Single Fragment or Bulk Soil DNA Metabarcoding: Which is Better for Characterizing Biological Taxa Found in Surface Soils for Sample Separation?


Abstract
1. Introduction
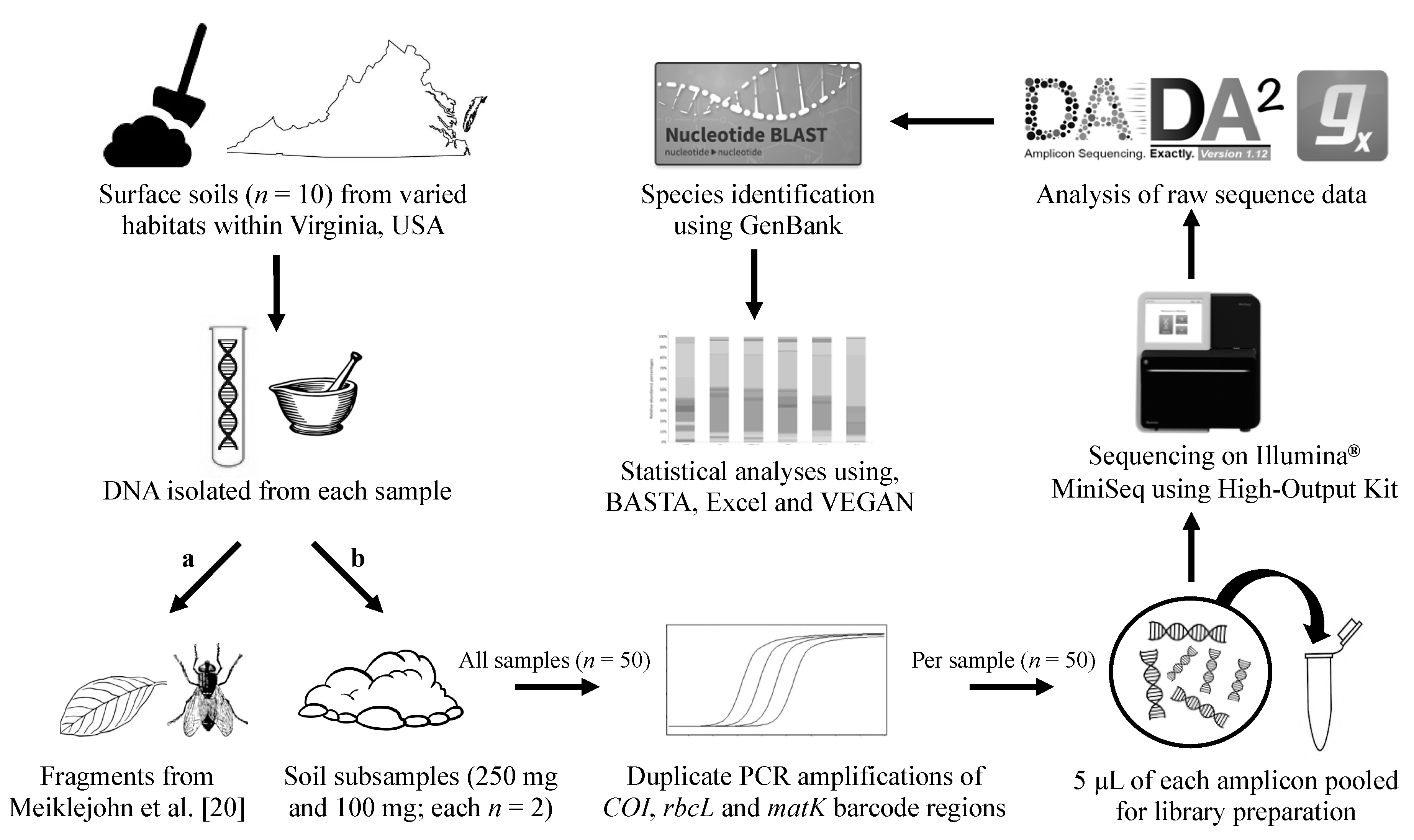
2. Materials and Methods
2.1. Soil Samples
2.2. Individual Fragment DNA Extractions and Pooling
2.3. Bulk Soil DNA Extraction and Quantification
2.4. Amplification and Quantification
2.5. Library Preparation
2.6. Sequencing Using the Illumina® MiniSeq
2.7. Processing Massively Parallel Sequencing Data
2.8. Searching Sequence Data against GenBank
2.9. Data Analysis
3. Results and Discussion
3.1. Total Genomic DNA Yields from Bulk Soil
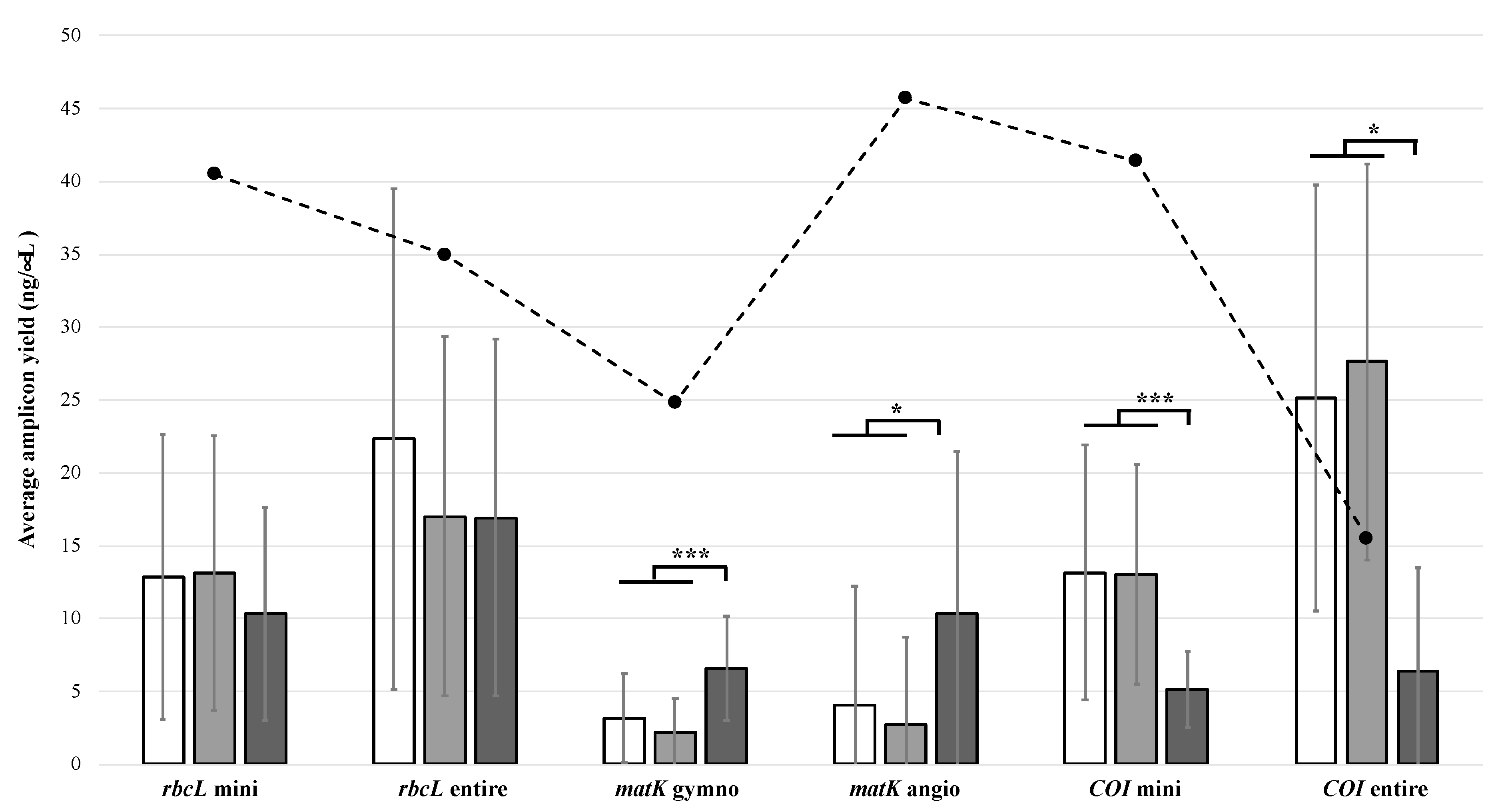
3.2. Polymerase Chain Reaction Amplification
3.3. Barcode Sequence Recovery
3.4. Unique and Target Sequences
3.5. Comparing DNA Barcoding to DNA Metabarcoding
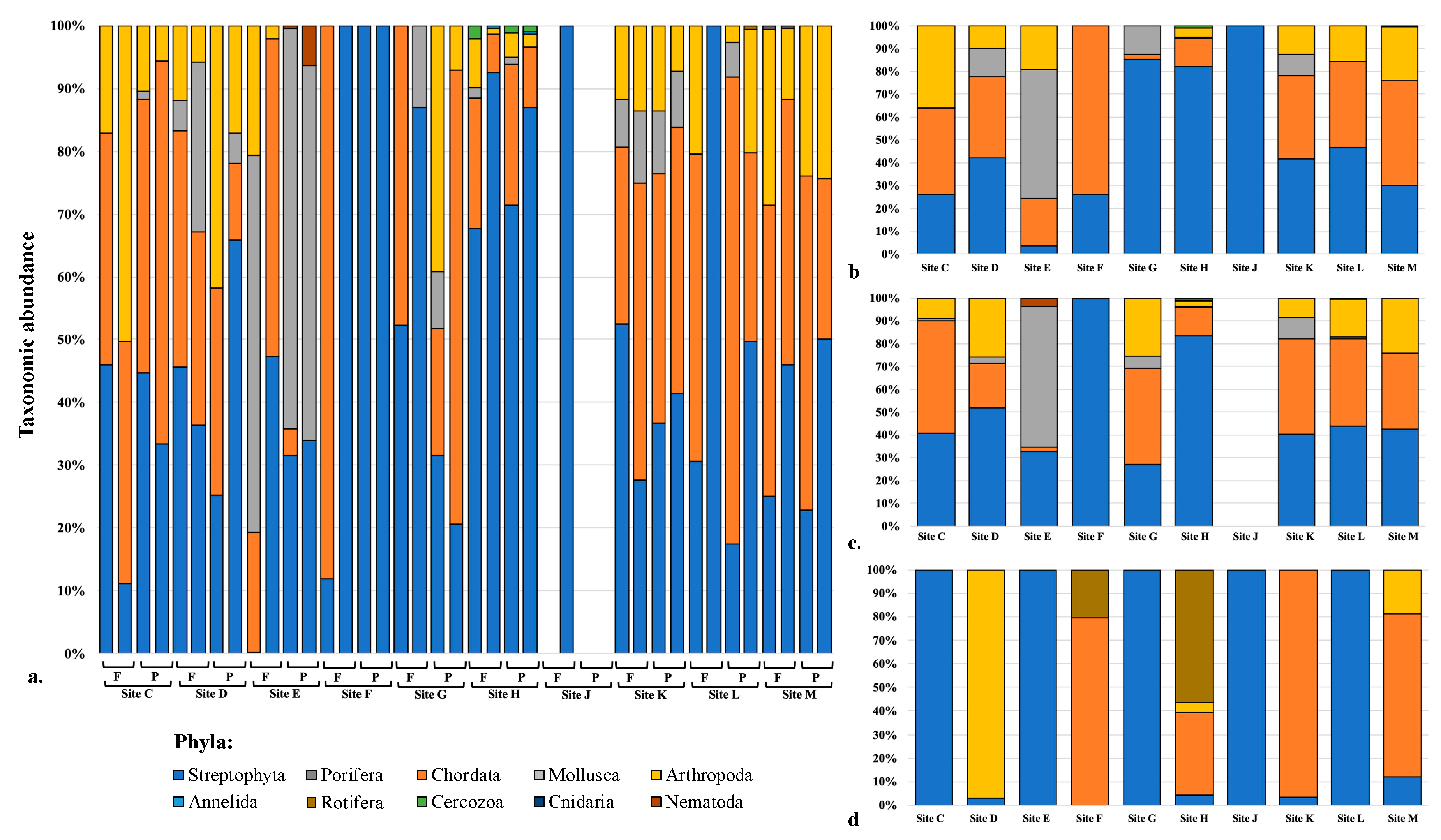
3.6. Taxon Abundance and Utility for Sample Site Discrimination
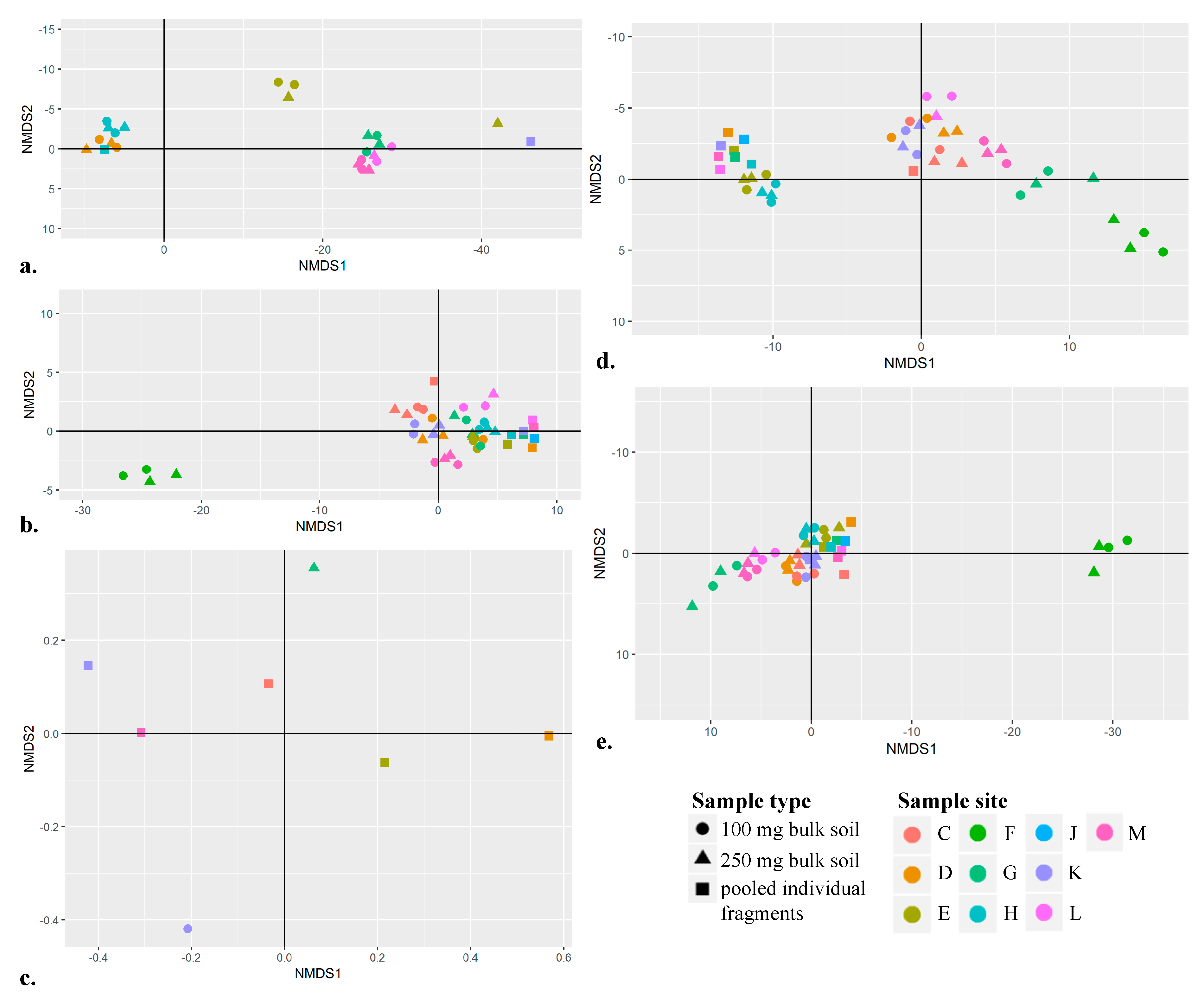
3.7. Using Non-Metric Multidimensional Scaling Plots to Spatially Separate Sample Sites
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Dawson, L.A.; Mayes, R.W. Criminal and environmental soil forensics: Soil as physical evidence in forensic investigations. In Introduction to Environmental Forensics, 3rd ed.; Ritz, K., Dawson, L., Miller, D., Eds.; Springer: New York, NY, USA, 2015; pp. 457–486. [Google Scholar]
- Newton, P.N.; Fernandez, F.M.; Plancon, A.; Mildenhall, D.C.; Green, M.D.; Ziyong, L.; Christophel, E.M.; Phanouvong, S.; Howells, S.; McIntosh, E.; et al. Collaborative epidemiological investigation into the criminal fake artesunate trade in South East Asia. PLoS Med. 2008, 5, e32. [Google Scholar] [CrossRef] [PubMed]
- Bowen, A.M.; Caven, E.A. Forensic provenance investigations of soil and sediment samples. Geol. Soc. Lond. Special Publ. 2013, 384, 9–25. [Google Scholar] [CrossRef]
- Bowen, A.M. Forensic applications of foraminifera. Microscope 2010, 58, 3–18. [Google Scholar]
