Mitochondrial Introgression, Color Pattern Variation, and Severe Demographic Bottlenecks in Three Species of Malagasy Poison Frogs, Genus Mantella

 ,
,  ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Tissue Sampling, DNA Extraction, and Amplification
2.2. Sequence Alignment, Statistics, and Phasing
2.3. Phylogenetic Analysis
2.4. Haplotype Network Reconstruction
2.5. Microsatellite Analyses
2.6. Population Structure
2.7. Demographic History
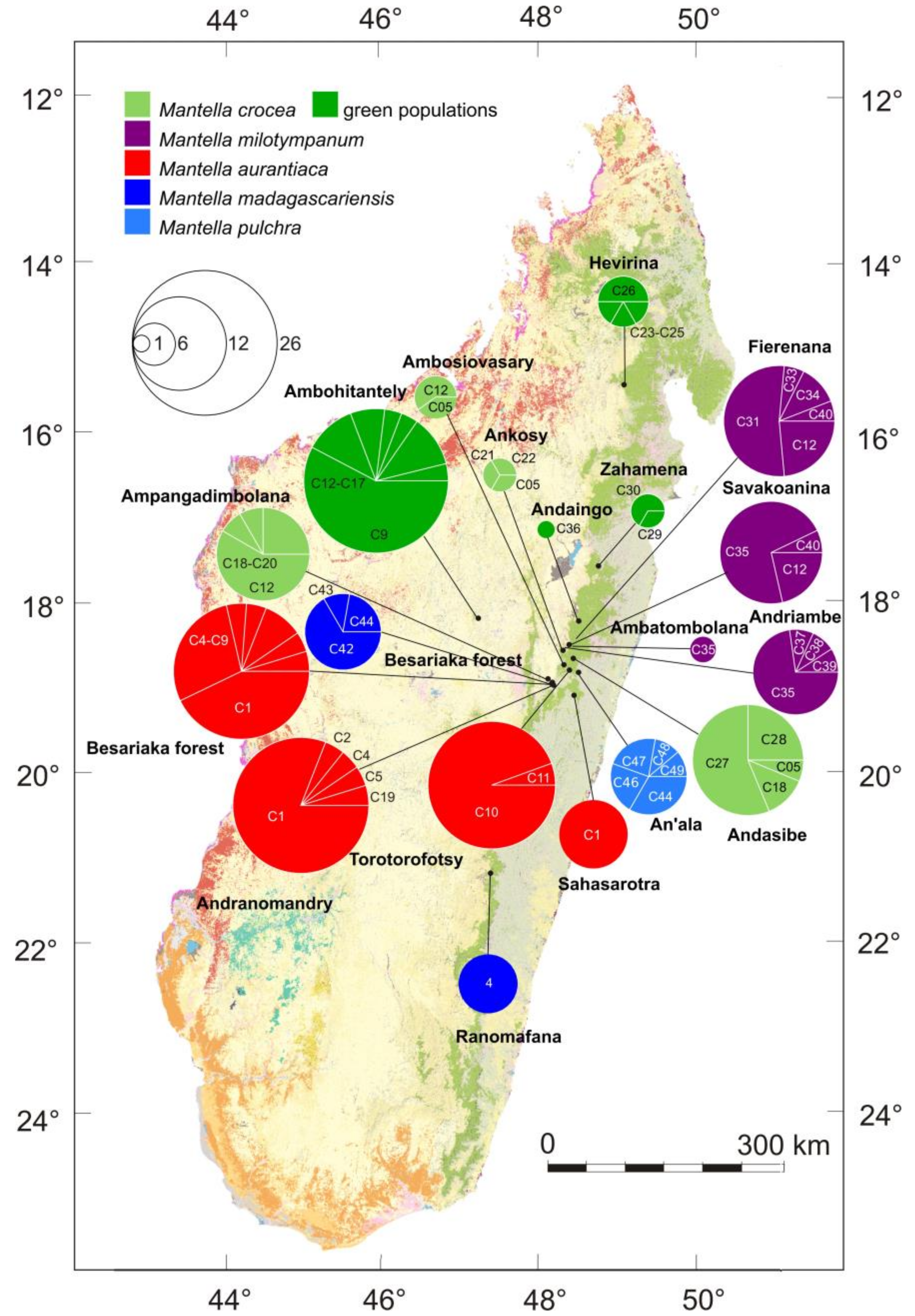
3. Results
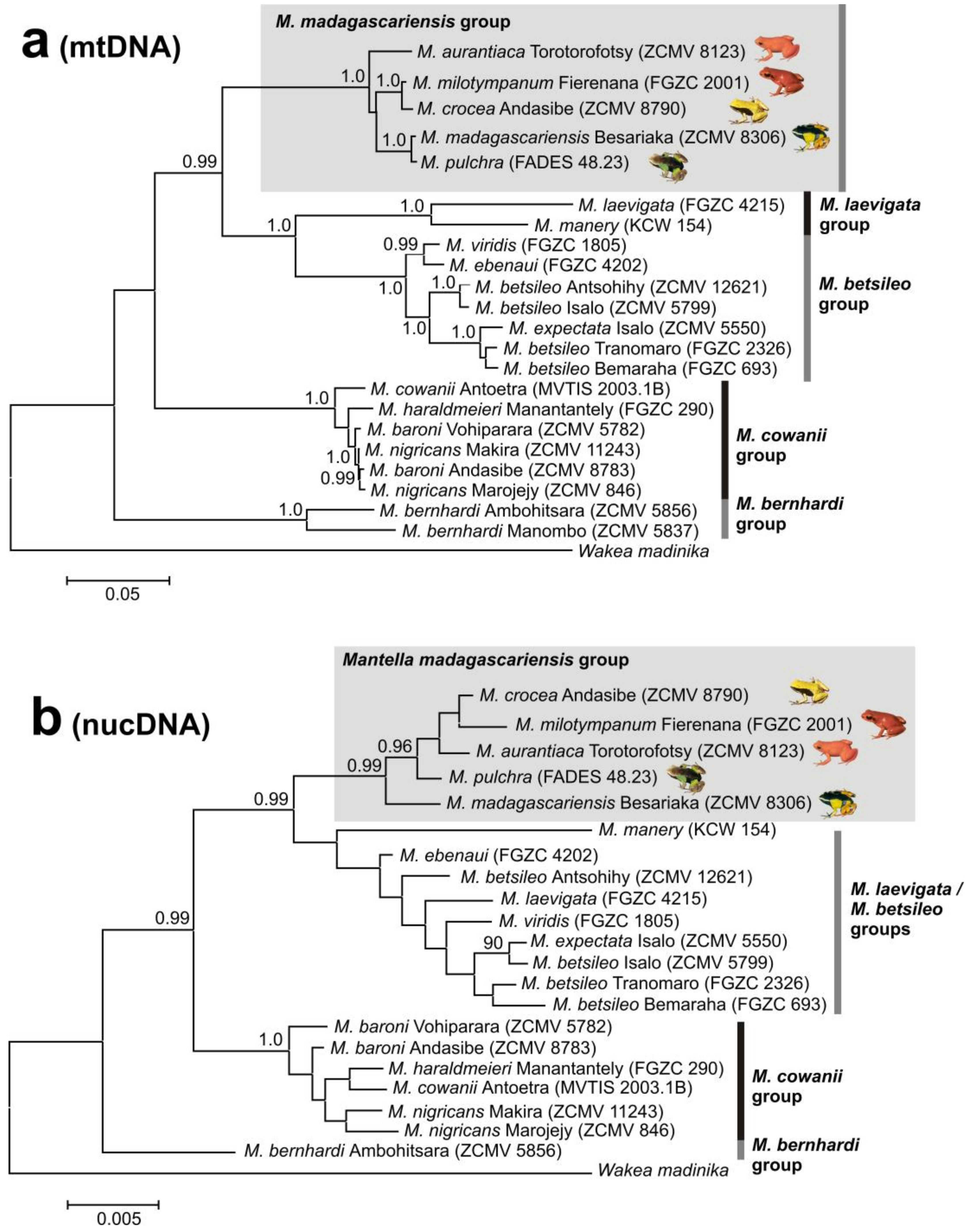
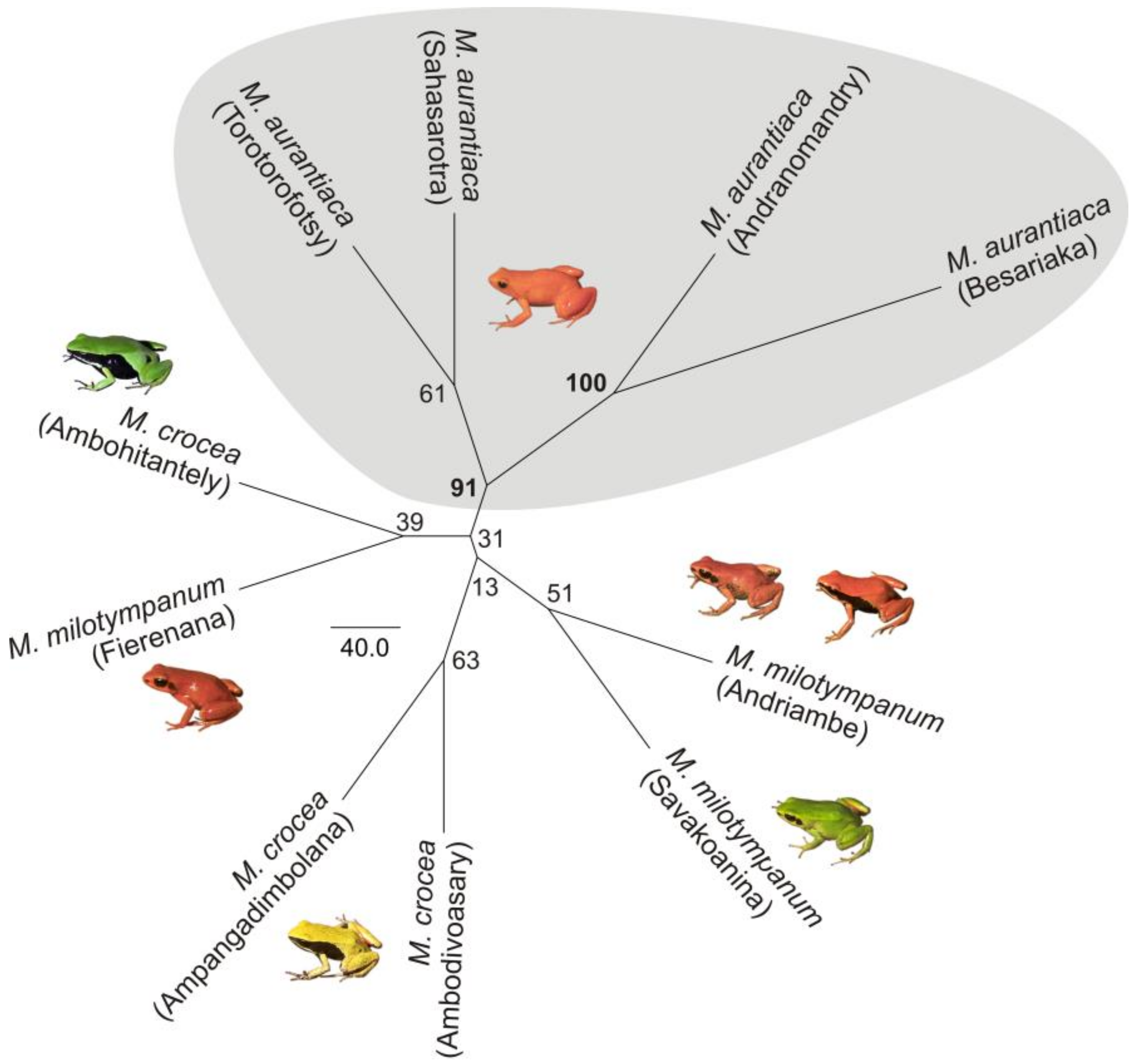
3.1. Monophyly of the M. madagascariensis Group
3.2. DNA Sequence Diversity and Differentiation within the M. madagascariensis Group
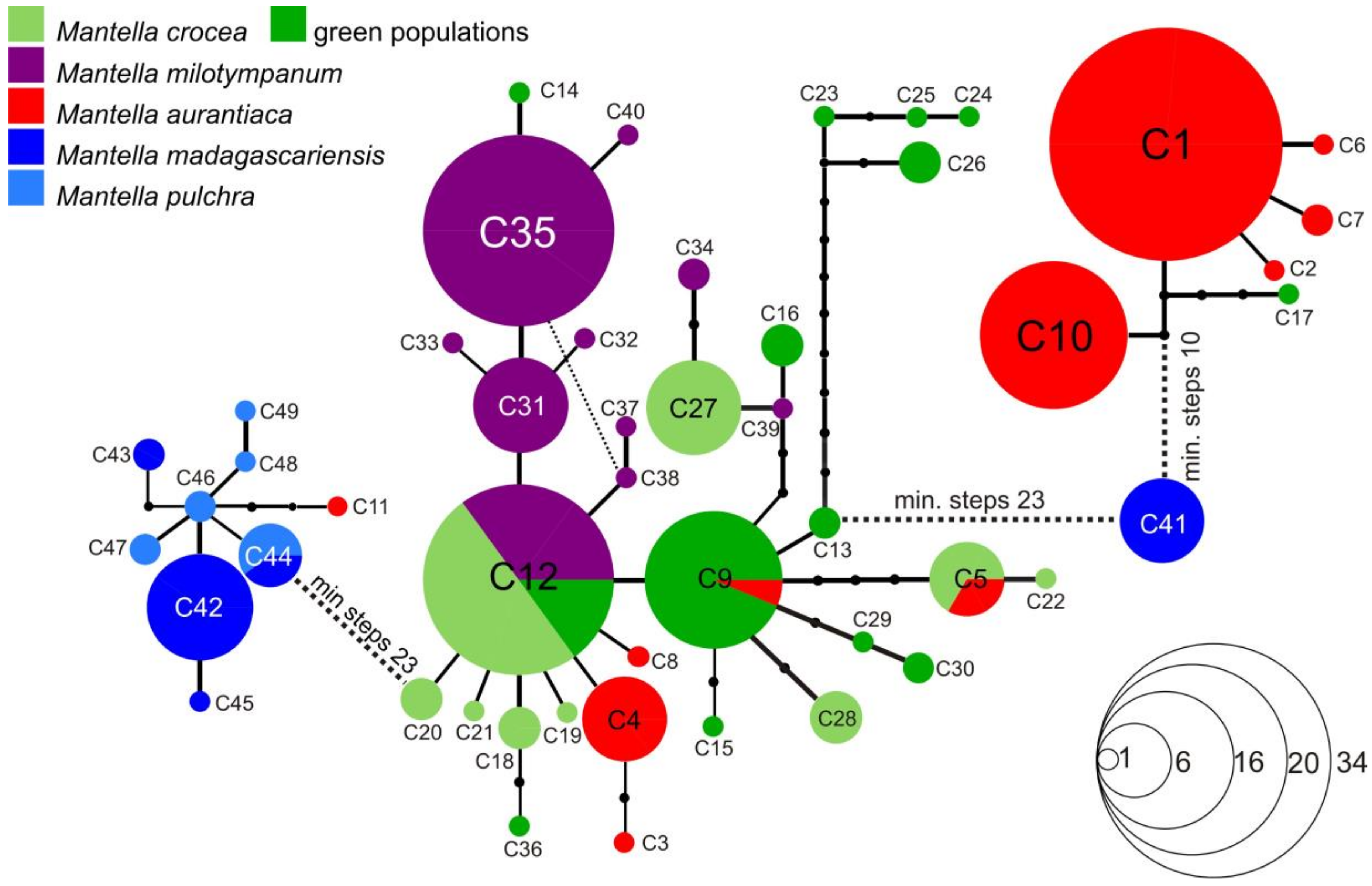
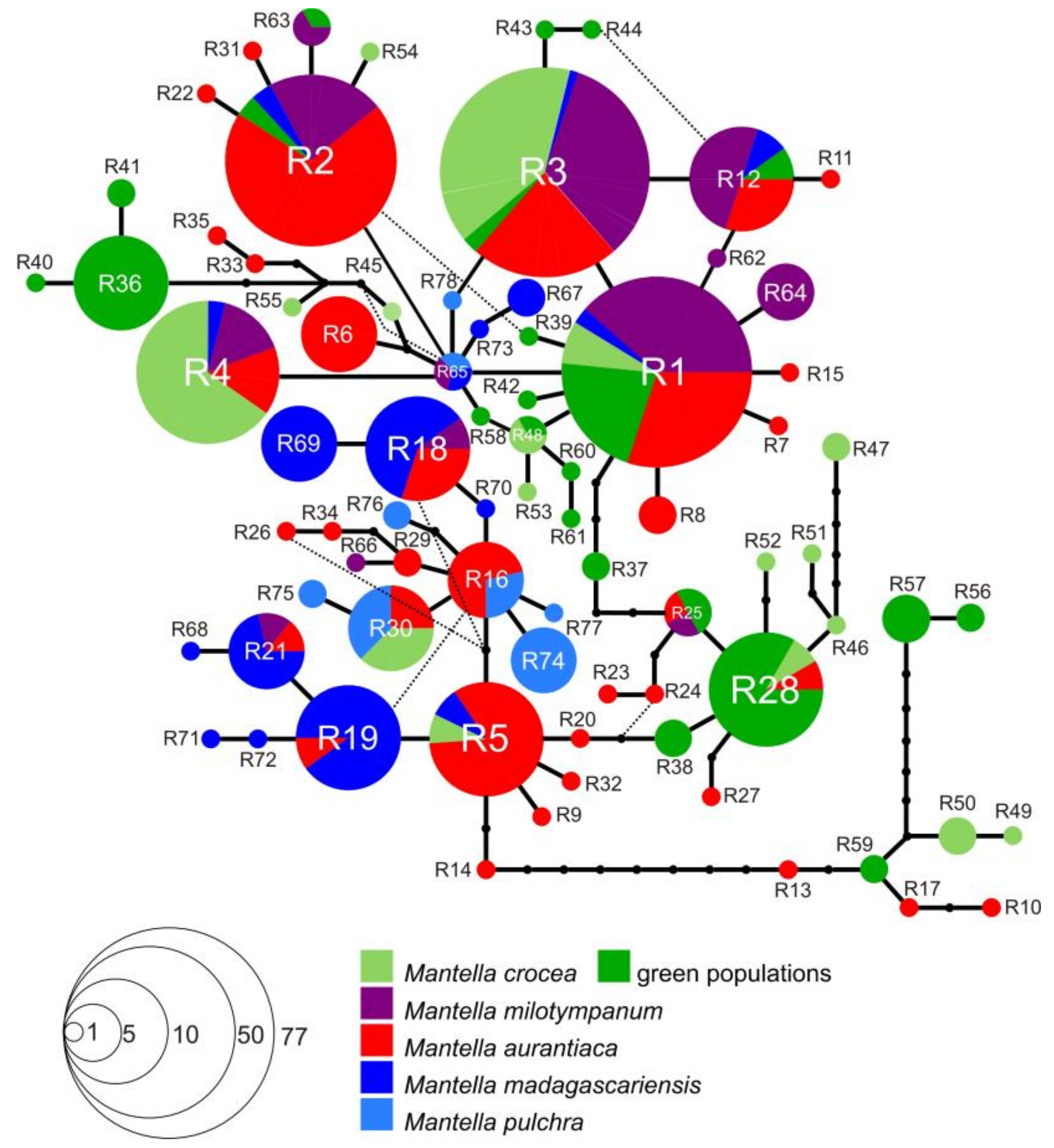
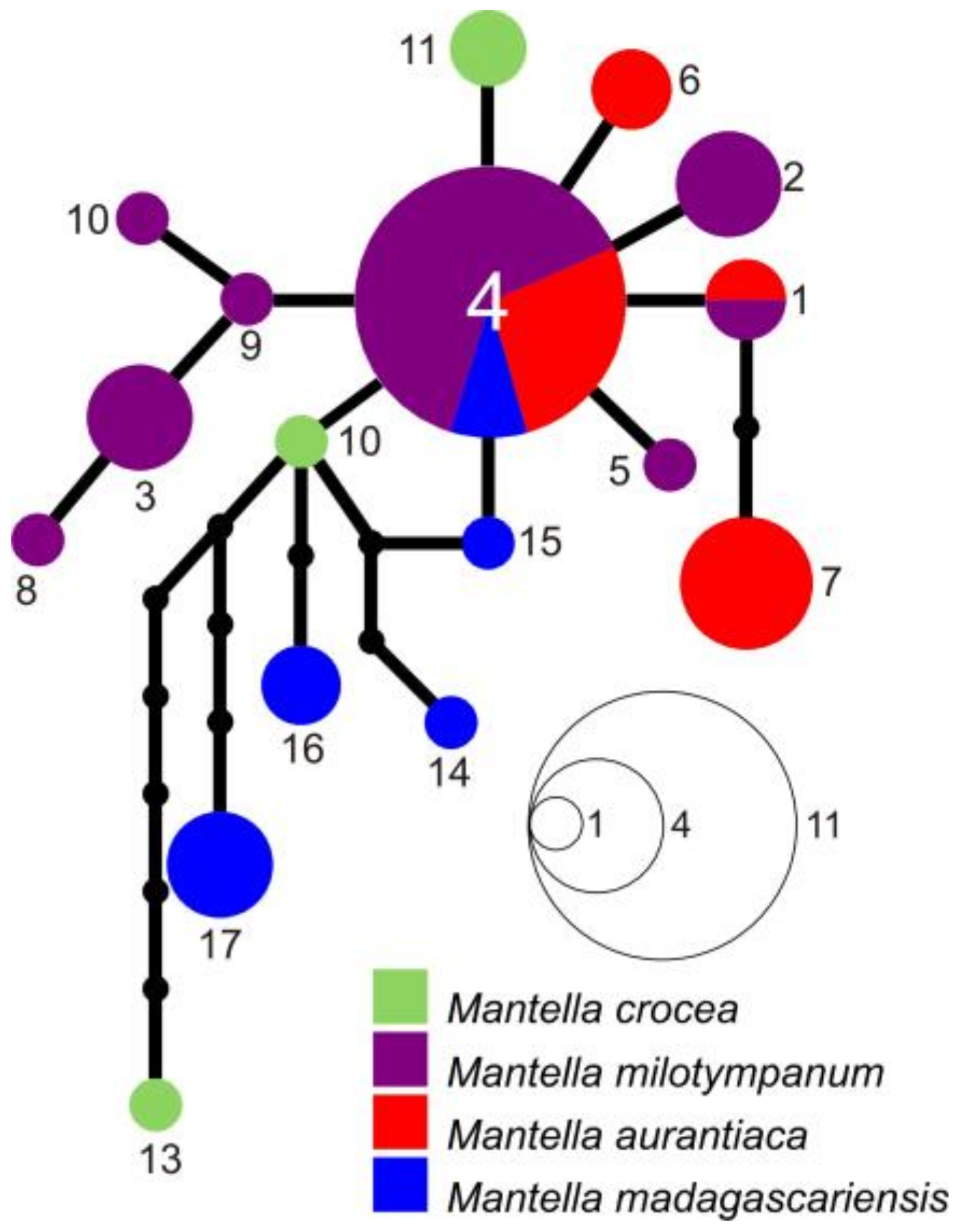
3.3. Haplotype Networks
3.4. Population Genetic Variation in Microsatellites
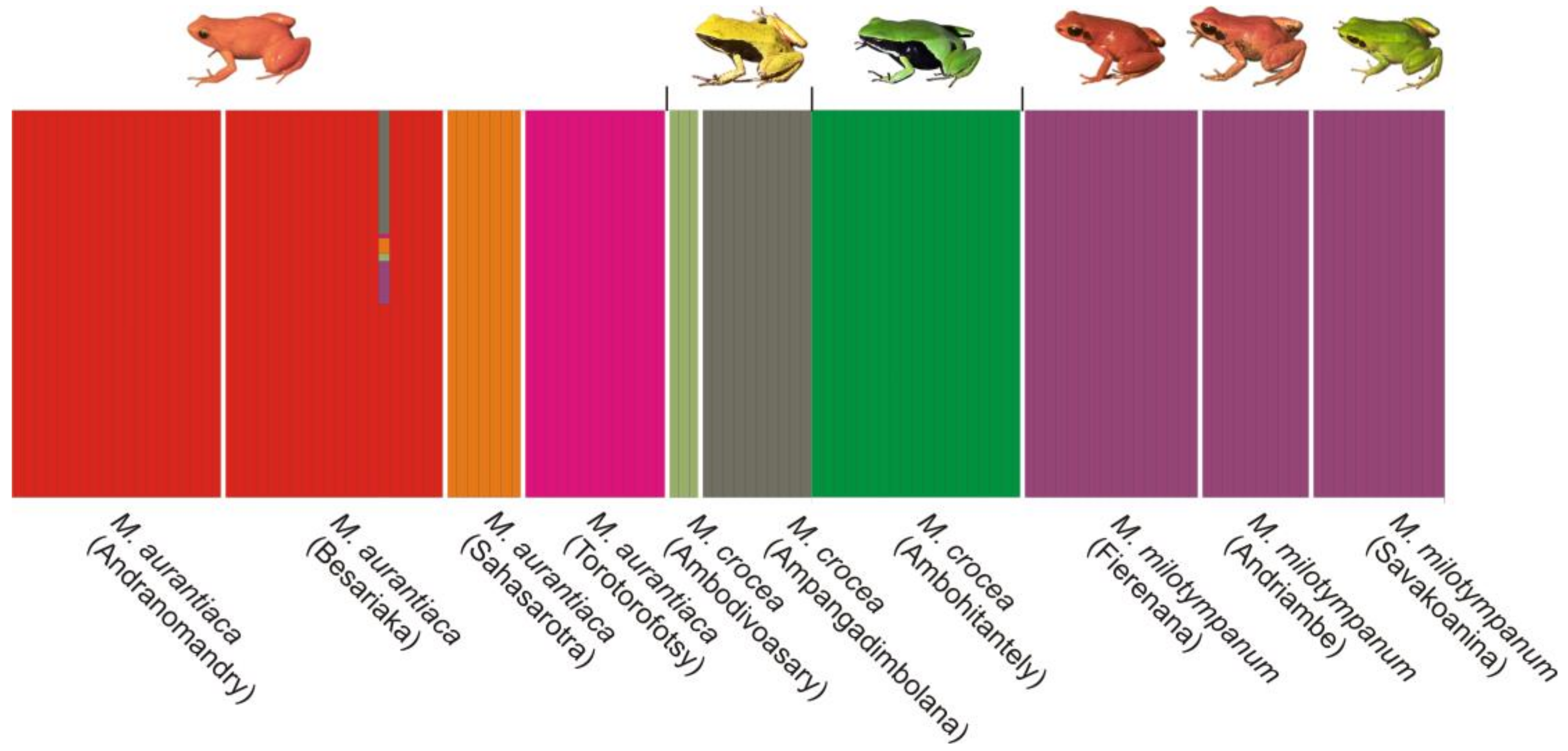
3.5. Population Structure
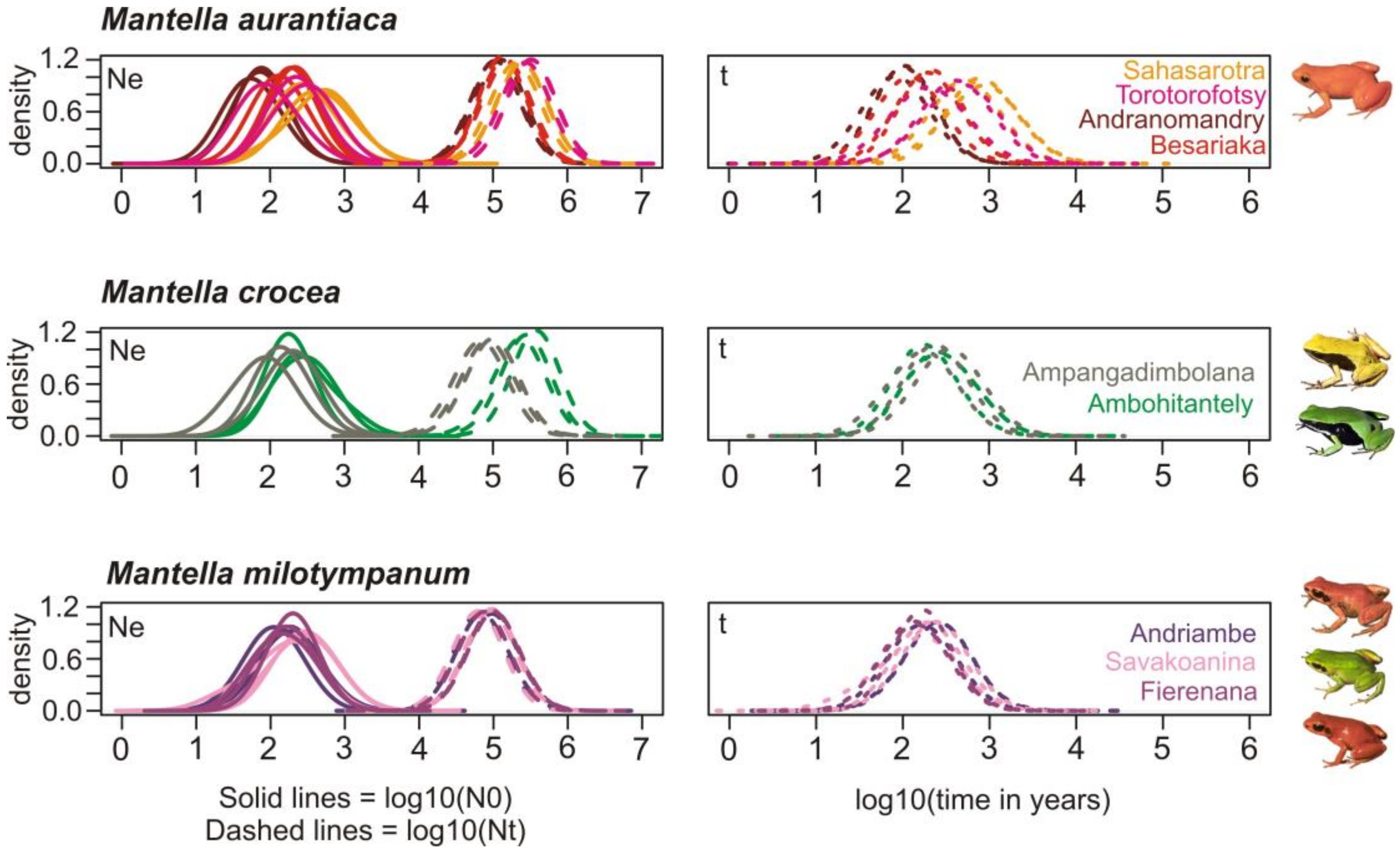
3.6. Demographic History
4. Discussion
4.1. M. aurantiaca, M. crocea and M. milotympanum—Separate Species or Color Morphotypes?
4.2. Taxonomic Conclusions
4.3. Mitochondrial Introgression in the Northern Populations of M. madagascariensis?
4.4. The Effect of Deforestation on the Demographic History of the Swamp Forest Species
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Goodman, S.M.; Benstead, J. The Natural History of Madagascar, 1st ed.; The University of Chicago Press: Chicago, IL, USA, 2004; ISBN 0226303071. [Google Scholar]
- Glaw, F.; Vences, M. A Fieldguide to the Amphibians and Reptiles of Madagascar, 3rd ed.; Vences & Glaw Verlag: Cologne, Germany, 2007; ISBN 392944903X. [Google Scholar]
- Crottini, A.; Madsen, O.; Poux, C.; Strauß, A.; Vieites, D.R.; Vences, M. Vertebrate time-tree elucidates the biogeographic pattern of a major biotic change around the K-T boundary in Madagascar. Proc. Natl. Acad. Sci. USA 2012, 109, 5358–5363. [Google Scholar] [CrossRef]
