On the Population Dynamics of Junk: A Review on the Population Genomics of Transposable Elements



Abstract
1. Introduction
2. Transposable Elements: Classification and Mechanisms of Transposition
3. How Population Dynamics and Intrinsic Properties of Genomes Shape TEs Polymorphisms
3.1. The Role of Purifying Selection and Demography
3.2. Non-Equilibrium between Transposition and Loss
3.3. Transposition and Variable Rates of Recombination
3.4. Coevolutionary Dynamics
4. Transposable Elements as a Source of Adaptation
4.1. Evidence for Positive Selection on TEs and SNPs
4.2. Quantifying Positive Selection on TEs
4.3. Studying Balancing Selection on TEs
4.4. Limitations and Future Improvements
5. The Role of Selfish Elements in Genomic Conflicts: Impact in Natural Populations
6. Future Directions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sotero-Caio, C.G.; Platt, R.N.; Suh, A.; Ray, D.A. Evolution and diversity of transposable elements in vertebrate genomes. Genome Biol. Evol. 2017, 9, 161–177. [Google Scholar] [CrossRef] [PubMed]
- Chuong, E.B.; Elde, N.C.; Feschotte, C. Regulatory activities of transposable elements: From conflicts to benefits. Nat. Rev. Genet. 2017, 18, 71–86. [Google Scholar] [CrossRef] [PubMed]
- Song, M.J.; Schaack, S. Evolutionary Conflict between Mobile DNA and Host Genomes. Am. Nat. 2018, 192, 263–273. [Google Scholar] [CrossRef] [PubMed]
- Charlesworth, B.; Charlesworth, D. The Population Genetics of Transposable Elements. Genet. Res. 1983, 42, 1–27. [Google Scholar] [CrossRef]
- Barron, M.G.; Fiston-Lavier, A.-S.; Petrov, D.A.; Gonzalez, J. Population Genomics of Transposable Elements in Drosophila. Annu. Rev. Genet. 2014, 48, 561–581. [Google Scholar] [CrossRef] [PubMed]
- Bergland, A.O.; Tobler, R.; Gonzalez, J.; Schmidt, P.; Petrov, D. Secondary contact and local adaptation contribute to genome-wide patterns of clinal variation in Drosophila melanogaster. Mol. Ecol. 2016, 25, 1157–1174. [Google Scholar] [CrossRef] [PubMed]
- Lockton, S.; Ross-Ibarra, J.; Gaut, B.S. Demography and weak selection drive patterns of transposable element diversity in natural populations of Arabidopsis lyrata. Proc. Natl. Acad. Sci. USA 2008, 105, 13965–13970. [Google Scholar] [CrossRef] [PubMed]
- Biémont, C. A brief history of the status of transposable elements: From junk DNA to major players in evolution. Genetics 2010, 186, 1085–1093. [Google Scholar] [CrossRef] [PubMed]
- Villanueva-Cañas, J.L.; Rech, G.E.; de Cara, M.A.R.; González, J. Beyond SNPs: how to detect selection on transposable element insertions. Methods Ecol. Evol. 2017, 8, 728–737. [Google Scholar] [CrossRef]
- Hoban, S.; Kelley, J.L.; Lotterhos, K.E.; Antolin, M.F.; Bradburd, G.; Lowry, D.B.; Poss, M.L.; Reed, L.K.; Storfer, A.; Whitlock, M.C. Finding the Genomic Basis of Local Adaptation: Pitfalls, Practical Solutions, and Future Directions. Am. Nat. 2016, 188, 379–397. [Google Scholar] [CrossRef]
- Doolittle, W.F.; Sapienza, C. Selfish genes, the phenotype paradigm and genome evolution. Nature 1980, 284, 601–603. [Google Scholar] [CrossRef] [PubMed]
- Blumenstiel, J.P.; Chen, X.; He, M.; Bergman, C.M. An age-of-allele test of neutrality for transposable element insertions. Genetics 2014, 196, 523–538. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Sutherland, H.G.; Martin, D.I.; Whitelaw, E. Epigenetic inheritance at the agouti locus in the mouse. Nat. Genet. 1999, 23, 314–318. [Google Scholar] [CrossRef] [PubMed]
- Stuart, T.; Eichten, S.R.; Cahn, J.; Karpievitch, Y.V.; Borevitz, J.O.; Lister, R. Population scale mapping of transposable element diversity reveals links to gene regulation and epigenomic variation. Elife 2016, 5, 1–27. [Google Scholar] [CrossRef] [PubMed]
- Tollis, M.; Boissinot, S. The evolutionary dynamics of transposable elements in eukaryote genomes. In Genome Dynamics; MA, G.-R., Ed.; Karger: Basel, Switzerland, 2012; pp. 68–91. ISBN 9783318021509. [Google Scholar]
- Mobile DNA III; Craig, N., Chandler, M., Gellert, M., Lambowitz, A., Rice, P., Sandmeyer, S., Eds.; American Society for Microbiology (ASM): Washington, DC, USA, 2015. [Google Scholar]
- Bourque, G.; Burns, K.H.; Gehring, M.; Gorbunova, V.; Seluanov, A.; Hammell, M.; Imbeault, M.; Izsvák, Z.; Levin, H.L.; Macfarlan, T.S.; et al. Ten things you should know about transposable elements. Genome Biol. 2018, 19, 199. [Google Scholar] [CrossRef]
- Luan, D.D.; Korman, M.H.; Jakubczak, J.L.; Eickbush, T.H. Reverse transcription of R2Bm RNA is primed by a nick at the chromosomal target site: A mechanism for non-LTR retrotransposition. Cell 1993, 72, 595–605. [Google Scholar] [CrossRef]
- Dewannieux, M.; Esnault, C.; Heidmann, T. LINE-mediated retrotransposition of marked Alu sequences. Nat. Genet. 2003, 35, 41–48. [Google Scholar] [CrossRef]
- Malik, H.S.; Burke, W.D.; Eickbush, T.H. The age and evolution of non-LTR retrotransposable elements. Mol. Biol. Evol. 1999, 16, 793–805. [Google Scholar] [CrossRef]
- Kordiš, D.; Lovšin, N.; Gubenšek, F. Phylogenomic analysis of the L1 retrotransposons in Deuterostomia. Syst. Biol. 2006, 55, 886–901. [Google Scholar] [CrossRef]
- Waters, P.D.; Dobigny, G.; Waddell, P.J.; Robinson, T.J. Evolutionary history of LINE-1 in the major clades of placental mammals. PLoS ONE 2007, 2. [Google Scholar] [CrossRef]
- Kordis, D. Unusual horizontal transfer of a long interspersed nuclear element between distant vertebrate classes. Proc. Natl. Acad. Sci. USA 1998, 95, 10704–10709. [Google Scholar] [CrossRef] [PubMed]