- Brown, T.; Smith, A.; Elmhurst, O. The combined use of pollen and soil analyses in a search and subsequent murder investigation. J. Forensic Sci. 2002, 47, 614–618. [Google Scholar] [CrossRef] [PubMed]
- Peabody, A.J. Diatoms in forensic science. J. Forensic Sci. Soc. 1977, 17, 81–87. [Google Scholar] [CrossRef]
- Siver, P.; Lord, W.; McCarthy, D. Forensic limnology: The use of freshwater algal community ecology to link suspects to an aquatic crime scene in southern New England. J. Forensic Sci. 1994, 39, 847–853. [Google Scholar] [CrossRef]
- Bryant, V.M.; Jones, J.G.; Mildenhall, D.C. Forensic palynology in the United States of America. Palynology 1990, 14, 193–208. [Google Scholar] [CrossRef]
- Andersen, K.; Bird, K.L.; Rasmussen, M.; Haile, J.; Breuning-Madsen, H.; Kjær, K.H.; Orlando, L.; Gilbert, M.T.P.; Willerslev, E. Meta-Barcoding of ‘dirt’ DNA from soil reflects vertebrate biodiversity. Mol. Ecol. 2012, 21, 1966–1979. [Google Scholar] [CrossRef] [PubMed]
- Bienert, F.; De Danieli, S.; Miquel, C.; Coissac, E.; Poillot, C.; Brun, J.; Taberlet, P. Tracking earthworm communities from soil DNA. Mol. Ecol. 2012, 21, 2017–2030. [Google Scholar] [CrossRef] [PubMed]
- Epp, L.S.; Boessenkool, S.; Bellemain, E.P.; Haile, J.; Esposito, A.; Riaz, T.; Erséus, C.; Gusarov, V.I.; Edwards, M.E.; Johnsen, A.; et al. New environmental metabarcodes for analysing soil DNA: potential for studying past and present ecosystems. Mol. Ecol. 2012, 21, 1821–1833. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Prud’Homme, S.M.; Campione, E.; Roy, J.; Miquel, C.; Shehzad, W.; Gielly, L.; Rioux, D.; Choler, P.; Clément, J.; et al. Soil sampling and isolation of extracellular DNA from large amount of starting material suitable for metabarcoding studies. Mol. Ecol. 2012, 21, 1816–1820. [Google Scholar] [CrossRef] [PubMed]
- Oliverio, A.M.; Gan, H.; Wickings, K.; Fierer, N. A DNA metabarcoding approach to characterize soil arthropod communities. Soil Biol. Biochem. 2018, 125, 37–43. [Google Scholar] [CrossRef]