- Wollenberg, K.C.; Vieites, D.R.; Glaw, F.; Vences, M. Speciation in little: The role of range and body size in the diversification of Malagasy mantellid frogs. BMC Evol. Biol. 2011, 11, 217. [Google Scholar] [CrossRef]
- Scherz, M.D.; Vences, M.; Rakotoarison, A.; Andreone, F.; Köhler, J.; Glaw, F.; Crottini, A. Reconciling molecular phylogeny, morphological divergence and classification of Madagascan narrow-mouthed frogs (Amphibia: Microhylidae). Mol. Phylogenet. Evol. 2016, 100, 372–381. [Google Scholar] [CrossRef]
- Vieites, D.R.; Wollenberg, K.C.; Andreone, F.; Köhler, J.; Glaw, F.; Vences, M. Vast underestimation of Madagascar’s biodiversity evidenced by an integrative amphibian inventory. Proc. Natl. Acad. Sci. USA 2009, 106, 8267–8272. [Google Scholar] [CrossRef]
- Perl, R.G.B.; Nagy, Z.T.; Sonet, G.; Glaw, F.; Wollenberg, K.C.; Vences, M. DNA barcoding Madagascar’s amphibian fauna. Amphib.-Reptil. 2014, 35, 197–206. [Google Scholar] [CrossRef]
- Glaw, F.; Vences, M. A Fieldguide to the Amphibians and Reptiles of Madagascar, 2nd ed.; Vences & Glaw Verlag: Cologne, Germany, 1994; ISBN 3929449013. [Google Scholar]
- Köhler, J.; Vieites, D.R.; Bonett, R.M.; García, F.H.; Glaw, F.; Steinke, D.; Vences, M. New amphibians and global conservation: A boost in species discoveries in a highly endangered vertebrate group. BioScience 2005, 55, 693–696. [Google Scholar] [CrossRef]
- Vences, M.; Wollenberg, K.C.; Vieites, D.R.; Lees, D.C. Madagascar as a model region of species diversification. Trends Ecol. Evol. 2009, 24, 456–465. [Google Scholar] [CrossRef]
- Pabijan, M.; Wollenberg, K.C.; Vences, M. Small body size increases the regional differentiation of populations of tropical mantellid frogs (Anura: Mantellidae). J. Evol. Biol. 2012, 25, 2310–2324. [Google Scholar] [CrossRef]
- Wollenberg Valero, K.C.; Garcia-Porta, J.; Rodríguez, A.; Arias, M.; Shah, A.; Randrianiaina, R.D.; Brown, J.L.; Glaw, F.; Amat, F.; Künzel, S.; et al. Transcriptomic and macroevolutionary evidence for phenotypic uncoupling between frog life history phases. Nat. Commun. 2017, 8, 15213. [Google Scholar] [CrossRef]
