Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population

Abstract
1. Introduction
2. What Proportion of the Human Genome is Defined by Peri/Centromeric Satellite DNAs?
3. What is the Nature of Sequence Variation within a Single Satellite Array?
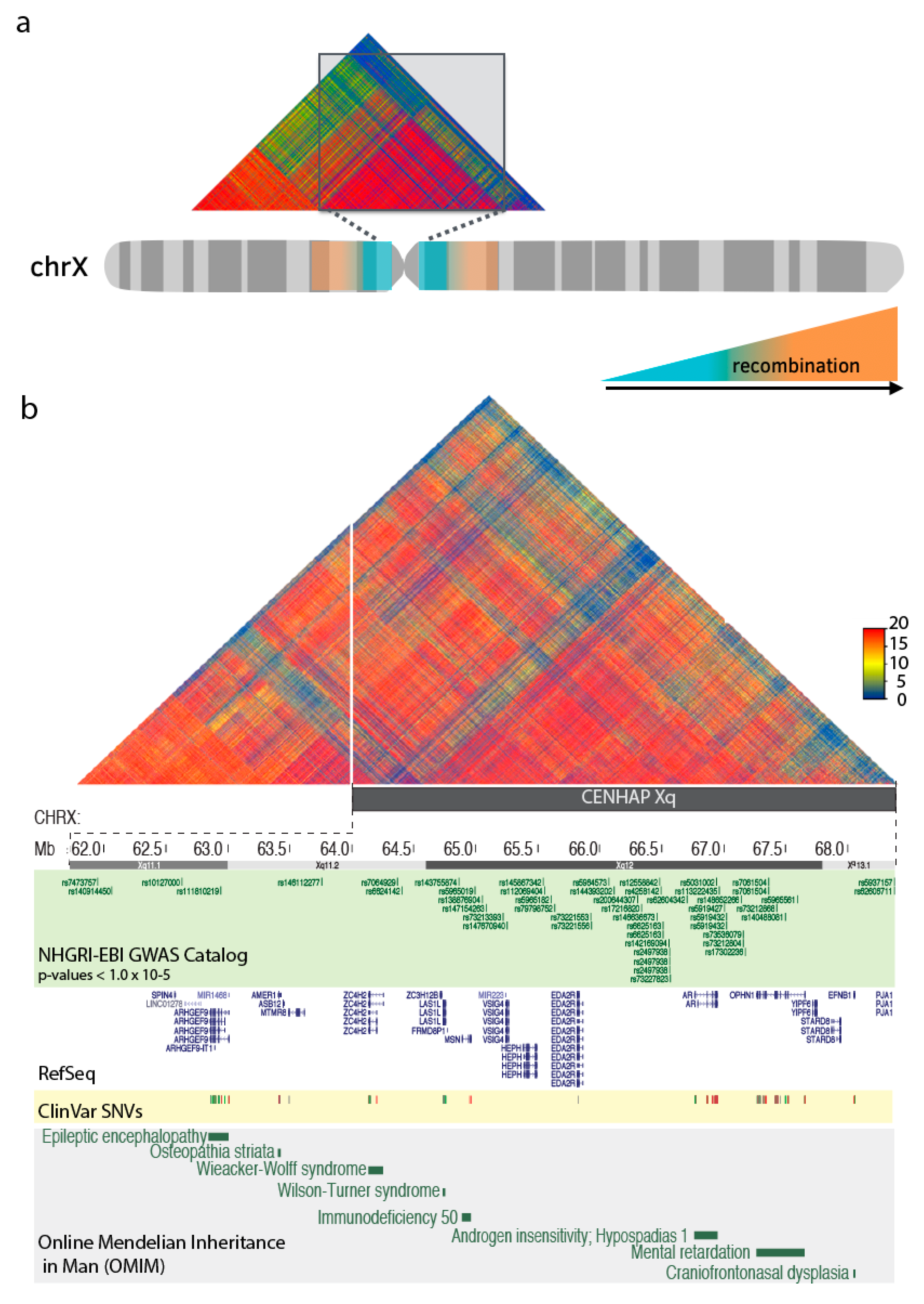
4. Centromeric Regions Span Variants Associated with Disease.
5. Concluding Remarks
Funding
Acknowledgments
Conflicts of Interest
References
- 1000 Genomes Project Consortium; Auton, A.; Brooks, L.D.; Durbin, R.M.; Garrison, E.P.; Kang, H.M.; Korbel, J.O.; Marchini, J.L.; McCarthy, S.; McVean, G.A.; et al. A global reference for human genetic variation. Nature 2015, 526, 68–74. [Google Scholar]
- Lander, E.S.; Linton, L.M.; Birren, B.; Nusbaum, C.; Zody, M.C.; Baldwin, J.; Devon, K.; Dewar, K.; Doyle, M.; FitzHugh, W.; et al. Initial sequencing and analysis of the human genome. Nature 2001, 409, 860–921. [Google Scholar] [PubMed]
- Venter, J.C.; Adams, M.D.; Myers, E.W.; Li, P.W.; Mural, R.J.; Sutton, G.G.; Smith, H.O.; Yandell, M.; Evans, C.A.; Holt, R.A.; et al. The sequence of the human genome. Science 2001, 291, 1304–1351. [Google Scholar] [CrossRef]
- Miga, K.H. Completing the human genome: The progress and challenge of satellite DNA assembly. Chromosome Res. 2015, 23, 421–426. [Google Scholar] [CrossRef]
- Jain, M.; Koren, S.; Miga, K.H.; Quick, J.; Rand, A.C.; Sasani, T.A.; Tyson, J.R.; Beggs, A.D.; Dilthey, A.T.; Fiddes, I.T.; et al. Nanopore sequencing and assembly of a human genome with ultra-long reads. Nat. Biotechnol. 2018, 36, 338. [Google Scholar] [CrossRef]
- Jain, M.; Olsen, H.E.; Turner, D.; Stoddart, D.; Paten, B.; Haussler, D.; Willard, H.F.; Akeson, M.; Miga, K.H. Linear assembly of a human centromere on the Y chromosome. Nat. Biotechnol. 2018, 36, 321–323. [Google Scholar] [CrossRef] [PubMed]
- Miga, K.H.; Newton, Y.; Jain, M.; Altemose, N.; Willard, H.F.; Kent, W.J. Centromere reference models for human chromosomes X and Y satellite arrays. Genome Res. 2014, 24, 697–707. [Google Scholar] [CrossRef] [PubMed]
- Black, E.M.; Giunta, S. Repetitive Fragile Sites: Centromere Satellite DNA As a Source of Genome Instability in Human Diseases. Genes 2018, 9, 615. [Google Scholar] [CrossRef] [PubMed]
- Ferreira, D.; Meles, S.; Escudeiro, A.; Mendes-da-Silva, A.; Adega, F.; Chaves, R. Satellite non-coding RNAs: The emerging players in cells, cellular pathways and cancer. Chromosome Res. 2015, 23, 479–493. [Google Scholar] [CrossRef]
- Enukashvily, N.I.; Donev, R.; Waisertreiger, I.S.-R.; Podgornaya, O.I. Human chromosome 1 satellite 3 DNA is decondensed, demethylated and transcribed in senescent cells and in A431 epithelial carcinoma cells. Cytogenet. Genome Res. 2007, 118, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Atkin, N.B.; Brito-Babapulle, V. Heterochromatin polymorphism and human cancer. Cancer Genet. Cytogenet. 1981, 3, 261–272. [Google Scholar] [CrossRef]
- Berger, R.; Bernheim, A.; Kristoffersson, U.; Mitelman, F.; Olsson, H. C-band heteromorphism in breast cancer patients. Cancer Genet. Cytogenet. 1985, 18, 37–42. [Google Scholar] [CrossRef]
- Sahin, F.I.; Yilmaz, Z.; Yuregir, O.O.; Bulakbasi, T.; Ozer, O.; Zeyneloglu, H.B. Chromosome heteromorphisms: An impact on infertility. J. Assist. Reprod. Genet. 2008, 25, 191–195. [Google Scholar] [CrossRef]
