Unique Mutational Spectrum of the GJB2 Gene and Its Pathogenic Contribution to Deafness in Tuvinians (Southern Siberia, Russia): A High Prevalence of Rare Variant c.516G>C (p.Trp172Cys)

 , , , , ,
, , , , ,  and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Control Sample
2.3. Ethics Statement
2.4. Mutation Analysis of the GJB2 Gene
2.5. Verification of Cis-Configuration of GJB2 Variants c.79G>A (p.Val27Ile) and c.341A>G (p.Glu114Gly)
2.6. Bioinformatics Prediction Tools
2.7. 3D Cx26 Molecule Structure Modelling
2.8. Statistical Methods
3. Results
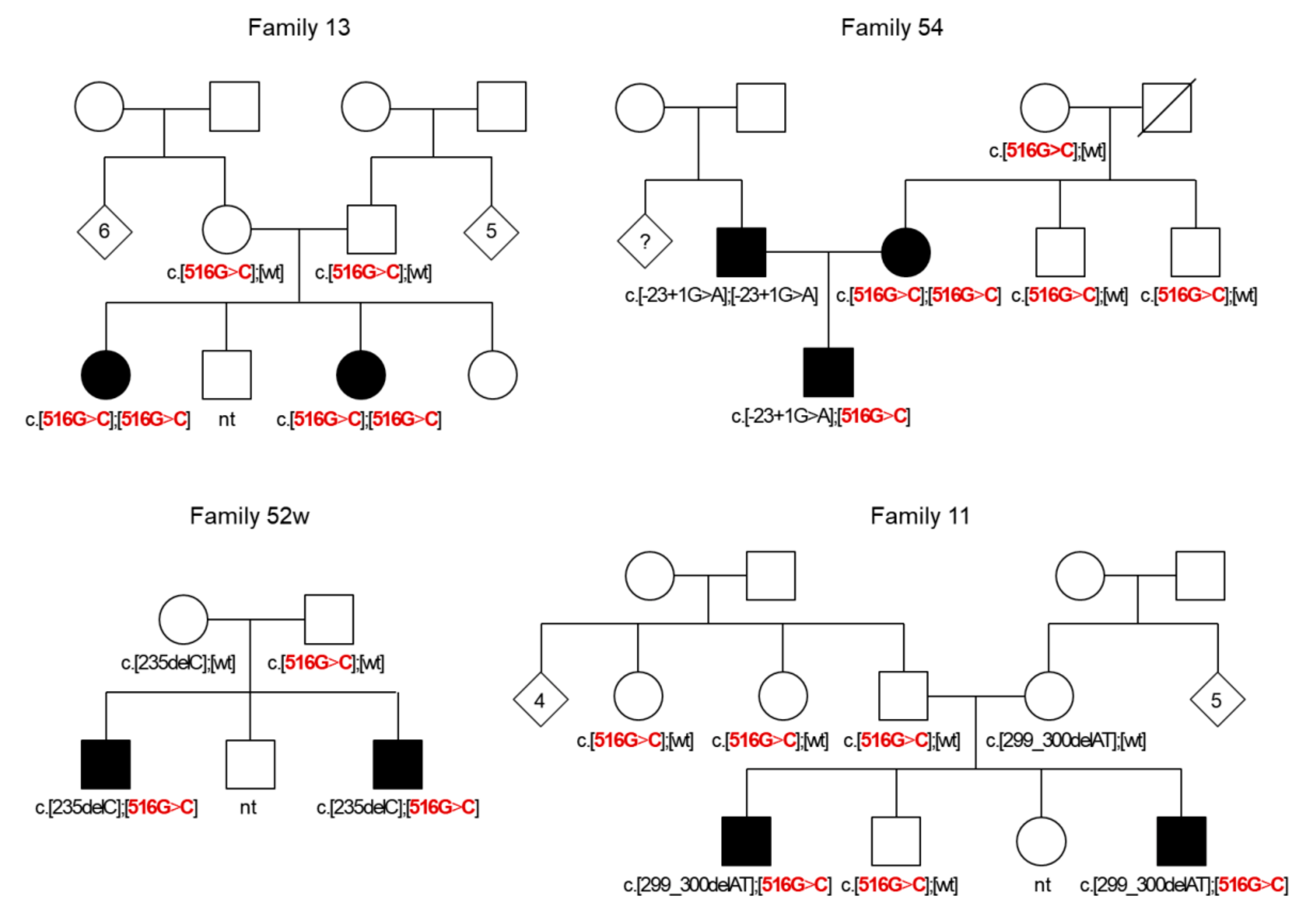
3.1. GJB2 Genotypes Observed in Patients and Control Sample
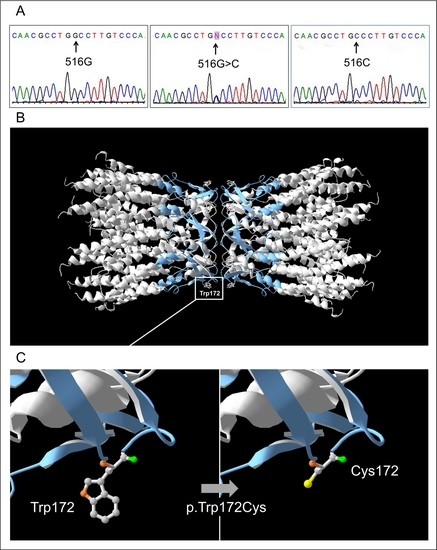
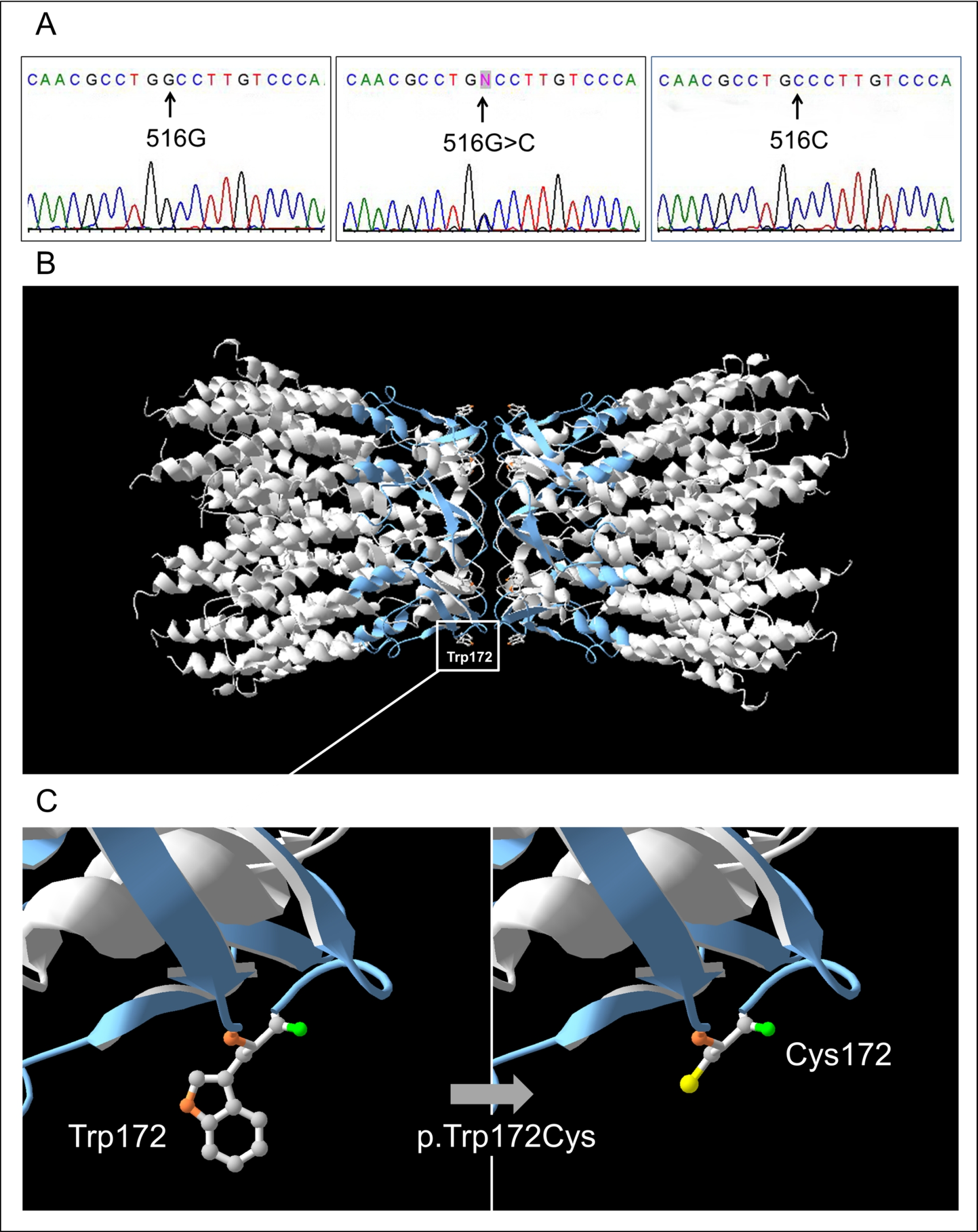
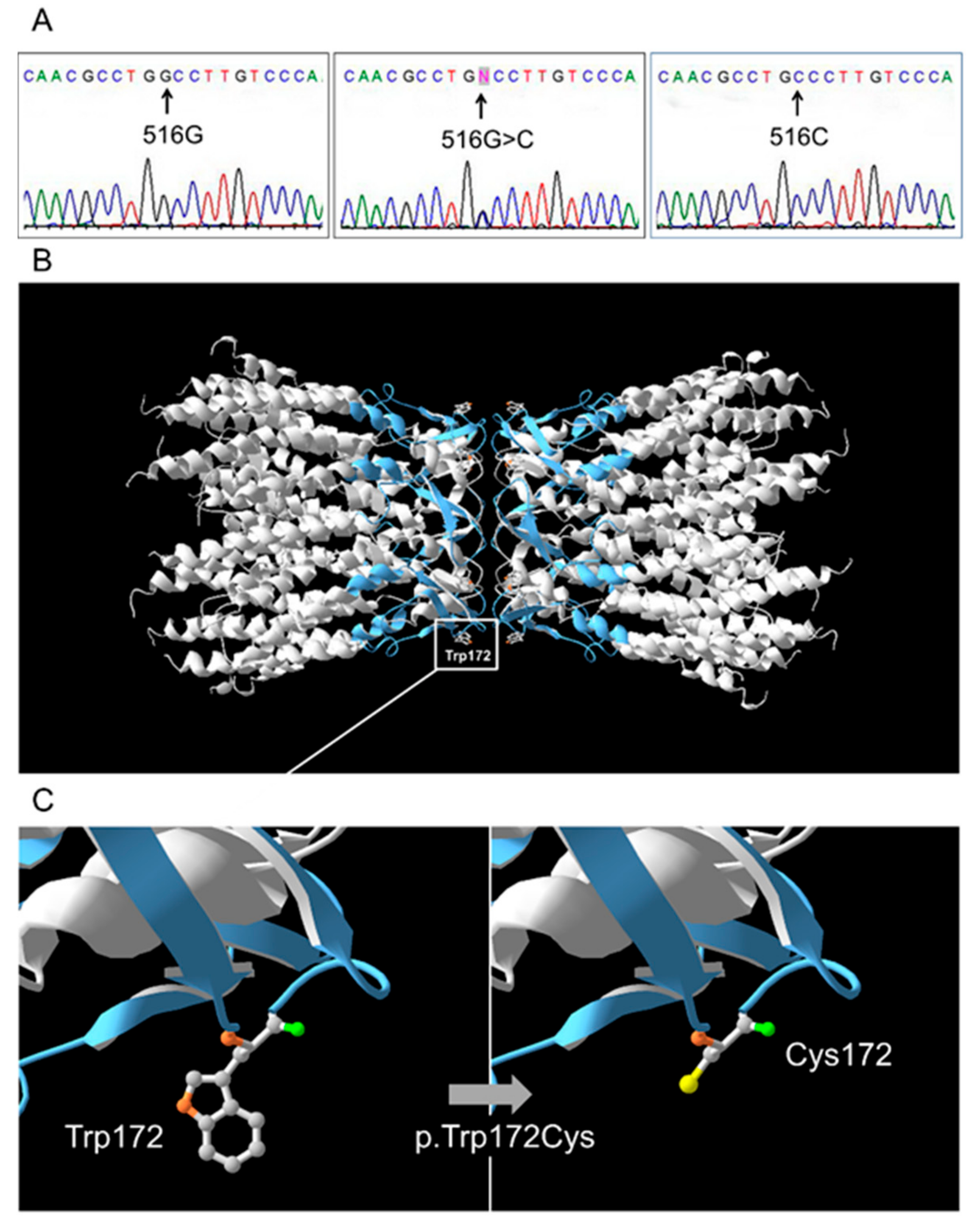
3.2. GJB2 Sequence Variations in Patients and Control Sample
3.3. Group of Patients with a Single GJB2 Pathogenic Variant
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Morton, C.C.; Nance, W.E. Newborn hearing screening—A silent revolution. N. Engl. J. Med. 2006, 354, 2151–2164. [Google Scholar] [CrossRef] [PubMed]
- Toriello, H.V.; Smith, S.D. Hereditary Hearing Loss and Its Syndromes, 3rd ed.; Oxford University Press: Oxford, UK, 2013; p. 756. [Google Scholar]
- Van Camp, G.; Smith, R.J.H. Hereditary Hearing Loss Homepage. Available online: https://hereditaryhearingloss.org (accessed on 4 April 2019).
- Del Castillo, F.J.; del Castillo, I. DFNB1 Non-syndromic hearing impairment: diversity of mutations and associated phenotypes. Front. Mol. Neurosci. 2017, 10, 428. [Google Scholar] [CrossRef] [PubMed]
- Bruzzone, R.; White, T.W.; Paul, D.L. Connections with connexins: The molecular basis of direct intercellular signaling. Eur. J. Biochem. 1996, 238, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Kenneson, A.; Van Naarden Braun, K.; Boyle, C. GJB2 (connexin 26) variants and nonsyndromic sensorineural hearing loss: A HuGE review. Genet. Med. 2002, 4, 258–274. [Google Scholar] [CrossRef] [PubMed]
- Chan, D.K.; Chang, K.W. GJB2-associated hearing loss: Systematic review of worldwide prevalence, genotype, and auditory phenotype: Systematic review of Cx-26-associated hearing loss. Laryngoscope 2014, 124, E34–E53. [Google Scholar] [CrossRef] [PubMed]
- Stenson, P.D.; Mort, M.; Ball, E.V.; Evans, K.; Hayden, M.; Heywood, S.; Hussain, M.; Phillips, A.D.; Cooper, D.N. The human gene mutation database: Towards a comprehensive repository of inherited mutation data for medical research, genetic diagnosis and next-generation sequencing studies. Hum. Genet. 2017, 136, 665–677. [Google Scholar] [CrossRef] [PubMed]
- Tsukada, K.; Nishio, S.Y.; Hattori, M.; Usami, S. Ethnic-specific spectrum of GJB2 and SLC26A4 mutations: Their origin and a literature review. Ann. Otol. Rhinol. Laryngol. 2015, 124, 61S–76S. [Google Scholar] [CrossRef] [PubMed]
- Gasparini, P.; Rabionet, R.; Barbujani, G.; Melchionda, S.; Petersen, M.; Brøndum-Nielsen, K.; Metspalu, A.; Oitmaa, E.; Pisano, M.; Fortina, P.; et al. High carrier frequency of the 35delG deafness mutation in European populations. Eur. J. Hum. Genet. 2000, 8, 19–23. [Google Scholar] [CrossRef] [PubMed]