- Marshall, J.C.; Bastiaans, E.; Caccone, A.; Camargo, A.; Morando, M.; Niemiller, M.L.; Pabijan, M.; Russello, M.A.; Sinervo, B.; Sites, J.W., Jr.; et al. Mechanisms of speciation in reptiles and amphibians: A synopsis. PeerJ Prepr. 2018, 6, e27279v1. [Google Scholar] [CrossRef]
- Orozco-Terwengel, P.; Andreone, F.; Louis, E., Jr.; Vences, M. Mitochondrial introgressive hybridization following a demographic expansion in the tomato frogs of Madagascar, genus Dyscophus. Mol. Ecol. 2013, 22, 6074–6090. [Google Scholar] [CrossRef]
- Crottini, A.; Chiari, Y.; Mercurio, V.; Meyer, A.; Vences, M.; Andreone, F. Into the canyons: The phylogeography of the Malagasy frogs Mantella expectata and Scaphiophryne gottlebei in the arid Isalo Massif, and its significance for conservation (Amphibia: Mantellidae and Microhylidae). Org. Divers. Evol. 2008, 8, 368–377. [Google Scholar] [CrossRef]
- Vieites, D.R.; Chiari, Y.; Vences, M.; Andreone, F.; Rabemananjara, F.C.E.; Bora, P.; Nieto-Román, S.; Meyer, A. Mitochondrial evidence for distinct phylogeographic units in the endangered Malagasy poison frog Mantella bernhardi. Mol. Ecol. 2006, 15, 1617–1625. [Google Scholar] [CrossRef]
- Rabemananjara, R.C.E.; Chiari, Y.; Ravoahangimalala Ramilijaona, O.; Vences, M. Evidence for recent gene flow between north-eastern and south-eastern Madagascan poison frogs from a phylogeography of the Mantella cowani group. Front. Zool. 2007, 4, 1. [Google Scholar] [CrossRef] [PubMed]
- Avise, J.C. Phylogeography: The History and Formation of Species; Harvard University Press: Cambridge, MA, USA, 2000; ISBN 9780674666382. [Google Scholar]
- Vences, M.; Wake, D.B. Speciation, species boundaries and phylogeography of amphibians. In Amphibian Biology; Heatwole, H.H., Tyler, M., Eds.; Surrey Beatty & Sons: Chipping Norton, Australia, 2007; Volume 7, ISBN 9780980311310. [Google Scholar]
- Zeisset, I.; Beebee, T.J.C. Amphibian phylogeography: A model for understanding historical aspects of species distributions. Heredity 2008, 101, 109–119. [Google Scholar] [CrossRef] [PubMed]
