1. Introduction
Over the last decade, the emergence of next-generation sequencing (NGS) technologies has catalyzed a proliferation of reference assemblies, including those of non-traditional model species (e.g., [
1]). However, only a small subset of such assemblies includes the Y-chromosome. This disparity is driven by the challenges associated with assembling Y-chromosome sequence, especially in eutherian mammals. One of the main issues is that the Y-chromosome contains a high proportion of repetitive sequences, which are difficult to assemble from short sequencing reads [
2,
3,
4,
5]. Sequencing projects specifically targeting the Y-chromosome often circumvent this problem using traditional methods, such as bacterial artificial chromosome (BAC) cloning or long-read Sanger sequencing technology [
6,
7,
8], but this dependence on more expensive technologies means that many of the advances made towards a reduction in the financial and time investment required for genome assembly do not extend to Y-chromosome assembly. While approaches to de novo assembly that utilize third-generation long-read sequencing technologies are emerging (e.g., [
9]), these approaches remain largely inaccessible to assembly projects targeting non-model species.
Work in several species has indicated that Y-chromosome information can be extracted from genomes sequenced with short-read technologies. De novo contigs constituting a partial assembly (186 Kbp) of the horse Y-chromosome were assembled using Roche 454 reads to conduct targeted resequencing of horse BAC clones that had been selected based on homology to Y-chromosome genes in other mammals [
10]. In the tongue sole, which is a flatfish with a 477-Mbp genome, scaffolds in the reference genome that corresponded to the constitutively haploid chromosome (W) were identified by sequencing the genomes of a homogametic (ZZ) and a heterogametic (ZW) fish at 212× coverage and by comparing the depth of coverage across the scaffolds between the male and female individuals [
11]. Using a similar approach, the 72,214 scaffolds comprising the Illumina-sequenced 2.3-Gbp polar bear reference genome [
12] were analyzed by comparing male-to-female depth of coverage across scaffolds and identifying scaffolds syntenic to Y-chromosome genes found in other eutherian mammals (human, dog, chimpanzee, and mouse) [
13]. This analysis identified 1.9 Mbp of putative Y-chromosome sequence across 112 scaffolds of the polar bear reference genome. In another de novo carnivore assembly, the grey wolf, comparison of male and female sequence coverage alongside known canine Y-linked genes similarly allowed for the identification of putative Y-chromosome scaffolds [
14]. These studies indicate that when a heterogametic individual is sequenced for de novo reference assembly, even when the assembly project uses short-read NGS technology, fragments (contigs and/or scaffolds) containing identifiable fragments of the constitutively haploid chromosome sequence are often produced.
The red fox (
Vulpes vulpes) is a non-traditional mammalian genomic model in which characterization of Y-chromosome diversity is of particular interest. The red fox is the world’s widest-spread terrestrial carnivore [
15,
16], and the species’ behavioral ecology results in males dispersing more widely than females [
17]. Mitochondrial DNA (mtDNA) haplotypes have been characterized in a number of populations to address a range of questions related to red fox population history and diversity (e.g., [
18,
19,
20,
21,
22,
23,
24]). However, because mtDNA is matrilineally inherited, mtDNA diversity alone may not accurately reflect genome-wide diversity [
25,
26]. Prior to the assembly of a red fox reference genome [
27], Y-chromosomal resources for the red fox were limited to two dog-derived microsatellite markers [
18].
Opportunities to develop Y-chromosome resources for the red fox expanded with the recent red fox genome sequencing project [
27], which produced 2.5 Gbp of sequence from a farm-bred male fox. The draft genome is organized in 676,878 scaffolds ranging in size from as large as 55.7 Mb to as small as 100 bp, with a scaffold N50 of 11.8 Mbp [
27]. Preliminary analysis [
28] of two scaffolds found to show higher synteny with the dog Y-chromosome than any other dog chromosome facilitated the development of 11 novel male-specific microsatellite markers that were used to conduct a preliminary analysis of patterns of diversity across red fox populations. Although the development of genomic resources for the red fox has focused primarily on experimentally bred tame and aggressive lines developed and maintained through the Russian Farm Fox Experiment at the Institute for Cytology and Genetics in Novosibirsk, Russia [
29], the preliminary analysis of inter-population diversity using the 13 microsatellite markers suggested that resources developed in farm-bred foxes are still useful for ascertaining variation in geographically diverse populations [
28].
The development of additional Y-chromosome resources for the red fox remains a priority. In particular, Y-chromosomal single nucleotide polymorphism (SNP) markers would provide higher resolution than microsatellite markers and allow for phylogenetic comparisons over longer timescales [
30]. Identifying the sequence of the red fox Y-chromosome would represent a significant step towards a dense SNP marker set for these analyses. The two known Y-chromosome scaffolds comprise only 1 Mbp of sequence, whereas the male-specific region of the Y-chromosome (MSY) is approximately 2.5 Mbp in many other carnivores [
6], suggesting that additional sequence may be present in the assembly. Likewise, the two Y-scaffolds contain only nine predicted genes [
27,
28], including only four of the 11 MSY genes consistently observed across carnivore species [
31]. Therefore, additional analysis is required to identify Y-chromosome sequence, including genes, present in the red fox draft genome.
The present analysis characterizes MSY sequence using three complementary approaches: analysis of gene content within scaffolds, identification of male-specific sequence motifs, and comparison of sequencing depth between males and females. The first approach, similarity between the scaffold sequence content and known Y-chromosome genes, has been used to identify MSY sequence in other mammals (e.g., [
10,
13,
14,
32]). MSY assemblies for two species closely related to the red fox, the cat (
Felis catus; KP081775.1) and dog (
Canis lupus familiaris; KP081776.1), are available [
6], with the red fox’s least common ancestors (LCA) with the cat estimated at 50–65 million years ago (MYA) and with the dog estimated at 9–15 MYA [
33,
34]. Many dog and cat Y-chromosome genes and protein sequences have been deposited in the databases maintained by the National Center for Biotechnology Information (NCBI) [
7,
35]. These sequences can therefore be used as probes to identify scaffolds in the draft genome that are likely to contain MSY sequence.
Two additional methods are used to complement the syntenic analysis. These methods are not restricted to regions that contain genes, but instead examine sex-specific patterns in whole genome resequencing data (WGS) mapped onto the reference assembly. Specifically, 15 male and 15 female red foxes bred on the same farm as the reference genome donor fox were resequenced at a depth of 2.5× per fox [
27]. The WGS data were analyzed to identify scaffolds likely to contain MSY sequence based on two metrics: sequence motifs exclusive to males and therefore likely to be derived from the MSY, and differences in sequencing depth in the heterogametic (male) versus homogametic (female) individuals. For the first metric, Carvalho and Clark [
36] developed software to identify male-specific sequence by fragmenting the scaffolds into
k-mers and tabulating
k-mer frequency in the male and female resequencing data. For the second, copy number variation (CNV) was characterized with CNV-seq [
37] to identify differences in sequence coverage of the scaffolds in male and female sequencing data. Analyzing the scaffolds along these two axes facilitates the identification of the scaffolds most likely to contain Y-chromosome sequence and thus provides an approach to identify MSY sequence bioinformatically. Used together, these approaches represent a consilience-oriented approach to the identification of MSY sequence from fragments assembled with short-read NGS technologies.