- Mahdieh, N.; Rabbani, B. Statistical study of 35delG mutation of GJB2 gene: A meta-analysis of carrier frequency. Int. J. Audiol. 2009, 48, 363–370. [Google Scholar] [CrossRef]
- Liu, X.; Xia, X.; Ke, X.; Ouyang, X.; Du, L.; Liu, Y.; Angeli, S.; Telischi, F.; Nance, W.; Balkany, T.; et al. The prevalence of connexin 26 (GJB2) mutations in the Chinese population. Hum. Genet. 2002, 111, 394–397. [Google Scholar] [CrossRef]
- Ohtsuka, A.; Yuge, I.; Kimura, S.; Namba, A.; Abe, S. GJB2 deafness gene shows a specific spectrum of mutations in Japan, including a frequent founder mutation. Hum. Genet. 2003, 112, 329–333. [Google Scholar] [PubMed]
- Yao, J.; Lu, Y.; Wei, Q.; Cao, X.; Xing, G. A systematic review and meta-analysis of 235delC mutation of GJB2 gene. J. Transl. Med. 2012, 10, 136. [Google Scholar] [CrossRef]
- Morell, R.J.; Kim, H.J.; Hood, L.J.; Goforth, L.; Friderici, K.; Fisher, R.; van Camp, G.; Berlin, C.I.; Oddoux, C.; Ostrer, H.; et al. Mutations in the Connexin 26 Gene (GJB2) among Ashkenazi Jews with nonsyndromic recessive deafness. N. Engl. J. Med. 1998, 339, 1500–1505. [Google Scholar] [CrossRef] [PubMed]
- Hamelmann, C.; Amedofu, G.K.; Albrecht, K.; Muntau, B.; Gelhaus, A.; Brobby, G.W.; Horstmann, R.D. Pattern of connexin 26 (GJB2) mutations causing sensorineural hearing impairment in Ghana. Hum. Mutat. 2001, 18, 84–85. [Google Scholar] [CrossRef] [PubMed]
- RamShankar, M.; Girirajan, S.; Dagan, O.; Ravi Shankar, H.M.; Jalvi, R.; Rangasayee, R.; Avraham, K.B.; Anand, A. Contribution of connexin26 (GJB2) mutations and founder effect to non-syndromic hearing loss in India. J. Med. Genet. 2003, 40, e68. [Google Scholar] [CrossRef] [PubMed]
- Minárik, G.; Ferák, V.; Feráková, E.; Ficek, A.; Poláková, H.; Kádasi, Ľ. High frequency of GJB2 mutation W24X among Slovak Romany (Gypsy) patients with non-syndromic hearing loss (NSHL). Gen. Physiol. Biophys. 2003, 22, 549–556. [Google Scholar] [PubMed]
- Álvarez, A.; del Castillo, I.; Villamar, M.; Aguirre, L.A.; González-Neira, A.; López-Nevot, A.; Moreno-Pelayo, M.A.; Moreno, F. High prevalence of theW24X mutation in the gene encoding connexin-26 (GJB2) in Spanish Romani (gypsies) with autosomal recessive non-syndromic hearing loss. Am. J. Med. Genet. A 2005, 137A, 255–258. [Google Scholar] [CrossRef] [PubMed]
- Wattanasirichaigoon, D.; Limwongse, C.; Jariengprasert, C.; Yenchitsomanus, P.; Tocharoenthanaphol, C.; Thongnoppakhun, W.; Thawil, C.; Charoenpipop, D.; Pho-iam, T.; Thongpradit, S.; et al. High prevalence of V37I genetic variant in the connexin-26 (GJB2) gene among non-syndromic hearing-impaired and control Thai individuals: High prevalence of V37I among Thai subjects. Clin. Genet. 2004, 66, 452–460. [Google Scholar] [CrossRef]
- Barashkov, N.A.; Dzhemileva, L.U.; Fedorova, S.A.; Teryutin, F.M.; Posukh, O.L.; Fedotova, E.E.; Lobov, S.L.; Khusnutdinova, E.K. Autosomal recessive deafness 1A (DFNB1A) in Yakut population isolate in Eastern Siberia: Extensive accumulation of the splice site mutation IVS1+1G>A in GJB2 gene as a result of founder effect. J. Hum. Genet. 2011, 56, 631–639. [Google Scholar] [CrossRef]
- Carranza, C.; Menendez, I.; Herrera, M.; Castellanos, P.; Amado, C.; Maldonado, F.; Rosales, L.; Escobar, N.; Guerra, M.; Alvarez, D.; et al. A Mayan founder mutation is a common cause of deafness in Guatemala. Clin. Genet. 2016, 89, 461–465. [Google Scholar] [CrossRef]
