Simplified Transformation of Ostreococcus tauri Using Polyethylene Glycol

 and
and
Abstract

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction
2. Materials and Methods
2.1. Algal Culture
2.2. Flow Cytometry
2.3. DNA Preparation
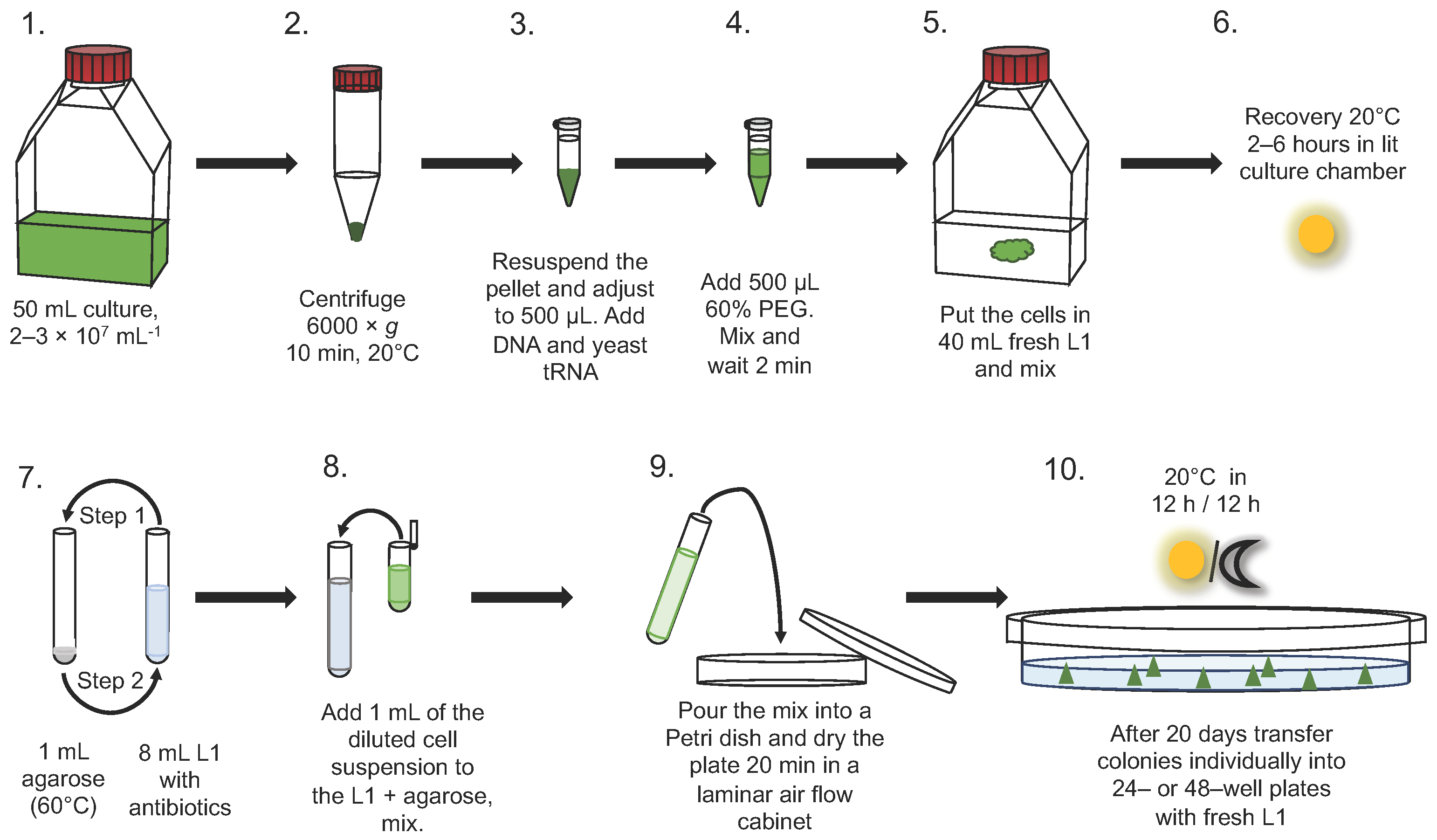
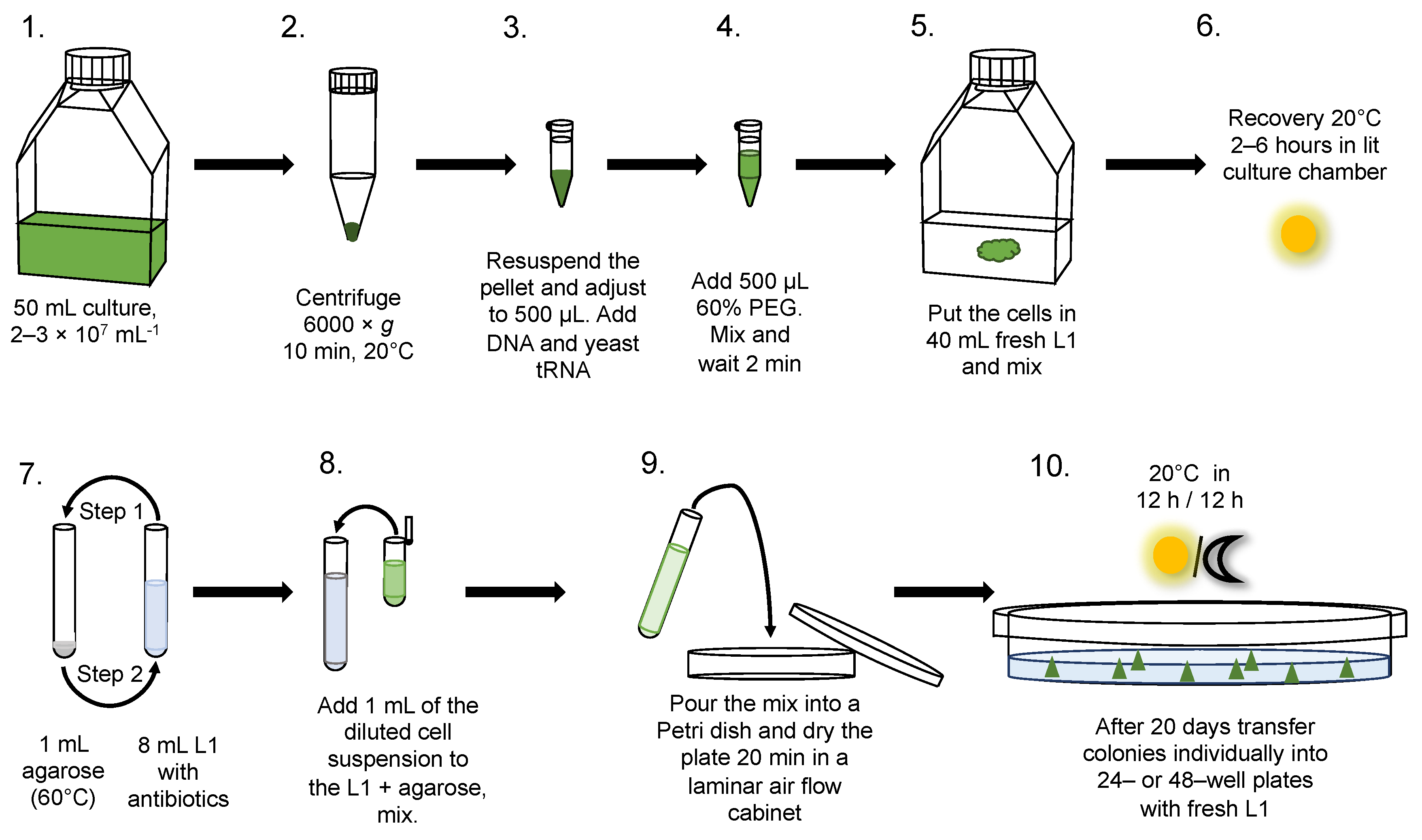
2.4. Transformation
2.5. Selection on Plates in Semi-Solid Medium
2.6. Luciferase Assays
2.7. Pulsed-Field Gel Electrophoresis
3. Results
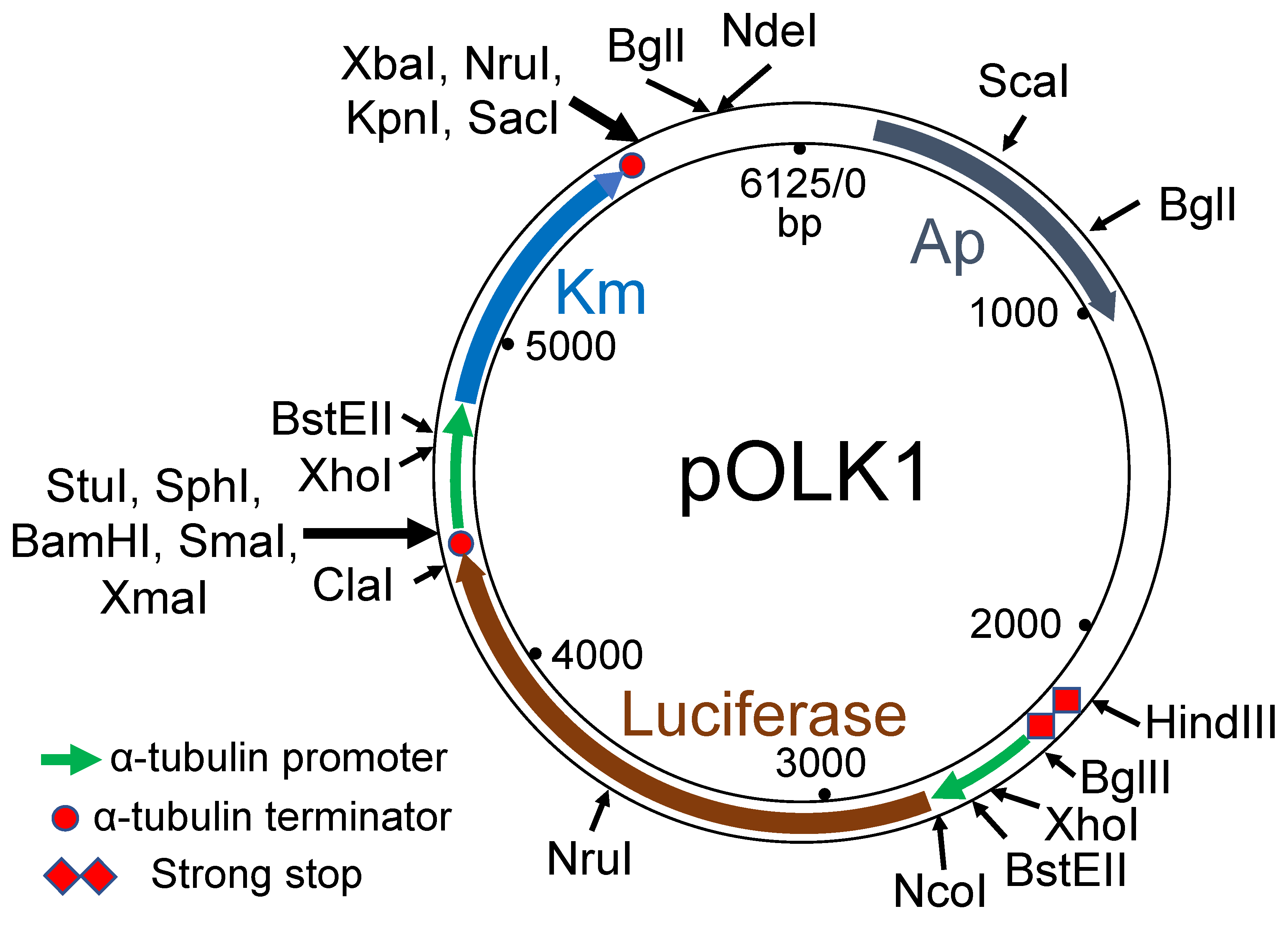
3.1. Choice of Selectable Markers for Transformation
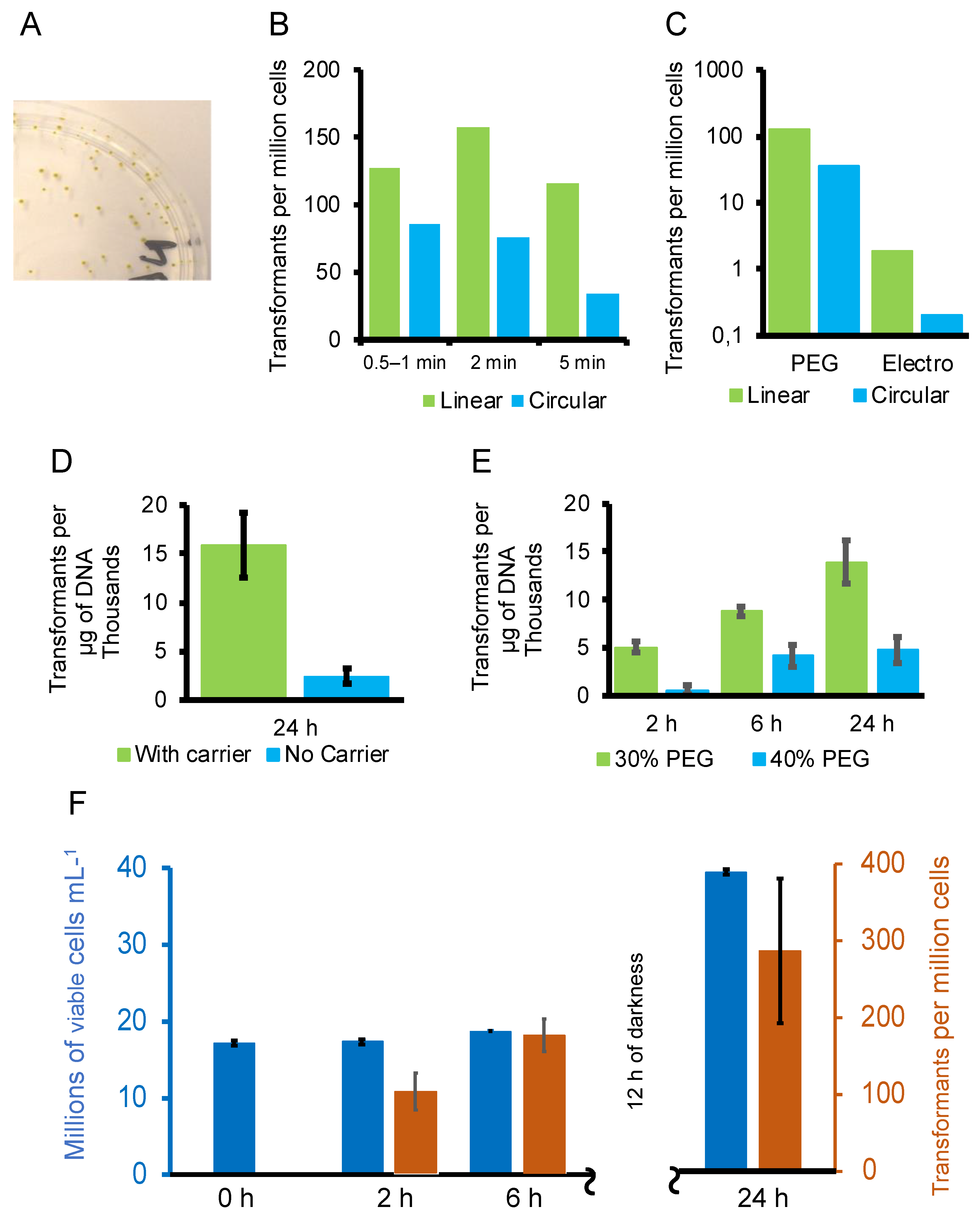
3.2. Optimisation of the Transformation Procedure
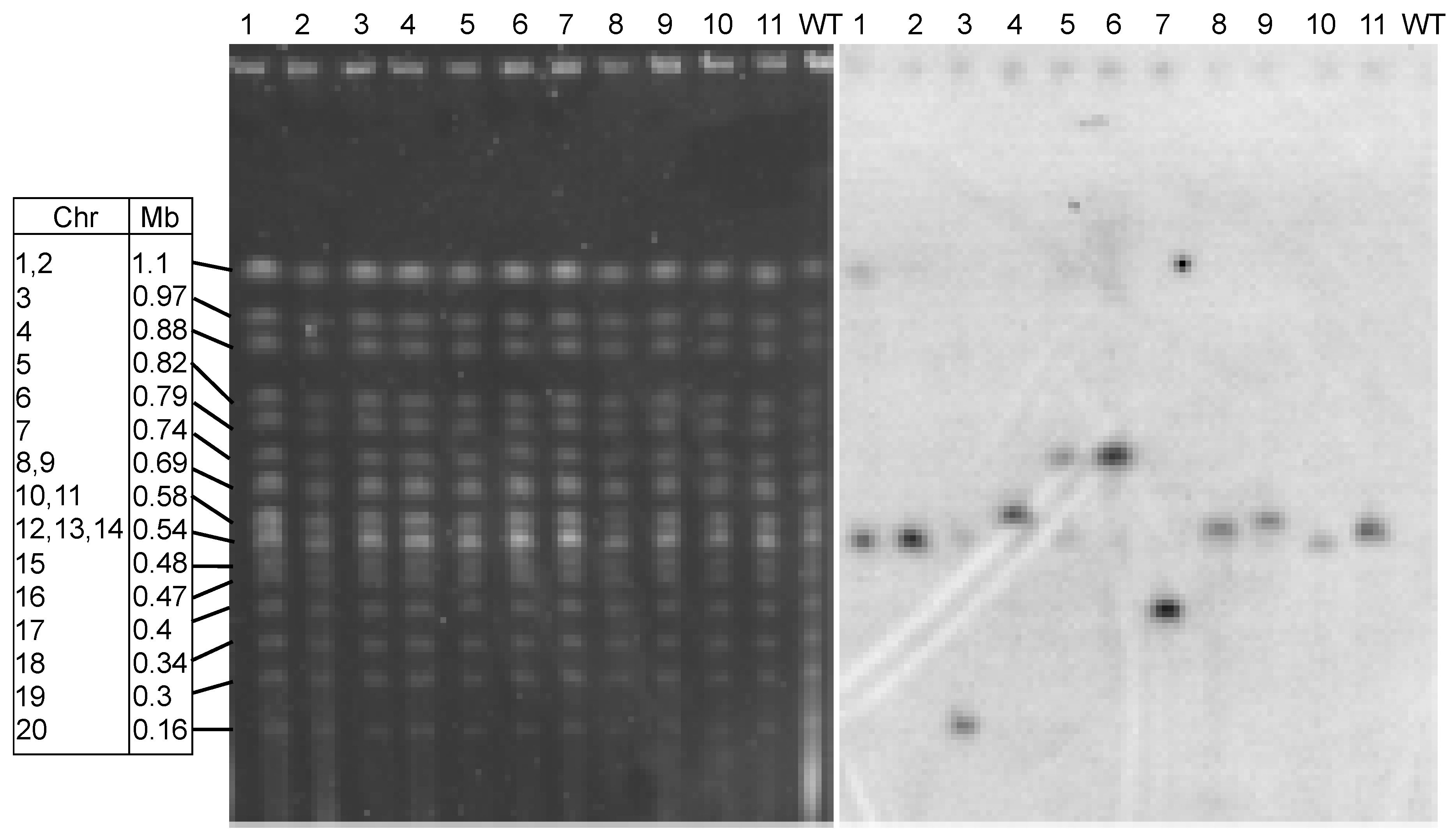
3.3. Integration of Transforming DNA is Random
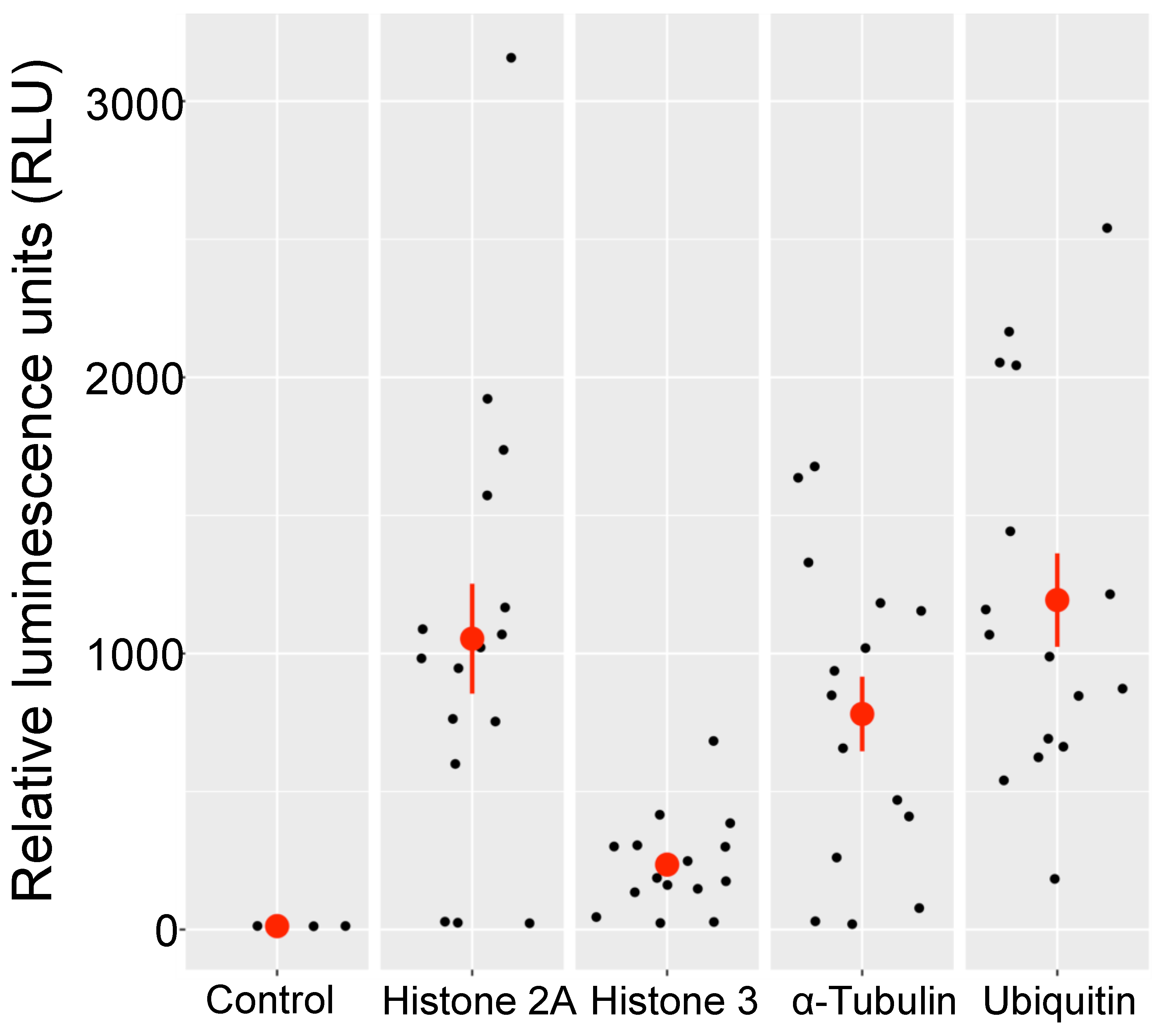
3.4. Transgene Expression from Integrated DNA
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Vaulot, D.; Le Gall, F.; Marie, D.; Guillou, L.; Partensky, F. The Roscoff Culture Collection (RCC): A collection dedicated to marine picoplankton. Nova Hedwig. 2004, 79, 49–70. [Google Scholar] [CrossRef]
- Leliaert, F.; Smith, D.R.; Moreau, H.; Herron, M.D.; Verbruggen, H.; Delwiche, C.F.; De Clerck, O. Phylogeny and Molecular Evolution of the Green Algae. Crit. Rev. Plant Sci. 2012, 31, 1–46. [Google Scholar] [CrossRef]
- Palenik, B.; Grimwood, J.; Aerts, A.; Rouze, P.; Salamov, A.; Putnam, N.; Dupont, C.; Jorgensen, R.; Derelle, E.; Rombauts, S.; et al. The tiny eukaryote Ostreococcus provides genomic insights into the paradox of plankton speciation. Proc. Natl. Acad. Sci. USA 2007, 104, 7705–7710. [Google Scholar] [CrossRef]
- Derelle, E.; Ferraz, C.; Rombauts, S.; Rouzé, P.; Worden, A.Z.; Robbens, S.; Partensky, F.; Degroeve, S.; Echeynié, S.; Cooke, R.; et al. Genome analysis of the smallest free-living eukaryote Ostreococcus tauri unveils many unique features. Proc. Natl. Acad. Sci. USA 2006, 103, 11647–11652. [Google Scholar] [CrossRef] [PubMed]
- Moreau, H.; Verhelst, B.; Couloux, A.; Derelle, E.; Rombauts, S.; Grimsley, N.; Bel, M.V.; Poulain, J.; Katinka, M.; Hohmann-Marriott, M.F.; et al. Gene functionalities and genome structure in Bathycoccus prasinos reflect cellular specializations at the base of the green lineage. Genome Biol. 2012, 13, R74. [Google Scholar] [CrossRef] [PubMed]
- Worden, A.Z.; Lee, J.-H.; Mock, T.; Rouze, P.; Simmons, M.P.; Aerts, A.L.; Allen, A.E.; Cuvelier, M.L.; Derelle, E.; Everett, M.V.; et al. Green Evolution and Dynamic Adaptations Revealed by Genomes of the Marine Picoeukaryotes Micromonas. Science 2009, 324, 268–272. [Google Scholar] [CrossRef] [PubMed]