- Crottini, A.; Brown, J.L.; Mercurio, V.; Glaw, F.; Vences, M.; Andreone, F. Phylogeography of the poison frog Mantella viridis (Amphibia: Mantellidae) reveals chromatic and genetic differentiation across ecotones in northern Madagascar. J. Zool. Syst. Evol. Res. 2012, 50, 305–314. [Google Scholar] [CrossRef]
- Chiari, Y.; Vences, M.; Vieites, D.R.; Rabemananjara, F.C.E.; Bora, P.; Ramilijaona Ravoahangimalala, O.; Meyer, A. New evidence for parallel evolution of colour patterns in Malagasy poison frogs (Mantella). Mol. Ecol. 2004, 13, 3763–3774. [Google Scholar] [CrossRef] [PubMed]
- Vences, M.; Chiari, Y.; Raharivololoniaina, L.; Meyer, A. High mitochondrial diversity within and among populations of Malagasy poison frogs. Mol. Phylogenet. Evol. 2004, 30, 295–307. [Google Scholar] [CrossRef]
- Rabemananjara, F.C.E.; Crottini, A.; Chiari, Y.; Anderone, F.; Glaw, F.; Duguet, R.; Bora, P.; Ravoahangimalala Ramilijaona, O.; Vences, M. Molecular systematics of Malagasy poison frogs in the Mantella betsileo and M. laevigata species groups. Zootaxa 2007, 1501, 31–44. [Google Scholar] [CrossRef]
- Cocca, W.; Rosa, G.M.; Andreone, F.; Aprea, G.; Eusebio Bergò, P.; Mattioli, F.; Mercurio, V.; Randrianirina, J.E.; Rosado, D.; Vences, M.; et al. The herpetofauna (Amphibia, Crocodylia, Squamata, Testudines) of the Isalo Massif, Southwest Madagascar: Combining morphological, molecular and museum data. Salamandra 2018, 54, 178–200. [Google Scholar]
- Daly, J.W.; Highet, R.J.; Myers, C.W. Occurrence of skin alkaloids in non-dendrobatid frogs from Brazil (Bufonidae), Australia (Myobatrachidae) and Madagascar (Mantellinae). Toxicon 1984, 22, 905–919. [Google Scholar] [CrossRef]
- Vences, M.; Glaw, F.; Böhme, W. Evolutionary correlates of microphagy in alkaloid-containing frogs (Amphibia: Anura). Zoologischer Anzeiger 1998, 236, 217–230. [Google Scholar]
- Tarvin, R.D.; Santos, J.C.; O’Connell, L.A.; Zakon, H.H.; Cannatella, D.C. Convergent substitutions in a sodium channel suggest multiple origins of toxin resistance in poison frogs. Mol. Biol. Evol. 2016, 33, 1068–1081. [Google Scholar] [CrossRef]