- Vainshtein, S.I.; Mannay-ool, M.H. History of Tyva, 2nd ed.; Science: Novosibirsk, Russia, 2001. (In Russian) [Google Scholar]
- Mongush, M.V. Tuvans of Mongolia and China. Int. J. Cent. Asian Stud. 1996, 1, 225–243. [Google Scholar]
- Bady-Khoo, M.S.; Posukh, O.L.; Zorkoltseva, I.V.; Skidanova, O.V.; Barashkov, N.A.; Omzar, O.S.; Mongush, R.S.; Bamba, O.M.; Tukar, V.M.; Zytsar, M.V.; et al. Study of hereditary hearing loss in the Republic of Tuva. I. Epidemiology of hearing impairments in the Republic of Tuva. Med. Genetika 2014, 13, 17–26. (In Russian) [Google Scholar]
- Nance, W.E.; Liu, X.-Z.; Pandya, A. Relation between choice of partner and high frequency of connexin-26 deafness. Lancet 2000, 356, 500–501. [Google Scholar] [CrossRef]
- Del Castillo, I.; Villamar, M.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Álvarez, A.; Tellería, D.; Menéndez, I.; Moreno, F. A deletion involving the Connexin 30 Gene in nonsyndromic hearing impairment. N. Engl. J. Med. 2002, 346, 243–249. [Google Scholar] [CrossRef] [PubMed]
- Brown, K.K.; Rehm, H.L. Molecular diagnosis of hearing loss. Curr. Protoc. Hum. Genet. 2012, 72, 9.16.1–9.16.16. [Google Scholar] [CrossRef] [PubMed]
- Matos, T.D.; Caria, H.; Simoes-Teixeira, H.; Aasen, T.; Nickel, R.; Jagger, D.J.; O’Neill, A.; Kelsell, D.P.; Fialho, G. A novel hearing loss-related mutation occurring in the GJB2 basal promoter. J. Med. Genet. 2007, 44, 721–725. [Google Scholar] [CrossRef] [PubMed]
- Sirmaci, A.; Akcayoz-Duman, D.; Tekin, M. The c.IVS1+1G>A mutation in the GJB2 gene is prevalent and large deletions involving the GJB6 gene are not present in the Turkish population. J. Genet. 2006, 85, 213–216. [Google Scholar] [CrossRef] [PubMed]
- Adzhubei, I.A.; Schmidt, S.; Peshkin, L.; Ramensky, V.E.; Gerasimova, A.; Bork, P.; Kondrashov, A.S.; Sunyaev, S.R. A method and server for predicting damaging missense mutations. Nat. Methods 2010, 7, 248–249. [Google Scholar] [CrossRef] [PubMed]
- Choi, Y.; Chan, A.P. PROVEAN web server: A tool to predict the functional effect of amino acid substitutions and indels. Bioinformatics 2015, 31, 2745–2747. [Google Scholar] [CrossRef]
- Tang, H.; Thomas, P.D. PANTHER-PSEP: Predicting disease-causing genetic variants using position-specific evolutionary preservation. Bioinformatics 2016, 32, 2230–2232. [Google Scholar] [CrossRef]
- Schwarz, J.M.; Cooper, D.N.; Schuelke, M.; Seelow, D. MutationTaster2: Mutation prediction for the deep-sequencing age. Nat. Methods 2014, 11, 361–362. [Google Scholar] [CrossRef] [PubMed]
- Shihab, H.A.; Gough, J.; Cooper, D.N.; Stenson, P.D.; Barker, G.L.A.; Edwards, K.J.; Day, I.N.M.; Gaunt, T.R. Predicting the functional, molecular, and phenotypic consequences of amino acid substitutions using hidden markov models. Hum. Mutat. 2013, 34, 57–65. [Google Scholar] [CrossRef] [PubMed]
- Guex, N.; Peitsch, M.C. SWISS-MODEL and the Swiss-PdbViewer: An environment for comparative protein modeling. Electrophoresis 1997, 18, 2714–2723. [Google Scholar] [CrossRef] [PubMed]