- Ivancevic, A.M.; Kortschak, R.D.; Bertozzi, T.; Adelson, D.L. Horizontal transfer of BovB and L1 retrotransposons in eukaryotes. Genome Biol. 2018, 19, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Schaack, S.; Gilbert, C.; Feschotte, C. Promiscuous DNA: Horizontal transfer of transposable elements and why it matters for eukaryotic evolution. Trends Ecol. Evol. 2010, 25, 537–546. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, C.; Bello, X.; Maside, X. Widespread evidence for horizontal transfer of transposable elements across Drosophila genomes. Genome Biol. 2009, 10. [Google Scholar] [CrossRef] [PubMed]
- Reiss, D.; Mialdea, G.; Miele, V.; de Vienne, D.; Peccoud, J.; Gilbert, C.; Duret, L.; Charlat, S. Global survey of mobile DNA horizontal transfer in arthropods reveals Lepidoptera as a prime hotspot. PLoS Genet. 2019, 15, e1007965. [Google Scholar] [CrossRef] [PubMed]
- Pace, J.K., II; Feschotte, C. The evolutionary history of human DNA transposons: Evidence for intense activity in the primate lineage. Genome Res. 2007, 17, 422–432. [Google Scholar] [CrossRef] [PubMed]
- Thomas, J.; Schaack, S.; Pritham, E.J. Pervasive horizontal transfer of rolling-circle transposons among animals. Genome Biol. Evol. 2010, 2, 656–664. [Google Scholar] [CrossRef]
- Gilbert, C.; Hernandez, S.S.; Flores-Benabib, J.; Smith, E.N.; Feschotte, C. Rampant horizontal transfer of SPIN transposons in squamate reptiles. Mol. Biol. Evol. 2012, 29, 503–515. [Google Scholar] [CrossRef][Green Version]
- Novick, P.; Smith, J.; Ray, D.; Boissinot, S. Independent and parallel lateral transfer of DNA transposons in tetrapod genomes. Gene 2010, 449, 85–94. [Google Scholar] [CrossRef]
- Ribet, D.; Harper, F.; Dupressoir, A.; Dewannieux, M.; Pierron, G.; Heidmann, T. An infectious progenitor for the murine IAP retrotransposon: Emergence of an intracellular genetic parasite from an ancient retrovirus. Genome Res. 2008, 18, 597–609. [Google Scholar] [CrossRef]
- Gifford, R.; Tristem, M. The evolution, distribution and diversity of endogenous retroviruses. Virus Genes 2003, 26, 291–316. [Google Scholar] [CrossRef] [PubMed]
- Nelson, K.E.; Peterson, J.; Gardner, M.J.; Mungall, C.; White, O.; Angiuoli, S.; Shallom, S.J.; Selengut, J.; Rutherford, K.; Nene, V.; et al. Genome sequence of the human malaria parasite Plasmodium falciparum. Nature 2002, 419, 498–511. [Google Scholar]
- Carlton, J.M.; Hirt, R.P.; Silva, J.C.; Delcher, A.L.; Schatz, M.; Zhao, Q.; Wortman, J.R.; Bidwell, S.L.; Alsmark, U.C.M.; Besteiro, S.; et al. Draft Genome Sequence of the Sexually Transmitted Pathogen Trichomonas vaginalis. Science 2007, 315, 207–213. [Google Scholar] [CrossRef] [PubMed]
- Schnable, P.S.; Page, S.E.E.L.; Pasternak, S.; Liang, C.; Zhang, J.; Fulton, L.; Graves, T.A.; Minx, P.; Reily, A.D.; Courtney, L.; et al. The B73 Maize Genome: Complexity, Diversity, and Dynamics. Science 2012, 326, 1112–1115. [Google Scholar] [CrossRef] [PubMed]
- The Arabidopsis Genome Initiative. Analysis of the genome sequence of the flowering plant Arabidopsis thaliana. Nature 2000, 408, 796–815. [Google Scholar] [CrossRef] [PubMed]
- Chalopin, D.; Naville, M.; Plard, F.; Galiana, D.; Volff, J.N. Comparative analysis of transposable elements highlights mobilome diversity and evolution in vertebrates. Genome Biol. Evol. 2015, 7, 567–580. [Google Scholar] [CrossRef]
- Furano, A.V.; Duvernell, D.D.; Boissinot, S. L1 (LINE-1) retrotransposon diversity differs dramatically between mammals and fish. Trends Genet. 2004, 20, 9–14. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, S.; Sookdeo, A. The Evolution of Line-1 in Vertebrates. Genome Biol. Evol. 2016, 8, 3485–3507. [Google Scholar] [CrossRef]
- Qian, Y.; Mancini-DiNardo, D.; Judkins, T.; Cox, H.C.; Brown, K.; Elias, M.; Singh, N.; Daniels, C.; Holladay, J.; Coffee, B.; et al. Identification of pathogenic retrotransposon insertions in cancer predisposition genes. Cancer Genet. 2017, 216–217, 159–169. [Google Scholar] [CrossRef]
- Green, P.M.; Bagnall, R.D.; Waseem, N.H.; Giannelli, F. Haemophilia A mutations in the UK: Results of screening one-third of the population. Br. J. Haematol. 2008, 143, 115–128. [Google Scholar] [CrossRef]
- Hancks, D.C.; Kazazian, H.H. Roles for retrotransposon insertions in human disease. Mob. DNA 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R.; Betancourt, A.J.; Schlötterer, C. Sequencing of pooled DNA samples (Pool-Seq) uncovers complex dynamics of transposable element insertions in Drosophila melanogaster. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.A.; Fiston-Lavier, A.-S.; Lipatov, M.; Lenkov, K.; Gonzalez, J. Population Genomics of Transposable Elements in Drosophila melanogaster. Mol. Biol. Evol. 2011, 28, 1633–1644. [Google Scholar] [CrossRef] [PubMed]
- Stritt, C.; Gordon, S.P.; Wicker, T.; Vogel, J.P.; Roulin, A.C. Recent activity in expanding populations and purifying selection have shaped transposable element landscapes across natural accessions of the mediterranean grass Brachypodium distachyon. Genome Biol. Evol. 2018, 10, 304–318. [Google Scholar] [CrossRef] [PubMed]
- Hazzouri, K.M.; Mohajer, A.; Dejak, S.I.; Otto, S.P.; Wright, S.I. Contrasting patterns of transposable-element insertion polymorphism and nucleotide diversity in autotetraploid and allotetraploid Arabidopsis species. Genetics 2008, 179, 581–592. [Google Scholar] [CrossRef]
- González, J.; Macpherson, J.M.; Messer, P.W.; Petrov, D.A. Inferring the strength of selection in Drosophila under complex demographic models. Mol. Biol. Evol. 2009, 26, 513–526. [Google Scholar] [CrossRef]