- Rabemananjara, F.C.E.; Ravoahangimalala Ramilijaona, O.; Raminosoa, N.R.; Andreone, F.; Bora, P.; Carpenter, A.I.; Glaw, F.; Razafindrabe, T.; Vallan, D.; Vieites, D.R.; et al. Malagasy poison frogs in the pet trade: A survey of levels of exploitation in the species in the genus Mantella. In A Conservation Strategy for the Amphibians of Madagascar; Andreone, F., Randriamahazo, H., Eds.; Museo Regionale di Scienze Naturali: Torino, Italy, 2008; pp. 277–300. ISBN 9788886041720. [Google Scholar]
- Carpenter, A.I.; Andreone, F.; Moore, R.D.; Griffiths, R.A. A review of the international trade in amphibians:the types, levels and dynamics of trade in CITES-listed species. Oryx 2014, 48, 565–574. [Google Scholar] [CrossRef]
- Vences, M.; Hille, A.; Glaw, F. Allozyme differentiation in the genus Mantella (Amphibia: Anura: Mantellinae). Folia Zool. 1998, 47, 261–274. [Google Scholar]
- Rabemananjara, F.C.E.; Bora, P.; Razafindrabe, T.; Randriamitso, E.; Ravoahangimalala Ramilijaona, O.; Raminosoa, N.R.; Rakotondravony, D.; Vieites, D.R.; Vences, M. Rapid assessments of population sizes in ten species of Malagasy poison frogs, genus Mantella. In A Conservation Strategy for the Amphibians of Madagascar; Andreone, F., Randriamahazo, H., Eds.; Museo Regionale di Scienze Naturali: Torino, Italy, 2008; ISBN 9788886041720. [Google Scholar]
- Tessa, G.; Mattioli, F.; Mercurio, V.; Andreone, F. Egg numbers and fecundity traits in nine species of Mantella poison frogs from arid grasslands and rainforests of Madagascar (Anura: Mantellidae). Madag. Conserv. Dev. 2009, 4, 113–119. [Google Scholar] [CrossRef]
- Vieites, D.R.; Rabemananjara, F.E.C.; Bora, P.; Razafimahatratra, B.; Ramilijaona Ravoahangimalala, O.; Vences, M. Distribution and population density of the black-eared Malagasy poison frog, Mantella milotympanum Staniszewski, 1996 (Amphibia: Mantellidae). In African Biodiversity: Molecules, Organisms, Ecosystems, Proceedings of the 5th International Symposium on Tropical Biology, Museum Koenig, Bonn, Germaby, 5–8 May 2004; Huber, B.A., Sinclair, B.J., Lampe, K.H., Eds.; Springer: Berlin, Germany, 2005; pp. 197–204. ISBN 978-0-387-24320-7. [Google Scholar]
