Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain?

{kind=link}
{kind=link}
Abstract
1. Introduction
2. Hormone Signaling at Birth Defines Sex Differences in Brain Function
3. Regulation of Gene Expression in the Brain
3.1. DNA Methylation
3.2. Histone Modifications
3.3. Genome Organization
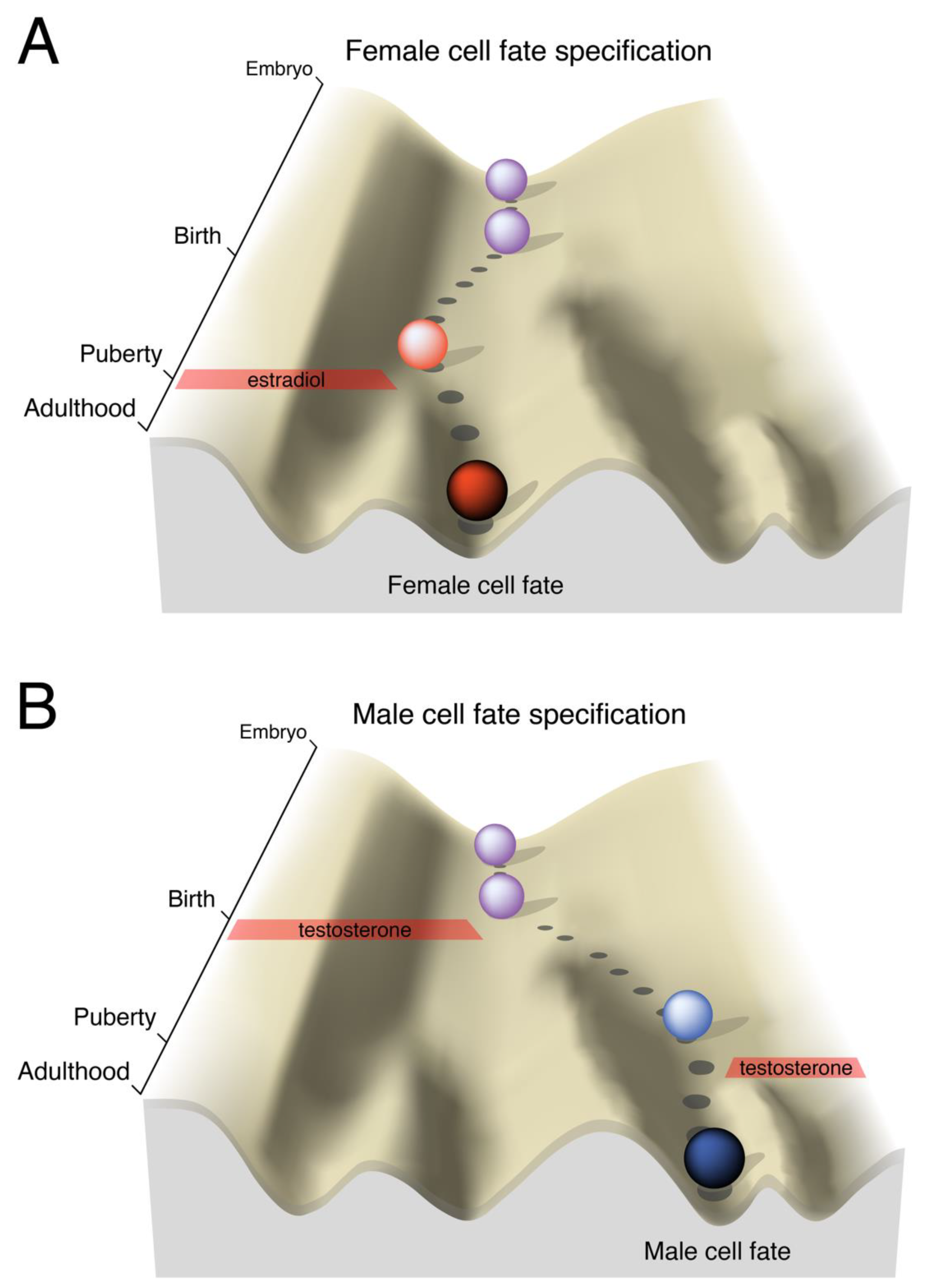
4. Sexual Differentiation of the Brain is Developmental Programming
5. Sex Differences in Gene Regulation May Underlie Sex Differences in Disease Susceptibility
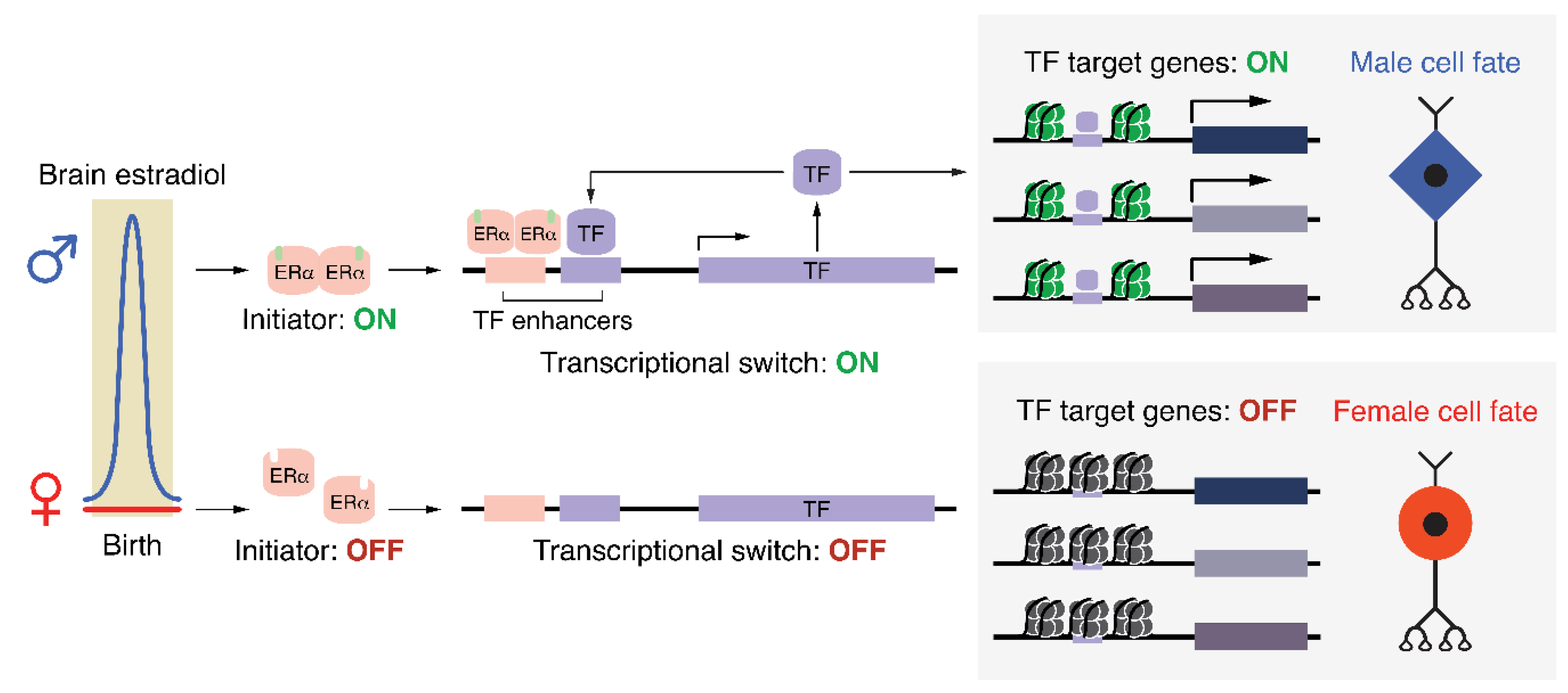
6. Towards a bona fide Epigenetic Mechanism Underlying Sex Differences in the Brain
Funding
Conflicts of Interest
References
- Berger, S.L.; Kouzarides, T.; Shiekhattar, R.; Shilatifard, A. An operational definition of epigenetics. Genes Dev. 2009, 23, 781–783. [Google Scholar] [CrossRef] [PubMed]
- Ptashne, M. Epigenetics: Core misconcept. Proc. Natl. Acad. Sci. USA 2013, 110, 7101–7103. [Google Scholar] [CrossRef] [PubMed]
- Henikoff, S.; Greally, J.M. Epigenetics, cellular memory and gene regulation. Curr. Biol. 2016, 26, R644–R648. [Google Scholar] [CrossRef] [PubMed]
- Gilbert, S.F. Commentary: “The epigenotype” by C.H. Waddington. Int. J. Epidemiol. 2012, 41, 20–23. [Google Scholar] [CrossRef] [PubMed]
- Hershey, A.D.; Chase, M. Independent functions of viral protein and nucleic acid in growth of bacteriophage. J. Gen. Physiol. 1952, 36, 39–56. [Google Scholar] [CrossRef] [PubMed]
- Waddington, C.H. Organisers and Genes (1940), by Conrad Hal Waddington; The University Press: Dhaka, Bangladesh, 1940. [Google Scholar]
- Waddington, C.H. The Strategy of the Genes; Routledge: Abingdon-on-Thames, UK, 1957. [Google Scholar]
- Phoenix, C.H.; Goy, R.W.; Gerall, A.A.; Young, W.C. Organizing action of prenatally administered testosterone propionate on the tissues mediating mating behavior in the female guinea pig. Endocrinology 1959, 504, 369–382. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Wright, C.L.; Schwarz, J.M. New tricks by an old dogma: Mechanisms of the Organizational/Activational Hypothesis of steroid-mediated sexual differentiation of brain and behavior. Horm. Behav. 2009, 55, 655–665. [Google Scholar] [CrossRef]
- Balthazart, J.; Ball, G.F. New insights into the regulation and function of brain estrogen synthase (aromatase). Trends Neurosci. 1998, 21, 243–249. [Google Scholar] [CrossRef]
- Clarkson, J.; Herbison, A.E. Hypothalamic control of the male neonatal testosterone surge. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150115. [Google Scholar] [CrossRef]
- McKenna, N.J.; O’Malley, B.W. Combinatorial control of gene expression by nuclear receptors and coregulators. Cell 2002, 108, 465–474. [Google Scholar] [CrossRef]
- Woolley, C.S. Acute Effects of Estrogen on Neuronal Physiology. Annu. Rev. Pharmacol. Toxicol. 2007, 47, 657–680. [Google Scholar] [CrossRef] [PubMed]
- Micevych, P.E.; Kelly, M.J. Membrane estrogen receptor regulation of hypothalamic function. Neuroendocrinology 2012, 96, 103–110. [Google Scholar] [CrossRef] [PubMed]
- Heimovics, S.A.; Trainor, B.C.; Soma, K.K. Rapid Effects of Estradiol on Aggression in Birds and Mice: The Fast and the Furious. Integr. Comp. Biol. 2015, 55, 281–293. [Google Scholar] [CrossRef] [PubMed]
- Stincic, T.L.; Rønnekleiv, O.K.; Kelly, M.J. Diverse actions of estradiol on anorexigenic and orexigenic hypothalamic arcuate neurons. Horm. Behav. 2018, 104, 146–155. [Google Scholar] [CrossRef] [PubMed]
- Gegenhuber, B.; Tollkuhn, J. Signatures of sex: Sex differences in gene expression in the vertebrate brain. Wiley Interdiscip. Rev. Dev. Biol. 2019, e348. [Google Scholar] [CrossRef] [PubMed]
- Ogawa, S.; Lubahn, D.B.; Korach, K.S.; Pfaff, D.W. Behavioral effects of estrogen receptor gene disruption in male mice. Proc. Natl. Acad. Sci. USA 1997, 94, 1476–1481. [Google Scholar] [CrossRef] [PubMed]
- Wersinger, S.R.; Sannen, K.; Villalba, C.; Lubahn, D.B.; Rissman, E.F.; De Vries, G.J. Masculine sexual behavior is disrupted in male and female mice lacking a functional estrogen receptor alpha gene. Horm. Behav. 1997, 32, 176–183. [Google Scholar] [CrossRef]
- Rissman, E.F.; Wersinger, S.R.; Fugger, H.N.; Foster, T.C. Sex with knockout models: Behavioral studies of estrogen receptor. Brain Res. 1999, 835, 80–90. [Google Scholar] [CrossRef]
- Scordalakes, E.M.; Rissman, E.F. Aggression in Male Mice Lacking Functional Estrogen Receptor alpha. Behav. Neurosci. 2003, 117, 38–45. [Google Scholar] [CrossRef]
- Wu, M.V.; Tollkuhn, J. Estrogen receptor alpha is required in GABAergic, but not glutamatergic, neurons to masculinize behavior. Horm. Behav. 2017, 95, 3–12. [Google Scholar] [CrossRef]
- McCarthy, M.M. Estradiol and the developing brain. Physiol. Rev. 2008, 88, 91–124. [Google Scholar] [CrossRef]