- Blanc-Mathieu, R.; Verhelst, B.; Derelle, E.; Rombauts, S.; Bouget, F.-Y.; Carré, I.; Château, A.; Eyre-Walker, A.; Grimsley, N.; Moreau, H.; et al. An improved genome of the model marine alga Ostreococcus tauri unfolds by assessing Illumina de novo assemblies. BMC Genomics 2014, 15, 1103. [Google Scholar] [CrossRef]
- Yau, S.; Krasovec, M.; Rombauts, S.; Groussin, M.; Benites, L.F.; Vancaester, E.; Aury, J.-M.; Derelle, E.; Desdevises, Y.; Escande, M.-L.; et al. Virus-host coexistence in phytoplankton through the genomic lens. bioRxiv 2019, 513622. [Google Scholar]
- Jancek, S.; Gourbiere, S.; Moreau, H.; Piganeau, G. Clues about the Genetic Basis of Adaptation Emerge from Comparing the Proteomes of Two Ostreococcus Ecotypes (Chlorophyta, Prasinophyceae). Mol. Biol. Evol. 2008, 25, 2293–2300. [Google Scholar] [CrossRef]
- Wood, V.; Gwilliam, R.; Rajandream, M.-A.; Lyne, M.; Lyne, R.; Stewart, A.; Sgouros, J.; Peat, N.; Hayles, J.; Baker, S.; et al. The genome sequence of Schizosaccharomyces pombe. Nature 2002, 415, 871–880. [Google Scholar] [CrossRef]
- Lang, D.; Ullrich, K.K.; Murat, F.; Fuchs, J.; Jenkins, J.; Haas, F.B.; Piednoel, M.; Gundlach, H.; Bel, M.V.; Meyberg, R.; et al. The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J. 2018, 93, 515–533. [Google Scholar] [CrossRef] [PubMed]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef] [PubMed]
- Haberer, G.; Young, S.; Bharti, A.K.; Gundlach, H.; Raymond, C.; Fuks, G.; Butler, E.; Wing, R.A.; Rounsley, S.; Birren, B.; et al. Structure and architecture of the maize genome. Plant Physiol. 2005, 139, 1612–1624. [Google Scholar] [CrossRef]
- Corellou, F.; Camasses, A.; Ligat, L.; Peaucellier, G.; Bouget, F.Y. Atypical regulation of a green lineage-specific B-type cyclin-dependent kinase. Plant Physiol. 2005, 138, 1627–1636. [Google Scholar] [CrossRef] [PubMed]
- Lozano, J.-C.; Schatt, P.; Botebol, H.; Vergé, V.; Lesuisse, E.; Blain, S.; Carré, I.A.; Bouget, F.-Y. Efficient gene targeting and removal of foreign DNA by homologous recombination in the picoeukaryote Ostreococcus. Plant J. 2014, 78, 1073–1083. [Google Scholar] [CrossRef] [PubMed]
- van Ooijen, G.; Hindle, M.; Martin, S.F.; Barrios-Llerena, M.; Sanchez, F.; Bouget, F.-Y.; O’Neill, J.S.; Le Bihan, T.; Millar, A.J. Functional Analysis of Casein Kinase 1 in a Minimal Circadian System. PLoS ONE 2013, 8, e70021. [Google Scholar] [CrossRef] [PubMed]