2. Materials and Methods
First, we sought to identify red fox orthologs of genes located on the MSY of dog and cat, which are two carnivores closely related to the red fox [
6,
7,
35,
38] (
Table 1). Most of these genes are X-degenerate, meaning they are thought to be derived from genes shared by the X- and Y-chromosomes in their ancestral state as a pair of homologous autosomes [
39], but some (e.g.,
TETY2 or
FLJ36031Y) were more recently transposed to the Y-chromosome from the X-chromosome or an autosome [
6,
7,
35] (
Table 1). Dog protein sequences or transcripts were downloaded, as available, from the NCBI Sequence Read Archive (SRA) for each of the genes on the dog MSY.
DYNG, which is a novel Y-chromosomal gene identified in dog [
6], was excluded at this stage due to the lack of a protein or mRNA sequence in NCBI SRA or other databases. Cat transcripts or nucleotide sequences were downloaded, as available, for the four genes present on the feline, but not the canine, MSY [
6] (
AMELY,
FLJ36031Y,
RPS4Y, and
TETY1) and for
EIF2S3Y, whose canine protein sequence has not been deposited.
Gene sequences were then mapped against all scaffolds longer than 5 Kbp in the draft red fox genome [
27] using translated BLAST (tblastn) or standard nucleotide BLAST (blastn), as appropriate, in the command line implementation of BLAST+ version 2.2.29 [
40]. A minimum e-value of 10
−5 was specified. Hits to autosomes and the X-chromosome were removed based on the chromosomal positions assigned to the scaffolds [
41] (
Table S1). Of the remaining hits, the best hit was determined to be the scaffold with the longest continuous stretch of query sequence mapping with greater than 90% (canine) or 80% (cat) identity. Hits from multiple scaffolds were included as long as each scaffold contained at least one hit meeting the percent identity criteria.
Next, the scaffolds matching one or more known carnivore MSY genes were examined to identify whether they also contained any predicted genes from the red fox draft annotation [
27]. Predicted gene sequences from the annotation that had been translated into protein sequences [
27] were compared to
C. l. familiaris sequences deposited in NCBI using the web browser version of tblastn. The best match was selected based on total score. When the best dog hit had a known MSY homolog, the positions of the dog-vs-fox and fox-vs-dog queries were compared to determine whether they overlapped.
Additionally, a recent analysis of the wolf (
Canis lupus lupus) Y-chromosome [
14] identified and provided reference positions for three genes not previously reported in dogs or cats (
TMSB4Y,
AP1S2Y, and
WWC3Y) along with a wolf ortholog of the dog gene
DYNG (
Table 2). The protein sequences of the dog X-chromosome genes paralogous to
AP1S2Y and
WWC3Y were downloaded from NCBI. For
TMSB4Y and
DYNG, the nucleotide sequence of the corresponding region was extracted from the wolf reference genome assembly [
42] using the approximate positions reported [
14] and compared to the red fox genome using blastn. Because the genes
AMELY,
FLJ36031Y, and
RPS4Y were not analyzed in the grey wolf Y-chromosome assembly [
14], the cat sequences of these genes were also compared to the grey wolf reference genome [
42] to evaluate whether these genes were present on wolf Y-linked scaffolds (
Table S2).
Table 1.
The 22 genes of interest for the fox male-specific region of the Y-chromosome (MSY) based on cat and dog. Genes were selected as probes based on their presence on the MSY of dogs and/or cats [
6,
7,
43]. Where a gene is present in one species and absent in the other, grey shading is used to highlight the derived state relative to other carnivores. The term ‘X-transposed’ denotes that
OFD1Y may have been recently transposed from the X to the Y chromosome [
44]. Evolutionary origins of genes in carnivores are based on analyses of the dog and cat Y-chromosomes [
6,
7,
35]. The gene
TXLNGY was previously called
CYorf15, and
UBE1Y is also referred to as
UBA1Y in the literature.
Table 1.
The 22 genes of interest for the fox male-specific region of the Y-chromosome (MSY) based on cat and dog. Genes were selected as probes based on their presence on the MSY of dogs and/or cats [
6,
7,
43]. Where a gene is present in one species and absent in the other, grey shading is used to highlight the derived state relative to other carnivores. The term ‘X-transposed’ denotes that
OFD1Y may have been recently transposed from the X to the Y chromosome [
44]. Evolutionary origins of genes in carnivores are based on analyses of the dog and cat Y-chromosomes [
6,
7,
35]. The gene
TXLNGY was previously called
CYorf15, and
UBE1Y is also referred to as
UBA1Y in the literature.
| Gene/Gene Family | Dog | Cat | Sequence Used | Sequence Species | Origin |
|---|
| AMELY | - | + | EU879968 | Cat | X-degenerate |
| BCORY1 | + | - | AGS47779 | Dog | X-degenerate |
| BCORY2 | + | - | AGS47770 | Dog | X-degenerate |
| CUL4BY | + | + | AGS47784 | Dog | X-degenerate |
| DDX3Y | + | + | JX964855 | Dog | X-degenerate |
| EIF2S3Y | + | + | EU879975 | Cat | X-degenerate |
| EIF1AY | + | + | AKI82173 | Dog | X-degenerate |
| FLJ36031Y | - | + | NP_001108352 | Cat | Ampliconic (autosome-derived) |
| HSFY | + | + | AKI82172 | Dog | Ampliconic (X-derived) |
| KDM5D | + | + | AGS47774.1 | Dog | X-degenerate |
| OFD1Y | + | + | AGS47782.1 | Dog | X-transposed |
| RPS4Y | - | + | EU879986 | Cat | X-degenerate |
| RBMYL | + | - | AKI82176 | Dog | X-degenerate |
| SRY | + | + | AAD40225 | Dog | X-degenerate |
| TETY1 | - | + | AZD12964.1 | Cat | Ampliconic (autosome-derived) |
| TETY2 | + | + | AGS47775 | Dog | Ampliconic (X-derived) |
| TSPY | + | + | AGS47785 | Dog | Ampliconic (X-derived) |
| TXLNGY | + | + | AKI82175.1 | Dog | Ampliconic (X-derived) |
| UBE1Y | + | + | AKI82178 | Dog | X-degenerate |
| USP9Y | + | + | AKI82171 | Dog | X-degenerate |
| UTY | + | + | NM_001284484 | Dog | X-degenerate |
| ZFY | + | + | JX964866 | Dog | X-degenerate |
In order to identify the position of the pseudoautosomal boundary in the red fox, whole genome resequencing reads from 30 farm-bred red foxes—drawn equally from each of three lines maintained at the Institute for Cytology and Genetics in Novosibirsk, Russia (NCBI BioProject PRJNA376561; [
27])—were aligned using Bowtie2 [
45] to a version of the dog reference genome assembly that was created by concatenating canFam3.1 [
46] and the dog Y-chromosome assembly (GenBank: KP081776.1; [
6]). Of these 30 foxes, 15 were male and 15 were female, and each fox was sequenced at approximately 2.5× coverage [
27]. Duplicates were marked at the level of the individual with Picard MarkDuplicates [
47] and then the alignments were pooled at the population level (experimental line) and recalibrated with RealignerTargetCreator and IndelRealigner in the Genome Analysis Toolkit version 3.7 [
48]. Data were combined across lines for all individuals of each sex, and depths were then tabulated for males and females separately using SAMTools depth [
49] for the X-chromosome only (-r chrX). Average depth was calculated for each sex in windows of 100 Kbp and 1 Mbp in Python 2.7 and then plotted in R [
50] with ggplot2 [
51].