- Posukh, O.; Pallares-Ruiz, N.; Tadinova, V.; Osipova, L.; Claustres, M.; Roux, A.-F. First molecular screening of deafness in the Altai Republic population. BMC Med. Genet. 2005, 6, 12. [Google Scholar] [CrossRef] [PubMed]
- Tekin, M.; Xia, X.-J.; Erdenetungalag, R.; Cengiz, F.B.; White, T.W.; Radnaabazar, J.; Dangaasuren, B.; Tastan, H.; Nance, W.E.; Pandya, A. GJB2 mutations in Mongolia: Complex alleles, low frequency, and reduced fitness of the deaf. Ann. Hum. Genet. 2010, 74, 155–164. [Google Scholar] [CrossRef] [PubMed]
- Maeda, S.; Nakagawa, S.; Suga, M.; Yamashita, E.; Oshima, A.; Fujiyoshi, Y.; Tsukihara, T. Structure of the connexin 26 gap junction channel at 3.5 Å resolution. Nature 2009, 458, 597–602. [Google Scholar] [CrossRef]
- Seeman, P.; Sakmaryová, I. High prevalence of the IVS 1 + 1 G to A/GJB2 mutation among Czech hearing impaired patients with monoallelic mutation in the coding region of GJB2: IVS1 + 1 G to A GJB2 mutation in Czech. Clin. Genet. 2006, 69, 410–413. [Google Scholar] [CrossRef]
- Yuan, Y.; Yu, F.; Wang, G.; Huang, S.; Yu, R.; Zhang, X.; Huang, D.; Han, D.; Dai, P. Prevalence of the GJB2 IVS1+1G > A mutation in Chinese hearing loss patients with monoallelic pathogenic mutation in the coding region of GJB2. J. Transl. Med. 2010, 8, 127. [Google Scholar] [CrossRef]
- Bazazzadegan, N.; Nikzat, N.; Fattahi, Z.; Nishimura, C.; Meyer, N.; Sahraian, S.; Jamali, P.; Babanejad, M.; Kashef, A.; Yazdan, H.; et al. The spectrum of GJB2 mutations in the Iranian population with non-syndromic hearing loss—A twelve year study. Int. J. Pediatr. Otorhinolaryngol. 2012, 76, 1164–1174. [Google Scholar] [CrossRef]
- Barashkov, N.A.; Pshennikova, V.G.; Posukh, O.L.; Teryutin, F.M.; Solovyev, A.V.; Klarov, L.A.; Romanov, G.P.; Gotovtsev, N.N.; Kozhevnikov, A.A.; Kirillina, E.V.; et al. Spectrum and frequency of the GJB2 gene pathogenic variants in a large cohort of patients with hearing impairment living in a subarctic region of Russia (the Sakha Republic). PLoS ONE 2016, 11, e0156300. [Google Scholar] [CrossRef]
- Erdenechuluun, J.; Lin, Y.-H.; Ganbat, K.; Bataakhuu, D.; Makhbal, Z.; Tsai, C.-Y.; Lin, Y.-H.; Chan, Y.-H.; Hsu, C.-J.; Hsu, W.-C.; et al. Unique spectra of deafness-associated mutations in Mongolians provide insights into the genetic relationships among Eurasian populations. PLoS ONE 2018, 13, e0209797. [Google Scholar] [CrossRef]
- Wu, C.-C.; Chen, P.-J.; Chiu, Y.-H.; Lu, Y.-C.; Wu, M.-C.; Hsu, C.-J. Prospective mutation screening of three common deafness genes in a large taiwanese cohort with idiopathic bilateral sensorineural hearing impairment reveals a difference in the results between families from hospitals and those from rehabilitation facilities. Audiol. Neurotol. 2008, 13, 172–181. [Google Scholar]
- Dai, P.; Yu, F.; Han, B.; Liu, X.; Wang, G.; Li, Q.; Yuan, Y.; Liu, X.; Huang, D.; Kang, D.; et al. GJB2 mutation spectrum in 2063 Chinese patients with nonsyndromic hearing impairment. J. Transl. Med. 2009, 7, 26. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ao, L.; Ding, H.; Zhang, D. Genetic frequencies related to severe or profound sensorineural hearing loss in Inner Mongolia Autonomous Region. Genet. Mol. Biol. 2016, 39, 567–572. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Putcha, G.V.; Bejjani, B.A.; Bleoo, S.; Booker, J.K.; Carey, J.C.; Carson, N.; Das, S.; Dempsey, M.A.; Gastier-Foster, J.M.; Greinwald, J.H.; et al. A multicenter study of the frequency and distribution of GJB2 and GJB6 mutations in a large North American cohort. Genet. Med. 2007, 9, 413–426. [Google Scholar] [CrossRef] [PubMed]