- Yoccoz, N.G.; Bråthen, K.A.; Gielly, L.; Haile, J.; Edwards, M.E.; Goslar, T.; Von Stedingk, H.; Brysting, A.K.; Coissac, E.; Pompanon, F.; et al. DNA from soil mirrors plant taxonomic and growth form diversity. Mol. Ecol. 2012, 21, 3647–3655. [Google Scholar] [CrossRef] [PubMed]
- Badgley, A.J.; Jesmock, E.M.; Foran, D.R. Time radically alters ex situ evidentiary soil 16S bacterial profiles produced via next-generation sequencing. J. Forensic Sci. 2018, 63, 1356–1365. [Google Scholar] [CrossRef] [PubMed]
- Habtom, H.; Demanèche, S.; Dawson, L.; Azulay, C.; Matan, O.; Robe, P.; Gafny, R.; Simonet, P.; Jurkevitch, E.; Pasternak, Z. Soil characterisation by bacterial community analysis for forensic applications: A quantitative comparison of environmental technologies. Forensic Sci. Int. Genet. 2016, 26, 21–29. [Google Scholar] [CrossRef] [PubMed]
- Jesmok, E.M.; Hopkins, J.M.; Foran, D.R. Next-generation sequencing of the bacterial 16S rRNA gene for forensic soil comparison: a feasibility study. J. Forensic Sci. 2016, 61, 607–617. [Google Scholar] [CrossRef] [PubMed]
- Demanèche, S.; Schauser, L.; Dawson, L.; Franqueville, L.; Simonet, P. Microbial soil community analyses for forensic science: application to a blind test. Forensic Sci. Int. 2017, 270, 153–158. [Google Scholar] [CrossRef] [PubMed]
- Habtom, H.; Pasternak, Z.; Matan, O.; Azlay, C.; Gafny, R.; Jurkevitch, E. Applying microbial biogeography in soil forensics. Forensic Sci. Int. Genet. 2019, 38, 195–203. [Google Scholar] [CrossRef]
- Meiklejohn, K.A.; Jackson, M.L.; Stern, L.A.; Robertson, J.M. A protocol for obtaining DNA barcodes from plant and insect fragments isolated from forensic-type soils. Int. J. Legal Med. 2018, 132, 1515–1526. [Google Scholar] [CrossRef]
- Young, J.M.; Weyrich, L.S.; Cooper, A. High-throughput sequencing of trace quantities of soil provides reproducible and discriminative fungal DNA profiles. J. Forensic Sci. 2016, 61, 478–484. [Google Scholar] [CrossRef]
- Young, J.M.; Weyrich, L.S.; Cooper, A. Forensic soil DNA analysis using high-throughput sequencing: A comparison of four molecular markers. Forensic Sci. Int. Genet. 2014, 13, 176–184. [Google Scholar] [CrossRef] [PubMed]