- Ting, D.T.; Lipson, D.; Paul, S.; Brannigan, B.W.; Akhavanfard, S.; Coffman, E.J.; Contino, G.; Deshpande, V.; Iafrate, A.J.; Letovsky, S.; et al. Aberrant overexpression of satellite repeats in pancreatic and other epithelial cancers. Science 2011, 331, 593–596. [Google Scholar] [CrossRef]
- Atkin, N.B.; Brito-Babapulle, V. Chromosome 1 heterochromatin variants and cancer: A reassessment. Cancer Genet. Cytogenet. 1985, 18, 325–331. [Google Scholar] [CrossRef]
- Wu, J.C.; Manuelidis, L. Sequence definition and organization of a human repeated DNA. J. Mol. Biol. 1980, 142, 363–386. [Google Scholar] [CrossRef]
- Lee, C.; Wevrick, R.; Fisher, R.B.; Ferguson-Smith, M.A.; Lin, C.C. Human centromeric DNAs. Hum. Genet. 1997, 100, 291–304. [Google Scholar] [CrossRef]
- Rudd, M.K.; Willard, H.F. Analysis of the centromeric regions of the human genome assembly. Trends Genet. 2004, 20, 529–533. [Google Scholar] [CrossRef]
- Eichler, E.E.; Clark, R.A.; She, X. An assessment of the sequence gaps: Unfinished business in a finished human genome. Nat. Rev. Genet. 2004, 5, 345–354. [Google Scholar] [CrossRef]
- Willard, H.F. Chromosome-specific organization of human alpha satellite DNA. Am. J. Hum. Genet. 1985, 37, 524–532. [Google Scholar]
- Waye, J.S.; Willard, H.F. Nucleotide sequence heterogeneity of alpha satellite repetitive DNA: A survey of alphoid sequences from different human chromosomes. Nucleic Acids Res. 1987, 15, 7549–7569. [Google Scholar] [CrossRef]
- Manuelidis, L.; Wu, J.C. Homology between human and simian repeated DNA. Nature 1978, 276, 92–94. [Google Scholar] [CrossRef]
- Hayden, K.E.; Strome, E.D.; Merrett, S.L.; Lee, H.-R.; Rudd, M.K.; Willard, H.F. Sequences associated with centromere competency in the human genome. Mol. Cell. Biol. 2013, 33, 763–772. [Google Scholar] [CrossRef]
- Wevrick, R.; Willard, H.F. Long-range organization of tandem arrays of alpha satellite DNA at the centromeres of human chromosomes: High-frequency array-length polymorphism and meiotic stability. Proc. Natl. Acad. Sci. USA 1989, 86, 9394–9398. [Google Scholar] [CrossRef]
- Mahtani, M.M.; Willard, H.F. Pulsed-field gel analysis of alpha-satellite DNA at the human X chromosome centromere: High-frequency polymorphisms and array size estimate. Genomics 1990, 7, 607–613. [Google Scholar] [CrossRef]
- Marçais, B.; Bellis, M.; Gérard, A.; Pagès, M.; Boublik, Y.; Roizès, G. Structural organization and polymorphism of the alpha satellite DNA sequences of chromosomes 13 and 21 as revealed by pulse field gel electrophoresis. Hum. Genet. 1991, 86, 311–316. [Google Scholar] [CrossRef]
- Jones, K.W.; Prosser, J.; Corneo, G.; Ginelli, E. The chromosomal location of human satellite DNA III. Chromosoma 1973, 42, 445–451. [Google Scholar] [CrossRef]
- Jones, K.W.; Corneo, G. Location of satellite and homogeneous DNA sequences on human chromosomes. Nat. New Biol. 1971, 233, 268–271. [Google Scholar] [CrossRef]
- Gosden, J.R.; Mitchell, A.R.; Buckland, R.A.; Clayton, R.P.; Evans, H.J. The location of four human satellite DNAs on human chromosomes. Exp. Cell Res. 1975, 92, 148–158. [Google Scholar] [CrossRef]
- Tagarro, I.; Fernández-Peralta, A.M.; González-Aguilera, J.J. Chromosomal localization of human satellites 2 and 3 by a FISH method using oligonucleotides as probes. Hum. Genet. 1994, 93, 383–388. [Google Scholar] [CrossRef]
- Altemose, N.; Miga, K.H.; Maggioni, M.; Willard, H.F. Genomic characterization of large heterochromatic gaps in the human genome assembly. PLoS Comput. Biol. 2014, 10, e1003628. [Google Scholar] [CrossRef]