- Choi, S.-Y.; Lee, K.Y.; Kim, H.-J.; Kim, H.-K.; Chang, Q.; Park, H.-J.; Jeon, C.-J.; Lin, X.; Bok, J.; Kim, U.-K. Functional evaluation of GJB2 variants in nonsyndromic hearing loss. Mol. Med. 2011, 17, 550–556. [Google Scholar] [CrossRef] [PubMed]
- Zhao, F.-F.; Ji, Y.-B.; Wang, D.-Y.; Lan, L.; Han, M.-K.; Li, Q.; Zhao, Y.; Rao, S.; Han, D.; Wang, Q.-J. Phenotype–genotype correlation in 295 Chinese deaf subjects with biallelic causative mutations in the GJB2 gene. Genet. Test. Mol. Biomark. 2011, 15, 619–625. [Google Scholar] [CrossRef]
- Chen, W.; Huang, Y.; Yang, X.; Duan, B.; Lu, P.; Wang, Y.; Xu, Z. The homozygote p.V27I/p.E114G variant of GJB2 is a putative indicator of nonsyndromic hearing loss in Chinese infants. Int. J. Pediatr. Otorhinolaryngol. 2016, 84, 48–51. [Google Scholar] [CrossRef]
- Choung, Y.H.; Moon, S.-K.; Park, H.-J. Functional study of GJB2 in hereditary hearing loss. Laryngoscope 2002, 112, 1667–1671. [Google Scholar] [CrossRef]
- Wilcox, S.A.; Saunders, K.; Osborn, A.H.; Arnold, A.; Wunderlich, J.; Kelly, T.; Collins, V.; Wilcox, L.J.; McKinlay Gardner, R.; Kamarinos, M.; et al. High frequency hearing loss correlated with mutations in the GJB2 gene. Hum. Genet. 2000, 106, 399–405. [Google Scholar] [CrossRef]
- Del Castillo, I.; Moreno-Pelayo, M.A.; del Castillo, F.J.; Brownstein, Z.; Marlin, S.; Adina, Q.; Cockburn, D.J.; Pandya, A.; Siemering, K.R.; Chamberlin, G.P.; et al. Prevalence and evolutionary origins of the del(GJB6-D13S1830) mutation in the DFNB1 locus in hearing-impaired subjects: A multicenter study. Am. J. Hum. Genet. 2003, 73, 1452–1458. [Google Scholar] [CrossRef] [PubMed]
- Azaiez, H.; Chamberlin, G.P.; Fischer, S.M.; Welp, C.L.; Prasad, S.D.; Taggart, R.T.; Castillo, I.; del Camp, G.V.; Smith, R.J.H. GJB2: The spectrum of deafness-causing allele variants and their phenotype. Hum. Mutat. 2004, 24, 305–311. [Google Scholar] [CrossRef] [PubMed]
- Angeli, S.I. Phenotype/genotype correlations in a DFNB1 cohort with ethnical diversity. Laryngoscope 2008, 118, 2014–2023. [Google Scholar] [CrossRef] [PubMed]
- Pollak, A.; Mueller-Malesinska, M.; Skorka, A.; Kostrzewa, G.; Oldak, M.; Korniszewski, L.; Skarzynski, H.; Ploski, R. GJB2 and hearing impairment: Promoter defects do not explain the excess of monoallelic mutations. J. Med. Genet. 2008, 45, 607–608. [Google Scholar] [CrossRef] [PubMed]
- Kashef, A.; Nikzat, N.; Bazzazadegan, N.; Fattahi, Z.; Sabbagh-Kermani, F.; Taghdiri, M.; Azadeh, B.; Mojahedi, F.; Khoshaeen, A.; Habibi, H.; et al. Finding mutation within non-coding region of GJB2 reveals its importance in genetic testing of Hearing Loss in Iranian population. Int. J. Pediatr. Otorhinolaryngol. 2015, 79, 136–138. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.F.; Warnecke, A.; Schöner-Heinisch, A.; Lesinski-Schiedat, A.; Maier, H.; Lenarz, T. Prevalence and audiological profiles of GJB2 mutations in a large collective of hearing impaired patients. Hear. Res. 2016, 333, 77–86. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.Y.; Kim, A.R.; Kim, N.K.D.; Lee, C.; Kim, M.Y.; Jeon, E.-H.; Park, W.-Y.; Choi, B.Y. Unraveling of enigmatic hearing-impaired GJB2 single heterozygotes by massive parallel sequencing: DFNB1 or not? Medicine (Baltimore) 2016, 95, e3029. [Google Scholar] [CrossRef]