Male-specific sequence motifs were then identified using a pipeline for comparing
k-mers across two groups [
36]. The pipeline in the Full Methods section of [
36] was followed to prepare the red fox reference genome version 2.2 [
27], which had been masked using RepeatMasker 4.0.5 [
52] with the carnivore repeat library, for analysis with the script YGS.pl described in [
36]. YGS.pl was then used to compare the male and female 18-mers to identify those that were single-copy (i.e., only one copy present in the reference genome), valid (i.e., found in the male sequencing reads) and unmatched (i.e., found in the male but not the female sequencing reads). In order to reduce computing time during this analysis, only scaffolds 1 Kbp or longer were analyzed. These 12,625 scaffolds comprise 96.1% of the complete draft genome sequence by length. Scaffolds found to have no valid single-copy
k-mers in the YGS.pl output were excluded from downstream analysis. The percent V_SC_UK (valid, single-copy k-mers unmatched in females) on each scaffold was normalized by calculating the standard score (i.e., by subtracting the mean and dividing by the standard deviation as estimated across all scaffolds).
In order to compare sequencing depth across the red fox draft genome between males and females, the male and female reads were aligned using Bowtie2 [
45] to the 676,878 scaffolds of the repeat-masked red fox reference genome, as described above. The bam files corresponding to the aligned reads from each individual were pooled by sex for downstream analysis. The overall depth of coverage was estimated for the males and females using SAMTools depth [
49]. CNV were analyzed using CNV-Seq [
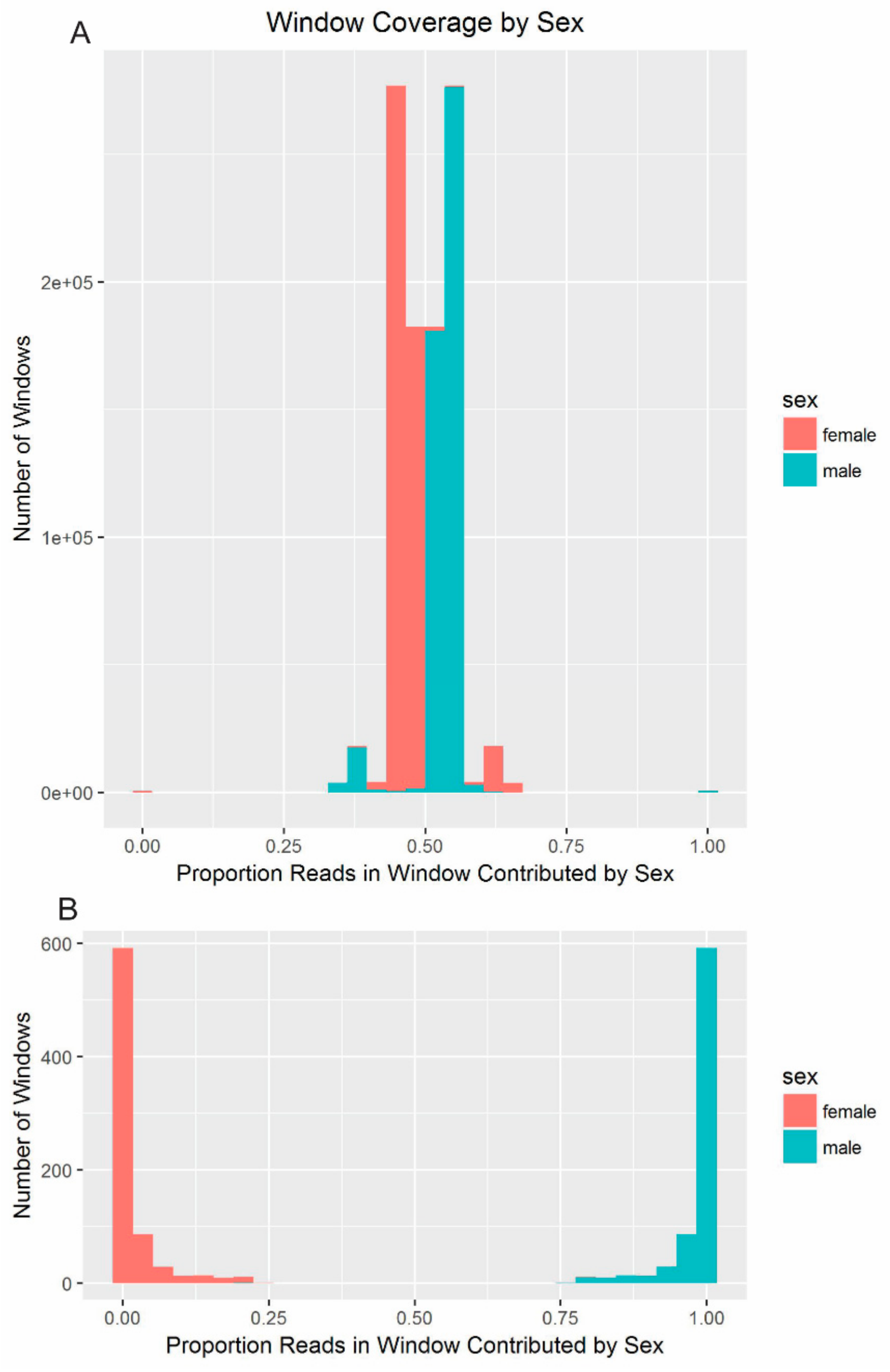
37] to identify differences in depth-of-coverage along the genome in the male and female resequencing data. CNV-Seq was run with the genome size set to 2,496,140,267 bp and the window size to 10,000 bp. The female data was used as the reference and the male data as the test data. CNV-Seq estimated the number of reads mapping to each 10,000-bp window along each masked scaffold, with 5000 bp of overlap between windows. Any window containing fewer than 100 reads, which corresponded to a coverage of less than 0.01×, was excluded from downstream analysis. For each window, the percentage of mapped reads that originated in the male resequencing data was estimated by dividing the number of reads mapping to the window in the male dataset by the total number of reads mapping to the window across both datasets. The percentages were again normalized to a standard score. All scaffolds shorter than 1 Kbp were dropped from the CNV-Seq output, as they been excluded from the analysis with YGS.pl.