- Prosser, J.; Frommer, M.; Paul, C.; Vincent, P.C. Sequence relationships of three human satellite DNAs. J. Mol. Biol. 1986, 187, 145–155. [Google Scholar] [CrossRef]
- Cooke, H. Repeated sequence specific to human males. Nature 1976, 262, 182–186. [Google Scholar] [CrossRef]
- Kunkel, L.M.; Smith, K.D.; Boyer, S.H.; Borgaonkar, D.S.; Wachtel, S.S.; Miller, O.J.; Breg, W.R.; Jones, H.W., Jr.; Rary, J.M. Analysis of human Y-chromosome-specific reiterated DNA in chromosome variants. Proc. Natl. Acad. Sci. USA 1977, 74, 1245–1249. [Google Scholar] [CrossRef]
- Nakahori, Y.; Mitani, K.; Yamada, M.; Nakagome, Y. A human Y-chromosome specific repeated DNA family (DYZ1) consists of a tandem array of pentanucleotides. Nucleic Acids Res. 1986, 14, 7569–7580. [Google Scholar] [CrossRef]
- Willard, H.F.; Waye, J.S. Chromosome-specific subsets of human alpha satellite DNA: Analysis of sequence divergence within and between chromosomal subsets and evidence for an ancestral pentameric repeat. J. Mol. Evol. 1987, 25, 207–214. [Google Scholar] [CrossRef]
- Alexandrov, I.; Kazakov, A.; Tumeneva, I.; Shepelev, V.; Yurov, Y. Alpha-satellite DNA of primates: Old and new families. Chromosoma 2001, 110, 253–266. [Google Scholar] [CrossRef]
- Jeanpierre, M.; Weil, D.; Gallano, P.; Creau-Goldberg, N.; Junien, C. The organization of two related subfamilies of a human tandemly repeated DNA is chromosome specific. Hum. Genet. 1985, 70, 302–310. [Google Scholar] [CrossRef]
- Sherman, R.M.; Forman, J.; Antonescu, V.; Puiu, D.; Daya, M.; Rafaels, N.; Boorgula, M.P.; Chavan, S.; Vergara, C.; Ortega, V.E.; et al. Assembly of a pan-genome from deep sequencing of 910 humans of African descent. Nat. Genet. 2019, 51, 30. [Google Scholar] [CrossRef]
- Schneider, V.A.; Graves-Lindsay, T.; Howe, K.; Bouk, N.; Chen, H.-C.; Kitts, P.A.; Murphy, T.D.; Pruitt, K.D.; Thibaud-Nissen, F.; Albracht, D.; et al. Evaluation of GRCh38 and de novo haploid genome assemblies demonstrates the enduring quality of the reference assembly. Genome Res. 2017, 27, 849–864. [Google Scholar] [CrossRef]
- Miga, K.H.; Eisenhart, C.; Kent, W.J. Utilizing mapping targets of sequences underrepresented in the reference assembly to reduce false positive alignments. Nucleic Acids Res. 2015, 43, e133. [Google Scholar] [CrossRef]
- Nechemia-Arbely, Y.; Fachinetti, D.; Miga, K.H.; Sekulic, N.; Soni, G.V.; Kim, D.H.; Wong, A.K.; Lee, A.Y.; Nguyen, K.; Dekker, C.; et al. Human centromeric CENP-A chromatin is a homotypic, octameric nucleosome at all cell cycle points. J. Cell Biol. 2017, 216, 607–621. [Google Scholar] [CrossRef]
- Levy, S.; Sutton, G.; Ng, P.C.; Feuk, L.; Halpern, A.L.; Walenz, B.P.; Axelrod, N.; Huang, J.; Kirkness, E.F.; Denisov, G.; et al. The diploid genome sequence of an individual human. PLoS Biol. 2007, 5, e254. [Google Scholar] [CrossRef]
- Audano, P.A.; Sulovari, A.; Graves-Lindsay, T.A.; Cantsilieris, S.; Sorensen, M.; Welch, A.E.; Dougherty, M.L.; Nelson, B.J.; Shah, A.; Dutcher, S.K.; et al. Characterizing the Major Structural Variant Alleles of the Human Genome. Cell 2019, 176, 663–675.e19. [Google Scholar] [CrossRef]
- Chaisson, M.J.P.; Wilson, R.K.; Eichler, E.E. Genetic variation and the de novo assembly of human genomes. Nat. Rev. Genet. 2015, 16, 627–640. [Google Scholar] [CrossRef] [PubMed]
- Marçais, B.; Charlieu, J.P.; Allain, B.; Brun, E.; Bellis, M.; Roizès, G. On the mode of evolution of alpha satellite DNA in human populations. J. Mol. Evol. 1991, 33, 42–48. [Google Scholar] [CrossRef] [PubMed]