- Parzefall, T.; Frohne, A.; Koenighofer, M.; Kirchnawy, A.; Streubel, B.; Schoefer, C.; Frei, K.; Lucas, T. Whole-exome sequencing to identify the cause of congenital sensorineural hearing loss in carriers of a heterozygous GJB2 mutation. Eur. Arch. Otorhinolaryngol. 2017, 274, 3619–3625. [Google Scholar] [CrossRef]
- Tu, Z.J.; Kiang, D.T. Mapping and characterization of the basal promoter of the human connexin26 gene. Biochim. Biophys. Acta—Gene Struct. Expr. 1998, 1443, 169–181. [Google Scholar] [CrossRef]
- Zoll, B.; Petersen, L.; Lange, K.; Gabriel, P.; Kiese-Himmel, C.; Rausch, P.; Berger, J.; Pasche, B.; Meins, M.; Gross, M.; et al. Evaluation of Cx26/GJB2 in German hearing impaired persons: Mutation spectrum and detection of disequilibrium between M34T (c.101T>C) and −493del10. Hum. Mutat. 2003, 21, 98. [Google Scholar] [CrossRef]
- Mani, R.S.; Ganapathy, A.; Jalvi, R.; Srikumari Srisailapathy, C.R.; Malhotra, V.; Chadha, S.; Agarwal, A.; Ramesh, A.; Rangasayee, R.R.; Anand, A. Functional consequences of novel connexin 26 mutations associated with hereditary hearing loss. Eur. J. Hum. Genet. 2009, 17, 502–509. [Google Scholar] [CrossRef] [PubMed]
- Matos, T.D.; Simões-Teixeira, H.; Caria, H.; Cascão, R.; Rosa, H.; O’Neill, A.; Dias, Ó.; Andrea, M.E.; Kelsell, D.P.; Fialho, G. Assessing noncoding sequence variants of GJB2 for hearing loss association. Genet. Res. Int. 2011, 2011, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Gandía, M.; del Castillo, F.J.; Rodríguez-Álvarez, F.J.; Garrido, G.; Villamar, M.; Calderón, M.; Moreno-Pelayo, M.A.; Moreno, F.; del Castillo, I. A novel splice-site mutation in the GJB2 gene causing mild postlingual hearing impairment. PLoS ONE 2013, 8, e73566. [Google Scholar] [CrossRef] [PubMed]
- del Castillo, F.J. A novel deletion involving the connexin-30 gene, del(GJB6-d13s1854), found in trans with mutations in the GJB2 gene (connexin-26) in subjects with DFNB1 non-syndromic hearing impairment. J. Med. Genet. 2005, 42, 588–594. [Google Scholar] [CrossRef] [PubMed]
- Feldmann, D.; Le Maréchal, C.; Jonard, L.; Thierry, P.; Czajka, C.; Couderc, R.; Ferec, C.; Denoyelle, F.; Marlin, S.; Fellmann, F. A new large deletion in the DFNB1 locus causes nonsyndromic hearing loss. Eur. J. Med. Genet. 2009, 52, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Wilch, E.; Azaiez, H.; Fisher, R.; Elfenbein, J.; Murgia, A.; Birkenhäger, R.; Bolz, H.; Da Silva-Costa, S.; del Castillo, I.; Haaf, T.; et al. A novel DFNB1 deletion allele supports the existence of a distant cis-regulatory region that controls GJB2 and GJB6 expression. Clin. Genet. 2010, 78, 267–274. [Google Scholar] [CrossRef] [PubMed]