- Bora, P.; Dolch, R.; Jenkins, R.; Jovanovic, O.; Rabemananjara, F.C.E.; Randrianirina, J.; Rafanomezantsoa, J.; Raharivololoniaina, L.; Ramilijaona, O.; Raminosoa, N.; et al. Geographical distribution of three species of Malagasy poison frogs of high conservation priority: Mantella aurantiaca, M. crocea and M. milotympanum. Herpetol. Notes 2008, 1, 39–48. [Google Scholar]
- Randrianavelona, R.; Rakotonoely, H.; Ratsimbazafy, J.; Jenkins, R.K.B. Conservation assessment of the critically endangered frog Mantella aurantiaca in Madagascar. Afr. J. Herpetol. 2010, 59, 65–78. [Google Scholar] [CrossRef]
- Piludu, N.; Dubos, N.; Razafimanahaka, J.H.; Razafindraibe, P.; Christian, J.; Jenkins, R.K.B. Distribution, threats and conservation of a Critically Endangered amphibian (Mantella aurantiaca) in Eastern Madagascar. Herpetol. Notes 2015, 8, 119–123. [Google Scholar]
- Schaefer, H.C.; Vences, M.; Veith, M. Molecular phylogeny of Malagasy poison frogs, genus Mantella (Anura: Mantellidae): Homoplastic evolution of color pattern in aposematic amphibians. Org. Divers. Evol. 2002, 2, 97–105. [Google Scholar] [CrossRef]
- Andriamaharavo, N.R.; Garraffo, H.M.; Spande, T.F.; Giddings, L.A.; Vieites, D.R.; Vences, M.; Saporito, R.A. Individual and geographic variation of skin alkaloids in three swamp-forest species of Madagascan poison frogs (Mantella). J. Chem. Ecol. 2015, 41, 837–847. [Google Scholar] [CrossRef]
- Woodhead, C.; Vences, M.; Vieites, D.R.; Gamboni, I.; Fisher, B.L.; Griffiths, R.A. Specialist or generalist? Feeding ecology of the Malagasy poison frog Mantella aurantiaca. Herpetol. J. 2007, 17, 225–236. [Google Scholar]
- International Union for Conservation of Nature. The IUCN Red List of Threatened Species. Version 2018-2. IUCN, Gland, Switzerland. Available online: http://www.iucnredlist.org (accessed on 1 February 2019).