- Caló, G.; Scheidegger, D.; Martínez-Noël, G.M.A.; Salerno, G.L. Ancient signal for nitrogen status sensing in the green lineage: Functional evidence of CDPK repertoire in Ostreococcus tauri. Plant Physiol. Biochem. 2017, 118, 377–384. [Google Scholar] [CrossRef] [PubMed]
- Botebol, H.; Lelandais, G.; Six, C.; Lesuisse, E.; Meng, A.; Bittner, L.; Lecrom, S.; Sutak, R.; Lozano, J.-C.; Schatt, P.; et al. Acclimation of a low iron adapted Ostreococcus strain to iron limitation through cell biomass lowering. Sci. Rep. 2017, 7, 327. [Google Scholar] [CrossRef] [PubMed]
- Hirth, M.; Liverani, S.; Mahlow, S.; Bouget, F.Y.; Pohnert, G.; Sasso, S. Metabolic profiling identifies trehalose as an abundant and diurnally fluctuating metabolite in the microalga Ostreococcus tauri. Metabolomics Off. J. Metabolomic Soc. Metabolomics 2017, 13, 68. [Google Scholar] [CrossRef] [PubMed]
- Grimsley, N.; Yau, S.; Piganeau, G.; Moreau, H. Typical Features of Genomes in the Mamiellophyceae. In Marine Protists; Ohtsuka, S., Suzaki, T., Horiguchi, T., Suzuki, N., Not, F., Eds.; Springer: Cambridge, MA, USA, 2015; pp. 107–127. ISBN 978-4-431-55129-4. [Google Scholar]
- Vannier, T.; Leconte, J.; Seeleuthner, Y.; Mondy, S.; Pelletier, E.; Aury, J.-M.; de Vargas, C.; Sieracki, M.; Iudicone, D.; Vaulot, D.; et al. Survey of the green picoalga Bathycoccus genomes in the global ocean. Sci. Rep. 2016, 6, 37900. [Google Scholar] [CrossRef]
- Allen, L.Z.; McCrow, J.P.; Ininbergs, K.; Dupont, C.L.; Badger, J.H.; Hoffman, J.M.; Ekman, M.; Allen, A.E.; Bergman, B.; Venter, J.C. The Baltic Sea Virome: Diversity and Transcriptional Activity of DNA and RNA Viruses. mSystems 2017, 2, e00125-16. [Google Scholar]
- Simmons, M.P.; Sudek, S.; Monier, A.; Limardo, A.J.; Jimenez, V.; Perle, C.R.; Elrod, V.A.; Pennington, J.T.; Worden, A.Z. Abundance and Biogeography of Picoprasinophyte Ecotypes and Other Phytoplankton in the Eastern North Pacific Ocean. Appl. Environ. Microbiol. 2016, 82, 1693–1705. [Google Scholar] [CrossRef]
- Tragin, M.; Vaulot, D. Novel diversity within marine Mamiellophyceae (Chlorophyta) unveiled by metabarcoding. bioRxiv 2018, 449298. [Google Scholar] [CrossRef]
- Tragin, M.; Vaulot, D. Green microalgae in marine coastal waters: The Ocean Sampling Day (OSD) dataset. Sci. Rep. 2018, 8, 14020. [Google Scholar] [CrossRef] [PubMed]