- Bliznetz, E.A.; Makienko, O.N.; Okuneva, E.G.; Markova, T.G.; Polyakov, A.V. New recurrent large deletion, encompassing both GJB2 and GJB6 genes, results in isolated sensorineural hearing impairment with autosomal recessive mode of inheritance. Russ. J. Genet. 2014, 50, 415–420. [Google Scholar] [CrossRef]
- Tayoun, A.N.A.; Mason-Suares, H.; Frisella, A.L.; Bowser, M.; Duffy, E.; Mahanta, L.; Funke, B.; Rehm, H.L.; Amr, S.S. Targeted droplet-digital PCR as a tool for novel deletion discovery at the DFNB1 locus. Hum. Mutat. 2016, 37, 119–126. [Google Scholar] [CrossRef]
- Pshennikova, V.G.; Barashkov, N.A.; Solovyev, A.V.; Romanov, G.P.; Diakonov, E.E.; Sazonov, N.N.; Morozov, I.V.; Bondar, A.A.; Posukh, O.L.; Dzhemileva, L.U.; et al. Analysis of GJB6 (Cx30) and GJB3 (Cx31) genes in deaf patients with monoallelic mutations in GJB2 (Cx26) gene in the Sakha Republic (Yakutia). Russ. J. Genet. 2017, 53, 688–697. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Posukh, O.L.; Institute of Cytology and Genetics, Novosibirsk, Russia. Personal Communication, 2019.
- Yang, X.-L.; Xu, B.-C.; Chen, X.-J.; Bian, P.-P.; Ma, J.-L.; Liu, X.W.; Zhang, Z.-W.; Wan, D.; Zhu, Y.-M.; Guo, Y.-F. Common molecular etiology of patients with nonsyndromic hearing loss in Tibetan, Tu nationality, and Mongolian patients in the northwest of China. Acta Otolaryngol. (Stockh.) 2013, 133, 930–934. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
{kind=link}
| GJB2 | Primers | Reference |
|---|---|---|
| GJB2 coding region (exon 2) and flanking sequences | 835-F: 5’-TGCTTGCTTACCCAGACTCA-3’ 835-R: 5’-CCTCATCCCTCTCATGCTGT-3’ | This study |
| or | ||
| F3044: 5’-AGTGCCTTTCAGCTAACGA-3’ R4242: 5’-GTGGCATCTGGAGTTTCACC-3’ | ||
| Upstream region of exon 1 (which includes the basal promoter), exon 1, donor splice site, and flanking intronic region | Ex1-F: 5’-TCTTTTCCAGAGCAAACCGC-3’ Ex1-R: 5’-CTGGGCAATGCGTTAAACTGG-3’ | [30] |
| Ex1-792-F: 5’-GCGTTCGTTCGGATTGGT-3’ Ex1-2239-R: 5’-CGGAAACAGACCCTCGTGAAGT-3’ | This study | |
| PF1: 5’- GGCTCAAAGGAACTAGGAGATCG-3’ PF2: 5’-CGTTCGTTCGGATTGGTGAG-3’PR1: 5’-CAGAAACGCCCGCTCCAGAA-3’ | [29] |
| GJB2 Genotypes * | Patients (n = 220) | Control Sample (n = 157) |
|---|---|---|
| GJB2 genotypes with biallelic recessive pathogenic variants | ||
| c.[516G>C];[516G>C] p.[Trp172Cys];[Trp172Cys] | 25 | - |
| c.[-23+1G>A];[-23+1G>A] p.[splice donor variant];[splice donor variant] | 9 | - |
| c.[-23+1G>A];[516G>C] p.[splice donor variant];[Trp172Cys] | 7 | - |
| c.[235delC];[516G>C] p.[Leu79Cysfs*3];[Trp172Cys] | 4 | - |
| c.[299_300delAT];[516G>C] p.[His100Argfs*14];[Trp172Cys] | 2 | - |
| c.[-23+1G>A];[299_300delAT] p.[splice donor variant];[His100Argfs*14] | 1 | - |
| c.[109G>A];[235delC] p.[Val37Ile];[Leu79Cysfs*3] | 1 | - |
| Total | 49 (22.3%) | - |
| GJB2 genotypes with a single recessive pathogenic variant | ||
| c.[-23+1G>A];[wt] p.[splice donor variant];[wt] | 5 | 3 |
| c.[516G>C];[wt] p.[Trp172Cys];[wt] | 9 | 6 |
| c.[-23+1G>A];[79G>A] p.[splice donor variant];[Val27Ile] | 1 | 3 |
| c.[79G>A];[516G>C] p.[Val27Ile];[Trp172Cys] | 1 | 0 |
| c.[235delC];[wt] p.[Leu79Cysfs*3];[wt] | 1 | 0 |
| c.[109G>A];[wt] p.[Val37Ile];[wt] | 1 | 2 |
| c.[79G>A];[109G>A] p.[Val27Ile];[Val37Ile] | 0 | 1 |
| Total | 18 (8.2%) | 15 (9.6%) |
| GJB2 genotypes with benign variants | ||
