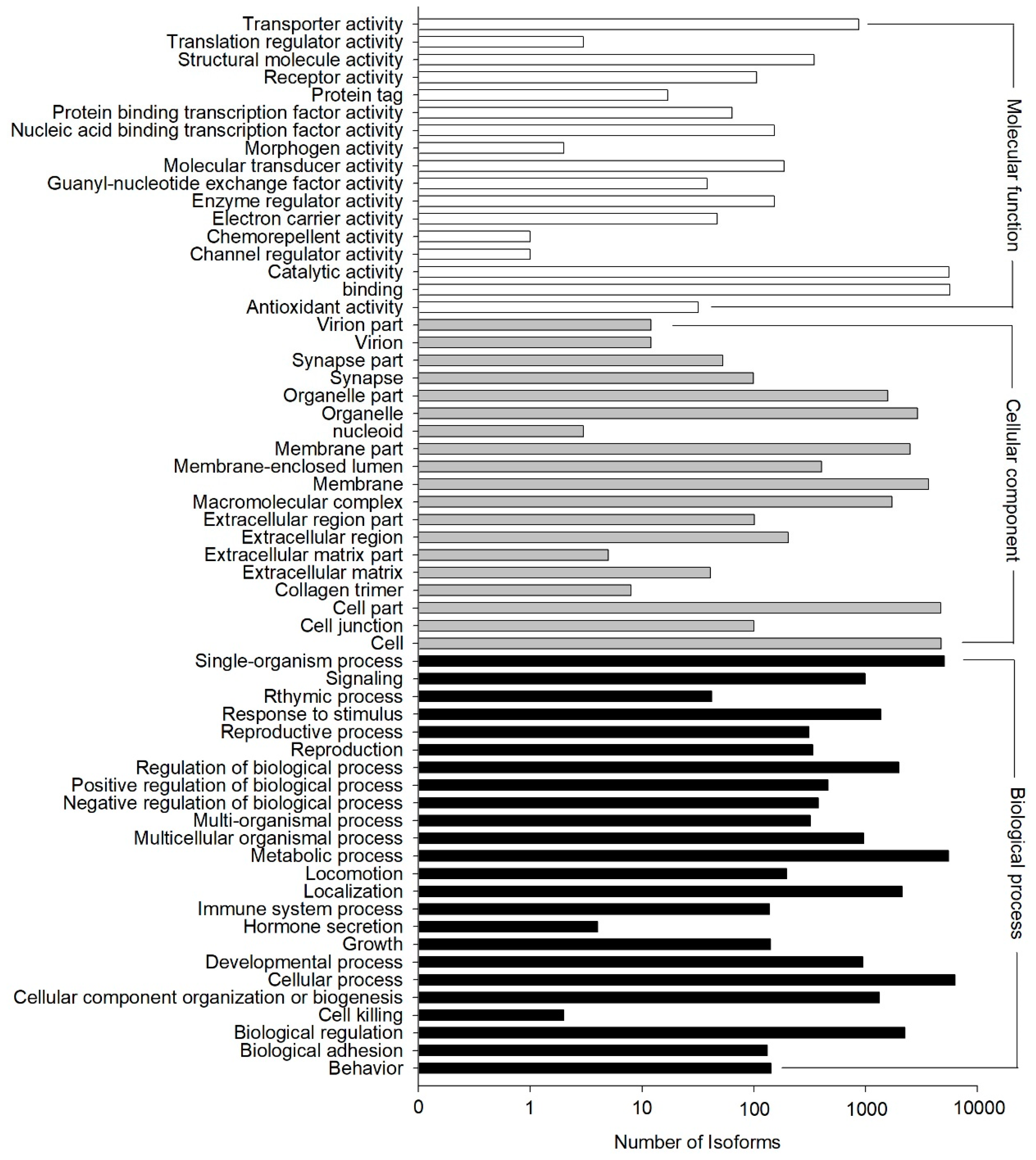
The GO annotation results showed that 11,880 out of the 33,238 NR hit isoforms could be assigned to 67,635 GO terms, with the three main categories of biological process, cellular component, and molecular function assigned 13,312, 22,878, and 31,445 terms, respectively (
Figure 4). GO terms were finally assigned into 60 functional groups, and the number of “cellular process” terms (6314 terms) was the largest for “biological process”, “cell” (4742 terms) for “cellular component”, and “binding” (5704 terms) for “molecular function.” In contrast, there were only two terms in the cluster of “cell killing”, which was among the main categories of “biological process”, “nucleoid” (three terms), the main category of “cellular component”, and both “channel regulator activity” (one term) and “chemorepellent activity” (one term) from the category of “molecular function”. (
Figure 4).