- Boissinot, S.; Davis, J.; Entezam, A.; Petrov, D.; Furano, A.V. Fitness cost of LINE-1 (L1) activity in humans. Proc. Natl. Acad. Sci. USA 2006, 103, 9590–9594. [Google Scholar] [CrossRef]
- Xue, A.T.; Ruggiero, R.P.; Hickerson, M.J.; Boissinot, S. Differential effect of selection against LINE retrotransposons among vertebrates inferred from whole-genome data and demographic modeling. Genome Biol. Evol. 2018, 10, 1265–1281. [Google Scholar] [CrossRef]
- Ruggiero, R.P.; Bourgeois, Y.; Boissinot, S. LINE Insertion Polymorphisms Are Abundant but at Low Frequencies across Populations of Anolis carolinensis. Front. Genet. 2017, 8, 1–14. [Google Scholar] [CrossRef]
- Quadrana, L.; Silveira, A.B.; Mayhew, G.F.; LeBlanc, C.; Martienssen, R.A.; Jeddeloh, J.A.; Colot, V. The Arabidopsis thaliana mobilome and its impact at the species level. Elife 2016, 5, 1–25. [Google Scholar] [CrossRef]
- Olivares, M.; Alonso, C.; López, M.C. The open reading frame 1 of the L1Tc retrotransposon of Trypanosoma cruzi codes for a protein with apurinic-apyrimidinic nuclease activity. J. Biol. Chem. 1997, 272, 25224–25228. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Conte, C.; Dastugue, B.; Vaury, C. Promoter competition as a mechanism of transcriptional interference mediated by retrotransposons. EMBO J. 2002, 21, 3908–3916. [Google Scholar] [CrossRef] [PubMed]
- Langley, C.H.; Montgomery, E.A.; Hudson, R.; Kaplan, N.; Charlesworth, B. On the role of unequal exchange in the containment of transposable element copy number. Genet. Res. 1988, 52, 223–235. [Google Scholar] [CrossRef] [PubMed]
- Dolgin, E.S.; Charlesworth, B. The effects of recombination rate on the distribution and abundance of transposable elements. Genetics 2008, 178, 2169–2177. [Google Scholar] [CrossRef] [PubMed]
- Petrov, D.A.; Aminetzach, Y.T.; Davis, J.C.; Bensasson, D.; Hirsh, A.E. Size matters: Non-LTR retrotransposable elements and ectopic recombination in Drosophila. Mol. Biol. Evol. 2003, 20, 880–892. [Google Scholar] [CrossRef] [PubMed]
- Cordaux, R.; Lee, J.; Dinoso, L.; Batzer, M.A. Recently integrated Alu retrotransposons are essentially neutral residents of the human genome. Gene 2006, 373, 138–144. [Google Scholar] [CrossRef] [PubMed]
- Song, M.; Boissinot, S. Selection against LINE-1 retrotransposons results principally from their ability to mediate ectopic recombination. Gene 2007, 390, 206–213. [Google Scholar] [CrossRef] [PubMed]
- Nam, K.; Ellegren, H. Recombination drives vertebrate genome contraction. PLoS Genet. 2012, 8. [Google Scholar] [CrossRef]
- Charlesworth, B. The organization and evolution of the human Y chromosome. Genome Biol. 2003, 4. [Google Scholar] [CrossRef] [PubMed]
- Boissinot, S.; Entezam, A.; Furano, A. V Selection Against Deleterious LINE-1-Containing Loci in the Human Lineage. Mol. Biol. 2001, 18, 926–935. [Google Scholar] [CrossRef]
- Montgomery, E.; Charlesworth, B.; Langley, C. A test for the role of natural selection in the stabilization of transposable element copy number in a population of Drosophila melanogaster. Genet Res. 1987, 49, 31–41. [Google Scholar] [CrossRef]
- Montgomery, E.A.; Huang, S.M.; Langley, C.H.; Judd, B.H. Chromosome rearrangement by ectopic recombination in Drosophila melanogaster: Genome structure and evolution. Genetics 1991, 129, 1085–1098. [Google Scholar]
- Le Rouzic, A.; Boutin, T.S.; Capy, P. Long-term evolution of transposable elements. Proc. Natl. Acad. Sci. USA 2007, 104, 19375–19380. [Google Scholar] [CrossRef]
- Charlesworth, B.; Sniegowski, P.; Stephan, W. The evolutionary dynamics of repetitive DNA in eukaryotes. Nature 1994, 371, 215–220. [Google Scholar] [CrossRef] [PubMed]
- Ross-Ibarra, J.; Wright, S.I.; Foxe, J.P.; Kawabe, A.; DeRose-Wilson, L.; Gos, G.; Charlesworth, D.; Gaut, B.S. Patterns of polymorphism and demographic history in natural populations of Arabidopsis lyrata. PLoS ONE 2008, 3. [Google Scholar] [CrossRef]
- García Guerreiro, M.P.; Chávez-Sandoval, B.E.; Balanyà, J.; Serra, L.; Fontdevila, A. Distribution of the transposable elements bilbo and gypsy in original and colonizing populations of Drosophila subobscura. BMC Evol. Biol. 2008, 8. [Google Scholar] [CrossRef] [PubMed]
- Blass, E.; Bell, M.; Boissinot, S. Accumulation and rapid decay of non-LTR retrotransposons in the genome of the three-spine stickleback. Genome Biol. Evol. 2012, 4, 687–702. [Google Scholar] [CrossRef] [PubMed]
- Tollis, M.; Boissinot, S. Lizards and LINEs: Selection and demography affect the fate of L1 retrotransposons in the genome of the green anole (Anolis carolinensis). Genome Biol. Evol. 2013, 5, 1754–1768. [Google Scholar] [CrossRef]
- Lynch, M.; Conery, J.S. The Origins of Genome Complexity. Science 2003, 302, 1401–1404. [Google Scholar] [CrossRef]
- Vieira, C.; Lepetit, D.; Dumont, S.; Biémont, C. Wake up of transposable elements following Drosophila simulans worldwide colonization. Mol. Biol. Evol. 1999, 16, 1251–1255. [Google Scholar] [CrossRef]
- Piegu, B.; Guyot, R.; Picault, N.; Roulin, A.; Saniyal, A.; Kim, H.; Collura, K.; Brar, D.S.; Jackson, S.; Wing, R.A.; et al. Doubling genome size without polyploidization: Dynamics of retrotransposition-driven genomic expansions in Oryza australiensis, a wild relative of rice. Genome Res. 2006, 16, 1262–1269. [Google Scholar] [CrossRef] [PubMed]
- Manthey, J.D.; Moyle, R.G.; Boissinot, S. Multiple and independent phases of transposable element amplification in the genomes of piciformes (woodpeckers and allies). Genome Biol. Evol. 2018, 10, 1445–1456. [Google Scholar] [CrossRef] [PubMed]