| c.[79G>A];[wt] p.[Val27Ile];[wt] | 27 | 31 |
| c.[79G>A;341A>G];[wt] p.[Val27Ile;Glu114Gly];[wt] | 15 | 12 |
| c.[79G>A];[79G>A] p.[Val27Ile];[Val27Ile] | 7 | 6 |
| c.[79G>A];[79G>A;341A>G] p.[Val27Ile];[Val27Ile;Glu114Gly] | 1 | 2 |
| c.[79G>A;341A>G];[571T>C] p.[Val27Ile;Glu114Gly];[Phe191Leu] | 1 | 0 |
| c.[79G>A];[571T>C] p.[Val27Ile];[Phe191Leu] | 1 | 1 |
| c.[457G>A];[wt] p.[Val153Ile];[wt] | 1 | 0 |
| c.[608T>C];[wt] p.[Ile203Thr];[wt] | 1 | 0 |
| c.[79G>A;341A>G];[457G>A] p.[Val27Ile;Glu114Gly];[Val153Ile] | 0 | 1 |
| Total | 54 (24.5%) | 53 (33.7%) |
| GJB2 genotype [wt];[wt] | 99 (45.0%) | 89 (56.7%) |
| GJB2 Gene Sequence Variations | dbSNP ID | Patients (n = 184) * Number of Alleles/Frequency | Control Sample (n = 157) Number of Alleles/Frequency |
|---|---|---|---|
| GJB2 pathogenic variants | |||
| c.516G>C (p.Trp172Cys) | not presented | 51/0.1386 | 6/0.019 |
| c.-23+1G>A | rs80338940 | 30/0.0815 | 6/0.019 |
| c.235delC | rs80338943 | 5/0.0136 | 0/0.0 |
| c.299_300delAT | rs111033204 | 2/0.0054 | 0/0.0 |
| c.109G>A (p.Val37Ile) | rs72474224 | 2/0.0054 | 3/0.0096 |
| Total | 90/0.2446 | 15/0.0478 | |
| GJB2 benign variants | |||
| c.79G>A (p.Val27Ile) | rs2274084 | 41/0.1141 | 50/0.1592 |
| c.[79G>A;341A>G] (p.[Val27Ile;Glu114Gly]) | rs2274084 + rs2274083 | 15/0.0408 | 15/0.0478 |
| c.571T>C (p.Phe191Leu) | rs397516878 | 2/0.0054 | 1/0.0032 |
| c.457G>A (p.Val153Ile) | rs111033186 | 1/0.0027 | 1/0.0032 |
| c.608T>C (p.Ile203Thr) | rs76838169 | 1/0.0027 | 0/0.0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Posukh, O.L.; Zytsar, M.V.; Bady-Khoo, M.S.; Danilchenko, V.Y.; Maslova, E.A.; Barashkov, N.A.; Bondar, A.A.; Morozov, I.V.; Maximov, V.N.; Voevoda, M.I. Unique Mutational Spectrum of the GJB2 Gene and Its Pathogenic Contribution to Deafness in Tuvinians (Southern Siberia, Russia): A High Prevalence of Rare Variant c.516G>C (p.Trp172Cys). Genes 2019, 10, 429. https://doi.org/10.3390/genes10060429
Posukh OL, Zytsar MV, Bady-Khoo MS, Danilchenko VY, Maslova EA, Barashkov NA, Bondar AA, Morozov IV, Maximov VN, Voevoda MI. Unique Mutational Spectrum of the GJB2 Gene and Its Pathogenic Contribution to Deafness in Tuvinians (Southern Siberia, Russia): A High Prevalence of Rare Variant c.516G>C (p.Trp172Cys). Genes. 2019; 10(6):429. https://doi.org/10.3390/genes10060429
Chicago/Turabian StylePosukh, Olga L., Marina V. Zytsar, Marita S. Bady-Khoo, Valeria Yu. Danilchenko, Ekaterina A. Maslova, Nikolay A. Barashkov, Alexander A. Bondar, Igor V. Morozov, Vladimir N. Maximov, and Michael I. Voevoda. 2019. "Unique Mutational Spectrum of the GJB2 Gene and Its Pathogenic Contribution to Deafness in Tuvinians (Southern Siberia, Russia): A High Prevalence of Rare Variant c.516G>C (p.Trp172Cys)" Genes 10, no. 6: 429. https://doi.org/10.3390/genes10060429
APA StylePosukh, O. L., Zytsar, M. V., Bady-Khoo, M. S., Danilchenko, V. Y., Maslova, E. A., Barashkov, N. A., Bondar, A. A., Morozov, I. V., Maximov, V. N., & Voevoda, M. I. (2019). Unique Mutational Spectrum of the GJB2 Gene and Its Pathogenic Contribution to Deafness in Tuvinians (Southern Siberia, Russia): A High Prevalence of Rare Variant c.516G>C (p.Trp172Cys). Genes, 10(6), 429. https://doi.org/10.3390/genes10060429