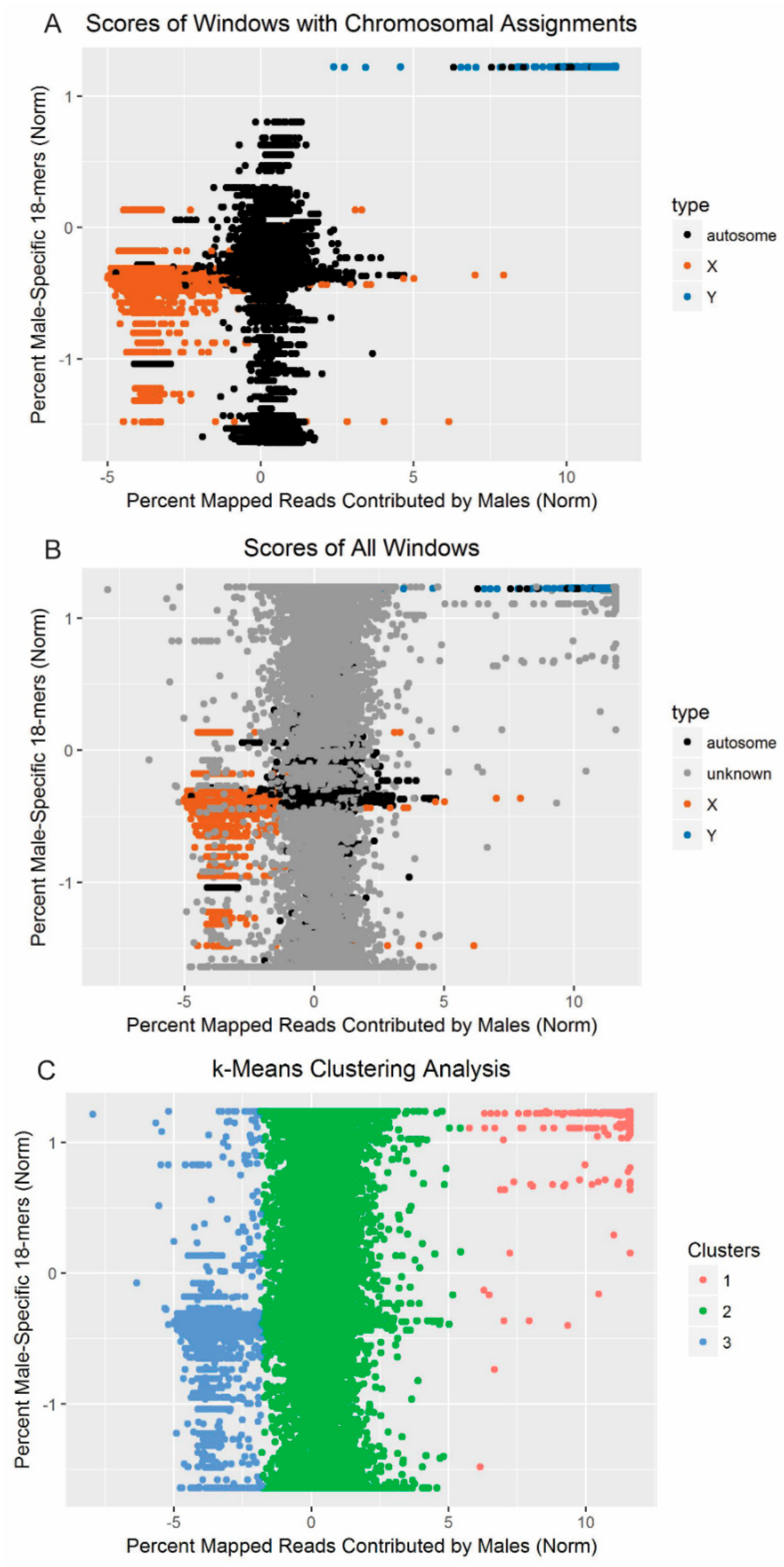
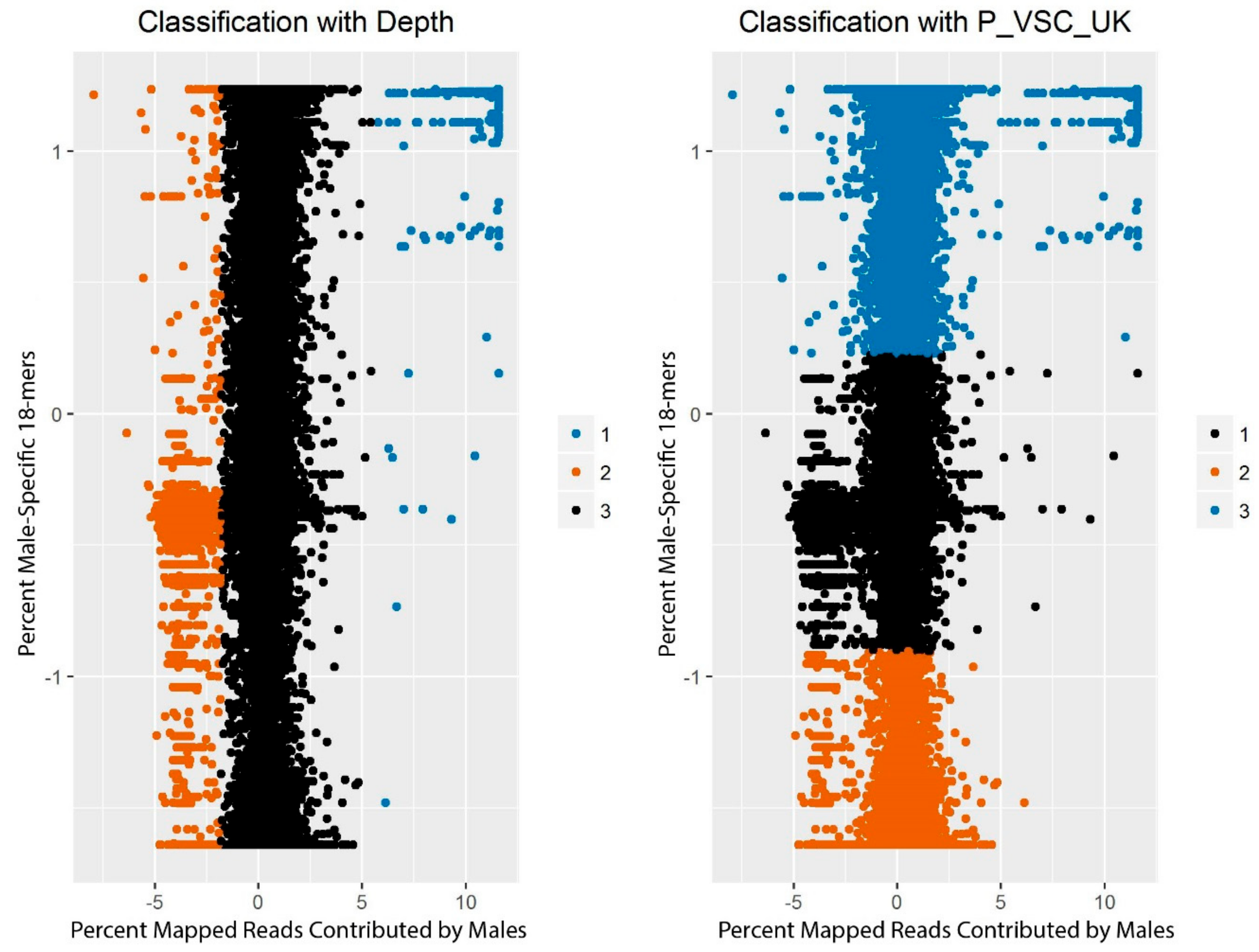
The scores corresponding to each window from CNV-Seq (sex-based depth) and from YGS.pl (male-specificity of 18-mers) were plotted, first, for only the windows on scaffolds with
a priori chromosomal origins assigned [
27,
28,
41], and then for all windows. Given that scaffolds containing sequence from the X-chromosome, autosomes, and the Y-chromosome were expected to form distinct clusters, we applied the
k-means clustering algorithm [
53] to the data with
k, or the number of centers, set to 3.
k-means clustering is an unsupervised machine learning algorithm that assigns individual data points to one of
k clusters and adjusts the positions of the centers of the clusters to minimize the point-to-center distance across all points. The particular implementation used here was R’s native
k-means clustering function [
54]. Clustering was conducted on a matrix containing, for each window, the standardized percent of reads mapping to the window that came from the male resequencing data (as identified with CNV-Seq) and the standardized percent of
k-mers on the scaffold that were valid, single-copy, and unmatched in the female reads (as identified with YGS.pl). The maximum number of sets of random centers to be selected (nstart) was set to 100.