- Hernández-Ruiz, M.; Barber-Lluch, E.; Prieto, A.; Álvarez-Salgado, X.A.; Logares, R.; Teira, E. Seasonal succession of small planktonic eukaryotes inhabiting surface waters of a coastal upwelling system. Environ. Microbiol. 2018, 20, 2955–2973. [Google Scholar] [CrossRef] [PubMed]
- Corellou, F.; Schwartz, C.; Motta, J.P.; Djouani-Tahri, E.; Sanchez, F.; Bouget, F.Y. Clocks in the Green Lineage: Comparative Functional Analysis of the Circadian Architecture of the Picoeukaryote Ostreococcus. Plant Cell 2009, 21, 3436–3449. [Google Scholar] [CrossRef]
- Velmurugan, N.; Deka, D. Transformation techniques for metabolic engineering of diatoms and haptophytes: Current state and prospects. Appl. Microbiol. Biotechnol. 2018, 102, 4255–4267. [Google Scholar] [CrossRef] [PubMed]
- Mini, P.; Demurtas, O.C.; Valentini, S.; Pallara, P.; Aprea, G.; Ferrante, P.; Giuliano, G. Agrobacterium-mediated and electroporation-mediated transformation of Chlamydomonas reinhardtii: A comparative study. BMC Biotechnol. 2018, 18, 11. [Google Scholar] [CrossRef] [PubMed]
- Djouani-Tahri, E.B.; Sanchez, F.; Lozano, J.-C.; Bouget, F.-Y. A Phosphate-Regulated Promoter for Fine-Tuned and Reversible Overexpression in Ostreococcus: Application to Circadian Clock Functional Analysis. PLoS ONE 2011, 6, e28471. [Google Scholar] [CrossRef] [PubMed]
- Sanchez, S.; Bouget, F.-Y.; Sanchez, F.; Garnier, L. Functional genomics approaches to study the involvement of transcription factors in the microalgae Ostreococcus tauri circadian clock. Comp. Biochem. Physiol. A Mol. Integr. Physiol. 2009, 153, S207. [Google Scholar] [CrossRef]
- Moulager, M.; Corellou, F.; Vergé, V.; Escande, M.-L.; Bouget, F.-Y. Integration of light signals by the retinoblastoma pathway in the control of S phase entry in the picophytoplanktonic cell Ostreococcus. PLoS Genet. 2010, 6, e1000957. [Google Scholar] [CrossRef] [PubMed]
- Botebol, H.; Lesuisse, E.; Šuták, R.; Six, C.; Lozano, J.-C.; Schatt, P.; Vergé, V.; Kirilovsky, A.; Morrissey, J.; Léger, T.; et al. Central role for ferritin in the day/night regulation of iron homeostasis in marine phytoplankton. Proc. Natl. Acad. Sci. USA 2015, 112, 14652–14657. [Google Scholar] [CrossRef] [PubMed]
- van Ooijen, G.; Knox, K.; Kis, K.; Bouget, F.Y.; Millar, A.J. Genomic transformation of the picoeukaryote Ostreococcus tauri. J. Vis. Exp. JoVE J. Vis. Exp. JoVE 2012, e4074. [Google Scholar] [CrossRef] [PubMed]
- Hansen, G.; Wright, M.S. Recent advances in the transformation of plants. Trends Plant Sci. 1999, 4, 226–231. [Google Scholar] [CrossRef]