- Turano, A.; Osborne, B.F.; Schwarz, J.M. Sexual Differentiation and Sex Differences in Neural Development. Curr. Top. Behav. Neurosci. 2018, 1–42. [Google Scholar] [CrossRef]
- Forger, N.G.; Rosen, G.J.; Waters, E.M.; Jacob, D.; Simerly, R.B.; de Vries, G.J. Deletion of Bax eliminates sex differences in the mouse forebrain. Proc. Natl. Acad. Sci. USA 2004, 101, 13666–13671. [Google Scholar] [CrossRef]
- Wu, M.V.; Manoli, D.S.; Fraser, E.J.; Coats, J.K.; Tollkuhn, J.; Honda, S.-I.; Harada, N.; Shah, N.M. Estrogen masculinizes neural pathways and sex-specific behaviors. Cell 2009, 139, 61–72. [Google Scholar] [CrossRef] [PubMed]
- Kelly, D.A.; Varnum, M.M.; Krentzel, A.A.; Krug, S.; Forger, N.G. Differential control of sex differences in estrogen receptor α in the bed nucleus of the stria terminalis and anteroventral periventricular nucleus. Endocrinology 2013, 154, 3836–3846. [Google Scholar] [CrossRef] [PubMed]
- Beach, F.A. Historical origins of modern research on hormones and behavior. Horm. Behav. 1981, 15, 325–376. [Google Scholar] [CrossRef]
- Lonstein, J.S.; Gammie, S.C. Sensory, hormonal, and neural control of maternal aggression. Neurosci. Biobehav. Rev. 2002, 26, 869–888. [Google Scholar] [CrossRef]
- Hashikawa, K.; Hashikawa, Y.; Lischinsky, J.; Lin, D. The Neural Mechanisms of Sexually Dimorphic Aggressive Behaviors. Trends Genet. 2018, 34, 755–776. [Google Scholar] [CrossRef] [PubMed]
- Ishii, K.K.; Touhara, K. Neural circuits regulating sexual behaviors via the olfactory system in mice. Neurosci. Res. 2019, 140, 59–76. [Google Scholar] [CrossRef] [PubMed]
- Bangasser, D.A.; Wicks, B. Sex-specific mechanisms for responding to stress. J. Neurosci. Res. 2017, 95, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Laman-Maharg, A.; Trainor, B.C. Stress, sex, and motivated behaviors. J. Neurosci. Res. 2017, 95, 83–92. [Google Scholar] [CrossRef] [PubMed]
- Becker, J.B.; Chartoff, E. Sex differences in neural mechanisms mediating reward and addiction. Neuropsychopharmacology 2019, 44, 166–183. [Google Scholar] [CrossRef] [PubMed]
- McCarthy, M.M.; Auger, A.P.; Bale, T.L.; De Vries, G.J.; Dunn, G.A.; Forger, N.G.; Murray, E.K.; Nugent, B.M.; Schwarz, J.M.; Wilson, M.E. The epigenetics of sex differences in the brain. J. Neurosci. 2009, 29, 12815–12823. [Google Scholar] [CrossRef] [PubMed]
- Bale, T.L.; Baram, T.Z.; Brown, A.S.; Goldstein, J.M.; Insel, T.R.; McCarthy, M.M.; Nemeroff, C.B.; Reyes, T.M.; Simerly, R.B.; Susser, E.S.; et al. Early life programming and neurodevelopmental disorders. Biol. Psychiatry 2010, 68, 314–319. [Google Scholar] [CrossRef] [PubMed]
- Dulac, C. Brain function and chromatin plasticity. Nature 2010, 465, 728–735. [Google Scholar] [CrossRef] [PubMed]
- Gagnidze, K.; Weil, Z.M.; Pfaff, D.W. Histone modifications proposed to regulate sexual differentiation of brain and behavior. Bioessays 2010, 32, 932–939. [Google Scholar] [CrossRef] [PubMed]
- Auger, A.P.; Jessen, H.M.; Edelmann, M.N. Epigenetic organization of brain sex differences and juvenile social play behavior. Horm. Behav. 2011, 59, 358–363. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Nugent, B.M.; McCarthy, M.M. Epigenetic underpinnings of developmental sex differences in the brain. Neuroendocrinology 2011, 93, 150–158. [Google Scholar] [CrossRef] [PubMed]
- Matsuda, K.I.; Mori, H.; Kawata, M. Epigenetic mechanisms are involved in sexual differentiation of the brain. Rev. Endocr. Metab. Disord. 2012, 13, 163–171. [Google Scholar] [CrossRef] [PubMed]
- Hodes, G.E.; Walker, D.M.; Labonté, B.; Nestler, E.J.; Russo, S.J. Understanding the epigenetic basis of sex differences in depression. J. Neurosci. Res. 2017, 95, 692–702. [Google Scholar] [CrossRef] [PubMed]
- Ratnu, V.S.; Emami, M.R.; Bredy, T.W. Genetic and epigenetic factors underlying sex differences in the regulation of gene expression in the brain. J. Neurosci. Res. 2017, 95, 301–310. [Google Scholar] [CrossRef] [PubMed]
- Forger, N.G. Past, present and future of epigenetics in brain sexual differentiation. J. Neuroendocrinol. 2018, 30, e12492. [Google Scholar] [CrossRef] [PubMed]
- Manoli, D.S.; Tollkuhn, J. Gene regulatory mechanisms underlying sex differences in brain development and psychiatric disease. Ann. N. Y. Acad. Sci. 2018, 1420, 26–45. [Google Scholar] [CrossRef] [PubMed]
- Jaenisch, R.; Bird, A. Epigenetic regulation of gene expression: How the genome integrates intrinsic and environmental signals. Nat. Genet. 2003, 33, 245–254. [Google Scholar] [CrossRef] [PubMed]
- Shilatifard, A. Chromatin modifications by methylation and ubiquitination: Implications in the regulation of gene expression. Annu. Rev. Biochem. 2006, 75, 243–269. [Google Scholar] [CrossRef] [PubMed]
- Gibcus, J.H.; Dekker, J. The hierarchy of the 3D genome. Mol. Cell 2013, 49, 773–782. [Google Scholar] [CrossRef]