- De Boer, J.G.; Yazawa, R.; Davidson, W.S.; Koop, B.F. Bursts and horizontal evolution of DNA transposons in the speciation of pseudotetraploid salmonids. BMC Genomics 2007, 8, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hellen, E.H.B.; Brookfield, J.F.Y. The diversity of class II transposable elements in mammalian genomes has arisen from ancestral phylogenetic splits during ancient waves of proliferation through the genome. Mol. Biol. Evol. 2013, 30, 100–108. [Google Scholar] [CrossRef] [PubMed]
- Hellen, E.H.B.; Brookfield, J.F.Y. Transposable element invasions. Mob. Genet. Elements 2013, 3, e23920. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Bergman, C.M.; Bensasson, D. Recent LTR retrotransposon insertion contrasts with waves of non-LTR insertion since speciation in Drosophila melanogaster. Proc. Natl. Acad. Sci. USA 2007, 104, 11340–11345. [Google Scholar] [CrossRef]
- Haller, B.C.; Messer, P.W. SLiM 3: Forward Genetic Simulations Beyond the Wright-Fisher Model. Mol. Biol. Evol. 2019, 36, 632–637. [Google Scholar] [CrossRef] [PubMed]
- Kent, T.V.; Uzunović, J.; Wright, S.I. Coevolution between transposable elements and recombination. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372. [Google Scholar] [CrossRef]
- Choi, K.; Zhao, X.; Kelly, K.A.; Venn, O.; Higgins, J.D.; Yelina, N.E.; Hardcastle, T.J.; Ziolkowski, P.A.; Copenhaver, G.P.; Franklin, F.C.H.; et al. Arabidopsis meiotic crossover hot spots overlap with H2A.Z nucleosomes at gene promoters. Nat. Genet. 2013, 45, 1327–1336. [Google Scholar] [CrossRef]
- Myers, S.; Bottolo, L.; Freeman, C.; McVean, G.; Donnelly, P. A Fine-Scale Map of Recombination Rates and Hotspots Across the Human Genome. Science 2005, 310, 321–324. [Google Scholar] [CrossRef]
- Hill, W.G.; Robertson, A. Local effects of limited recombination. Genet. Res. 1966, 8, 269–294. [Google Scholar] [CrossRef] [PubMed]
- Felsenstein, J. The evolution advantage of recombination. Genetics 1974, 78, 737–756. [Google Scholar] [PubMed]
- Kawakami, T.; Mugal, C.F.; Suh, A.; Nater, A.; Burri, R.; Smeds, L.; Ellegren, H. Whole-genome patterns of linkage disequilibrium across flycatcher populations clarify the causes and consequences of fine-scale recombination rate variation in birds. Mol. Ecol. 2017, 26, 4158–4172. [Google Scholar] [CrossRef] [PubMed]
- Jensen-Seaman, M.I.; Furey, T.S.; Payseur, B.A.; Lu, Y.; Roskin, K.M.; Chen, C.F.; Thomas, M.A.; Haussler, D.; Jacob, H.J. Comparative recombination rates in the rat, mouse, and human genomes. Genome Res. 2004, 14, 528–538. [Google Scholar] [CrossRef] [PubMed]
- Bartolomé, C.; Maside, X.; Charlesworth, B. On the abundance and distribution of transposable elements in the genome of Drosophila melanogaster. Mol. Biol. Evol. 2002, 19, 926–937. [Google Scholar] [CrossRef] [PubMed]
- Rizzon, C.; Marais, G.; Gouy, M.; Biémont, C. Recombination rate and the distribution of transposable elements in the Drosophila melanogaster genome. Genome Res. 2002, 12, 400–407. [Google Scholar] [CrossRef] [PubMed]
- Myers, S.; Freeman, C.; Auton, A.; Donnelly, P.; McVean, G. A common sequence motif associated with recombination hot spots and genome instability in humans. Nat. Genet. 2008, 40, 1124–1129. [Google Scholar] [CrossRef]
- Campos-Sánchez, R.; Cremona, M.A.; Pini, A.; Chiaromonte, F.; Makova, K.D. Integration and Fixation Preferences of Human and Mouse Endogenous Retroviruses Uncovered with Functional Data Analysis. PLoS Comput. Biol. 2016, 12, 1–41. [Google Scholar] [CrossRef]
- Duret, L.; Marais, G.; Biemont, C. Transposons but not retrotransposons are located preferentially in regions of high recombination rate in Caenorhabditis elegans. Genetics 2000, 156, 1661–1669. [Google Scholar]
- Csilléry, K.; Blum, M.G.B.; Gaggiotti, O.E.; François, O. Approximate Bayesian Computation (ABC) in practice. Trends Ecol. Evol. 2010, 25, 410–418. [Google Scholar] [CrossRef]
- Ågren, J.A.; Wright, S.I. Co-evolution between transposable elements and their hosts: A major factor in genome size evolution? Chromosom. Res. 2011, 19, 777–786. [Google Scholar] [CrossRef] [PubMed]
- Goodier, J.L. Restricting retrotransposons: A review. Mob. DNA 2016, 7. [Google Scholar] [CrossRef] [PubMed]
- Arias, J.F.; Koyama, T.; Kinomoto, M.; Tokunaga, K. Retroelements versus APOBEC3 family members: No great escape from the magnificent seven. Front. Microbiol. 2012, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Koito, A.; Ikeda, T. Intrinsic immunity against retrotransposons by APOBEC cytidine deaminases. Front. Microbiol. 2013, 4, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Lindič, N.; Budič, M.; Petan, T.; Knisbacher, B.A.; Levanon, E.Y.; Lovšin, N. Differential inhibition of LINE1 and LINE2 retrotransposition by vertebrate AID/APOBEC proteins. Retrovirology 2013, 10, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Yoder, J.A.; Walsh, C.P.; Bestor, T.H. Cytosine methylation and the ecology of intragenomic parasites. Trends Genet. 1997, 13, 335–340. [Google Scholar] [CrossRef]
- Huda, A.; Mariño-Ramírez, L.; Jordan, I.K. Epigenetic histone modifications of human transposable elements: Genome defense versus exaptation. Mob. DNA 2010, 1, 1–12. [Google Scholar] [CrossRef]
- Cheng, C.; Tarutani, Y.; Miyao, A.; Ito, T.; Yamazaki, M.; Sakai, H.; Fukai, E.; Hirochika, H. Loss of function mutations in the rice chromomethylase OsCMT3a cause a burst of transposition. Plant J. 2015, 83, 1069–1081. [Google Scholar] [CrossRef]