- Young, J.M.; Weyrich, L.S.; Breen, J.; Macdonald, L.M.; Cooper, A. Predicting the origin of soil evidence: High throughput eukaryote sequencing and MIR spectroscopy applied to a crime scene scenario. Forensic Sci. Int. 2015, 251, 22–31. [Google Scholar] [CrossRef] [PubMed]
- Lauber, C.L.; Ramirez, K.S.; Aanderud, Z.; Lennon, J.; Fierer, N. Temporal variability in soil microbial communities across land-use types. ISME J. 2013, 7, 1641–1650. [Google Scholar] [CrossRef] [PubMed]
- Kraaijeveld, K.; Weger, L.A.; Ventayol García, M.; Buermans, H.; Frank, J.; Hiemstra, P.S.; Dunnen, J.T. Efficient and sensitive identification and quantification of airborne pollen using next-generation DNA sequencing. Mol. Ecol. Resour. 2015, 15, 8–16. [Google Scholar] [CrossRef] [PubMed]
- Pornon, A.; Andalo, C.; Burrus, M.; Escaravage, N. DNA metabarcoding data unveils invisible pollination networks. Sci. Rep. 2017, 7, 16828. [Google Scholar] [CrossRef] [PubMed]
- Fahner, N.A.; Shokralla, S.; Baird, D.J.; Hajibabaei, M. Large-scale monitoring of plants through environmental DNA metabarcoding of soil: recovery, resolution, and annotation of four DNA markers. PLoS ONE 2016, 11, e0157505. [Google Scholar] [CrossRef] [PubMed]
- Taberlet, P.; Coissac, E.; Pompanon, F.; Gielly, L.; Miquel, C.; Valentini, A.; Vermat, T.; Corthier, G.; Brochmann, C.; Willerslev, E. Power and limitations of the chloroplast trnL (UAA) intron for plant DNA barcoding. Nucl. Acids Res. 2006, 35, e14. [Google Scholar] [CrossRef]
- Craine, J.; Barberán, A.; Lynch, R.; Menninger, H.; Dunn, R.; Fierer, N. Molecular analysis of environmental plant DNA in house dust across the United States. Aerobiologia 2017, 33, 71–86. [Google Scholar] [CrossRef]
- Cheng, T.; Xu, C.; Lei, L.; Li, C.; Zhang, Y.; Zhou, S. Barcoding the kingdom plantae: new PCR primers for ITS regions of plants with improved universality and specificity. Mol. Ecol. Resour. 2016, 16, 138–149. [Google Scholar] [CrossRef]