- Abel, S.; Theologis, A. Transient transformation of Arabidopsis leaf protoplasts: A versatile experimental system to study gene expression. Plant J. 1994, 5, 421–427. [Google Scholar] [CrossRef]
- Ohnuma, M.; Yokoyama, T.; Inouye, T.; Sekine, Y.; Tanaka, K. Polyethylene Glycol (PEG)-Mediated Transient Gene Expression in a Red Alga, Cyanidioschyzon merolae 10D. Plant Cell Physiol. 2008, 49, 117–120. [Google Scholar] [CrossRef] [PubMed]
- Endo, H.; Yoshida, M.; Uji, T.; Saga, N.; Inoue, K.; Nagasawa, H. Stable Nuclear Transformation System for the Coccolithophorid Alga Pleurochrysis carterae. Sci. Rep. 2016, 6, 22252. [Google Scholar] [CrossRef]
- Yang, B.; Liu, J.; Liu, B.; Sun, P.; Ma, X.; Jiang, Y.; Wei, D.; Chen, F. Development of a stable genetic system for Chlorella vulgaris—A promising green alga for CO2 biomitigation. Algal Res. 2015, 12, 134–141. [Google Scholar] [CrossRef]
- Miyoshi, D.; Sugimoto, N. Molecular crowding effects on structure and stability of DNA. Biochimie 2008, 90, 1040–1051. [Google Scholar] [CrossRef]
- Chebotareva, N.A.; Kurganov, B.I.; Livanova, N.B. Biochemical effects of molecular crowding. Biochem. Mosc. 2004, 69, 1239. [Google Scholar] [CrossRef]
- Kawai, S.; Hashimoto, W.; Murata, K. Transformation of Saccharomyces cerevisiae and other fungi. Bioeng. Bugs 2010, 1, 395–403. [Google Scholar] [CrossRef]
- Chen, P.; Liu, H.-H.; Cui, R.; Zhang, Z.-L.; Pang, D.-W.; Xie, Z.-X.; Zheng, H.-Z.; Lu, Z.-X.; Tong, H. Visualized investigation of yeast transformation induced with Li+ and polyethylene glycol. Talanta 2008, 77, 262–268. [Google Scholar] [CrossRef]
- Kawai, S.; Pham, T.A.; Nguyen, H.T.; Nankai, H.; Utsumi, T.; Fukuda, Y.; Murata, K. Molecular insights on DNA delivery into Saccharomyces cerevisiae. Biochem. Biophys. Res. Commun. 2004, 317, 100–107. [Google Scholar] [CrossRef] [PubMed]
- Ohnuma, M.; Yokoyama, T.; Inouye, T.; Sekine, Y.; Kuroiwa, T.; Tanaka, K. Optimization of polyethylene glycol (PEG)-mediated DNA introduction conditions for transient gene expression in the unicellular red alga Cyanidioschyzon merolae. J. Gen. Appl. Microbiol. 2014, 60, 156–159. [Google Scholar] [CrossRef] [PubMed]
- Keller, M.D.; Selvin, R.C.; Claus, W.; Guillard, R.R.L. Media for the culture of oceanic ultraphytoplankton. J. Phycol. 1987, 23, 633–638. [Google Scholar] [CrossRef]
- Trask, B.J.; van den Engh, G.J.; Elgershuizen, J.H. Analysis of phytoplankton by flow cytometry. Cytometry 1982, 2, 258–264. [Google Scholar] [CrossRef]