- Douglass, K.; Zinke, J. Forging ahead by land and by sea: Archaeology and Paleoclimate Reconstruction in Madagascar. Afr. Archaeol. Rev. 2015, 32, 267–299. [Google Scholar] [CrossRef]
- Anderson, A.; Clark, G.; Haberle, S.; Higham, T.; Nowak-Kemp, M.; Prendergast, A.; Radimilahy, C.; Rakotozafy, L.M.; Ramilisonina; Schwenninger, J.-L.; et al. New evidence of megafaunal bone damage indicates late colonization of Madagascar. PLoS ONE 2018, 13, e0204368. [Google Scholar] [CrossRef] [PubMed]
- Hauswaldt, S.J.; Crottini, A.; Vences, M. Nine microsatellite loci for the Malagasy Poison Frogs (Mantella). Conserv. Genet. Resour. 2009, 1, 269–271. [Google Scholar] [CrossRef]
- Bruford, M.W.; Hanotte, O.; Brookfield, J.F.Y.; Burke, T. Single locus and multilocus DNA fingerprint. In Molecular Genetic Analysis of Populations: A Practical Approach; Hoelzel, A.R., Ed.; IRL Press: Oxford, UK, 1992; pp. 225–270. [Google Scholar]
- Kumar, S.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis version 7.0 for bigger datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [PubMed]
- Stephens, M.; Smith, N.J.; Donnelly, P. A new statistical method for haplotype reconstruction from population data. Am. J. Hum. Genet. 2001, 68, 978–989. [Google Scholar] [CrossRef] [PubMed]
- Rozas, J.; Ferrer-Mata, A.; Sánchez-DelBarrio, J.C.; Guirao-Rico, S.; Librado, P.; Ramos-Onsins, S.E.; Sánchez-Gracia, A. DnaSP 6: DNA sequence polymorphism analysis of large datasets. Mol. Biol. Evol. 2017, 34, 3299–3302. [Google Scholar] [CrossRef] [PubMed]
- Tajima, F. The effect of change in population size on DNA polymorphism. Genetics 1989, 123, 597–601. [Google Scholar]
- Nei, M. Molecular Evolutionary Genetics, 1st ed.; Columbia University Press: New York, NY, USA, 1987; ISBN 9780231063210. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar]
- Darriba, D.; Taboada, G.L.; Doallo, R.; Posada, D. jModelTest 2: More models, new heuristics and parallel computing. Nat. Methods 2012, 9, 772. [Google Scholar] [CrossRef]
- Lanfear, R.; Calcott, B.; Ho, S.Y.W.; Guindon, S. PartitionFinder: Combined selection of partitioning schemes and substitution models for phylogenetic analyses. Mol. Biol. Evol. 2012, 29, 1695–1701. [Google Scholar] [CrossRef]
- Ronquist, F.; Teslenko, M.; Van der Mark, P.; Ayres, D.L.; Darling, A.; Höhna, S.; Larget, B.; Liu, L.; Suchard, M.A.; Huelsenbeck, J.P. MrBayes 3.2: Efficient Bayesian phylogenetic inference and model choice across a large model space. Syst. Biol. 2012, 61, 539–542. [Google Scholar] [CrossRef]
- Bruen, T.C.; Philippe, H.; Bryant, D. A simple and robust statistical test for detecting the presence of recombination. Genetics 2006, 172, 2665–2681. [Google Scholar] [CrossRef]
- Huson, D.H.; Bryant, D. Application of phylogenetic networks in evolutionary studies. Mol. Biol. Evol. 2006, 23, 254–267. [Google Scholar] [CrossRef]
- Clement, M.; Posada, D.; Crandall, K.A. TCS: A computer program to estimate gene genealogies. Mol. Ecol. 2000, 9, 1657–1659. [Google Scholar] [CrossRef]
- Templeton, A.R.; Crandall, K.A.; Sing, C.F. A cladistic analysis of phenotypic associations with haplotypes inferred from restriction endonuclease mapping and DNA sequence data. III. Cladogram estimation. Genetics 1992, 132, 619–633. [Google Scholar]
- Posada, D.; Crandall, K.A. Intraspecific phylogenetics: Trees grafting into networks. Trends Ecol. Evol. 2001, 16, 37–45. [Google Scholar] [CrossRef]
- Pfenninger, M.; Posada, D. Phylogeographic history of the land snail Candidula unifasciata (Poiret 1801) (Helicellinae, Stylommatophora): Fragmentation, corridor migration and secondary contact. Evolution 2002, 56, 1776–1788. [Google Scholar] [CrossRef]
- Van Oosterhout, C.; Hutchinson, W.F.; Shipley, P.; Wills, D.P.M. MICRO-CHECKER: Software for identifying and correcting genotyping errors in microsatellite data. Mol. Ecol. Notes 2004, 4, 535–538. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. Arlequin Suite ver 3.5, a new series of programs to perform population genetics analyses under Linux and Windows. Mol. Ecol. Resour. 2010, 10, 564–567. [Google Scholar] [CrossRef] [PubMed]
- Dieringer, D.; Schloetter, C. Microsatellite analyser (MSA): A platform independent analysis tool for large microsatellite data sets. Mol. Ecol. Notes 2003, 3, 167–169. [Google Scholar] [CrossRef]
- Raymond, M.; Rousset, F. Genepop (Version 1.2): Population-genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Corander, J.; Marttinen, P. Bayesian identification of admixture events using multilocus molecular markers. Mol. Ecol. 2006, 15, 2833–2843. [Google Scholar] [CrossRef]
- Felsenstein, J. PHYLIP (Phylogeny Inference Package), version 3.61; Department of Genome Sciences, University of Washington; Distributed by the author; Seattle, WA, USA, 1993. [Google Scholar]
- Bowcock, A.M.; Ruiz-Linares, A.; Tomfohrde, J.; Minch, E.; Kidd, J.R.; Cavalli-Sforza, L.L. High resolution of human evolutionary trees with polymorphic microsatellites despite a constraint in allele length. Nature 1994, 368, 455–457. [Google Scholar] [CrossRef]
- Wilson, G.A.; Rannala, B. Bayesian inference of recent migration rates using multilocus genotypes. Genetics 2003, 163, 1177–1191. [Google Scholar]
- Suchard, M.A.; Lemey, P.; Baele, G.; Ayres, D.L.; Drummond, A.J.; Rambaut, A. Bayesian phylogenetic and phylodynamic data integration using BEAST 1.10. Virus Evol. 2018, 4, vey016. [Google Scholar] [CrossRef]
- Beaumont, M.A. Detecting population expansion and decline using microsatellites. Genetics 1999, 153, 2013–2029. [Google Scholar]
- Storz, J.F.; Beaumont, M.A. Testing for genetic evidence of population expansion and contraction: An empirical analysis of microsatellite DNA variation using a hierarchical Bayesian model. Evolution 2002, 56, 154–166. [Google Scholar] [CrossRef]
- Brinkmann, B.; Klintschar, M.; Neuhuber, F.; Huhne, J.; Rolf, B. Mutation rate in human microsatellites: Influence of the structure and length of the tandem repeat. Am. J. Hum. Genet. 1998, 62, 1408–1415. [Google Scholar] [CrossRef]
- Brohede, J.; Primmer, C.R.; Moller, A.; Ellegren, H. Heterogeneity in the rate and pattern of germline mutation at individual microsatellite loci. Nucleic Acids Res. 2002, 30, 1997–2003. [Google Scholar] [CrossRef] [PubMed]
- Bulut, Z.; McCormick, C.R.; Gopurenko, D.; Williams, R.N.; Bos, D.H.; DeWoody, J.A. Microsatellite mutation rates in the eastern tiger salamander (Ambystoma tigrinum tigrinum) differ 10-fold across loci. Genetica 2009, 136, 501–504. [Google Scholar] [CrossRef] [PubMed]