- Smith, G.P. Evolution of repeated DNA sequences by unequal crossover. Science 1976, 191, 528–535. [Google Scholar] [CrossRef]
- Lower, S.S.; McGurk, M.P.; Clark, A.G.; Barbash, D.A. Satellite DNA evolution: old ideas, new approaches. Curr. Opin. Genet. Dev. 2018, 49, 70–78. [Google Scholar] [CrossRef] [PubMed]
- Stults, D.M.; Killen, M.W.; Pierce, H.H.; Pierce, A.J. Genomic architecture and inheritance of human ribosomal RNA gene clusters. Genome Res. 2008, 18, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Kim, J.-H.; Dilthey, A.T.; Nagaraja, R.; Lee, H.-S.; Koren, S.; Dudekula, D.; Wood, W.H., III; Piao, Y.; Ogurtsov, A.Y.; Utani, K.; et al. Variation in human chromosome 21 ribosomal RNA genes characterized by TAR cloning and long-read sequencing. Nucleic Acids Res. 2018, 46, 6712–6725. [Google Scholar] [CrossRef]
- Warburton, P.E.; Willard, H.F. Interhomologue sequence variation of alpha satellite DNA from human chromosome 17: Evidence for concerted evolution along haplotypic lineages. J. Mol. Evol. 1995, 41, 1006–1015. [Google Scholar] [CrossRef]
- Willard, H.F.; Waye, J.S. Hierarchical order in chromosome-specific human alpha satellite DNA. Trends Genet. 1987, 3, 192–198. [Google Scholar] [CrossRef]
- Hayden, K.E. Human centromere genomics: Now it’s personal. Chromosome Res. 2012, 20, 621–633. [Google Scholar] [CrossRef] [PubMed]
- Pluta, A.F.; Saitoh, N.; Goldberg, I.; Earnshaw, W.C. Identification of a subdomain of CENP-B that is necessary and sufficient for localization to the human centromere. J. Cell Biol. 1992, 116, 1081–1093. [Google Scholar] [CrossRef]
- Hudson, D.F.; Fowler, K.J.; Earle, E.; Saffery, R.; Kalitsis, P.; Trowell, H.; Hill, J.; Wreford, N.G.; de Kretser, D.M.; Cancilla, M.R.; et al. Centromere protein B null mice are mitotically and meiotically normal but have lower body and testis weights. J. Cell Biol. 1998, 141, 309–319. [Google Scholar] [CrossRef]
- Warburton, P.E.; Waye, J.S.; Willard, H.F. Nonrandom localization of recombination events in human alpha satellite repeat unit variants: Implications for higher-order structural characteristics within centromeric heterochromatin. Mol. Cell. Biol. 1993, 13, 6520–6529. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Fachinetti, D.; Han, J.S.; McMahon, M.A.; Ly, P.; Abdullah, A.; Wong, A.J.; Cleveland, D.W. DNA Sequence-Specific Binding of CENP-B Enhances the Fidelity of Human Centromere Function. Dev. Cell 2015, 33, 314–327. [Google Scholar] [CrossRef] [PubMed]
- Waye, J.S.; Willard, H.F. Structure, organization, and sequence of alpha satellite DNA from human chromosome 17: Evidence for evolution by unequal crossing-over and an ancestral pentamer repeat shared with the human X chromosome. Mol. Cell. Biol. 1986, 6, 3156–3165. [Google Scholar] [CrossRef] [PubMed]
- Maloney, K.A.; Sullivan, L.L.; Matheny, J.E.; Strome, E.D.; Merrett, S.L.; Ferris, A.; Sullivan, B.A. Functional epialleles at an endogenous human centromere. Proc. Natl. Acad. Sci. USA 2012, 109, 13704–13709. [Google Scholar] [CrossRef]
- Aldrup-MacDonald, M.E.; Kuo, M.E.; Sullivan, L.L.; Chew, K.; Sullivan, B.A. Genomic variation within alpha satellite DNA influences centromere location on human chromosomes with metastable epialleles. Genome Res. 2016, 26, 1301–1311. [Google Scholar] [CrossRef]
- McNulty, S.M.; Sullivan, L.L.; Sullivan, B.A. Human Centromeres Produce Chromosome-Specific and Array-Specific Alpha Satellite Transcripts that Are Complexed with CENP-A and CENP-C. Dev. Cell 2017, 42, 226–240.e6. [Google Scholar] [CrossRef]