- Van Rij, R.P.; Berezikov, E. Small RNAs and the control of transposons and viruses in Drosophila. Trends Microbiol. 2009, 17, 163–171. [Google Scholar] [CrossRef]
- Prud’homme, N.; Gans, M.; Masson, M.; Terzian, C.; Bucheton, A. Flamenco, a gene controlling the gypsy retrovirus of Drosophila melanogaster. Genetics 1995, 139, 697–711. [Google Scholar]
- Goriaux, C.; Desset, S.; Renaud, Y.; Vaury, C.; Brasset, E. Transcriptional properties and splicing of the flamenco piRNA cluster. EMBO Rep. 2014, 15, 411–418. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R. Dynamics of transposable element invasions with piRNA clusters. Mol. Biol. Evol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Roessler, K.; Bousios, A.; Meca, E.; Gaut, B.S. Modeling Interactions between Transposable Elements and the Plant Epigenetic Response: A Surprising Reliance on Element Retention. Genome Biol. Evol. 2018, 10, 803–815. [Google Scholar] [CrossRef] [PubMed]
- Palmer, W.H.; Hadfield, J.D.; Obbard, D.J. RNA-Interference Pathways Display High Rates of Adaptive Protein Evolution in Multiple Invertebrates. Genetics 2018, 208, 1585–1599. [Google Scholar] [CrossRef] [PubMed]
- Simkin, A.; Wong, A.; Poh, Y.P.; Theurkauf, W.E.; Jensen, J.D. Recurrent and recent selective sweeps in the piRNA pathway. Evolution 2013, 67, 1081–1090. [Google Scholar] [CrossRef] [PubMed]
- Jacobs, F.M.J.; Greenberg, D.; Nguyen, N.; Haeussler, M.; Ewing, A.D.; Katzman, S.; Paten, B.; Salama, S.R.; Haussler, D. An evolutionary arms race between KRAB zinc-finger genes ZNF91/93 and SVA/L1 retrotransposons. Nature 2014, 516, 242–245. [Google Scholar] [CrossRef] [PubMed]
- Haasl, R.J.; Payseur, B.A. Fifteen years of genomewide scans for selection: Trends, lessons and unaddressed genetic sources of complication. Mol. Ecol. 2016, 25, 5–23. [Google Scholar] [CrossRef]
- Miller, W.J.; McDonald, J.F.; Nouaud, D.; Anxolabehere, D. Molecular domestication—More than a sporadic episode in evolution. Genetica 1999, 107, 197–207. [Google Scholar] [CrossRef]
- Jung, D.; Alt, F.W. Unraveling V(D)J Recombination: Insights into Gene Regulation. Cell 2004, 116, 299–311. [Google Scholar] [CrossRef]
- Oettinger, M.A.; Schatz, D.G.; Gorka, C.; Baltimore, D.; Oetringer, M.A. RAG-1 and RAG-2, Adjacent Genes That Synergistically Activate V(D)J Recombination. Science 1990, 248, 1517–1523. [Google Scholar] [CrossRef]
- Kapitonov, V.V.; Koonin, E.V. Evolution of the RAG1-RAG2 locus: Both proteins came from the same transposon. Biol. Direct 2015, 10, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Pardue, M.-L.; DeBaryshe, P.G. Retrotransposons that maintain chromosome ends. Proc. Natl. Acad. Sci. USA 2011, 108, 20317–20324. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.G.; Leek, C.; Levine, M.T. Recurrent Innovation at Genes Required for Telomere Integrity in Drosophila. Mol. Biol. Evol. 2017, 34, 467–482. [Google Scholar] [PubMed]
- Zaratiegui, M.; Vaughn, M.W.; Irvine, D.V.; Goto, D.; Watt, S.; Bähler, J.; Arcangioli, B.; Martienssen, R.A. CENP-B preserves genome integrity at replication forks paused by retrotransposon LTR. Nature 2011, 469, 112–115. [Google Scholar] [CrossRef] [PubMed]
- Gao, D.; Jiang, N.; Wing, R.A.; Jiang, J.; Jackson, S.A. Transposons play an important role in the evolution and diversification of centromeres among closely related species. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef] [PubMed]
- Capy, P.; Gasperi, G.; Biémont, C.; Bazin, C. Stress and transposable elements: Co-evolution or useful parasites? Heredity 2000, 85, 101–106. [Google Scholar] [CrossRef] [PubMed]
- Rey, O.; Danchin, E.; Mirouze, M.; Loot, C.; Blanchet, S. Adaptation to Global Change: A Transposable Element-Epigenetics Perspective. Trends Ecol. Evol. 2016, 31, 514–526. [Google Scholar] [CrossRef] [PubMed]
- Kalendar, R.; Tanskanen, J.; Immonen, S.; Nevo, E.; Schulman, A.H. Genome evolution of wild barley (Hordeum spontaneum) by BARE-1 retrotransposon dynamics in response to sharp microclimatic divergence. Proc. Natl. Acad. Sci. USA 2000, 97, 6603–6607. [Google Scholar] [CrossRef]
- Feiner, N. Accumulation of transposable elements in HOX gene clusters during adaptive radiation of Anolis lizards. Proc. Biol. Sci. 2016, 283. [Google Scholar] [CrossRef]
- Yang, L.; Bennetzen, J.L. Distribution, diversity, evolution, and survival of Helitrons in the maize genome. Proc. Natl. Acad. Sci. USA 2009, 106, 19922–19927. [Google Scholar] [CrossRef]
- Schrader, L.; Kim, J.W.; Ence, D.; Zimin, A.; Klein, A.; Wyschetzki, K.; Weichselgartner, T.; Kemena, C.; Stökl, J.; Schultner, E.; et al. Transposable element islands facilitate adaptation to novel environments in an invasive species. Nat. Commun. 2014, 5, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Hof, A.E.V.; Campagne, P.; Rigden, D.J.; Yung, C.J.; Lingley, J.; Quail, M.A.; Hall, N.; Darby, A.C.; Saccheri, I.J. The industrial melanism mutation in British peppered moths is a transposable element. Nature 2016, 534, 102–105. [Google Scholar] [CrossRef] [PubMed]
- González, J.; Petrov, D.A. The adaptive role of transposable elements in the Drosophila genome. Gene 2009, 448, 124–133. [Google Scholar] [CrossRef] [PubMed]
- González, J.; Karasov, T.L.; Messer, P.W.; Petrov, D.A. Genome-wide patterns of adaptation to temperate environments associated with transposable elements in Drosophila. PLoS Genet. 2010, 6, 33–35. [Google Scholar] [CrossRef] [PubMed]
- Ullastres, A.; Petit, N.; González, J. Exploring the phenotypic space and the evolutionary history of a natural mutation in drosophila melanogaster. Mol. Biol. Evol. 2015, 32, 1800–1814. [Google Scholar] [CrossRef] [PubMed]