- Li, E.; Zhang, Y. DNA methylation in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a019133. [Google Scholar] [CrossRef] [PubMed]
- Barlow, D.P.; Bartolomei, M.S. Genomic imprinting in mammals. Cold Spring Harb. Perspect. Biol. 2014, 6, a018382. [Google Scholar] [CrossRef]
- Bestor, T.H.; Bourc’his, D. Transposon silencing and imprint establishment in mammalian germ cells. Cold Spring Harb. Symp. Quant. Biol. 2004, 69, 381–387. [Google Scholar] [CrossRef]
- Chow, J.; Heard, E. X inactivation and the complexities of silencing a sex chromosome. Curr. Opin. Cell Biol. 2009, 21, 359–366. [Google Scholar] [CrossRef]
- Morris, M.J.; Monteggia, L.M. Role of DNA methylation and the DNA methyltransferases in learning and memory. Dialogues Clin. Neurosci. 2014, 16, 359–371. [Google Scholar]
- Chen, W.G. Derepression of BDNF Transcription Involves Calcium-Dependent Phosphorylation of MeCP2. Science 2003, 302, 885–889. [Google Scholar] [CrossRef]
- Martinowich, K.; Hattori, D.; Wu, H.; Fouse, S.; He, F.; Hu, Y.; Fan, G.; Sun, Y.E. DNA methylation-related chromatin remodeling in activity-dependent BDNF gene regulation. Science 2003, 302, 890–893. [Google Scholar] [CrossRef]
- Nelson, E.D.; Kavalali, E.T.; Monteggia, L.M. Activity-dependent suppression of miniature neurotransmission through the regulation of DNA methylation. J. Neurosci. 2008, 28, 395–406. [Google Scholar] [CrossRef]
- Guo, J.U.; Ma, D.K.; Mo, H.; Ball, M.P.; Jang, M.-H.; Bonaguidi, M.A.; Balazer, J.A.; Eaves, H.L.; Xie, B.; Ford, E.; et al. Neuronal activity modifies the DNA methylation landscape in the adult brain. Nat. Neurosci. 2011, 14, 1345–1351. [Google Scholar] [CrossRef]
- Law, J.A.; Jacobsen, S.E. Establishing, maintaining and modifying DNA methylation patterns in plants and animals. Nat. Rev. Genet. 2010, 11, 204–220. [Google Scholar] [CrossRef]
- Kriaucionis, S.; Heintz, N. The nuclear DNA base 5-hydroxymethylcytosine is present in Purkinje neurons and the brain. Science 2009, 324, 929–930. [Google Scholar] [CrossRef]
- Tahiliani, M.; Koh, K.P.; Shen, Y.; Pastor, W.A.; Bandukwala, H.; Brudno, Y.; Agarwal, S.; Iyer, L.M.; Liu, D.R.; Aravind, L.; et al. Conversion of 5-methylcytosine to 5-hydroxymethylcytosine in mammalian DNA by MLL partner TET1. Science 2009, 324, 930–935. [Google Scholar] [CrossRef]
- Wu, X.; Zhang, Y. TET-mediated active DNA demethylation: Mechanism, function and beyond. Nat. Rev. Genet. 2017, 18, 517–534. [Google Scholar] [CrossRef]
- Bayraktar, G.; Kreutz, M.R. The Role of Activity-Dependent DNA Demethylation in the Adult Brain and in Neurological Disorders. Front. Mol. Neurosci. 2018, 11, 169. [Google Scholar] [CrossRef]
- Lister, R.; Mukamel, E. a.; Nery, J.R.; Urich, M.; Puddifoot, C. a.; Johnson, N.D.; Lucero, J.; Huang, Y.; Dwork, A.J.; Schultz, M.D.; et al. Global Epigenomic Reconfiguration During Mammalian Brain Development. Science 2013, 341, 629–643. [Google Scholar] [CrossRef]
- He, Y.; Ecker, J.R. Non-CG Methylation in the Human Genome. Annu. Rev. Genomics Hum. Genet. 2015, 16, 55–77. [Google Scholar] [CrossRef]
- Stroud, H.; Su, S.C.; Hrvatin, S.; Greben, A.W.; Renthal, W.; Boxer, L.D.; Nagy, M.A.; Hochbaum, D.R.; Kinde, B.; Gabel, H.W.; et al. Early-Life Gene Expression in Neurons Modulates Lasting Epigenetic States. Cell 2017, 171, 1151–1164. [Google Scholar] [CrossRef]
- Kurian, J.R.; Olesen, K.M.; Auger, A.P. Sex differences in epigenetic regulation of the estrogen receptor-alpha promoter within the developing preoptic area. Endocrinology 2010, 151, 2297–2305. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Nugent, B.M.; McCarthy, M.M. Developmental and hormone-induced epigenetic changes to estrogen and progesterone receptor genes in brain are dynamic across the life span. Endocrinology 2010, 151, 4871–4881. [Google Scholar] [CrossRef]
- Westberry, J.M.; Trout, A.L.; Wilson, M.E. Epigenetic regulation of estrogen receptor alpha gene expression in the mouse cortex during early postnatal development. Endocrinology 2010, 151, 731–740. [Google Scholar] [CrossRef]
- Ghahramani, N.M.; Ngun, T.C.; Chen, P.-Y.; Tian, Y.; Krishnan, S.; Muir, S.; Rubbi, L.; Arnold, A.P.; de Vries, G.J.; Forger, N.G.; et al. The effects of perinatal testosterone exposure on the DNA methylome of the mouse brain are late-emerging. Biol. Sex Differ. 2014, 5, 8. [Google Scholar] [CrossRef]
- Nugent, B.M.; Wright, C.L.; Shetty, A.C.; Hodes, G.E.; Lenz, K.M.; Mahurkar, A.; Russo, S.J.; Devine, S.E.; Mccarthy, M.M. Brain feminization requires active repression of masculinization via DNA methylation. Nat. Neurosci. 2015, 18, 690. [Google Scholar] [CrossRef]
- Tobet, S.A.; Henderson, R.G.; Whiting, P.J.; Sieghart, W. Special relationship of gamma-aminobutyric acid to the ventromedial nucleus of the hypothalamus during embryonic development. J. Comp. Neurol. 1999, 405, 88–98. [Google Scholar] [CrossRef]