- Hall, L.L.; Byron, M.; Carone, D.M.; Whitfield, T.W.; Pouliot, G.P.; Fischer, A.; Jones, P.; Lawrence, J.B. Demethylated HSATII DNA and HSATII RNA Foci Sequester PRC1 and MeCP2 into Cancer-Specific Nuclear Bodies. Cell Rep. 2017, 18, 2943–2956. [Google Scholar] [CrossRef]
- Cobb, B.S.; Morales-Alcelay, S.; Kleiger, G.; Brown, K.E.; Fisher, A.G.; Smale, S.T. Targeting of Ikaros to pericentromeric heterochromatin by direct DNA binding. Genes Dev. 2000, 14, 2146–2160. [Google Scholar] [CrossRef]
- Nishibuchi, G.; Déjardin, J. The molecular basis of the organization of repetitive DNA-containing constitutive heterochromatin in mammals. Chromosome Res. 2017, 25, 77–87. [Google Scholar] [CrossRef]
- Delpu, Y.; McNamara, T.F.; Griffin, P.; Kaleem, S.; Narayan, S.; Schildkraut, C.; Miga, K.H.; Tahiliani, M. Chromosomal rearrangements at hypomethylated Satellite 2 sequences are associated with impaired replication efficiency and increased fork stalling. bioRxiv 2019. [Google Scholar] [CrossRef]
- Erliandri, I.; Fu, H.; Nakano, M.; Kim, J.-H.; Miga, K.H.; Liskovykh, M.; Earnshaw, W.C.; Masumoto, H.; Kouprina, N.; Aladjem, M.I.; et al. Replication of alpha-satellite DNA arrays in endogenous human centromeric regions and in human artificial chromosome. Nucleic Acids Res. 2014, 42, 11502–11516. [Google Scholar] [CrossRef]
- Bersani, F.; Lee, E.; Kharchenko, P.V.; Xu, A.W.; Liu, M.; Xega, K.; MacKenzie, O.C.; Brannigan, B.W.; Wittner, B.S.; Jung, H.; et al. Pericentromeric satellite repeat expansions through RNA-derived DNA intermediates in cancer. Proc. Natl. Acad. Sci. USA 2015, 112, 15148–15153. [Google Scholar] [CrossRef]
- Sevim, V.; Bashir, A.; Chin, C.-S.; Miga, K.H. Alpha-CENTAURI: Assessing novel centromeric repeat sequence variation with long read sequencing. Bioinformatics 2016, 32, 1921–1924. [Google Scholar] [CrossRef]
- Pathak, D.; Premi, S.; Srivastava, J.; Chandy, S.P.; Ali, S. Genomic instability of the DYZ1 repeat in patients with Y chromosome anomalies and males exposed to natural background radiation. DNA Res. 2006, 13, 103–109. [Google Scholar] [CrossRef]
- Rahman, M.M.; Bashamboo, A.; Prasad, A.; Pathak, D.; Ali, S. Organizational variation of DYZ1 repeat sequences on the human Y chromosome and its diagnostic potentials. DNA Cell Biol. 2004, 23, 561–571. [Google Scholar] [CrossRef]
- Oakey, R.; Tyler-Smith, C. Y chromosome DNA haplotyping suggests that most European and Asian men are descended from one of two males. Genomics 1990, 7, 325–330. [Google Scholar] [CrossRef]
- Edgar, R.C. MUSCLE: Multiple sequence alignment with high accuracy and high throughput. Nucleic Acids Res. 2004, 32, 1792–1797. [Google Scholar] [CrossRef]
- Schueler, M.G.; Higgins, A.W.; Rudd, M.K.; Gustashaw, K.; Willard, H.F. Genomic and genetic definition of a functional human centromere. Science 2001, 294, 109–115. [Google Scholar] [CrossRef]
- Langley, S.A.; Miga, K.; Karpen, G.H.; Langley, C.H. Haplotypes spanning centromeric regions reveal persistence of large blocks of archaic DNA. BioRxiv 2018. [Google Scholar] [CrossRef]
- She, X.; Horvath, J.E.; Jiang, Z.; Liu, G.; Furey, T.S.; Christ, L.; Clark, R.; Graves, T.; Gulden, C.L.; Alkan, C.; et al. The structure and evolution of centromeric transition regions within the human genome. Nature 2004, 430, 857–864. [Google Scholar] [CrossRef]