- Gasol, J.M.; Zweifel, U.L.; Peters, F.; Fuhrman, J.A.; Hagström, Å. Significance of Size and Nucleic Acid Content Heterogeneity as Measured by Flow Cytometry in Natural Planktonic Bacteria. Appl Env. Microbiol 1999, 65, 4475–4483. [Google Scholar]
- Ogden, R.C.; Adams, D.A. Electrophoresis in agarose and acrylamide gels. In Methods in Enzymology; Guide to Molecular Cloning Techniques; Academic Press: Cambridge, MA, USA, 1987; Volume 152, pp. 61–87. [Google Scholar]
- Tsao, S.G.S.; Brunk, C.F.; Pearlman, R.E. Hybridization of nucleic acids directly in agarose gels. Anal. Biochem. 1983, 131, 365–372. [Google Scholar] [CrossRef]
- Brugère, J.-F.; Cornillot, E.; Méténier, G.; Bensimon, A.; Vivarès, C.P. Encephalitozoon cuniculi (Microspora) genome: Physical map and evidence for telomere-associated rDNA units on all chromosomes. Nucleic Acids Res. 2000, 28, 2026–2033. [Google Scholar] [CrossRef] [PubMed]
- Mead, J.R.; Arrowood, M.J.; Current, W.L.; Sterling, C.R. Field inversion gel electrophoretic separation of Cryptosporidium spp. chromosome-sized DNA. J. Parasitol. 1988, 74, 366–369. [Google Scholar] [CrossRef] [PubMed]
- Derelle, E.; Ferraz, C.; Lagoda, P.; Eychenié, S.; Cooke, R.; Regad, F.; Sabau, X.; Courties, C.; Delseny, M.; Demaille, J.; et al. DNA Libraries for Sequencing the Genome of Ostreococcus tauri (chlorophyta, Prasinophyceae): The Smallest Free-Living Eukaryotic Cell. J. Phycol. 2002, 38, 1150–1156. [Google Scholar] [CrossRef]
- Altenburger, R.; Callies, R.; Grimme, L.H.; Leibfritz, D.; Mayer, A. The mode of action of glufosinate in algae: The role of uptake and nitrogen assimilation pathways. Pestic. Sci. 1995, 45, 305–310. [Google Scholar] [CrossRef]
- Jakobiak, T.; Mages, W.; Scharf, B.; Babinger, P.; Stark, K.; Schmitt, R. The Bacterial Paromomycin Resistance Gene, aphH, as a Dominant Selectable Marker in Volvox carteri. Protist 2004, 155, 381–393. [Google Scholar] [CrossRef]
- Meslet-Cladière, L.; Vallon, O. Novel Shuttle Markers for Nuclear Transformation of the Green Alga Chlamydomonas reinhardtii. Eukaryot. Cell 2011, 10, 1670–1678. [Google Scholar] [CrossRef]
- Huang, S.; Houghton, P.J. Resistance to Rapamycin: A Novel Anticancer Drug. Cancer Metastasis Rev. 2001, 20, 69–78. [Google Scholar] [CrossRef] [PubMed]
- Oka, A.; Sugisaki, H.; Takanami, M. Nucleotide sequence of the kanamycin resistance transposon Tn903. J. Mol. Biol. 1981, 147, 217–226. [Google Scholar] [CrossRef]