- DonCarlos, L.L.; McAbee, M.; Ramer-Quinn, D.S.; Stancik, D.M. Estrogen receptor mRNA levels in the preoptic area of neonatal rats are responsive to hormone manipulation. Brain Res. Dev. Brain Res. 1995, 84, 253–260. [Google Scholar] [CrossRef]
- DonCarlos, L.L.; Handa, R.J. Developmental profile of estrogen receptor mRNA in the preoptic area of male and female neonatal rats. Brain Res. Dev. Brain Res. 1994, 79, 283–289. [Google Scholar] [CrossRef]
- Xu, X.; Coats, J.K.; Yang, C.F.; Wang, A.; Ahmed, O.M.; Alvarado, M.; Izumi, T.; Shah, N.M. Modular Genetic Control of Sexually Dimorphic Behaviors. Cell 2012, 148, 596–607. [Google Scholar] [CrossRef]
- Westberry, J.M.; Prewitt, A.K.; Wilson, M.E. Epigenetic regulation of the estrogen receptor alpha promoter in the cerebral cortex following ischemia in male and female rats. Neuroscience 2008, 152, 982–989. [Google Scholar] [CrossRef]
- Wilson, M.E.; Westberry, J.M.; Prewitt, A.K. Dynamic regulation of estrogen receptor-alpha gene expression in the brain: A role for promoter methylation? Front. Neuroendocrinol. 2008, 29, 375–385. [Google Scholar] [CrossRef]
- Edelmann, M.N.; Auger, A.P. Epigenetic impact of simulated maternal grooming on estrogen receptor alpha within the developing amygdala. Brain Behav. Immun. 2011, 25, 1299–1304. [Google Scholar] [CrossRef]
- Wilson, M.E.; Westberry, J.M.; Trout, A.L. Estrogen receptor-alpha gene expression in the cortex: Sex differences during development and in adulthood. Horm. Behav. 2011, 59, 353–357. [Google Scholar] [CrossRef]
- Westberry, J.M.; Wilson, M.E. Regulation of estrogen receptor alpha gene expression in the mouse prefrontal cortex during early postnatal development. Neurogenetics 2012, 13, 159–167. [Google Scholar] [CrossRef]
- Kos, M.; Reid, G.; Denger, S.; Gannon, F. Minireview: Genomic organization of the human ERalpha gene promoter region. Mol. Endocrinol. 2001, 15, 2057–2063. [Google Scholar] [CrossRef]
- Maekawa, R.; Sato, S.; Okada, M.; Lee, L.; Tamura, I.; Jozaki, K.; Kajimura, T.; Asada, H.; Yamagata, Y.; Tamura, H.; et al. Tissue-Specific Expression of Estrogen Receptor 1 Is Regulated by DNA Methylation in a T-DMR. Mol. Endocrinol. 2016, 30, 335–347. [Google Scholar] [CrossRef]
- Hon, G.C.; Rajagopal, N.; Shen, Y.; McCleary, D.F.; Yue, F.; Dang, M.D.; Ren, B. Epigenetic memory at embryonic enhancers identified in DNA methylation maps from adult mouse tissues. Nat. Genet. 2013, 45, 1198–1206. [Google Scholar] [CrossRef]
- Stone, A.; Zotenko, E.; Locke, W.J.; Korbie, D.; Millar, E.K.A.; Pidsley, R.; Stirzaker, C.; Graham, P.; Trau, M.; Musgrove, E.A.; et al. DNA methylation of oestrogen-regulated enhancers defines endocrine sensitivity in breast cancer. Nat. Commun. 2015, 6, 7758. [Google Scholar] [CrossRef]
- Fleischer, T.; Tekpli, X.; Mathelier, A.; Wang, S.; Nebdal, D.; Dhakal, H.P.; Sahlberg, K.K.; Schlichting, E.; Oslo Breast Cancer Research Consortium (OSBREAC); Børresen-Dale, A.-L.; et al. DNA methylation at enhancers identifies distinct breast cancer lineages. Nat. Commun. 2017, 8, 1379. [Google Scholar] [CrossRef]
- Wang, L.; Ozark, P.A.; Smith, E.R.; Zhao, Z.; Marshall, S.A.; Rendleman, E.J.; Piunti, A.; Ryan, C.; Whelan, A.L.; Helmin, K.A.; et al. TET2 coactivates gene expression through demethylation of enhancers. Sci. Adv. 2018, 4, eaau6986. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Yun, M.; Wu, J.; Workman, J.L.; Li, B. Readers of histone modifications. Cell Res. 2011, 21, 564–578. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Yadav, T.; Quivy, J.-P.; Almouzni, G. Chromatin plasticity: A versatile landscape that underlies cell fate and identity. Science 2018, 361, 1332–1336. [Google Scholar] [CrossRef]
- McCarthy, M.M.; Nugent, B.M. At the frontier of epigenetics of brain sex differences. Front. Behav. Neurosci. 2015, 9, 221. [Google Scholar] [CrossRef]
- Perissi, V.; Rosenfeld, M.G. Controlling nuclear receptors: The circular logic of cofactor cycles. Nat. Rev. Mol. Cell Biol. 2005, 6, 542. [Google Scholar] [CrossRef]
- Foulds, C.E.; Feng, Q.; Ding, C.; Bailey, S.; Hunsaker, T.L.; Malovannaya, A.; Hamilton, R.A.; Gates, L.A.; Zhang, Z.; Li, C.; et al. Proteomic analysis of coregulators bound to ERα on DNA and nucleosomes reveals coregulator dynamics. Mol. Cell 2013, 51, 185–199. [Google Scholar] [CrossRef]
- Yi, P.; Wang, Z.; Feng, Q.; Pintilie, G.D.; Foulds, C.E.; Lanz, R.B.; Ludtke, S.J.; Schmid, M.F.; Chiu, W.; O’Malley, B.W. Structure of a biologically active estrogen receptor-coactivator complex on DNA. Mol. Cell 2015, 57, 1047–1058. [Google Scholar] [CrossRef]
- Apostolakis, E.M.; Ramamurphy, M.; Zhou, D.; Oñate, S.; O’Malley, B.W. Acute disruption of select steroid receptor coactivators prevents reproductive behavior in rats and unmasks genetic adaptation in knockout mice. Mol. Endocrinol. 2002, 16, 1511–1523. [Google Scholar] [CrossRef]