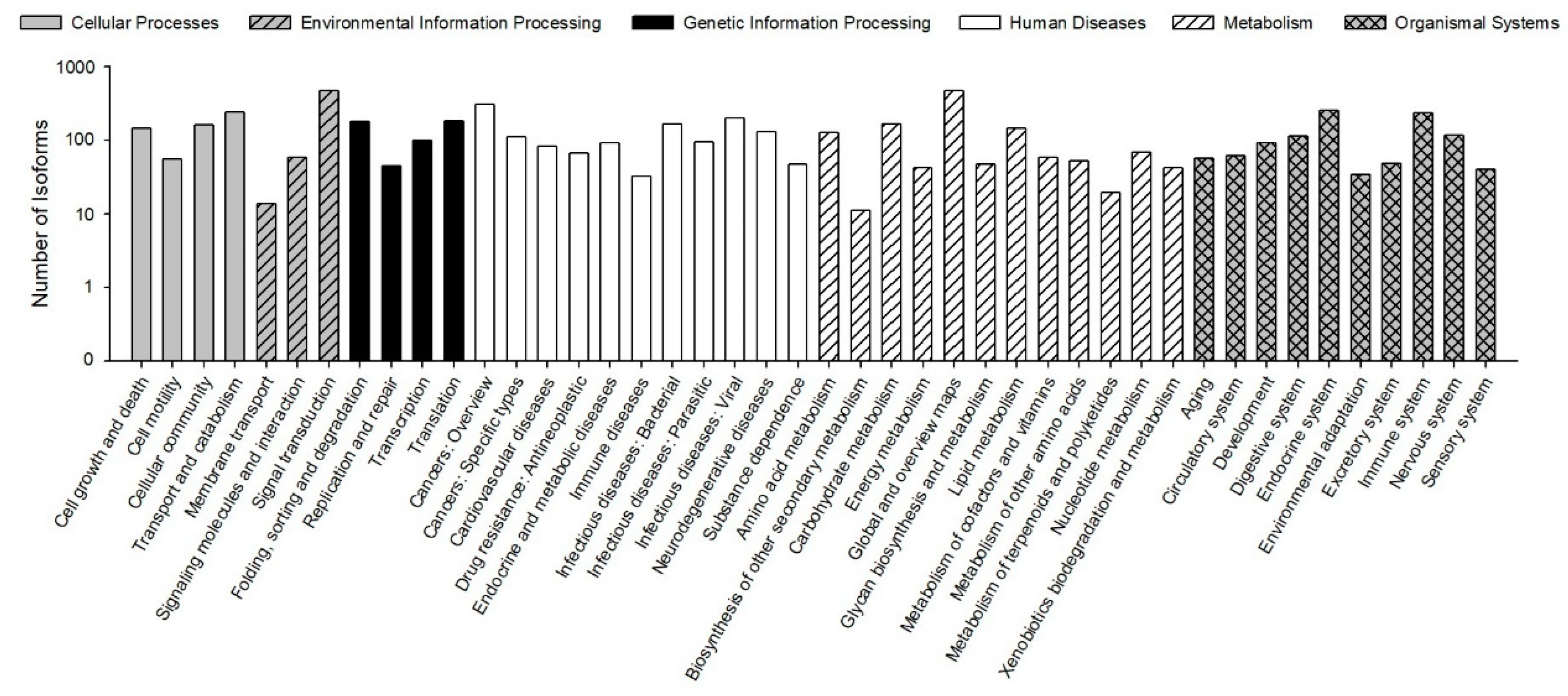
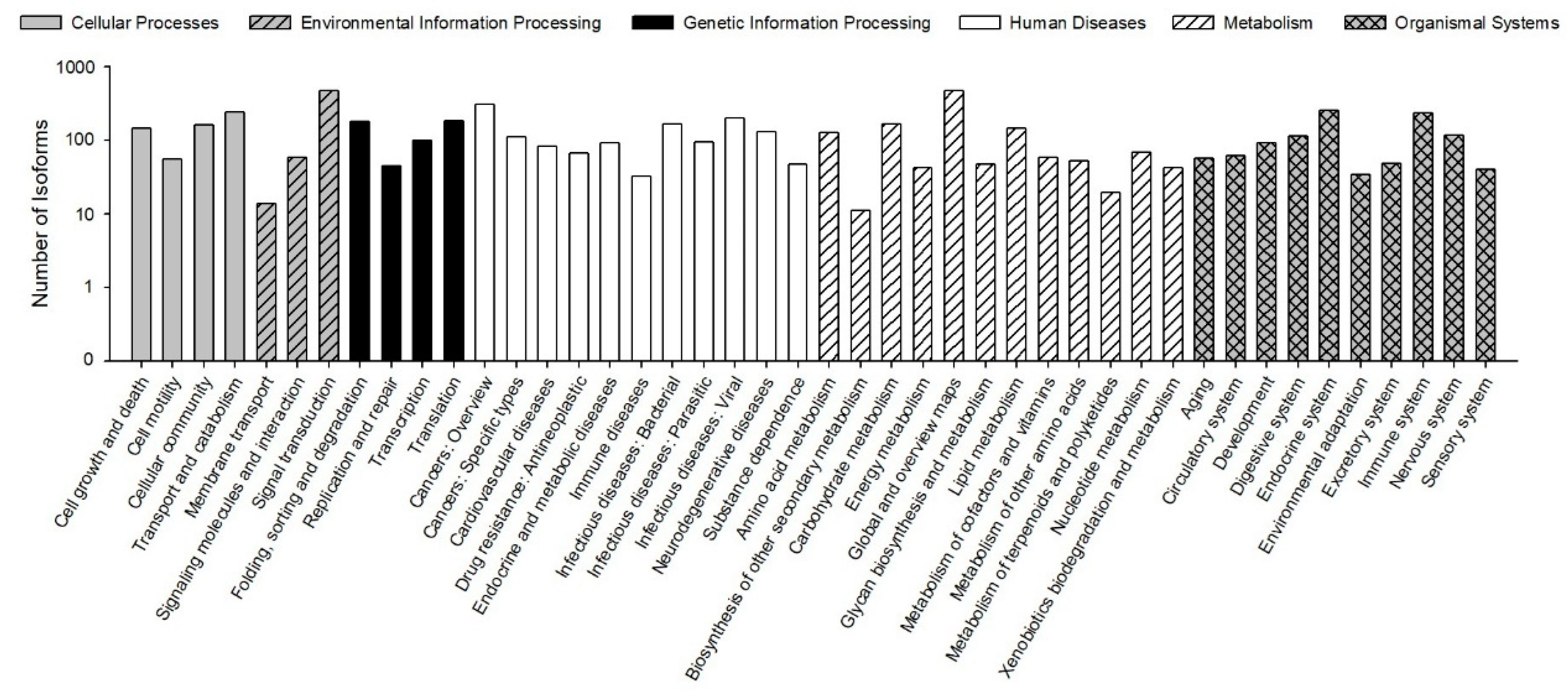
The KEGG pathways results showed that 30,610 (78.23% of 39,129) isoforms were mapped into different KEGG pathways. The pathways most assigned by the isoforms were “signal transduction” (4720, 8.92%), followed by “global and overview maps” (4672, 8.83%), and “cancers: overview” (3072, 5.80%) (
Figure 5). The KEGG pathway could provide basic information for isoforms involved in the specific processes and pathways during
B. odoriphaga research. For example, 428 isoforms were mapped into “xenobiotics biodegradation and metabolism,” and this pathway is closely related to insecticide detoxification and metabolism.