- Guio, L.; Barrõn, M.G.; González, J. The transposable element Bari-Jheh mediates oxidative stress response in Drosophila. Mol. Ecol. 2014, 23, 2020–2030. [Google Scholar] [CrossRef] [PubMed]
- Rech, G.E.; Bogaerts-Marquez, M.; Barron, M.G.; Merenciano, M.; Villanueva-Canas, J.L.; Horvath, V.; Fiston-Lavier, A.-S.; Luyten, I.; Venkataram, S.; Quesneville, H.; et al. Stress response, behavior, and development are shaped by transposable element-induced mutations in Drosophila. PLoS Genet. 2018, 15, e1007900. [Google Scholar] [CrossRef]
- González, J.; Macpherson, J.M.; Petrov, D.A. A recent adaptive transposable element insertion near highly conserved developmental loci in Drosophila melanogaster. Mol. Biol. Evol. 2009, 26, 1949–1961. [Google Scholar] [CrossRef]
- Rishishwar, L.; Wang, L.; Wang, J.; Yi, S.V.; Lachance, J.; Jordan, I.K. Evidence for positive selection on recent human transposable element insertions. Gene 2018, 675, 69–79. [Google Scholar] [CrossRef]
- Feschotte, C. Transposable elements and the evolution of regulatory networks. Nat. Rev. Genet. 2008, 9, 397–405. [Google Scholar] [CrossRef]
- Lotterhos, K.E.; Whitlock, M.C. Evaluation of demographic history and neutral parameterization on the performance of FST outlier tests. Mol. Ecol. 2014, 23, 2178–2192. [Google Scholar] [CrossRef] [PubMed]
- Sabeti, P.C.; Schaffner, S.F.; Fry, B.; Lohmueller, J.; Varilly, P.; Shamovsky, O.; Palma, A.; Mikkelsen, T.S.; Altshuler, D.; Lander, E.S. Positive natural selection in the human lineage. Science 2006, 312, 1614–1620. [Google Scholar] [CrossRef] [PubMed]
- Garud, N.R.; Messer, P.W.; Buzbas, E.O.; Petrov, D.A. Recent Selective Sweeps in North American Drosophila melanogaster Show Signatures of Soft Sweeps. PLoS Genet. 2015, 11, 1–32. [Google Scholar] [CrossRef] [PubMed]
- Ferrer-Admetlla, A.; Liang, M.; Korneliussen, T.; Nielsen, R. On detecting incomplete soft or hard selective sweeps using haplotype structure. Mol. Biol. Evol. 2014, 31, 1275–1291. [Google Scholar] [CrossRef] [PubMed]
- McCarroll, S.A.; Sabeti, P.C.; Frazer, K.A.; Varilly, P.; Fry, B.; Ballinger, D.G.; Lohmueller, J.; Cox, D.R.; Hostetter, E.; Hinds, D.A.; et al. Genome-wide detection and characterization of positive selection in human populations. Nature 2007, 449, 913–918. [Google Scholar]
- Gautier, M. Genome-Wide Scan for Adaptive Divergence and Association with Population-Specific Covariates. Genetics 2015, 201, 1555–1579. [Google Scholar] [CrossRef] [PubMed]
- Rasmussen, M.D.; Hubisz, M.J.; Gronau, I.; Siepel, A. Genome-Wide Inference of Ancestral Recombination Graphs. PLoS Genet. 2014, 10. [Google Scholar] [CrossRef]
- Schrider, D.R.; Kern, A.D. Supervised Machine Learning for Population Genetics: A New Paradigm. Trends Genet. 2018, 34, 301–312. [Google Scholar] [CrossRef]
- Schrider, D.R.; Kern, A.D. Machine Learning for Population Genetics: A New Paradigm. bioRxiv 2017, 206482. [Google Scholar] [CrossRef]
- Schrider, D.R.; Mendes, F.K.; Hahn, M.W.; Kern, A.D. Soft shoulders ahead: Spurious signatures of soft and partial selective sweeps result from linked hard sweeps. Genetics 2015, 200, 267–284. [Google Scholar] [CrossRef]
- Messer, P.W.; Petrov, D.A. Population genomics of rapid adaptation by soft selective sweeps. Trends Ecol. Evol. 2013, 28, 659–669. [Google Scholar] [CrossRef] [PubMed]
- Kern, A.D.; Schrider, D.R. diploS/HIC: An Updated Approach to Classifying Selective Sweeps. G3 2018, 8, 1959–1970. [Google Scholar] [CrossRef] [PubMed]
- Lee, K.M.; Coop, G. Distinguishing Among Modes of Convergent Adaptation Using Population Genomic Data. Genetics 2018, 207, 1591–1619. [Google Scholar] [CrossRef] [PubMed]
- Sellis, D.; Callahan, B.J.; Petrov, D.A.; Messer, P.W. Heterozygote advantage as a natural consequence of adaptation in diploids. Proc. Natl. Acad. Sci. USA 2011, 108, 20666–20671. [Google Scholar] [CrossRef] [PubMed]
- Siewert, K.M.; Voight, B.F. Detecting Long-Term Balancing Selection Using Allele Frequency Correlation. Mol. Biol. Evol. 2017, 34, 2996–3005. [Google Scholar] [CrossRef] [PubMed]
- DeGiorgio, M.; Lohmueller, K.E.; Nielsen, R. A model-based approach for identifying signatures of ancient balancing selection in genetic data. PLoS Genet. 2014, 10, e1004561. [Google Scholar] [CrossRef] [PubMed]
- Van Oosterhout, C. Transposons in the MHC: The Yin and Yang of the vertebrate immune system. Heredity 2009, 103, 190–191. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.; Zhang, B.; Xu, L.; Li, Q.; Jiang, F.; Yang, P.; Xu, Y.; Kang, L. Transposable Element-Mediated Balancing Selection at Hsp90 Underlies Embryo Developmental Variation. Mol. Biol. Evol. 2017, 34, 1127–1139. [Google Scholar] [CrossRef]
- van Oosterhout, C. A new theory of MHC evolution: Beyond selection on the immune genes. Proc. Biol. Sci. 2009, 276, 657–665. [Google Scholar] [CrossRef]
- Nicod, J.; Davies, R.W.; Cai, N.; Hassett, C.; Goodstadt, L.; Cosgrove, C.; Yee, B.K.; Lionikaite, V.; McIntyre, R.E.; Remme, C.A.; et al. Genome-wide association of multiple complex traits in outbred mice by ultra-low-coverage sequencing. Nat. Genet. 2016, 48, 912–918. [Google Scholar] [CrossRef]
- Gardner, E.J.; Lam, V.K.; Harris, D.N.; Chuang, N.T.; Scott, E.C.; Pittard, W.S.; Mills, R.E.; 1000 Genomes Project Consortium; Devine, S.E. The Mobile Element Locator Tool (MELT): Population-scale mobile element discovery and biology. Genome Res. 2017, 27, 1916–1929. [Google Scholar] [CrossRef] [PubMed]