The clusters identified by
k-means clustering were then evaluated to determine how likely they were to represent the three expected classes of chromosomes. Some scaffolds had been previously assigned to a position on the X-chromosome or autosomes [
41] or identified as likely to contain Y-chromosome sequence (
Table 3; [
27,
28]). These scaffolds were used to examine whether the clusters consistently contained scaffolds with the same chromosomal origin. The number of windows assigned to each cluster was also tabulated for each scaffold. If more than 15% but less than 85% of the windows on a scaffold were assigned to a cluster, the scaffold was evaluated manually.
The scaffolds assigned to the Y-chromosome using this process were then examined to ensure that all metrics were consistent with what was expected for the Y-chromosome. SAMTools depth was used to estimate the sequencing depth along the putative Y-chromosome scaffolds for the male and female WGS reads mapped to the fox reference assembly. Depth along each scaffold was visualized with ggplot2, as described above, in windows of 5 Kbp or 10% of scaffold length, whichever was smaller.
The next step was to reassemble the putative male-specific sequence using a different alignment algorithm than was used in the red fox reference genome assembly project. The alignment of the male resequencing data to the fox genome, as described above, was filtered using SAMTools view with the -L parameter to extract only the reads that mapped to putative Y-scaffolds. The 15 libraries from the genome project [
27], which provide 93.9× coverage from a single male fox (BioProject PRJNA378561), were then aligned to the original draft genome (vv2.2; [
27]) with the program Burrows-Wheeler Aligner (BWA) [
55]. The alignments were cleaned with SAMTools fixmate [
49] and then the reads mapping to the putative Y-chromosome scaffolds were extracted from each alignment using SAMTools view with the -L parameter. All of the alignments were then sorted by read name (using the -n parameter) with SAMTools sort and extracted into paired end fastq files using BEDTools bamToFastq [
56]. Each of the fastq files containing paired-end reads from the genome project was cleaned to remove duplicate reads using the functions dedupe and reformat from BBMap version 38.35 [
57].
ABySS version 2.1.5 [
58,
59] was then used to assemble the reads, with the paired-end reads from the genome assembly project and pooled male WGS data provided for assembly of the contigs (lib) and the mate-pair libraries from the genome project for scaffolding contigs (mp). Per the ABySS 2.0 manual, the program was tested with values of
k (
k-mer size) to optimize for N50 and assembly size. The values of
k tested ranged from 50 to 96.
The ABySS assembly and putative Y-chromosome draft genome scaffolds were then compared: first, to each other (percent identity = 95%; filtering = one-to-one); then to the dog Y-chromosome assembly (percent identity = 90%; filtering = one-to-one); and finally to the cat Y-chromosome assembly (percent identity = 80%; filtering = one-to-one) using MashMap [
60]. The results of the inter-assembly alignments were visualized with MashMap’s visualization script, generateDotPlot. MashMap’s output was also visualized in Evolution Highway [
61].
4. Discussion
Although the red fox reference assembly was developed with short-read Illumina sequencing technology, we demonstrate here that the data produced by the project [
27] is sufficient for the in silico identification of Y-chromosome sequence. The red fox genome project sequenced a male donor at 94× to assemble a 2.2-Gbp genome organized in 676,878 scaffolds and then sequenced an additional 30 foxes (15 male and 15 female) at approximately 2.5× per individual. Together, these resources made it possible to identify 171 scaffolds in the assembly constituting at least 1.7 Mbp of likely MSY sequence and containing 24 genes found on the Y-chromosomes of other carnivores.
Traditionally, Y-chromosome assembly projects targeting eutherian mammals have either flow-sorted chromosomes or used targeted BAC clones to amplify Y-chromosome sequence in vitro [
6,
8,
10,
63]. Such studies have also typically used long-read Sanger technologies for sequencing. While the emergence of affordable long-read next- and third-generation sequencing technologies is expected to benefit projects seeking to develop Y-chromosome assemblies for non-traditional mammalian models, analyses of species such as the polar bear [
13], wolf [
14], and now the red fox provide evidence that short-read technologies can also be leveraged for bioinformatic Y-chromosome sequence identification.
Previous studies have differentiated potential Y sequence based on the ratio of female and male sequences mapping to a sequence fragment [
11,
13,
64]. However, the assumption that female reads will not map to male-specific sequences is not always robust to misassembly in the reference genome [
65]. Additionally, the presence of highly amplified Y-chromosome sequence can confound efforts to use sex differences in sequence coverage to identify likely Y-scaffolds. In the present analysis, in order to reduce the effects of these potential sources of error, we included a second metric to assess Y-chromosome specificity by deconstructing the scaffolds into 18-bp sequence motifs (18-mers) that were counted in the male and female resequenced reads. All the same, the disproportionate influence of sequencing depth on cluster assignment means that Y-scaffolds containing male-specific sequence motifs but showing similar depth of coverage across males and females may not have been detected in the present analysis (
Appendix B). This limitation means that novel ampliconic or multicopy Y-chromosome genes could remain undetected in the red fox, and therefore future efforts to characterize the gene content of the red fox Y-chromosome should utilize approaches that do not rely on differences in depth of sequence coverage (e.g., as in the analyses conducted by [
7]).
In the present analysis, two methods were used in conjunction to differentiate scaffolds likely to belong to different types of chromosomes. Unsupervised learning revealed three clusters consistent with the Y-chromosome, autosomes, and X-chromosome, with windows from 176 distinct scaffolds assigned to the cluster consistent with the Y-chromosome. This clustering method was very effective in identifying thresholds that discriminated different types of chromosomes without requiring empirical threshold adjustment, in contrast to other approaches [
64,
65]. Only 1.6% of windows assigned to the Y-chromosome cluster by the algorithm were ultimately removed during quality control, and assignments to all three clusters were in almost complete agreement with previous synteny-based analyses [
27,
28,
41] (
Figure 4). Re-assembly of the reads mapping to the putative Y-scaffolds also recapitulated the sequence content of the scaffolds themselves (
Figure S1). The results therefore indicate that this multi-pronged approach worked to select only those scaffolds most likely to contain Y-chromosome sequence.
Research in well-developed mammalian genomic models such as dog and cat [
6,
7,
35] facilitated the identification of red fox orthologs of carnivore MSY genes. Out of 18 dog MSY genes analyzed, 17 were identified in putative Y-linked red fox scaffolds, including all 11 of the core carnivore MSY genes [
31]. The only dog protein that was not assigned a position in the fox scaffolds was
TSPY.