- Chen, S.; Yao, H.; Han, J.; Liu, C.; Song, J.; Shi, L.; Zhu, Y.; Ma, X.; Gao, T.; Pang, X.; et al. Validation of the ITS2 region as a novel DNA barcode for identifying medicinal plant species. PLoS ONE 2010, 5, e8613. [Google Scholar] [CrossRef]
- Moorhouse-Gann, R.J.; Dunn, J.C.; de Vere, N.; Goder, M.; Cole, N.; Hipperson, H.; Symondson, W.O.C. New universal ITS2 primers for high-resolution herbivory analyses using DNA metabarcoding in both tropical and temperate zones. Sci. Rep. 2018, 8, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Arulandhu, A.J.; Staats, M.; Hagelaar, R.; Voorhuijzen, M.M.; Prins, T.W.; Scholtens, I.; Costessi, A.; Duijsings, D.; Rechenmann, F.; Gaspar, F.B.; et al. Development and validation of a multi-locus DNA metabarcoding method to identify endangered species in complex samples. Gigascience 2017, 6, 1–18. [Google Scholar] [CrossRef] [PubMed]
- Xin, T.; Xu, Z.; Jia, J.; Leon, C.; Hu, S.; Lin, Y.; Ragupathy, S.; Song, J.; Newmaster, S.G. Biomonitoring for traditional herbal medicinal products using DNA metabarcoding and single molecule, real-time sequencing. Acta Pharm Sin. B 2018, 8, 488–497. [Google Scholar] [CrossRef] [PubMed]
- Meusnier, I.; Singer, G.A.C.; Landry, J.; Hickey, D.A.; Hebert, P.D.N.; Hajibabaei, M. A universal DNA mini-barcode for biodiversity analysis. BMC Genomics 2008, 9, 214. [Google Scholar] [CrossRef] [PubMed]
- Nelson, L.A.; Wallman, J.F.; Dowton, M. Using COI barcodes to identify forensically and medically important blowflies. Med. Vet. Entomol. 2007, 21, 44–52. [Google Scholar] [CrossRef] [PubMed]
- Boumans, L.; Tierno, D.F. Introgression and species demarcation in Western European Leuctra Fusca (Linnaeus, 1758) and L. Digitata Kempny, 1899 (Plecoptera: Leuctridae). Aquat Insects 2016, 37, 115–126. [Google Scholar] [CrossRef]
- Zhan, A.; Bailey, S.A.; Heath, D.D.; Macisaac, H.J. Performance comparison of genetic markers for high-throughput sequencing-based biodiversity assessment in complex communities. Mol. Ecol. Resour. 2014, 14, 1049–1059. [Google Scholar] [CrossRef] [PubMed]