- Molenda, H.A.; Griffin, A.L.; Auger, A.P.; McCarthy, M.M.; Tetel, M.J. Nuclear receptor coactivators modulate hormone-dependent gene expression in brain and female reproductive behavior in rats. Endocrinology 2002, 143, 436–444. [Google Scholar] [CrossRef]
- Murray, E.K.; Hien, A.; de Vries, G.J.; Forger, N.G. Epigenetic control of sexual differentiation of the bed nucleus of the stria terminalis. Endocrinology 2009, 150, 4241–4247. [Google Scholar] [CrossRef]
- Matsuda, K.I.; Mori, H.; Nugent, B.M.; Pfaff, D.W.; McCarthy, M.M.; Kawata, M. Histone deacetylation during brain development is essential for permanent masculinization of sexual behavior. Endocrinology 2011, 152, 2760–2767. [Google Scholar] [CrossRef]
- Shen, E.Y.; Ahern, T.H.; Cheung, I.; Straubhaar, J.; Dincer, A.; Houston, I.; de Vries, G.J.; Akbarian, S.; Forger, N.G. Epigenetics and sex differences in the brain: A genome-wide comparison of histone-3 lysine-4 trimethylation (H3K4me3) in male and female mice. Exp. Neurol. 2015, 268, 21–29. [Google Scholar] [CrossRef]
- Reinberg, D.; Vales, L.D. Chromatin domains rich in inheritance. Science 2018, 361, 33–34. [Google Scholar] [CrossRef]
- Barth, T.K.; Imhof, A. Fast signals and slow marks: The dynamics of histone modifications. Trends Biochem. Sci. 2010, 35, 618–626. [Google Scholar] [CrossRef]
- .Creyghton, M.P.; Cheng, A.W.; Welstead, G.G.; Kooistra, T.; Carey, B.W.; Steine, E.J.; Hanna, J.; Lodato, M.A.; Frampton, G.M.; Sharp, P.A.; et al. Histone H3K27ac separates active from poised enhancers and predicts developmental state. Proc. Natl. Acad. Sci USA 2010, 107, 21931–21936. [Google Scholar] [CrossRef]
- Rada-Iglesias, A.; Bajpai, R.; Swigut, T.; Brugmann S a Flynn R a Wysocka, J. A unique chromatin signature uncovers early developmental enhancers in humans. Nature 2011, 470, 279–283. [Google Scholar] [CrossRef]
- Gallegos, D.A.; Chan, U.; Chen, L.-F.; West, A.E. Chromatin Regulation of Neuronal Maturation and Plasticity. Trends Neurosci. 2018, 41, 311–324. [Google Scholar] [CrossRef]
- Boyer, L.A.; Plath, K.; Zeitlinger, J.; Brambrink, T.; Medeiros, L.A.; Lee, T.I.; Levine, S.S.; Wernig, M.; Tajonar, A.; Ray, M.K.; et al. Polycomb complexes repress developmental regulators in murine embryonic stem cells. Nature 2006, 441, 349–353. [Google Scholar] [CrossRef]
- Jadhav, U.; Nalapareddy, K.; Saxena, M.; O’Neill, N.K.; Pinello, L.; Yuan, G.-C.; Orkin, S.H.; Shivdasani, R.A. Acquired Tissue-Specific Promoter Bivalency Is a Basis for PRC2 Necessity in Adult Cells. Cell 2016, 165, 1389–1400. [Google Scholar] [CrossRef]
- Lomvardas, S.; Maniatis, T. Histone and DNA Modifications as Regulators of Neuronal Development and Function. Cold Spring Harb. Perspect. Biol. 2016, 8, a024208. [Google Scholar] [CrossRef]
- Feng, X.; Juan, A.H.; Wang, H.A.; Ko, K.D.; Zare, H.; Sartorelli, V. Polycomb Ezh2 controls the fate of GABAergic neurons in the embryonic cerebellum. Development 2016, 143, 1971–1980. [Google Scholar] [CrossRef]
- von Schimmelmann, M.; Feinberg, P.A.; Sullivan, J.M.; Ku, S.M.; Badimon, A.; Duff, M.K.; Wang, Z.; Lachmann, A.; Dewell, S.; Ma’ayan, A.; et al. Polycomb repressive complex 2 (PRC2) silences genes responsible for neurodegeneration. Nat. Neurosci. 2016, 19, 1321–1330. [Google Scholar] [CrossRef]
- Södersten, E.; Toskas, K.; Rraklli, V.; Tiklova, K.; Björklund, Å.K.; Ringnér, M.; Perlmann, T.; Holmberg, J. A comprehensive map coupling histone modifications with gene regulation in adult dopaminergic and serotonergic neurons. Nat. Commun. 2018, 9, 1226. [Google Scholar] [CrossRef]
- Mahfouz, A.; Lelieveldt, B.P.F.; Grefhorst, A.; van Weert, L.T.C.M.; Mol, I.M.; Sips, H.C.M.; van den Heuvel, J.K.; Datson, N.A.; Visser, J.A.; Reinders, M.J.T.; et al. Genome-wide coexpression of steroid receptors in the mouse brain: Identifying signaling pathways and functionally coordinated regions. Proc. Natl. Acad. Sci. USA 2016, 113, 2738–2743. [Google Scholar] [CrossRef]
- Bickmore, W.A.; Van Steensel, B. Genome architecture: Domain organization of interphase chromosomes. Cell 2013, 152, 1270–1284. [Google Scholar] [CrossRef]
- Dekker, J.; Heard, E. Structural and functional diversity of Topologically Associating Domains. FEBS Lett. 2015, 589, 2877–2884. [Google Scholar] [CrossRef]
- Alexander, J.M.; Lomvardas, S. Nuclear architecture as an epigenetic regulator of neural development and function. Neuroscience 2014, 264, 39–50. [Google Scholar] [CrossRef]
- Medrano-Fernández, A.; Barco, A. Nuclear organization and 3D chromatin architecture in cognition and neuropsychiatric disorders. Mol. Brain 2016, 9, 83. [Google Scholar] [CrossRef]
- Watson, L.A.; Tsai, L.H. In the loop: How chromatin topology links genome structure to function in mechanisms underlying learning and memory. Curr. Opin. Neurobiol. 2017, 43, 48–56. [Google Scholar] [CrossRef]