TSPY is X-degenerate, but BLAST analysis of the dog protein sequence against the red fox genome failed to identify any scaffolds meeting all mapping criteria, including an X-chromosomal copy (
Table S1). This result is notable because
TSPY is one of six genes found, either active or as a pseudogene, across the full range of eutherian mammalian taxa [
6,
44]. The analysis of MSY genes in the short-read-assembled polar bear reference genome also failed to identify a location for this gene [
13]. Given that assumptions of parsimony would be violated by a loss of this gene in both red foxes and polar bears given its presence on the dog Y-chromosome, the more likely explanation is that multicopy, X-degenerate genes such as
TSPY are particularly difficult to assemble from short-read sequencing. Despite the limitations preventing the identification of
TSPY, these findings suggest that the gene content of the red fox MSY is very similar to that of the dog. Though this result is not surprising given the relatively recent divergence of dog and fox 6 to 9 million years ago, the human and chimp Y-chromosomes show significant divergence despite a similar estimated divergence date of 6 MYA [
66].
However, there are also some ways in which the genes identified on the red fox MSY differ from those reported in dog. This study independently verified the presence of a Y-chromosomal paralog of
WWC3, previously reported only on the wolf MSY [
14].
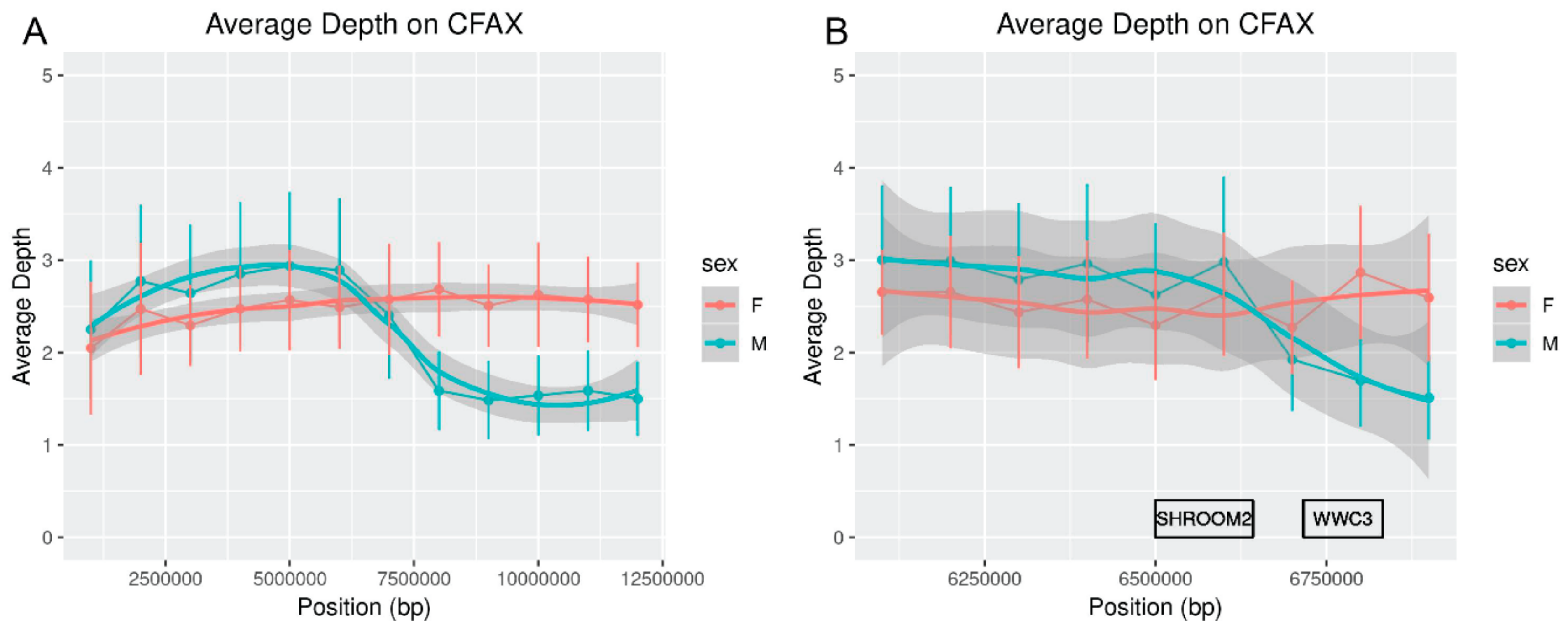
WWC3Y was present in the predicted annotation of red fox scaffold310. Analysis of depth of sequencing near the X-chromosomal gene
WWC3 indicated that it is located near the pseudoautosomal boundary but also supported the existence of a male-specific copy (
Figure 1). Lack of a strict pseudoautosomal boundary could explain for how this gene arose on the MSY in some canids and suggests that it could constitute a potential region of interest for studies of genetic diversity on the canid sex chromosomes [
67]. However, another predicted gene on the Y-linked scaffold292 was not homologous to any known carnivore MSY genes. This prediction based on
RPS20, a gene found on dog chromosome 20.
RPS4Y, a gene in the same gene family found on the cat MSY, mapped to a position less than 400 Kbp away on the same scaffold. However, the nucleotide sequence of the predicted gene is very similar to a region of the dog MSY assembly [
6]. Whether the gene is an artifact of annotation or a functional gene in the red fox is currently unknown.
Additionally, two of four genes found on the cat but not the dog MSY, including
RPS4Y, were identified in the red fox Y-scaffolds. This finding suggests that
AMELY and
RPS4Y may have been lost in dogs subsequent to divergence from red foxes 6 to 9 million years ago. However, using the same BLAST criteria as described for the cat–fox comparison to compare cat gene sequences to the wolf reference genome [
42] revealed that one exon of
AMELY does map to a Y-linked wolf scaffold [
14] (
Table S2). This result suggests that the loss of
AMELY in dogs may be very recent.