- Janjua, S.; Fakhar-I-Abbas William, K.; Malik, I.U.; Mehr, J. DNA mini-barcoding for wildlife trade Control: a case study on identification of highly processed animal materials. Mitochondrial DNA Part. A Mapp. Seq. Anal. 2017, 28, 544–546. [Google Scholar] [CrossRef]
- Khan, F.M.; William, K.; Aruge, S.; Janjua, S.; Shah, S.A. Illegal product manufacturing and exportation from Pakistan: revealing the factuality of highly processed wildlife skin samples via DNA mini-barcoding. Nucleosides Nucleotides Nucl. Acids 2018, 37, 179–185. [Google Scholar] [CrossRef]
- Dopheide, A.; Xie, D.; Buckley, T.R.; Drummond, A.J.; Newcomb, R.D. Impacts of DNA extraction and PCR on DNA metabarcoding estimates of soil biodiversity. Methods Ecol. Evol. 2019, 10, 120–133. [Google Scholar] [CrossRef]
- Akbari, M.; Doré Hansen, M.; Halgunset, J.; Skorpen, F.; Krokan, H.E. Low copy number DNA template can render polymerase chain reaction error prone in a sequence-dependent manner. J. Mol. Diagn 2005, 7, 36–39. [Google Scholar] [CrossRef]
- Cummings, S.M.; McMullan, M.; Joyce, D.A.; van Oosterhout, C. Solutions for PCR, cloning and sequencing Errors in population genetic analysis. Conserv. Genet. 2010, 11, 1095–1097. [Google Scholar] [CrossRef]
- Weusten, J.; Herbergs, J. A stochastic model of the processes in PCR based amplification of STR DNA in forensic applications. Forensic Sci. Int. Genet. 2012, 6, 17–25. [Google Scholar] [CrossRef] [PubMed]
- Fazekas, A.J.; Kuzmina, M.L.; Newmaster, S.G.; Hollingsworth, P.M. DNA Barcoding Methods for Land Plants. In DNA Barcodes: Methods and Protocols; Kress, J.W., Erickson, D.L., Eds.; Springer: New York, NY, USA, 2012; pp. 223–252. [Google Scholar]
- Li, Y.; Gao, L.; Poudel, R.C.; Li, D.; Forrest, A. High universality of matK primers for barcoding gymnosperms. J. Syst. Evol. 2011, 49, 169–175. [Google Scholar] [CrossRef]
- Levin, R.A.; Wagner, W.L.; Hoch, P.C.; Nepokroeff, M.; Pires, J.C.; Zimmer, E.A.; Sytsma, K.J. Family-level relationships of Onagraceae based on chloroplast rbcL and ndhF data. Am. J. Bot. 2003, 90, 107–115. [Google Scholar] [CrossRef] [PubMed]
- Little, D.P. A DNA mini-barcode for land plants. Mol. Ecol. Resour. 2014, 14, 437–446. [Google Scholar] [CrossRef]