- Rajarajan, P.; Gil, S.E.; Brennand, K.J.; Akbarian, S. Spatial genome organization and cognition. Nat. Rev. Neurosci. 2016, 17, 681–691. [Google Scholar] [CrossRef]
- Rajarajan, P.; Borrman, T.; Liao, W.; Schrode, N.; Flaherty, E.; Casiño, C.; Powell, S.; Yashaswini, C.; LaMarca, E.A.; Kassim, B.; et al. Neuron-specific signatures in the chromosomal connectome associated with schizophrenia risk. Science 2018, 362, eaat4311. [Google Scholar] [CrossRef]
- Sun, J.H.; Zhou, L.; Emerson, D.J.; Phyo, S.A.; Titus, K.R.; Gong, W.; Gilgenast, T.G.; Beagan, J.A.; Davidson, B.L.; Tassone, F.; et al. Disease-Associated Short Tandem Repeats Co-localize with Chromatin Domain Boundaries. Cell 2018, 175, 224–238.e15. [Google Scholar] [CrossRef]
- Demontis, D.; Walters, R.K.; Martin, J.; Mattheisen, M.; Als, T.D.; Agerbo, E.; Baldursson, G.; Belliveau, R.; Bybjerg-Grauholm, J.; Bækvad-Hansen, M.; et al. Discovery of the first genome-wide significant risk loci for attention deficit/hyperactivity disorder. Nat. Genet. 2019, 51, 63–75. [Google Scholar] [CrossRef]
- Fudenberg, G.; Pollard, K.S. Chromatin features constrain structural variation across evolutionary timescales. Proc. Natl. Acad. Sci. USA 2019, 116, 2175–2180. [Google Scholar] [CrossRef]
- Wang, Q.; Carroll, J.S.; Brown, M. Spatial and temporal recruitment of androgen receptor and its coactivators involves chromosomal looping and polymerase tracking. Mol. Cell 2005, 19, 631–642. [Google Scholar] [CrossRef]
- Hsu, P.Y.; Hsu, H.K.; Singer, G.A.C.; Yan, P.S.; Rodriguez, B.A.T.; Liu, J.C.; Weng, Y.I.; Deatherage, D.E.; Chen, Z.; Pereira, J.S.; et al. Estrogen-mediated epigenetic repression of large chromosomal regions through DNA looping. Genome Res. 2010, 20, 733–744. [Google Scholar] [CrossRef]
- Fullwood, M.J.; Liu, M.H.; Pan, Y.F.; Liu, J.; Xu, H.; Mohamed, Y.B.; Orlov, Y.L.; Velkov, S.; Ho, A.; Mei, P.H.; et al. An oestrogen-receptor-alpha-bound human chromatin interactome. Nature 2009, 462, 58–64. [Google Scholar] [CrossRef]
- Ross-Innes, C.S.; Brown, G.D.; Carroll, J.S. A co-ordinated interaction between CTCF and ER in breast cancer cells. BMC Genomics 2011, 12, 593. [Google Scholar] [CrossRef]
- Hsieh, C.-L.; Fei, T.; Chen, Y.; Li, T.; Gao, Y.; Wang, X.; Sun, T.; Sweeney, C.J.; Lee, G.-S.M.; Chen, S.; et al. Enhancer RNAs participate in androgen receptor-driven looping that selectively enhances gene activation. Proc. Natl. Acad. Sci. USA 2014, 111, 7319–7324. [Google Scholar] [CrossRef]
- Panigrahi, A.K.; Foulds, C.E.; Lanz, R.B.; Hamilton, R.A.; Yi, P.; Lonard, D.M.; Tsai, M.-J.; Tsai, S.Y.; O’Malley, B.W. SRC-3 Coactivator Governs Dynamic Estrogen-Induced Chromatin Looping Interactions during Transcription. Mol. Cell 2018, 70, 679–694.e7. [Google Scholar] [CrossRef]
- Le Dily, F.; Vidal, E.; Cuartero, Y.; Quilez, J.; Nacht, A.S.; Vicent, G.P.; Carbonell-Caballero, J.; Sharma, P.; Villanueva-Cañas, J.L.; Ferrari, R.; et al. Hormone-control regions mediate steroid receptor-dependent genome organization. Genome Res. 2019, 29, 29–39. [Google Scholar] [CrossRef]
- Rafique, S.; Thomas, J.S.; Sproul, D.; Bickmore, W.A. Estrogen-induced chromatin decondensation and nuclear re-organization linked to regional epigenetic regulation in breast cancer. Genome Biol. 2015, 16, 145. [Google Scholar] [CrossRef]
- Jubb, A.W.; Boyle, S.; Hume, D.A.; Bickmore, W.A. Glucocorticoid Receptor Binding Induces Rapid and Prolonged Large-Scale Chromatin Decompaction at Multiple Target Loci. Cell Rep. 2017, 21, 3022–3031. [Google Scholar] [CrossRef]
- Le Dily, F.; Beato, M. Signaling by steroid hormones in the 3D nuclear space. Int. J. Mol. Sci. 2018, 19, 306. [Google Scholar] [CrossRef]
- Wiench, M.; Miranda, T.B.; Hager, G.L. Control of nuclear receptor function by local chromatin structure. FEBS J. 2011, 278, 2211–2230. [Google Scholar] [CrossRef]
- Quintin, J.; Le Péron, C.; Palierne, G.; Bizot, M.; Cunha, S.; Sérandour, A.A.; Avner, S.; Henry, C.; Percevault, F.; Belaud-Rotureau, M.-A.; et al. Dynamic estrogen receptor interactomes control estrogen-responsive trefoil Factor (TFF) locus cell-specific activities. Mol. Cell Biol. 2014, 34, 2418–2436. [Google Scholar] [CrossRef]
- Jones, K.J.; Pfaff, D.W.; McEwen, B.S. Early estrogen-induced nuclear changes in rat hypothalamic ventromedial neurons: An ultrastructural and morphometric analysis. J. Comp. Neurol. 1985, 239, 255–266. [Google Scholar] [CrossRef]
- Bonasio, R.; Tu, S.; Reinberg, D. Molecular signals of epigenetic states. Science 2010, 330, 612–616. [Google Scholar] [CrossRef]
- Weintraub, H.; Davis, R.; Krause, S.M.; Benezra, R.; Rupp, R.; Hollenberg, S.; Zhuang, Y.; Lassar, A. The myoD Gene Family: Nodal Point During Specification of the Muscle Cell Lineage. Nature 1987, 335, 155. [Google Scholar] [CrossRef]