- Pruitt, K.D.; Tatusova, T.; Maglott, D.R. NCBI Reference Sequence (RefSeq): A curated non-redundant sequence database of genomes, transcripts and proteins. Nucleic Acids Res. 2005, 33, D501–D504. [Google Scholar] [CrossRef]
- Amberger, J.; Bocchini, C.A.; Scott, A.F.; Hamosh, A. McKusick’s Online Mendelian Inheritance in Man (OMIM®). Nucleic Acids Res. 2009, 37, D793–D796. [Google Scholar] [CrossRef]
- Hamosh, A.; Scott, A.F.; Amberger, J.S.; Bocchini, C.A.; McKusick, V.A. Online Mendelian Inheritance in Man (OMIM), a knowledgebase of human genes and genetic disorders. Nucleic Acids Res. 2005, 33, D514–D517. [Google Scholar] [CrossRef]
- Landrum, M.J.; Lee, J.M.; Benson, M.; Brown, G.; Chao, C.; Chitipiralla, S.; Gu, B.; Hart, J.; Hoffman, D.; Hoover, J.; et al. ClinVar: Public archive of interpretations of clinically relevant variants. Nucleic Acids Res. 2016, 44, D862–D868. [Google Scholar] [CrossRef]
- Hindorff, L.A.; Sethupathy, P.; Junkins, H.A.; Ramos, E.M.; Mehta, J.P.; Collins, F.S.; Manolio, T.A. Potential etiologic and functional implications of genome-wide association loci for human diseases and traits. Proc. Natl. Acad. Sci. USA 2009, 106, 9362–9367. [Google Scholar] [CrossRef]
- Loh, P.-R.; Genovese, G.; Handsaker, R.E.; Finucane, H.K.; Reshef, Y.A.; Palamara, P.F.; Birmann, B.M.; Talkowski, M.E.; Bakhoum, S.F.; McCarroll, S.A.; et al. Insights into clonal haematopoiesis from 8,342 mosaic chromosomal alterations. Nature 2018, 559, 350–355. [Google Scholar] [CrossRef]
- Reich, D.; Patterson, N.; De Jager, P.L.; McDonald, G.J.; Waliszewska, A.; Tandon, A.; Lincoln, R.R.; DeLoa, C.; Fruhan, S.A.; Cabre, P.; et al. A whole-genome admixture scan finds a candidate locus for multiple sclerosis susceptibility. Nat. Genet. 2005, 37, 1113–1118. [Google Scholar] [CrossRef] [PubMed]
- Karolchik, D.; Hinrichs, A.S.; Furey, T.S.; Roskin, K.M.; Sugnet, C.W.; Haussler, D.; Kent, W.J. The UCSC Table Browser data retrieval tool. Nucleic Acids Res. 2004, 32, D493–D496. [Google Scholar] [CrossRef]
- Barra, V.; Fachinetti, D. The dark side of centromeres: types, causes and consequences of structural abnormalities implicating centromeric DNA. Nat. Commun. 2018, 9, 4340. [Google Scholar] [CrossRef]
- Levy-Sakin, M.; Pastor, S.; Mostovoy, Y.; Li, L.; Leung, A.K.Y.; McCaffrey, J.; Young, E.; Lam, E.T.; Hastie, A.R.; Wong, K.H.Y.; et al. Genome maps across 26 human populations reveal population-specific patterns of structural variation. Nat. Commun. 2019, 10, 1025. [Google Scholar] [CrossRef]
- Michailidou, K.; Lindström, S.; Dennis, J.; Beesley, J.; Hui, S.; Kar, S.; Lemaçon, A.; Soucy, P.; Glubb, D.; Rostamianfar, A.; et al. Association analysis identifies 65 new breast cancer risk loci. Nature 2017, 551, 92–94. [Google Scholar] [CrossRef]
- O’Donnell, P.H.; Stark, A.L.; Gamazon, E.R.; Wheeler, H.E.; McIlwee, B.E.; Gorsic, L.; Im, H.K.; Huang, R.S.; Cox, N.J.; Dolan, M.E. Identification of novel germline polymorphisms governing capecitabine sensitivity. Cancer 2012, 118, 4063–4073. [Google Scholar] [CrossRef] [PubMed]
- Moore, K.N.; Tritchler, D.; Kaufman, K.M.; Lankes, H.; Quinn, M.C.J.; Ovarian Cancer Association Consortium; Van Le, L.; Berchuck, A.; Backes, F.J.; Tewari, K.S.; et al. Genome-wide association study evaluating single-nucleotide polymorphisms and outcomes in patients with advanced stage serous ovarian or primary peritoneal cancer: An NRG Oncology/Gynecologic Oncology Group study. Gynecol. Oncol. 2017, 147, 396–401. [Google Scholar] [CrossRef] [PubMed]