- Wen, Y.J.; Zhang, H.; Ni, Y.L.; Huang, B.; Zhang, J.; Feng, J.Y.; Wang, S.B.; Dunwell, J.M.; Zhang, Y.M.; Wu, R. Methodological implementation of mixed linear models in multi-locus genome-wide association studies. Brief. Bioinform. 2018, 19, 700–712. [Google Scholar] [CrossRef]
- Rishishwar, L.; Mariño-Ramírez, L.; Jordan, I.K. Benchmarking computational tools for polymorphic transposable element detection. Brief. Bioinform. 2017, 18, 908–918. [Google Scholar] [CrossRef] [PubMed]
- Kofler, R.; Gómez-Sánchez, D.; Schlötterer, C. PoPoolationTE2: Comparative Population Genomics of Transposable Elements Using Pool-Seq. Mol. Biol. Evol. 2016, 33, 2759–2764. [Google Scholar] [CrossRef] [PubMed]
- Fiston-Lavier, A.S.; Barrón, M.G.; Petrov, D.A.; González, J. T-lex2: Genotyping, frequency estimation and re-annotation of transposable elements using single or pooled next-generation sequencing data. Nucleic Acids Res. 2015, 43. [Google Scholar] [CrossRef] [PubMed]
- Santander, C.G.; Gambron, P.; Marchi, E.; Karamitros, T.; Katzourakis, A.; Magiorkinis, G. STEAK: A specific tool for transposable elements and retrovirus detection in high-throughput sequencing data. Virus Evol. 2017, 3, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Rahman, R.; Chirn, G.W.; Kanodia, A.; Sytnikova, Y.A.; Brembs, B.; Bergman, C.M.; Lau, N.C. Unique transposon landscapes are pervasive across Drosophila melanogaster genomes. Nucleic Acids Res. 2015, 43, 10655–10672. [Google Scholar] [CrossRef] [PubMed]
- Disdero, E.; Filée, J. LoRTE: Detecting transposon-induced genomic variants using low coverage PacBio long read sequences. Mob. DNA 2017, 8, 4–9. [Google Scholar] [CrossRef]
- Jiang, C.; Chen, C.; Huang, Z.; Liu, R.; Verdier, J. ITIS, a bioinformatics tool for accurate identification of transposon insertion sites using next-generation sequencing data. BMC Bioinformatics 2015, 16, 1–8. [Google Scholar] [CrossRef][Green Version]
- Zhuang, J.; Wang, J.; Theurkauf, W.; Weng, Z. TEMP: A computational method for analyzing transposable element polymorphism in populations. Nucleic Acids Res. 2014, 42, 6826–6838. [Google Scholar] [CrossRef]
- Thung, D.T.; de Ligt, J.; Vissers, L.E.M.; Steehouwer, M.; Kroon, M.; de Vries, P.; Slagboom, E.P.; Ye, K.; Veltman, J.A.; Hehir-Kwa, J.Y. Mobster: Accurate detection of mobile element insertions in next generation sequencing data. Genome Biol. 2014, 15, 488. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Lee, W.P.; Ward, A.; Walker, J.A.; Konkel, M.K.; Batzer, M.A.; Marth, G.T. Tangram: A comprehensive toolbox for mobile element insertion detection. BMC Genomics 2014, 15, 1–15. [Google Scholar] [CrossRef] [PubMed]
- Keane, T.M.; Wong, K.; Adams, D.J. RetroSeq: Transposable element discovery from next-generation sequencing data. Bioinformatics 2013, 29, 389–390. [Google Scholar] [CrossRef] [PubMed]
- Chen, J.; Wrightsman, T.R.; Wessler, S.R.; Stajich, J.E. RelocaTE2: A high resolution transposable element insertion site mapping tool for population resequencing. PeerJ 2017, 5, e2942. [Google Scholar] [CrossRef] [PubMed]
- Nelson, M.G.; Linheiro, R.S.; Bergman, C.M. McClintock: An Integrated Pipeline for Detecting Transposable Element Insertions in Whole-Genome Shotgun Sequencing Data. G3 2017, 7, 2763–2778. [Google Scholar] [CrossRef] [PubMed]
- Seehausen, O.; Butlin, R.K.; Keller, I.; Wagner, C.E.; Boughman, J.W.; Hohenlohe, P.A.; Peichel, C.L.; Saetre, G.-P.; Bank, C.; Brannstrom, A.; et al. Genomics and the origin of species. Nat. Rev. Genet. 2014, 15, 176–192. [Google Scholar] [CrossRef] [PubMed]
- Butlin, R.K.; Smadja, C.M. Coupling, Reinforcement, and Speciation. Am. Nat. 2017, 191, 155–172. [Google Scholar] [CrossRef]
- Jangam, D.; Feschotte, C.; Betrán, E. Transposable Element Domestication As an Adaptation to Evolutionary Conflicts. Trends Genet. 2017, 33, 817–831. [Google Scholar] [CrossRef]
- Lindholm, A.K.; Dyer, K.A.; Firman, R.C.; Fishman, L.; Forstmeier, W.; Holman, L.; Johannesson, H.; Knief, U.; Kokko, H.; Larracuente, A.M.; et al. The Ecology and Evolutionary Dynamics of Meiotic Drive. Trends Ecol. Evol. 2016, 31, 315–326. [Google Scholar] [CrossRef]
- Gardner, A.; Úbeda, F. The meaning of intragenomic conflict. Nat. Ecol. Evol. 2017, 1, 1807–1815. [Google Scholar] [CrossRef]
- Crespi, B.; Nosil, P. Conflictual speciation: Species formation via genomic conflict. Trends Ecol. Evol. 2013, 28, 48–57. [Google Scholar] [CrossRef] [PubMed]
- Serrato-Capuchina, A.; Matute, D.R. The role of transposable elements in speciation. Genes 2018, 9, 254. [Google Scholar] [CrossRef] [PubMed]
- Daniels, S.B.; Peterson, K.R.; Strausbaugh, L.D.; Kidwell, M.G.; Chovnik, A. Evidence for horizontal transmission of the P transposable element between Drosophila species. Genetics 1990, 124, 339–355. [Google Scholar]
- Kidwell, M.G. Hybrid dysgenesis in Drosophila melanogaster: The relationship between the P–M and I–R interaction systems. Genet. Res. 1979, 33, 205–217. [Google Scholar] [CrossRef]
- Kimura, K.; Kidwell, M.G. Differences in P element population dynamics between the sibling species Drosophila melanogaster and Drosophila simulans. Genet. Res. 1994, 63, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Yoshitake, Y.; Inomata, N.; Sano, M.; Kato, Y.; Itoh, M. The P element invaded rapidly and caused hybrid dysgenesis in natural populations of Drosophila simulans in Japan. Ecol. Evol. 2018, 8, 9590–9599. [Google Scholar] [CrossRef] [PubMed]
- Hill, T.; Schlötterer, C.; Betancourt, A.J. Hybrid Dysgenesis in Drosophila simulans Associated with a Rapid Invasion of the P-Element. PLoS Genet. 2016, 12, 1–17. [Google Scholar]