- Blau, H.M.; Baltimore, D. Differentiation requires continuous regulation. J. Cell Biol. 1991, 112, 781–783. [Google Scholar] [CrossRef]
- Chen, Z.F.; Paquette, A.J.; Anderson, D.J. NRSF/REST is required in vivo for repression of multiple neuronal target genes during embryogenesis. Nat. Genet. 1998, 20, 136–142. [Google Scholar] [CrossRef]
- Sánchez Alvarado, A.; Yamanaka, S. Rethinking differentiation: Stem cells, regeneration, and plasticity. Cell 2014, 157, 110–119. [Google Scholar] [CrossRef]
- Hobert, O. Regulation of terminal differentiation programs in the nervous system. Annu. Rev. Cell Dev. Biol. 2011, 27, 681–696. [Google Scholar] [CrossRef]
- Hobert, O. Terminal Selectors of Neuronal Identity. Curr. Top. Dev. Biol. 2016, 116, 455–475. [Google Scholar]
- Marchetti, G.; Tavosanis, G. Steroid Hormone Ecdysone Signaling Specifies Mushroom Body Neuron Sequential Fate via Chinmo. Curr. Biol. 2017, 27, 3017–3024. [Google Scholar] [CrossRef]
- Syed, M.H.; Mark, B.; Doe, C.Q. Steroid hormone induction of temporal gene expression in Drosophila brain neuroblasts generates neuronal and glial diversity. Elife 2017, 6, e26287. [Google Scholar] [CrossRef]
- MacLusky, N.J.; Naftolin, F. Sexual Differentiation of the Central Nervous System. Science 1981, 211, 1294–1303. [Google Scholar] [CrossRef]
- Workman, A.D.; Charvet, C.J.; Clancy, B.; Darlington, R.B.; Finlay, B.L. Modeling transformations of neurodevelopmental sequences across mammalian species. J. Neurosci. 2013, 33, 7368–7383. [Google Scholar] [CrossRef]
- Hines, M. Prenatal testosterone and gender-related behaviour. Eur. J. Endocrinol. 2006, 155, S115–S121. [Google Scholar] [CrossRef][Green Version]
- Hines, M. Sex-related variation in human behavior and the brain. Trends Cogn. Sci. 2010, 14, 448–456. [Google Scholar] [CrossRef]
- Berenbaum, S.A.; Bryk, K.L.K.; Beltz, A.M. Early androgen effects on spatial and mechanical abilities: Evidence from congenital adrenal hyperplasia. Behav. Neurosci. 2012, 126, 86–96. [Google Scholar] [CrossRef]
- Hines, M.; Pasterski, V.; Spencer, D.; Neufeld, S.; Patalay, P.; Hindmarsh, P.C.; Hughes, I.A.; Acerini, C.L. Prenatal androgen exposure alters girls’ responses to information indicating gender-appropriate behaviour. Philos. Trans. R. Soc. B Biol. Sci. 2016, 371, 20150125. [Google Scholar] [CrossRef]
- Häfner, H. Gender differences in schizophrenia. Psychoneuroendocrinology 2003, 28, 17–54. [Google Scholar] [CrossRef]
- Schulz, K.M.; Sisk, C.L. The organizing actions of adolescent gonadal steroid hormones on brain and behavioral development. Neurosci. Biobehav. Rev. 2016, 70, 148–158. [Google Scholar] [CrossRef]
- Becker, J.B.; Berkley, K.J.; Geary, N.; Hampson, E.; Herman, J.P.; Young, E. Sex Differences in the Brain: From Genes to Behavior; Oxford University Press: Oxford, UK, 2007. [Google Scholar]
- McCarthy, M.M.; Arnold, A.P.; Ball, G.F.; Blaustein, J.D.; De Vries, G.J. Sex differences in the brain: The not so inconvenient truth. J. Neurosci. 2012, 32, 2241–2247. [Google Scholar] [CrossRef]
- Werling, D.M. The role of sex-differential biology in risk for autism spectrum disorder. Biol. Sex Differ. 2016, 7, 58. [Google Scholar] [CrossRef]
- Doan, R.N.; Bae, B.-I.; Cubelos, B.; Chang, C.; Hossain, A.A.; Al-Saad, S.; Mukaddes, N.M.; Oner, O.; Al-Saffar, M.; Balkhy, S.; et al. Mutations in Human Accelerated Regions Disrupt Cognition and Social Behavior. Cell 2016, 167, 341–354.e12. [Google Scholar] [CrossRef]
- Parikshak, N.N.; Swarup, V.; Belgard, T.G.; Irimia, M.; Ramaswami, G.; Gandal, M.J.; Hartl, C.; Leppa, V.; Ubieta, L.; de la, T.; Huang, J.; et al. Genome-wide changes in lncRNA, splicing, and regional gene expression patterns in autism. Nature 2016, 540, 423–427. [Google Scholar] [CrossRef]
- Girdhar, K.; Hoffman, G.E.; Jiang, Y.; Brown, L.; Kundakovic, M.; Hauberg, M.E.; Francoeur, N.J.; Wang, Y.-C.; Shah, H.; Kavanagh, D.H.; et al. Cell-specific histone modification maps in the human frontal lobe link schizophrenia risk to the neuronal epigenome. Nat. Neurosci. 2018, 21, 1126–1136. [Google Scholar] [CrossRef]
- Li, M.; Santpere, G.; Imamura Kawasawa, Y.; Evgrafov, O.V.; Gulden, F.O.; Pochareddy, S.; Sunkin, S.M.; Li, Z.; Shin, Y.; Zhu, Y.; et al. Integrative functional genomic analysis of human brain development and neuropsychiatric risks. Science 2018, 362, eaat7615. [Google Scholar] [CrossRef]
- de la Torre-Ubieta, L.; Stein, J.L.; Won, H.; Opland, C.K.; Liang, D.; Lu, D.; Geschwind, D.H. The Dynamic Landscape of Open Chromatin during Human Cortical Neurogenesis. Cell 2018, 172, 289–304.e18. [Google Scholar] [CrossRef]
- Short, P.J.; McRae, J.F.; Gallone, G.; Sifrim, A.; Won, H.; Geschwind, D.H.; Wright, C.F.; Firth, H.V.; FitzPatrick, D.R.; Barrett, J.C.; et al. De novo mutations in regulatory elements in neurodevelopmental disorders. Nature 2018, 555, 611–616. [Google Scholar] [CrossRef]