- Hajibabaei, M.; Smith, M.A.; Janzen, D.H.; Rodriguez, J.J.; Whitfield, J.B.; Hebert, P.D.N. A minimalist barcode can identify a specimen whose DNA is degraded. Mol. Ecol. Notes 2006, 6, 959–964. [Google Scholar] [CrossRef]
- Min, X.J.; Hickey, D.A. Assessing the effect of varying sequence length on DNA barcoding of fungi. Mol Ecol. Notes 2007, 7, 365–373. [Google Scholar] [CrossRef]
- Chaves, P.B.; Graeff, V.G.; Lion, M.B.; Oliveira, L.R.; Eizirik, E. DNA barcoding meets molecular scatology: short mtDNA sequences for standardized species assignment of carnivore noninvasive samples. Mol. Ecol. Resour. 2012, 12, 18–35. [Google Scholar] [CrossRef]
- Grzywacz, A.; Wyborska, D.; Piwczyński, M. DNA barcoding allows identification of European Fanniidae (Diptera) of forensic interest. Forensic Sci. Int. 2017, 278, 106–114. [Google Scholar] [CrossRef]
- Callahan, B.J.; McMurdie, P.J.; Rosen, M.J.; Han, A.W.; Johnson, A.J.A.; Holmes, S.P. DADA2: High-resolution sample inference from Illumina amplicon data. Nat. Methods 2016, 13, 581–583. [Google Scholar] [CrossRef] [PubMed]
- Afgan, E.; Baker, D.; Batut, B.; van den Beek, M.; Bouvier, D.; Cech, M.; Chilton, J.; Clements, D.; Coraor, N.; Gruning, B.A.; et al. The galaxy platform for accessible, reproducible and collaborative biomedical analyses: 2018 update. Nucleic Acids Res. 2018, 46, W544. [Google Scholar] [CrossRef] [PubMed]
- Chamberlain, S.A.; Szocs, E. Taxize: taxonomic search and retrieval in R. F1000Res 2013, 2, 191. [Google Scholar] [CrossRef] [PubMed]
- Kahlke, T.; Ralph, P.J. BASTA – Taxonomic classification of sequences and sequence bins using last common ancestor estimations. Methods Ecol. Evol. 2019, 10, 100–103. [Google Scholar] [CrossRef]
- Oksanen, J.; Blanchet, F.G.; Friendly, M.; Kindt, R.; Legendre, P.; McGlinn, D.; Minchin, P.R.; O’Hara, R.B.; Simpson, G.L.; Solymos, P.; et al. Vegan: Community Ecology Package. R package version 2.5-3; 2018; Available online: https://cran.r-project.org/web/packages/vegan/index.html (accessed on 1 March 2019).