- Brooks, S.P.; Gelman, A. General methods for monitoring convergence of iterative simulations. J. Comput. Graph. Stat. 1998, 7, 434–455. [Google Scholar]
- Plummer, M.; Best, N.; Cowles, K.; Vines, K. CODA: Convergence diagnosis and output analysis for MCMC. R News 2006, 6, 7–11. [Google Scholar]
- R Development Core Team. R: A Language and Environment for Statistical Computing; R Foundation for Statistical Computing: Vienna, Austria, 2009. [Google Scholar]
- Belkhir, K.; Borsa, P.; Chikhi, L.; Raufaste, N.; Bonhomme, F. GENETIX4.05, Logiciel sous Windows TM pour la génétique des Populations. Laboratoire génome, Populations, Interactions, CNRS UMR. 5000; Université de Montpellier II: Montpellier, France, 2004. [Google Scholar]
- Odierna, G.; Vences, M.; Aprea, G.; Lötters, S.; Andreone, F. Chromosome data for Malagasy poison frogs (Amphibia: Ranidae: Mantella) and their bearing on taxonomy and phylogeny. Zool. Sci. 2001, 18, 505–514. [Google Scholar] [CrossRef][Green Version]
- Fouquet, A.; Gilles, A.; Vences, M.; Marty, C.; Blanc, M.; Gemmell, N.J. Underestimation of species richness in Neotropical frogs revealed by mtDNA analyses. PLoS ONE 2007, 2, e1109. [Google Scholar] [CrossRef]
- Pinna, P.H.; Fernandes, D.S.; Passos, P. If you choose not to decide you still have made a choice. Bionomina 2018, 13, 65–68. [Google Scholar] [CrossRef][Green Version]
- Klonoski, K.; Bi, K.; Rosenblum, E.B. Phenotypic and genetic diversity in aposematic Malagasy poison frogs (genus Mantella). Ecol. Evol. 2019, 9, 2725–2742. [Google Scholar] [CrossRef]
- Albertson, R.C.; Markert, J.A.; Danley, P.D.; Kocher, T.D. Phylogeny of a rapidly evolving clade: The cichlid fishes of Lake Malawi, East Africa. Proc. Natl. Acad. Sci. USA 1999, 96, 5107–5110. [Google Scholar] [CrossRef]
- Keinan, A.; Clark, A.G. Recent explosive human population growth has resulted in an excess of rare genetic variants. Science 2012, 336, 740–743. [Google Scholar] [CrossRef]
- Salmona, J.; Heller, R.; Quéméré, E.; Chikhi, L. Climate change and human colonization triggered habitat loss and fragmentation in Madagascar. Mol. Ecol. 2017, 26, 5203–5222. [Google Scholar] [CrossRef] [PubMed]
- Burney, D.A.; Burney, L.P.; Godfrey, L.R.; Jungers, W.L.; Goodman, S.M.; Wright, H.T.; Jull, A.J.T. A chronology for late prehistoric Madagascar. J. Hum. Evol. 2004, 47, 25–63. [Google Scholar] [CrossRef]
- Burns, S.J.; Godfrey, L.R.; Faina, P.; McGee, D.; Hardt, B.; Ranivoharimanana, L.; Randrianasy, J. Rapid human-induced landscape transformation in Madagascar at the end of the first millennium of the Common Era. Quat. Sci. Rev. 2016, 134, 92–99. [Google Scholar] [CrossRef]
- Jarosz, L. Defining and explaining tropical deforestation: Shifting cultivation and population growth in colonial Madagascar (1896–1940). Econ. Geogr. 1993, 69, 366–379. [Google Scholar] [CrossRef]
- Harper, G.J.; Steininger, M.K.; Tucker, C.J.; Juhn, D.; Hawkins, F. Fifty years of deforestation and forest fragmentation in Madagascar. Environ. Conserv. 2007, 34, 325–333. [Google Scholar] [CrossRef]
- Schwitzer, C.; Chikhi, L.; Donati, G.; Irwin, M.; Johnson, S.E.; Mittermeier, R.A.; Peacock, H.; Ratsimbazafy, J.; Razafindramanana, J.; Louis, E.E., Jr.; et al. Protecting lemurs–response. Science 2014, 344, 358–360. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Taxon | Locality | n | ANAP | HO | HE | FIS |
|---|---|---|---|---|---|---|
| M. aurantiaca | Andranomandry | 19 | 10.4 | 0.7 | 0.87 | 0.18 |
| Besariaka | 20 | 12.78 | 0.7 | 0.86 | 0.18 | |
| Sahasarotra | 7 | 6.11 | 0.55 | 0.77 | 0.25 | |
| Torotorofotsy | 13 | 8.78 | 0.59 | 0.79 | 0.22 | |
| M. crocea | Ambodivoasary | 3 | 3.44 | 0.59 | 0.76 | 0.12 |
| Ambohitantely | 19 | 11.89 | 0.69 | 0.88 | 0.21 | |
| Ampangadimbolana | 10 | 6.78 | 0.46 | 0.83 | 0.45 | |
| M. milotympanum | Fierenana-Sahamarolambo | 16 | 10.67 | 0.69 | 0.88 | 0.21 |
| Andriambe | 10 | 7.33 | 0.69 | 0.85 | 0.18 | |
| Savakoanina | 12 | 9.56 | 0.74 | 0.87 | 0.14 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | 9 | 10 | ||
|---|---|---|---|---|---|---|---|---|---|---|---|
| 1 | Aur—Andranomandry | 0 | 0.02 | 0.05 | 0.06 | 0.07 | 0.03 | 0.06 | 0.04 | 0.05 | 0.05 |
| 2 | aur—Besariaka | n.s. | 0 | 0.05 | 0.05 | 0.08 | 0.04 | 0.05 | 0.05 | 0.07 | 0.07 |
| 3 | aur—Sahasarotra | n.s. | 0.0045 | 0 | 0.06 | 0.14 | 0.08 | 0.08 | 0.07 | 0.09 | 0.10 |
| 4 | aur—Torotorofotsy | 0.0045 | 0.0045 | 0.0045 | 0 | 0.11 | 0.07 | 0.07 | 0.07 | 0.10 | 0.10 |
| 5 | croc—Ambodivoasary | n.s. | 0.0045 | n.s. | n.s. | 0 | 0.06 | 0.06 | 0.09 | 0.09 | 0.11 |
| 6 | croc—Ambohitantely | 0.0045 | 0.0045 | 0.0045 | 0.0045 | 0.0045 | 0 | 0.05 | 0.04 | 0.06 | 0.05 |
| 7 | croc—Ampangadimbolana | 0.0045 | 0.0045 | n.s. | 0.0045 | n.s. | 0.0045 | 0 | 0.04 | 0.07 | 0.05 |
| 8 | mil—Fierenana-Sahamarolambo | 0.0045 | 0.0045 | 0.0045 | 0.0045 | 0.0045 | 0.0045 | n.s. | 0 | 0.05 | 0.04 |
| 9 | mil—Andriambe | 0.0045 | 0.0045 | 0.0045 | 0.0045 | n.s. | 0.0045 | 0.0045 | 0.0045 | 0 | 0.04 |
| 10 | mil—Savakoanina | 0.0045 | 0.0045 | 0.0045 | 0.0045 | n.s. | 0.0045 | 0.0045 | 0.0045 | n.s. | 0 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Crottini, A.; Orozco-terWengel, P.; Rabemananjara, F.C.E.; Hauswaldt, J.S.; Vences, M. Mitochondrial Introgression, Color Pattern Variation, and Severe Demographic Bottlenecks in Three Species of Malagasy Poison Frogs, Genus Mantella. Genes 2019, 10, 317. https://doi.org/10.3390/genes10040317
Crottini A, Orozco-terWengel P, Rabemananjara FCE, Hauswaldt JS, Vences M. Mitochondrial Introgression, Color Pattern Variation, and Severe Demographic Bottlenecks in Three Species of Malagasy Poison Frogs, Genus Mantella. Genes. 2019; 10(4):317. https://doi.org/10.3390/genes10040317
Chicago/Turabian StyleCrottini, Angelica, Pablo Orozco-terWengel, Falitiana C. E. Rabemananjara, J. Susanne Hauswaldt, and Miguel Vences. 2019. "Mitochondrial Introgression, Color Pattern Variation, and Severe Demographic Bottlenecks in Three Species of Malagasy Poison Frogs, Genus Mantella" Genes 10, no. 4: 317. https://doi.org/10.3390/genes10040317
APA StyleCrottini, A., Orozco-terWengel, P., Rabemananjara, F. C. E., Hauswaldt, J. S., & Vences, M. (2019). Mitochondrial Introgression, Color Pattern Variation, and Severe Demographic Bottlenecks in Three Species of Malagasy Poison Frogs, Genus Mantella. Genes, 10(4), 317. https://doi.org/10.3390/genes10040317