- Ow, D.W.; Wet, J.R.D.; Helinski, D.R.; Howell, S.H.; Wood, K.V.; Deluca, M. Transient and Stable Expression of the Firefly Luciferase Gene in Plant Cells and Transgenic Plants. Science 1986, 234, 856–859. [Google Scholar] [CrossRef]
- Rice, P.; Longden, I.; Bleasby, A. EMBOSS: The European Molecular Biology Open Software Suite. Trends Genet. 2000, 16, 276–277. [Google Scholar] [CrossRef]
- Sterck, L.; Billiau, K.; Abeel, T.; Rouzé, P.; Van de Peer, Y. ORCAE: Online resource for community annotation of eukaryotes. Nat. Methods 2012, 9, 1041. [Google Scholar] [CrossRef] [PubMed]
- Michely, S.; Toulza, E.; Subirana, L.; John, U.; Cognat, V.; Maréchal-Drouard, L.; Grimsley, N.; Moreau, H.; Piganeau, G. Evolution of Codon Usage in the Smallest Photosynthetic Eukaryotes and Their Giant Viruses. Genome Biol. Evol. 2013, 5, 848–859. [Google Scholar] [CrossRef] [PubMed]
- Kianianmomeni, A.; Hallmann, A. Validation of reference genes for quantitative gene expression studies in Volvox carteri using real-time RT-PCR. Mol. Biol. Rep. 2013, 40, 6691–6699. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Wu, G.; Huang, X.; Liu, S.; Cong, B. Validation of housekeeping genes for gene expression studies in an ice alga Chlamydomonas during freezing acclimation. Extremophiles 2012, 16, 419–425. [Google Scholar] [CrossRef] [PubMed]
- Le Bail, A.; Dittami, S.M.; de Franco, P.-O.; Rousvoal, S.; Cock, M.J.; Tonon, T.; Charrier, B. Normalisation genes for expression analyses in the brown alga model Ectocarpus siliculosus. Bmc Mol. Biol. 2008, 9. [Google Scholar] [CrossRef]
- Derelle, E.; Yau, S.; Moreau, H.; Grimsley, N.H. Prasinovirus Attack of Ostreococcus Is Furtive by Day but Savage by Night. J. Virol. 2018, 92, e01703-17. [Google Scholar]
- Farinas, B.; Mary, C.; Manes, C.L.D.; Bhaud, Y.; Peaucellier, G.; Moreau, H. Natural synchronisation for the study of cell division in the green unicellular alga Ostreococcus tauri. Plant Mol. Biol. 2006, 60, 277–292. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez, F.; Derelle, E.; Guillou, L.; Le Gall, F.; Vaulot, D.; Moreau, H. Ecotype diversity in the marine picoeukaryote Ostreococcus (Chlorophyta, Prasinophyceae). Environ. Microbiol. 2005, 7, 853–859. [Google Scholar] [CrossRef]
- Weissgerber, T.L.; Milic, N.M.; Winham, S.J.; Garovic, V.D. Beyond Bar and Line Graphs: Time for a New Data Presentation Paradigm. PLOS Biol. 2015, 13, e1002128. [Google Scholar] [CrossRef]