- Wickman, H.; Chang, W.; Henry, L.; Pedersen, T.L.; Takahashi, K.; Wilke, C.; Woo, K. Ggplot2: Elegant graphics for data analysis, version 3.1.1; Springer-Verlag: New York, NY, USA, 2016. [Google Scholar]
- Shaw, J.L.A.; Clarke, L.J.; Wedderburn, S.D.; Barnes, T.C.; Weyrich, L.S.; Cooper, A. Comparison of environmental DNA metabarcoding and conventional fish survey methods in a river system. Biol. Cons 2016, 197, 131–138. [Google Scholar] [CrossRef]
- Zinger, L.; Chave, J.; Coissac, E.; Iribar, A.; Louisanna, E.; Manzi, S.; Schilling, V.; Schimann, H.; Sommeria-Klein, G.; Taberlet, P. Extracellular DNA extraction is a fast, cheap and reliable alternative for multi-taxa surveys based on soil DNA. Soil Biol. Biochem. 2016, 96, 16–19. [Google Scholar] [CrossRef]
- Watts, C.; Dopheide, A.; Holdaway, R.; Davis, C.; Wood, J.; Thornburrow, D.; Dickie, I.A. DNA metabarcoding as a tool for invertebrate community monitoring: a case study comparison with conventional techniques. Aust. Entomol. 2019. [Google Scholar] [CrossRef]
- Valentini, A.; Taberlet, P.; Miaud, C.; Civade, R.; Herder, J.; Thomsen, P.F.; Bellemain, E.; Besnard, A.; Coissac, E.; Boyer, F.; et al. Next-generation monitoring of aquatic biodiversity using environmental DNA metabarcoding. Mol. Ecol. 2016, 25, 929–942. [Google Scholar] [CrossRef]
- Osborne, C.A.; Zwart, A.B.; Broadhurst, L.M.; Young, A.G.; Richardson, A.E. The influence of sampling strategies and spatial variation on the detected soil bacterial communities under three different land-use types. FEMS Microbiol. Ecol. 2011, 78, 70–79. [Google Scholar] [CrossRef]
- Damaso, N.; Mendel, J.; Mendoza, M.; Wettberg, E.J.; Narasimhan, G.; Mills, D. Bioinformatics approach to assess the biogeographical patterns of soil communities: the utility for soil provenance. J. Forensic Sci. 2018, 63, 1033–1042. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Targeted Barcode Region and Primer Pairs (~amplicon length) | Pooled Plant Fragments (n = 10) | Pooled Insect Fragments (n = 10) | Bulk Soil (n = 40) |
|---|---|---|---|
| matK | |||
| - matK-KIM-1R/matK-KIM-3F (850 bp) [45] | ✓ | ✕ | ✓ |
| - GymF1A/GymR1A (850 bp) [46] | ✓ | ✕ | ✓ |
| rbcL | |||
| - rbcLa-F [47] /rbcLa-R [45] (590 bp) * | ✓ | ✕ | ✓ |
| - rbcL1/rbcLB (230 bp) [48] * | ✓ | ✕ | ✓ |
| COI | |||
| - LCO1490-L/HCO2198-L (658 bp) [36] * | ✕ | ✓ | ✓ |
| - uniminibarF1/uniminibarR1 (130 bp) [35] * | ✕ | ✓ | ✓ |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Boggs, L.M.; Scheible, M.K.R.; Machado, G.; Meiklejohn, K.A. Single Fragment or Bulk Soil DNA Metabarcoding: Which is Better for Characterizing Biological Taxa Found in Surface Soils for Sample Separation? Genes 2019, 10, 431. https://doi.org/10.3390/genes10060431
Boggs LM, Scheible MKR, Machado G, Meiklejohn KA. Single Fragment or Bulk Soil DNA Metabarcoding: Which is Better for Characterizing Biological Taxa Found in Surface Soils for Sample Separation? Genes. 2019; 10(6):431. https://doi.org/10.3390/genes10060431
Chicago/Turabian StyleBoggs, Laura M., Melissa K. R. Scheible, Gustavo Machado, and Kelly A. Meiklejohn. 2019. "Single Fragment or Bulk Soil DNA Metabarcoding: Which is Better for Characterizing Biological Taxa Found in Surface Soils for Sample Separation?" Genes 10, no. 6: 431. https://doi.org/10.3390/genes10060431
APA StyleBoggs, L. M., Scheible, M. K. R., Machado, G., & Meiklejohn, K. A. (2019). Single Fragment or Bulk Soil DNA Metabarcoding: Which is Better for Characterizing Biological Taxa Found in Surface Soils for Sample Separation? Genes, 10(6), 431. https://doi.org/10.3390/genes10060431