Aggregation of gene content analyses across multiple taxa can facilitate estimation of the timing of gene loss and gain within phylogenies [
31,
38]. Thus, analysis of the red fox MSY revealed additional insight into the timing of gene loss, and possibly gain, among carnivores, though additional analysis would allow for timescales to be estimated more precisely (
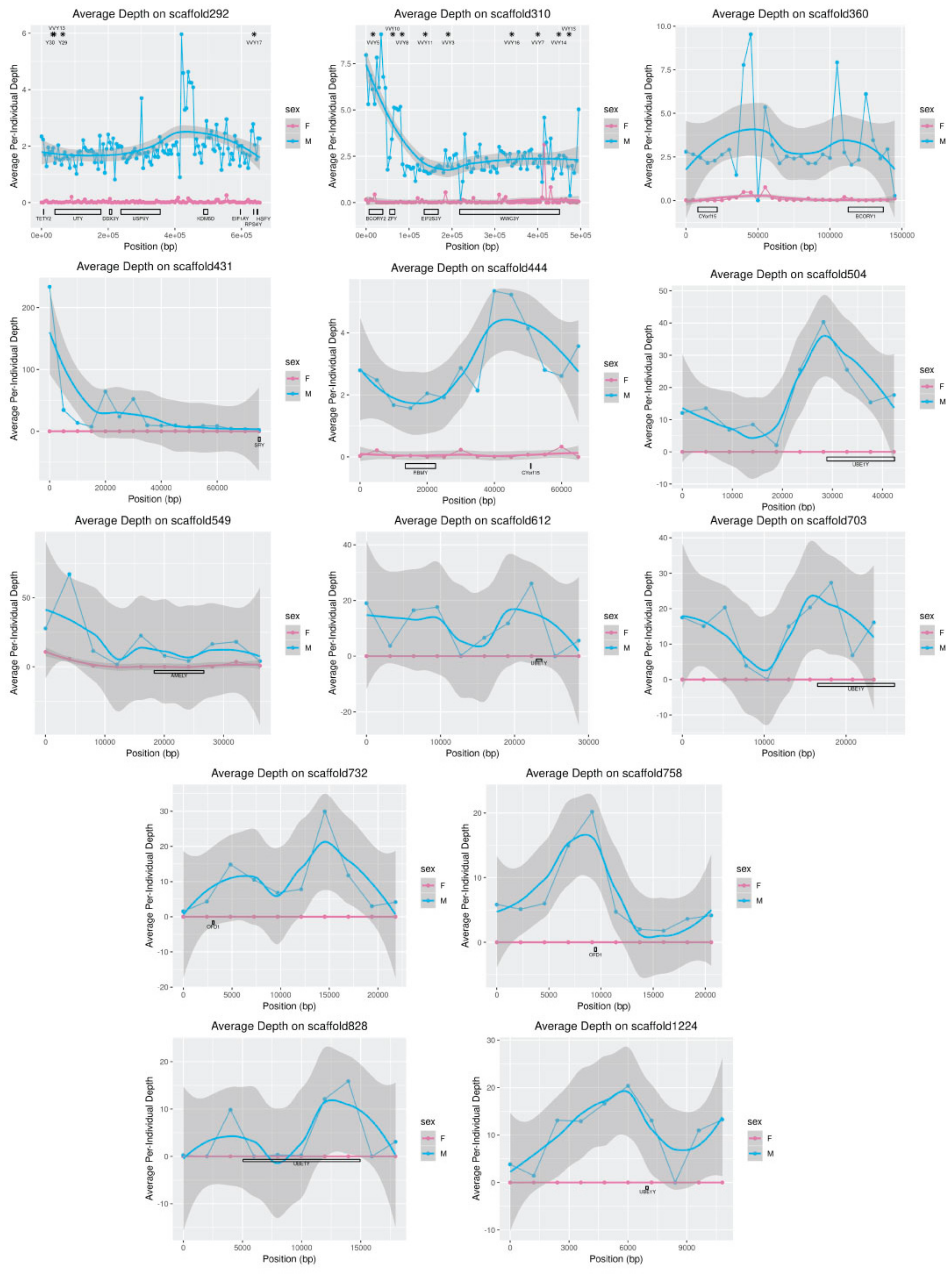
Table 6). Depth of coverage over MSY genes also offered insight into the timing of gene replication events in carnivores. Several copies of
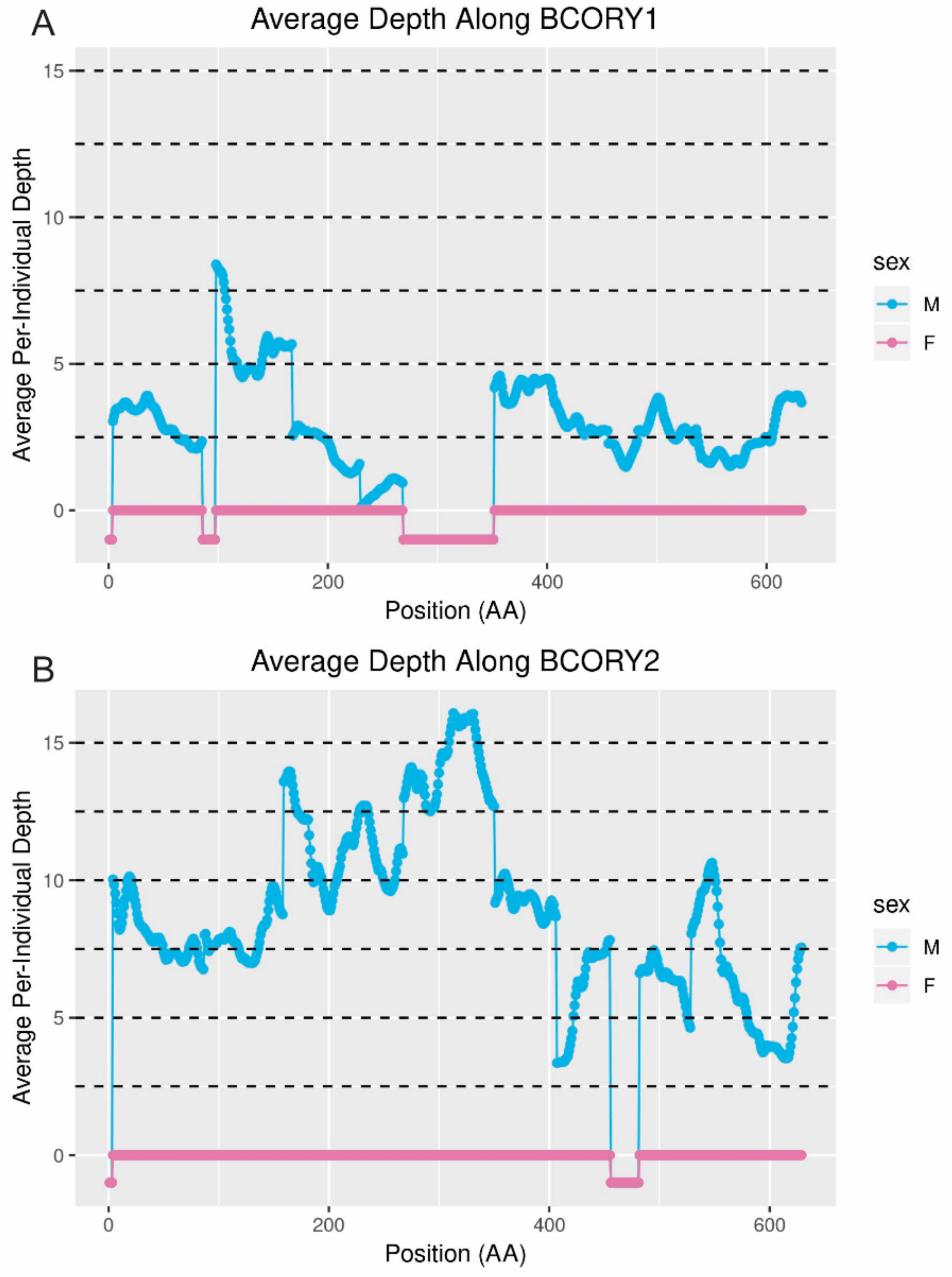
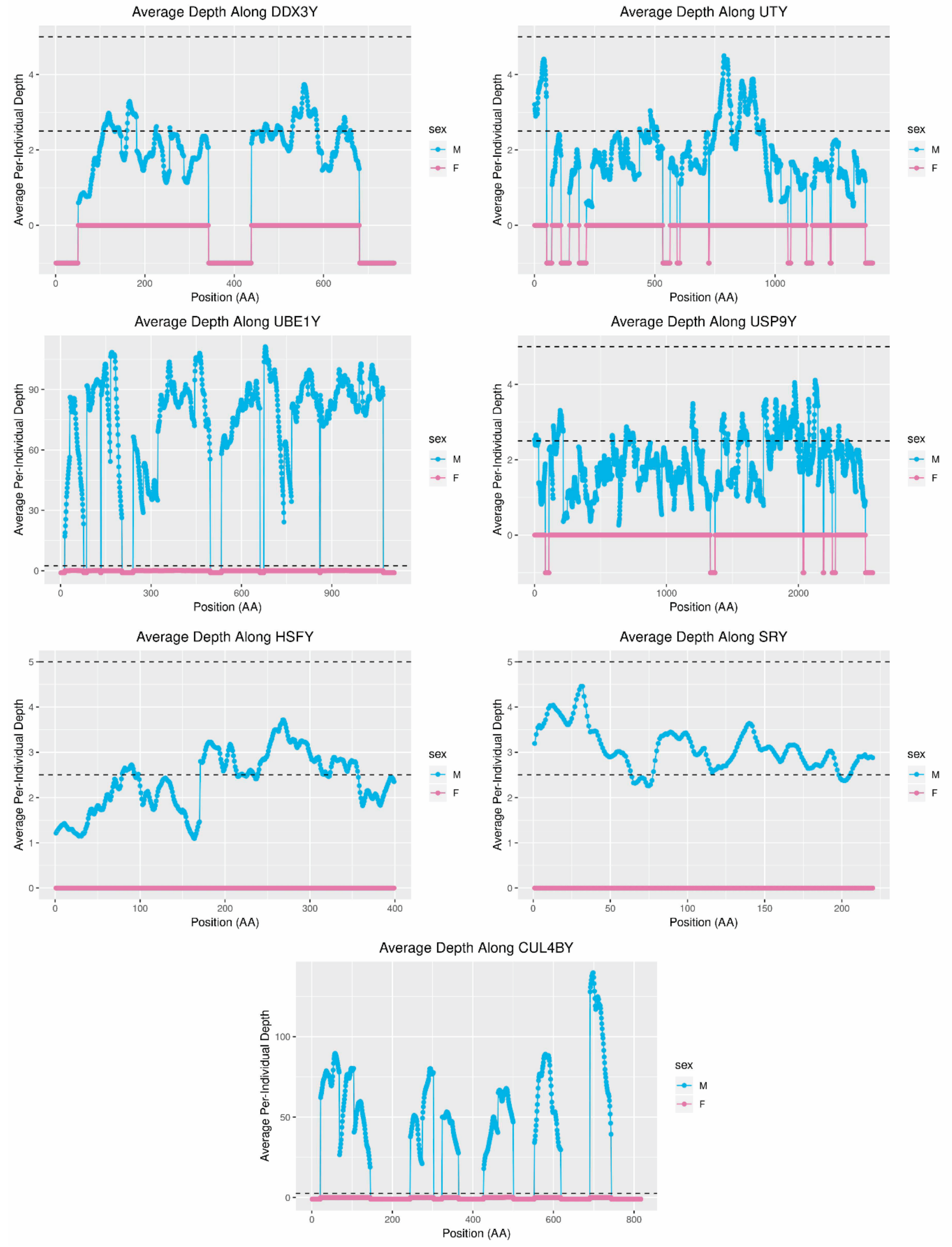
SRY are present in dogs and wolves [
6,
14], even though a single copy of this gene is more common across the mammalian phylogeny [
38]. In foxes,
SRY mapped to a single position in the red fox genome on scaffold431, and the depth of coverage in this region was consistent with one to two copies (
Figure 7). This finding suggests that replication of this gene, which is critical to sex determination, occurred recently in the dog/wolf lineage. In contrast, depth of sequencing suggested that
UBE1Y, which has been reported to be single copy in cats and dogs [
6,
38], may exist at as many as 36 copies in foxes. Interestingly, coverage of this gene in the grey wolf is consistent with two copies [
14] and in the horse is consistent with at least eight copies [
32,
44].
UBE1Y is expressed testis specifically in mice and horses [
32,
68] and has been hypothesized to play a role in male fertility via germ cell proliferation [
32]. Given that
UBE1Y has been reported to have a higher rate of evolution in carnivores than other mammalian clades [
31], further investigation into the phenotypic effects of its apparent replication in red foxes and/or wolves may be of interest. Present evidence of the gene content of the Y-chromosomes of species in
Pegasoferae (i.e., the clade containing odd-toed ungulates, bats, and carnivores [
69,
70,
71]) (
Table 6) suggests that some genes, such as
AP1S2Y,
RPS4Y,
TMSB4Y, and
WWC3Y, are either present in more species than has currently been ascertained, or have undergone multiple gain/loss events within Carnivora.
Although analysis of they-linked scaffolds in the de novo wolf assembly highlighted the possibility for X-degenerate genes and their Y-paralogs to be collapsed during assembly from short sequencing reads [
14], in foxes, this type of collapsing was observed only for putative segmental replications. Patterns of misassembly commonly caused by the algorithm used for assembly from short reads explains why 3.4 Mbp of sequence was provided to ABySS, but only 1.7 Mbp was assembled: when constructing long sequences from short reads, the assembler must determine whether two sequences that are close, but not exact, matches belong in different places (e.g., repetitive elements or segmental duplications) or the same place (e.g., heterozygosity or sequencing errors). Patterns of depth of coverage over the Y-scaffolds in males and females suggested that male-specific repeats are likely to be collapsed, especially on the shorter scaffolds. The increasing feasibility of incorporating long-read next-generation sequencing into projects such as this will allow for more accurate resolution of highly repetitive genomic regions such as the Y-chromosome, even in non-traditional models and wildlife species.