3.4. Insecticide Resistance and Metabolism-Related Isoforms
The functions annotated in the NR database related to cytochrome P450, glutathione transferase, carboxylesterase, trypsin, NADH dehydrogenase, catalase, sodium channel, acetylcholinesterase, superoxide dismutase, nicotinic acetylcholine receptor, and GABA receptor in this study and previous studies [
6] are listed in
Table 7. Less insecticide resistance and metabolism-related genes were generated in this study than in the results of the larval transcriptome (
Table 7), which may have been because the isoforms generated in this study could contain the full length of the genes, but those observed in previous studies only contained the partial sequence of the genes. There were 107 isoforms related to Cytochrome P450, which is a reasonable number for insect species because the number of P450 genes in insects ranges from 45 in
Apis mellifera to 204 in
C. quinquefasciatus, and there is an average of around 100 P450 genes in insect species [
24,
25,
26]. Usually, insects have one or two acetylcholinesterase genes, but based on previous
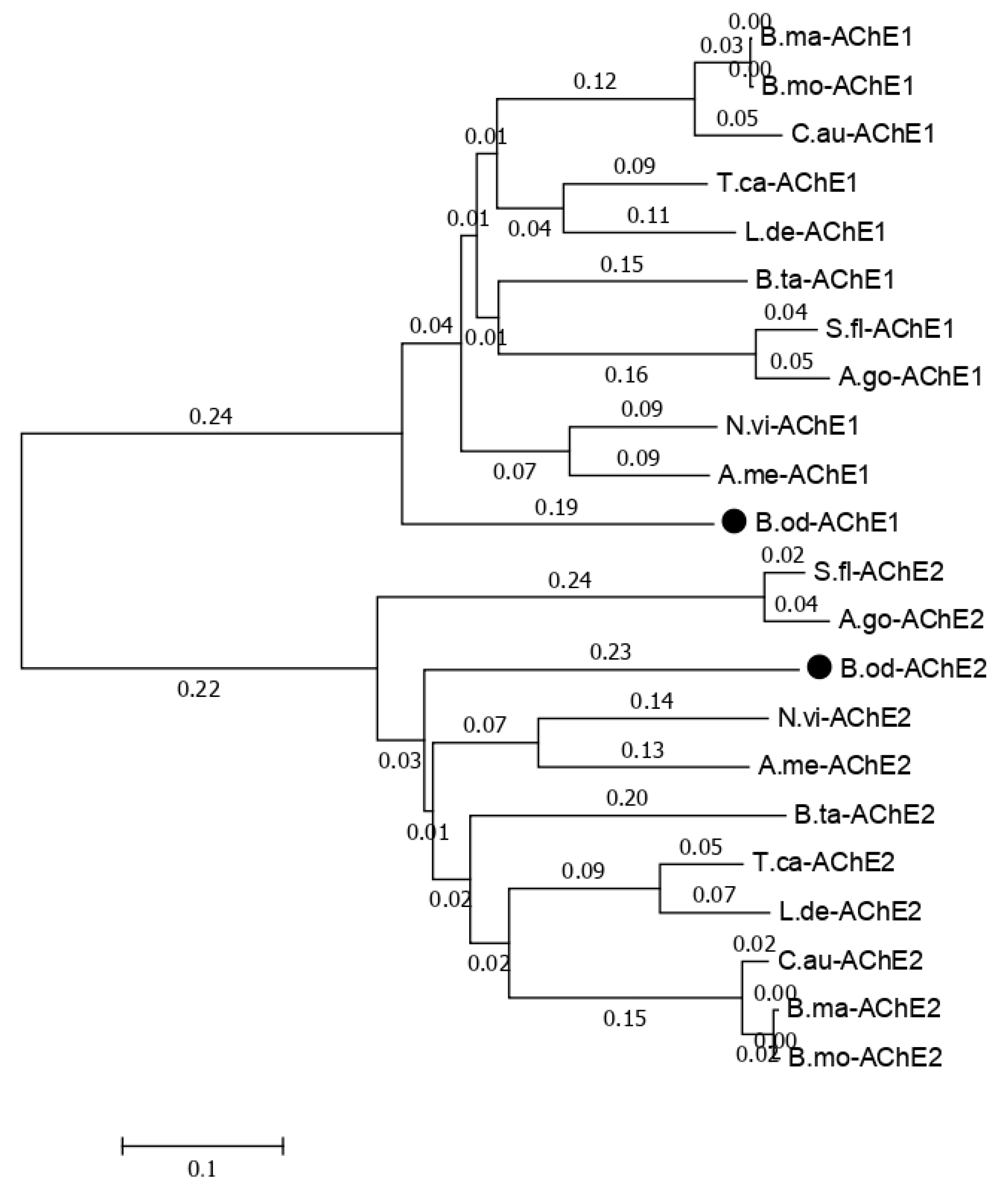
B. odoriphaga transcriptome data [
6], there are eight unigenes related to the acetylcholinesterase gene. In this study, we identified two isoforms of the acetylcholinesterase gene (
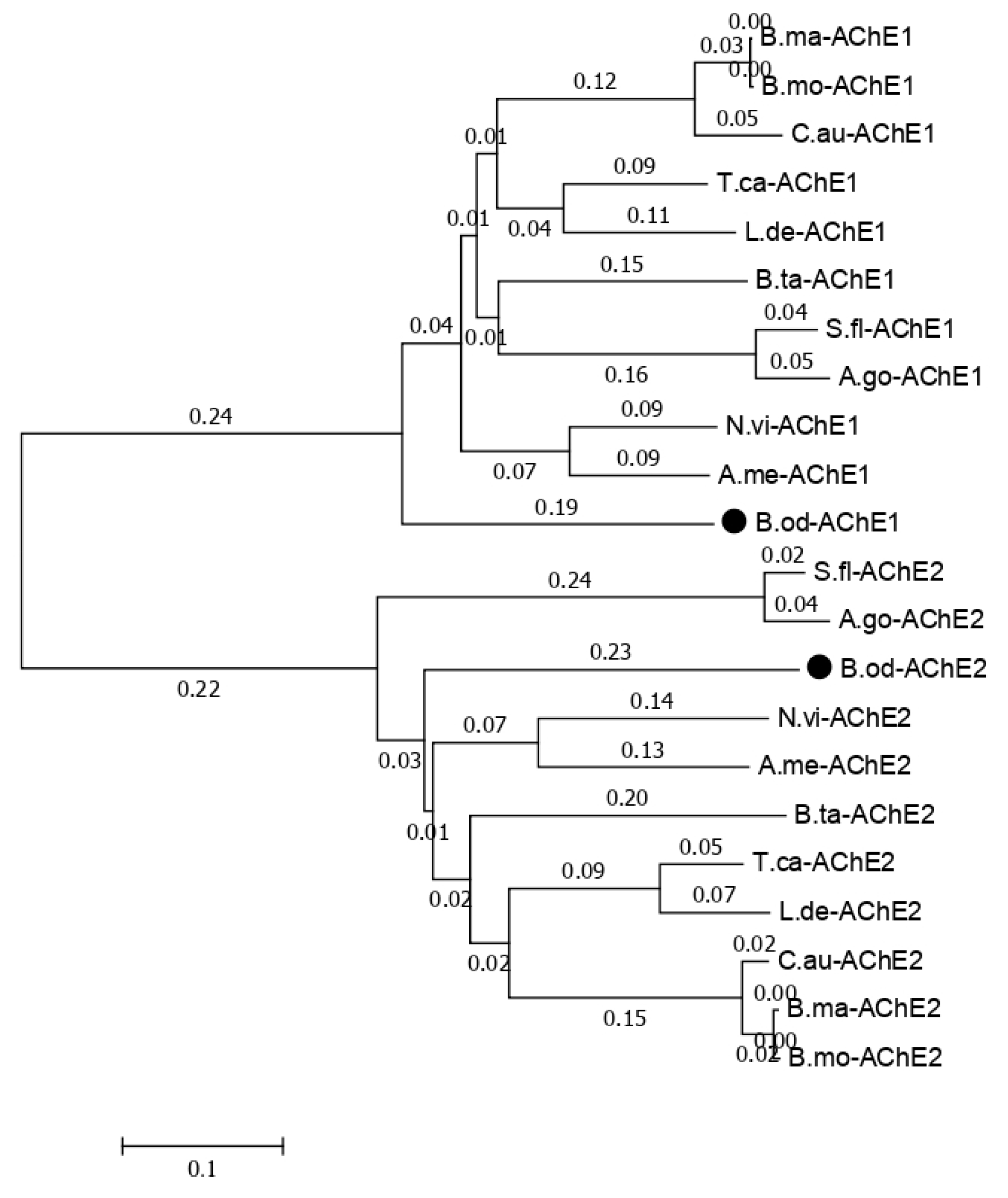
Figure S1). The phylogenic tree for the acetylcholinesterase-related isoforms with other insects that have two acetylcholinesterase genes shows that
B. odoriphaga contains two acetylcholinesterase genes (
Figure 7).



{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}