- Sullivan, P.F.; Agrawal, A.; Bulik, C.M.; Andreassen, O.A.; Børglum, A.D.; Breen, G.; Cichon, S.; Edenberg, H.J.; Faraone, S.V.; Gelernter, J.; et al. Psychiatric Genomics: An Update and an Agenda. Am. J. Psychiatry 2018, 175, 15–27. [Google Scholar] [CrossRef]
- Wang, D.; Liu, S.; Warrell, J.; Won, H.; Shi, X.; Navarro, F.C.P.; Clarke, D.; Gu, M.; Emani, P.; Yang, Y.T.; et al. Comprehensive functional genomic resource and integrative model for the human brain. Science 2018, 362, eaat8464. [Google Scholar] [CrossRef]
- Lim, E.T.; Uddin, M.; De Rubeis, S.; Chan, Y.; Kamumbu, A.S.; Zhang, X.; D, A.M.; Kim, S.N.; Sean Hill, R.; Goldberg, A.P.; et al. Rates, distribution and implications of postzygotic mosaic mutations in autism spectrum disorder. Nat. Neurosci. 2017, 20, 1217. [Google Scholar] [CrossRef]
- Yang, M.G.; West, A.E. Editing the Neuronal Genome: A CRISPR View of Chromatin Regulation in Neuronal Development, Function, and Plasticity. Yale J. Biol. Med. 2016, 89, 457–470. [Google Scholar]
- Savell, K.E.; Day, J.J. Applications of CRISPR/Cas9 in the Mammalian Central Nervous System. Yale J. Biol. Med. 2017, 90, 567–581. [Google Scholar]
- Savell, K.E.; Bach, S.V.; Zipperly, M.E.; Revanna, J.S.; Goska, N.A.; Tuscher, J.J.; Duke, C.G.; Sultan, F.A.; Burke, J.N.; Williams, D.; et al. A Neuron-Optimized CRISPR/dCas9 Activation System for Robust and Specific Gene Regulation. eNeuro 2019, 6. [Google Scholar] [CrossRef]
- Sabarís, G.; Laiker, I.; Preger-Ben Noon, E.; Frankel, N. Actors with Multiple Roles: Pleiotropic Enhancers and the Paradigm of Enhancer Modularity. Trends Genet. 2019, 35, 423–433. [Google Scholar] [CrossRef]
- Carleton, J.B.; Berrett, K.C.; Gertz, J. Multiplex Enhancer Interference Reveals Collaborative Control of Gene Regulation by Estrogen Receptor α-Bound Enhancers. Cell Syst. 2017, 5, 333–344.e5. [Google Scholar] [CrossRef]
- Hewitt, S.C.; Li, L.; Grimm, S.A.; Chen, Y.; Liu, L.; Li, Y.; Bushel, P.R.; Fargo, D.; Korach, K.S. Research resource: Whole-genome estrogen receptor α binding in mouse uterine tissue revealed by ChIP-seq. Mol. Endocrinol. 2012, 26, 887–898. [Google Scholar] [CrossRef]
- Jozwik, K.M.; Carroll, J.S. Pioneer factors in hormone-dependent cancers. Nat. Rev. Cancer 2012, 12, 381–385. [Google Scholar] [CrossRef]
- Gertz, J.; Savic, D.; Varley, K.E.; Partridge, E.C.; Safi, A.; Jain, P.; Cooper, G.M.; Reddy, T.E.; Crawford, G.E.; Myers, R.M. Distinct properties of cell-type-specific and shared transcription factor binding sites. Mol. Cell. 2013, 52, 25–36. [Google Scholar] [CrossRef]
- Gordon, F.K.; Vallaster, C.S.; Westerling, T.; Iyer, L.K.; Brown, M.; Schnitzler, G.R. Research resource: Aorta- and liver-specific ERα-binding patterns and gene regulation by estrogen. Mol. Endocrinol. 2014, 28, 1337–1351. [Google Scholar] [CrossRef]
- Yao, G.; Hu, S.; Yu, L.; Ru, Y.; Chen, C.D.; Liu, Q.; Zhang, Y. Genome-Wide Mapping of In Vivo ERα-Binding Sites in Male Mouse Efferent Ductules. Endocrinology 2017, 158, 3724–3737. [Google Scholar] [CrossRef][Green Version]
- Mohammed, H.; D’Santos, C.; Serandour, A.A.; Ali, H.R.; Brown, G.D.; Atkins, A.; Rueda, O.M.; Holmes, K.A.; Theodorou, V.; Robinson, J.L.L.; et al. Endogenous purification reveals GREB1 as a key estrogen receptor regulatory factor. Cell Rep. 2013, 3, 342–349. [Google Scholar] [CrossRef]
- Mo, A.; Mukamel, E.A.; Davis, F.P.; Luo, C.; Henry, G.L.; Picard, S.; Urich, M.A.; Nery, J.R.; Sejnowski, T.J.; Lister, R.; et al. Epigenomic Signatures of Neuronal Diversity in the Mammalian Brain. Neuron 2015, 86, 1369–1384. [Google Scholar] [CrossRef]
- Skene, P.J.; Henikoff, J.G.; Henikoff, S. Targeted in situ genome-wide profiling with high efficiency for low cell numbers. Nat. Protoc. 2018, 13, 1006–1019. [Google Scholar] [CrossRef]
- Hainer, S.J.; Bošković, A.; McCannell, K.N.; Rando, O.J.; Fazzio, T.G. Profiling of Pluripotency Factors in Single Cells and Early Embryos. Cell 2019, 177, 1319–1329. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Gegenhuber, B.; Tollkuhn, J. Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain? Genes 2019, 10, 432. https://doi.org/10.3390/genes10060432
Gegenhuber B, Tollkuhn J. Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain? Genes. 2019; 10(6):432. https://doi.org/10.3390/genes10060432
Chicago/Turabian StyleGegenhuber, Bruno, and Jessica Tollkuhn. 2019. "Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain?" Genes 10, no. 6: 432. https://doi.org/10.3390/genes10060432
APA StyleGegenhuber, B., & Tollkuhn, J. (2019). Sex Differences in the Epigenome: A Cause or Consequence of Sexual Differentiation of the Brain? Genes, 10(6), 432. https://doi.org/10.3390/genes10060432