- Hofer, P.; Hagmann, M.; Brezina, S.; Dolejsi, E.; Mach, K.; Leeb, G.; Baierl, A.; Buch, S.; Sutterlüty-Fall, H.; Karner-Hanusch, J.; et al. Bayesian and frequentist analysis of an Austrian genome-wide association study of colorectal cancer and advanced adenomas. Oncotarget 2017, 8, 98623–98634. [Google Scholar] [CrossRef]
- Deng, X.; Sabino, E.C.; Cunha-Neto, E.; Ribeiro, A.L.; Ianni, B.; Mady, C.; Busch, M.P.; Seielstad, M. REDSII Chagas Study Group from the NHLBI Retrovirus Epidemiology Donor Study-II Component International Genome wide association study (GWAS) of Chagas cardiomyopathy in Trypanosoma cruzi seropositive subjects. PLoS ONE 2013, 8, e79629. [Google Scholar] [CrossRef] [PubMed]
- Cordell, H.J.; Bentham, J.; Topf, A.; Zelenika, D.; Heath, S.; Mamasoula, C.; Cosgrove, C.; Blue, G.; Granados-Riveron, J.; Setchfield, K.; et al. Genome-wide association study of multiple congenital heart disease phenotypes identifies a susceptibility locus for atrial septal defect at chromosome 4p16. Nat. Genet. 2013, 45, 822–824. [Google Scholar] [CrossRef]
- van der Harst, P.; Verweij, N. Identification of 64 Novel Genetic Loci Provides an Expanded View on the Genetic Architecture of Coronary Artery Disease. Circ. Res. 2018, 122, 433–443. [Google Scholar] [CrossRef]
- Nagel, M.; Jansen, P.R.; Stringer, S.; Watanabe, K.; de Leeuw, C.A.; Bryois, J.; Savage, J.E.; Hammerschlag, A.R.; Skene, N.G.; Muñoz-Manchado, A.B.; et al. Meta-analysis of genome-wide association studies for neuroticism in 449,484 individuals identifies novel genetic loci and pathways. Nat. Genet. 2018, 50, 920–927. [Google Scholar] [CrossRef]
- Turley, P.; Walters, R.K.; Maghzian, O.; Okbay, A.; Lee, J.J.; Fontana, M.A.; Nguyen-Viet, T.A.; Wedow, R.; Zacher, M.; Furlotte, N.A.; et al. Multi-trait analysis of genome-wide association summary statistics using MTAG. Nat. Genet. 2018, 50, 229–237. [Google Scholar] [CrossRef]
- Herold, C.; Hooli, B.V.; Mullin, K.; Liu, T.; Roehr, J.T.; Mattheisen, M.; Parrado, A.R.; Bertram, L.; Lange, C.; Tanzi, R.E. Family-based association analyses of imputed genotypes reveal genome-wide significant association of Alzheimer’s disease with OSBPL6, PTPRG, and PDCL3. Mol. Psychiatry 2016, 21, 1608–1612. [Google Scholar] [CrossRef]
- Fung, H.-C.; Scholz, S.; Matarin, M.; Simón-Sánchez, J.; Hernandez, D.; Britton, A.; Gibbs, J.R.; Langefeld, C.; Stiegert, M.L.; Schymick, J.; et al. Genome-wide genotyping in Parkinson’s disease and neurologically normal controls: First stage analysis and public release of data. Lancet Neurol. 2006, 5, 911–916. [Google Scholar] [CrossRef]
- Goes, F.S.; McGrath, J.; Avramopoulos, D.; Wolyniec, P.; Pirooznia, M.; Ruczinski, I.; Nestadt, G.; Kenny, E.E.; Vacic, V.; Peters, I.; et al. Genome-wide association study of schizophrenia in Ashkenazi Jews. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2015, 168, 649–659. [Google Scholar] [CrossRef]
- Li, Z.; Chen, J.; Yu, H.; He, L.; Xu, Y.; Zhang, D.; Yi, Q.; Li, C.; Li, X.; Shen, J.; et al. Genome-wide association analysis identifies 30 new susceptibility loci for schizophrenia. Nat. Genet. 2017, 49, 1576–1583. [Google Scholar] [CrossRef]
- Beecham, G.W.; Hamilton, K.; Naj, A.C.; Martin, E.R.; Huentelman, M.; Myers, A.J.; Corneveaux, J.J.; Hardy, J.; Vonsattel, J.-P.; Younkin, S.G.; et al. Genome-wide association meta-analysis of neuropathologic features of Alzheimer’s disease and related dementias. PLoS Genet. 2014, 10, e1004606. [Google Scholar] [CrossRef]