- Feeney, K.A.; Hansen, L.L.; Putker, M.; Olivares-Yañez, C.; Day, J.; Eades, L.J.; Larrondo, L.F.; Hoyle, N.P.; O’Neill, J.S.; van Ooijen, G. Daily magnesium fluxes regulate cellular timekeeping and energy balance. Nature 2016, 532, 375–379. [Google Scholar] [CrossRef]
- Cogoni, C.; Macino, G. Post-transcriptional gene silencing across kingdoms. Curr. Opin. Genet. Dev. 2000, 10, 638–643. [Google Scholar] [CrossRef]
- Matzke, A.J.M.; Matzke, M.A. Position effects and epigenetic silencing of plant transgenes. Curr. Opin. Plant Biol. 1998, 1, 142–148. [Google Scholar] [CrossRef]
- Hobbs, S.L.A.; Warkentin, T.D.; DeLong, C.M.O. Transgene copy number can be positively or negatively associated with transgene expression. Plant Mol. Biol. 1993, 21, 17–26. [Google Scholar] [CrossRef] [PubMed]
- van der Krol, A.R.; Mur, L.A.; Beld, M.; Mol, J.N.; Stuitje, A.R. Flavonoid genes in petunia: Addition of a limited number of gene copies may lead to a suppression of gene expression. Plant Cell 1990, 2, 291–299. [Google Scholar] [CrossRef]
- Cerutti, H.; Ma, X.; Msanne, J.; Repas, T. RNA-Mediated Silencing in Algae: Biological Roles and Tools for Analysis of Gene Function. Eukaryot. Cell 2011, 10, 1164–1172. [Google Scholar] [CrossRef] [PubMed]
- Yau, S.; Hemon, C.; Derelle, E.; Moreau, H.; Piganeau, G.; Grimsley, N. A Viral Immunity Chromosome in the Marine Picoeukaryote. Ostreococcus Tauri. PLOS Pathog. 2016, 12, e1005965. [Google Scholar]
- Yau, S.; Caravello, G.; Fonvieille, N.; Desgranges, É.; Moreau, H.; Grimsley, N. Rapidity of Genomic Adaptations to Prasinovirus Infection in a Marine Microalga. Viruses 2018, 10, 441. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sanchez, F.; Geffroy, S.; Norest, M.; Yau, S.; Moreau, H.; Grimsley, N. Simplified Transformation of Ostreococcus tauri Using Polyethylene Glycol. Genes 2019, 10, 399. https://doi.org/10.3390/genes10050399
Sanchez F, Geffroy S, Norest M, Yau S, Moreau H, Grimsley N. Simplified Transformation of Ostreococcus tauri Using Polyethylene Glycol. Genes. 2019; 10(5):399. https://doi.org/10.3390/genes10050399
Chicago/Turabian StyleSanchez, Frédéric, Solène Geffroy, Manon Norest, Sheree Yau, Hervé Moreau, and Nigel Grimsley. 2019. "Simplified Transformation of Ostreococcus tauri Using Polyethylene Glycol" Genes 10, no. 5: 399. https://doi.org/10.3390/genes10050399
APA StyleSanchez, F., Geffroy, S., Norest, M., Yau, S., Moreau, H., & Grimsley, N. (2019). Simplified Transformation of Ostreococcus tauri Using Polyethylene Glycol. Genes, 10(5), 399. https://doi.org/10.3390/genes10050399