- Kofler, R.; Hill, T.; Nolte, V.; Betancourt, A.J.; Schlötterer, C. The recent invasion of natural Drosophila simulans populations by the P-element. Proc. Natl. Acad. Sci. USA 2015, 112, 6659–6663. [Google Scholar] [CrossRef]
- O’Neill, M.J.; O’Neill, R.J. Sex chromosome repeats tip the balance towards speciation. Mol. Ecol. 2018. [Google Scholar] [CrossRef]
- Brown, J.D.; O’Neill, R.J. Chromosomes, Conflict, and Epigenetics: Chromosomal Speciation Revisited. Annu. Rev. Genom. Hum. Genet. 2010, 11, 291–316. [Google Scholar] [CrossRef]
- Ellison, C.; Bachtrog, D. Dosage Compensation via Transposable Element Mediated Rewiring of a Regulatory Network. Science 2013, 342, 846–850. [Google Scholar] [CrossRef] [PubMed]
- Conrad, T.; Akhtar, A. Dosage compensation in Drosophila melanogaster: Epigenetic fine-tuning of chromosome-wide transcription. Nat. Rev. Genet. 2012, 13, 123–134. [Google Scholar] [CrossRef] [PubMed]
- Gay, L.; Crochet, P.-A.; Bell, D.A.; Lenormand, T. Comparing clines on molecular and phenotypic traits in hybrid zones: a window on tension zone models. Evolution 2008, 62, 2789–2806. [Google Scholar] [CrossRef] [PubMed]
- Lesecque, Y.; Glémin, S.; Lartillot, N.; Mouchiroud, D.; Duret, L. The Red Queen Model of Recombination Hotspots Evolution in the Light of Archaic and Modern Human Genomes. PLoS Genet. 2014, 10, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Bierne, N.; Welch, J.; Loire, E.; Bonhomme, F.; David, P. The coupling hypothesis: Why genome scans may fail to map local adaptation genes. Mol. Ecol. 2011, 20, 2044–2072. [Google Scholar] [CrossRef] [PubMed]
- Andrew, R.L.; Bernatchez, L.; Bonin, A.; Buerkle, C.A.; Carstens, B.C.; Emerson, B.C.; Garant, D.; Giraud, T.; Kane, N.C.; Rogers, S.M.; et al. A road map for molecular ecology. Mol. Ecol. 2013, 22, 2605–2626. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Li, H.; Jakobsson, M.; Li, S.; SjÖdin, P.; Lascoux, M. Joint analysis of demography and selection in population genetics: Where do we stand and where could we go? Mol. Ecol. 2012, 21, 28–44. [Google Scholar] [CrossRef] [PubMed]
- Orozco-terWengel, P. The devil is in the details: The effect of population structure on demographic inference. Heredity 2016, 116, 349–350. [Google Scholar] [CrossRef]
- Sattath, S.; Elyashiv, E.; Kolodny, O.; Rinott, Y.; Sella, G. Pervasive adaptive protein evolution apparent in diversity patterns around amino acid substitutions in drosophila simulans. PLoS Genet. 2011, 7. [Google Scholar] [CrossRef]
- Suh, A.; Smeds, L.; Ellegren, H. Abundant recent activity of retrovirus-like retrotransposons within and among flycatcher species implies a rich source of structural variation in songbird genomes. Mol. Ecol. 2018, 27, 99–111. [Google Scholar] [CrossRef]



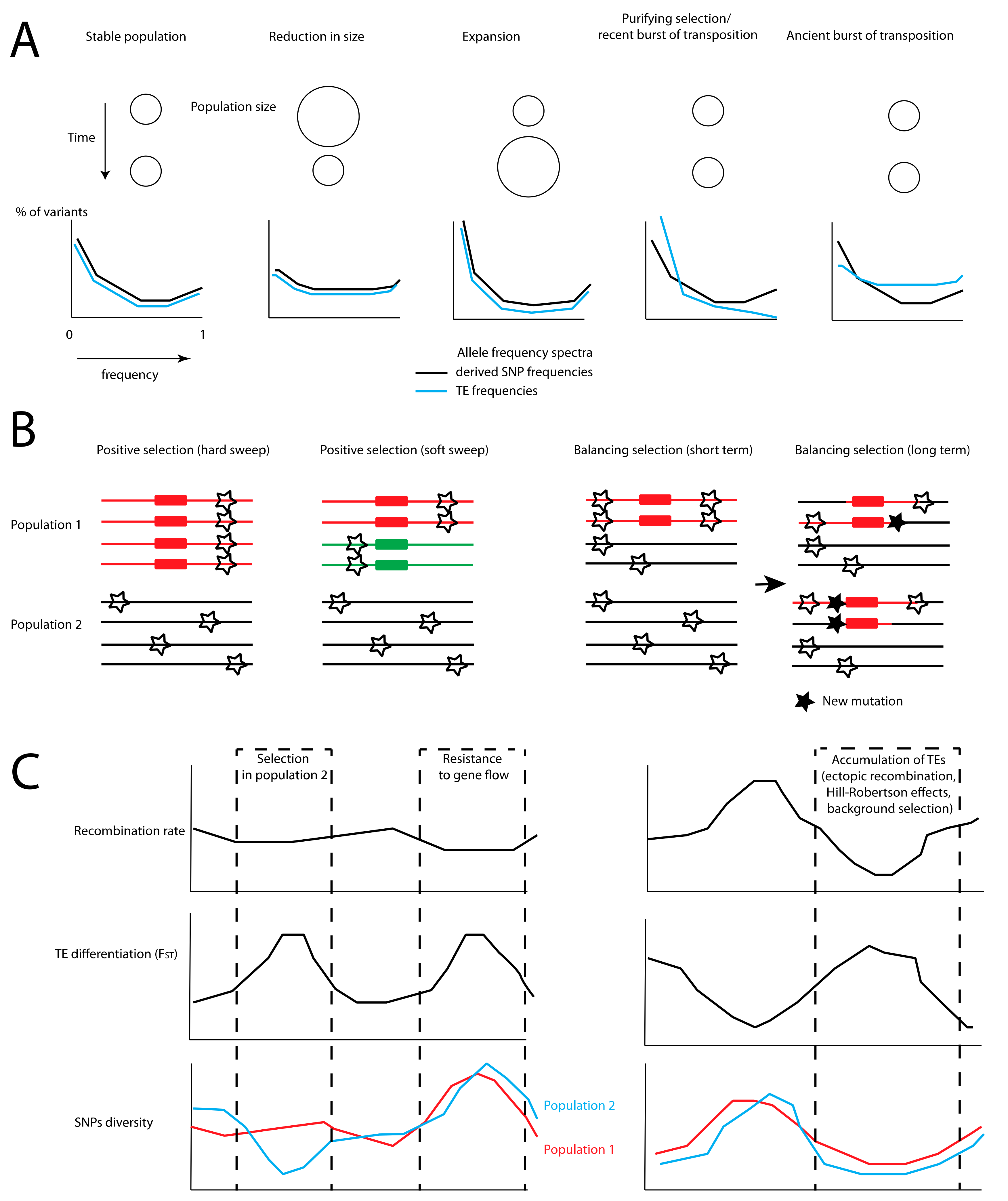{kind=link}
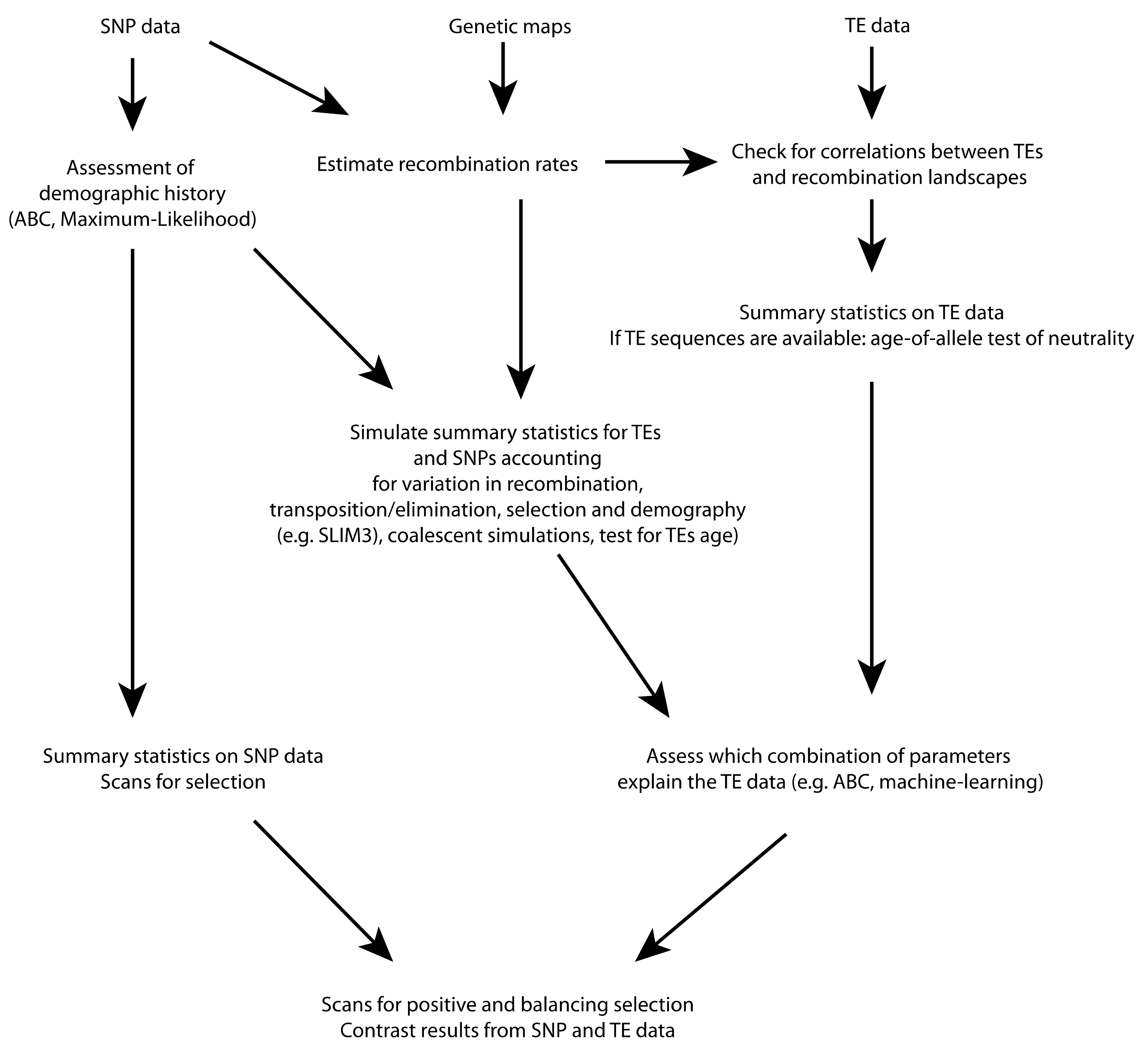{kind=link}
| Name of the Method | Purpose | Link | Reference |
|---|---|---|---|
| Popoolation_TE2 | TE detection in pooled designs | https://sourceforge.net/p/popoolation-te2/wiki/Home/ | [156] |
| T-LEX2 | Detection of polymorphic TEs from short reads | http://petrov.stanford.edu/cgi-bin/Tlex.html | [157] |
| STEAK | Detection of polymorphic TEs from short reads | https://github.com/applevir/STEAK | [158] |
| TIDAL | Detection of polymorphic TEs from short reads | http://www.bio.brandeis.edu/laulab/Tidal_Fly/Tidal_Fly_Home.html | [159] |
| MELT | Detection of polymorphic TEs from short reads | http://melt.igs.umaryland.edu/ | [153] |
| LoRTE | Detection of polymorphic TEs from PacBio sequencing | http://www.egce.cnrs-gif.fr/?p=6422 | [160] |
| ITIS | Detection of polymorphic TEs from short reads | https://github.com/Chuan-Jiang/ITIS | [161] |
| TEMP | Detection of polymorphic TEs from short reads | https://github.com/JialiUMassWengLab/TEMP | [162] |
| Mobster | Detection of polymorphic TEs from short reads | http://sourceforge.net/projects/mobster/ | [163] |
| Tangram | Detection of polymorphic TEs from short reads | https://github.com/jiantao/Tangram | [164] |
| RetroSeq | Detection of polymorphic TEs from short reads | https://github.com/tk2/RetroSeq | [165] |
| RelocaTE2 | Detection of polymorphic TEs from short reads | https://github.com/JinfengChen/RelocaTE2 | [166] |
| McClintock | Combination of several methods into a single pipeline | https://github.com/bergmanlab/mcclintock | [167] |
| Invade | Population genomics modeling (forward-in-time) incorporating coevolution with piRNA clusters | https://sourceforge.net/p/te-tools/code/HEAD/tree/sim3p/ | [104] |
| SLIM3 | Population genomics modeling (forward-in-time) | https://messerlab.org/slim/ | [79] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Bourgeois, Y.; Boissinot, S. On the Population Dynamics of Junk: A Review on the Population Genomics of Transposable Elements. Genes 2019, 10, 419. https://doi.org/10.3390/genes10060419
Bourgeois Y, Boissinot S. On the Population Dynamics of Junk: A Review on the Population Genomics of Transposable Elements. Genes. 2019; 10(6):419. https://doi.org/10.3390/genes10060419
Chicago/Turabian StyleBourgeois, Yann, and Stéphane Boissinot. 2019. "On the Population Dynamics of Junk: A Review on the Population Genomics of Transposable Elements" Genes 10, no. 6: 419. https://doi.org/10.3390/genes10060419
APA StyleBourgeois, Y., & Boissinot, S. (2019). On the Population Dynamics of Junk: A Review on the Population Genomics of Transposable Elements. Genes, 10(6), 419. https://doi.org/10.3390/genes10060419