In addition to the length of the assembly, short-read assembly can also influence the structure of sequence content within the assembly. Short scaffolds may be sequences that were erroneously excluded from larger contigs and scaffolds, and others may be orphaned by the collapsing of repetitive regions [
3,
4,
5]. For example,
UBE1Y and
CUL4BY, which are likely to be ampliconic in the red fox genome (
Figure 7;
Table 6), were fragmented across several scaffolds, suggesting heterogeneity across copies may have resulted in scaffold breakage [
2].
The opposite may have occurred on scaffold310: although, unlike in wolf, the genes
BCORY1 and
BCORY2 were assembled separately in the red fox scaffolds, depth in the region of scaffold310 containing
BCORY2 suggests that segmental replications were collapsed in this region of the assembly. A segmental replication in the region of
BCORY2 (
Figure 7) is consistent with previous findings that microsatellite markers in this region can carry up to three alleles per male [
28]. The presence of a segmental replication of this region was supported by sequence coverage of the nearby gene
ZFY (
Figure S2), which contains a microsatellite marker observed to carry up to two alleles per male [
28]. However, the fact that multi-allelic Y-chromosome microsatellite markers were observed only in males from the North American subspecies of red fox (
Vulpes vulpes fulva) suggests that these segmental replications, including additional copies of
BCORY2 and
ZFY, may have evolved on a short timescale and may distinguish European and North American red foxes. Similarly, rapid changes in segmental replications have been reported in grey wolf populations and haplogroups [
14].
With the red fox adding to the recent increase in the availability of comparative carnivore Y-chromosome resources, studies examining the timing and effect of Y-chromosome evolution, including replication events and gene loss and gain, have become increasingly feasible. The red fox sequence information presented here serves to refine the timing of differences in the Y-chromosome sequences of dogs and cats and also provides an outgroup for studies of Y-chromosome evolution during the evolution of dogs from wolves. These results support the set of Y-chromosome markers available for the red fox [
18,
28] and provide sequence that can be used for the development of additional tools for studying the evolution of the red fox Y-chromosome at a higher resolution and over longer timescales.

 ,
, 


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}