- Wang, K.-S.; Liu, X.-F.; Aragam, N. A genome-wide meta-analysis identifies novel loci associated with schizophrenia and bipolar disorder. Schizophr. Res. 2010, 124, 192–199. [Google Scholar] [CrossRef]
- Styrkarsdottir, U.; Halldorsson, B.V.; Gretarsdottir, S.; Gudbjartsson, D.F.; Walters, G.B.; Ingvarsson, T.; Jonsdottir, T.; Saemundsdottir, J.; Snorradóttir, S.; Center, J.R.; et al. New sequence variants associated with bone mineral density. Nat. Genet. 2009, 41, 15–17. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Zhou, Y.; Liu, S.; Song, X.; Yang, X.-Z.; Fan, Y.; Chen, W.; Akdemir, Z.C.; Yan, Z.; Zuo, Y.; et al. The coexistence of copy number variations (CNVs) and single nucleotide polymorphisms (SNPs) at a locus can result in distorted calculations of the significance in associating SNPs to disease. Hum. Genet. 2018, 137, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.Z.; van Sommeren, S.; Huang, H.; Ng, S.C.; Alberts, R.; Takahashi, A.; Ripke, S.; Lee, J.C.; Jostins, L.; Shah, T.; et al. Association analyses identify 38 susceptibility loci for inflammatory bowel disease and highlight shared genetic risk across populations. Nat. Genet. 2015, 47, 979–986. [Google Scholar] [CrossRef] [PubMed]
- Levine, D.M.; Ek, W.E.; Zhang, R.; Liu, X.; Onstad, L.; Sather, C.; Lao-Sirieix, P.; Gammon, M.D.; Corley, D.A.; Shaheen, N.J.; et al. A genome-wide association study identifies new susceptibility loci for esophageal adenocarcinoma and Barrett’s esophagus. Nat. Genet. 2013, 45, 1487–1493. [Google Scholar] [CrossRef] [PubMed]




{kind=link}
{kind=link}
{kind=link}
| Trait | SNPs | CEN adjacent (2Mb) Regions | Citation |
|---|---|---|---|
| Cancer | rs930395, rs2241024, rs142427110, rs35951924, rs199501877, rs11146838, rs6490525, rs2050203, rs7278690, rs35505947 | 4p12; 5p12; 5q11; 10p11; 13q12; 18p11; 19q11; 20p11; 21q11 | [86,87,88,89] |
| Cardiovascular disease | rs10132760, rs12186641, rs9367716, rs71566846, rs223290, rs144961578, rs3813127, rs1657346, rs1254531, rs10793514 | 5q11.2; 6p11.2; 6q11.1; 10q11.21; 14q11.2; 18q11.2 | [90,91,92] |
| Neurodegenerative diseases | rs11826064, rs13168838, rs62365447, rs140996952, rs1480597, rs10783624, rs7989524, rs6822736, rs13110633, rs2424635 | 4p11; 4q12; 5p12; 5q11.1; 6q11.1; 10q11; 11p11; 12q12; 13q12; 20p11 | [93,94,95,96,97,98,99,100] |
| Scoliosis/Bone Density (Spine) | rs8111296, rs11652527, rs1436931, rs6061081, rs17599071, rs10136383, rs9288898, rs10772040, rs4562194, rs810967, rs6050182, rs6511621, rs11229654, rs6551418, rs1006899 | 3p11.1; 3q11.2; 6q12; 7q11.21; 10q11.21; 11q11; 12p11.21; 14q11.2; 17q11.2; 19p12; 20p11.21; 21q11.2 | [101,102] |
| Digestive system disease | rs4243971, rs2342002, rs4800353, rs6058869, rs6087990 | 6q11.1; 18q11.2; 20q11.21 | [103,104] |
© 2019 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Miga, K.H. Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population. Genes 2019, 10, 352. https://doi.org/10.3390/genes10050352
Miga KH. Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population. Genes. 2019; 10(5):352. https://doi.org/10.3390/genes10050352
Chicago/Turabian StyleMiga, Karen H. 2019. "Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population" Genes 10, no. 5: 352. https://doi.org/10.3390/genes10050352
APA StyleMiga, K. H. (2019). Centromeric Satellite DNAs: Hidden Sequence Variation in the Human Population. Genes, 10(5), 352. https://doi.org/10.3390/genes10050352