The Elaboration of miRNA Regulation and Gene Regulatory Networks in Plant–Microbe Interactions



Abstract
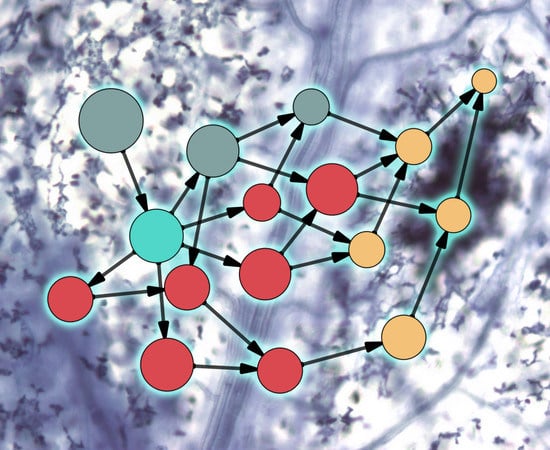
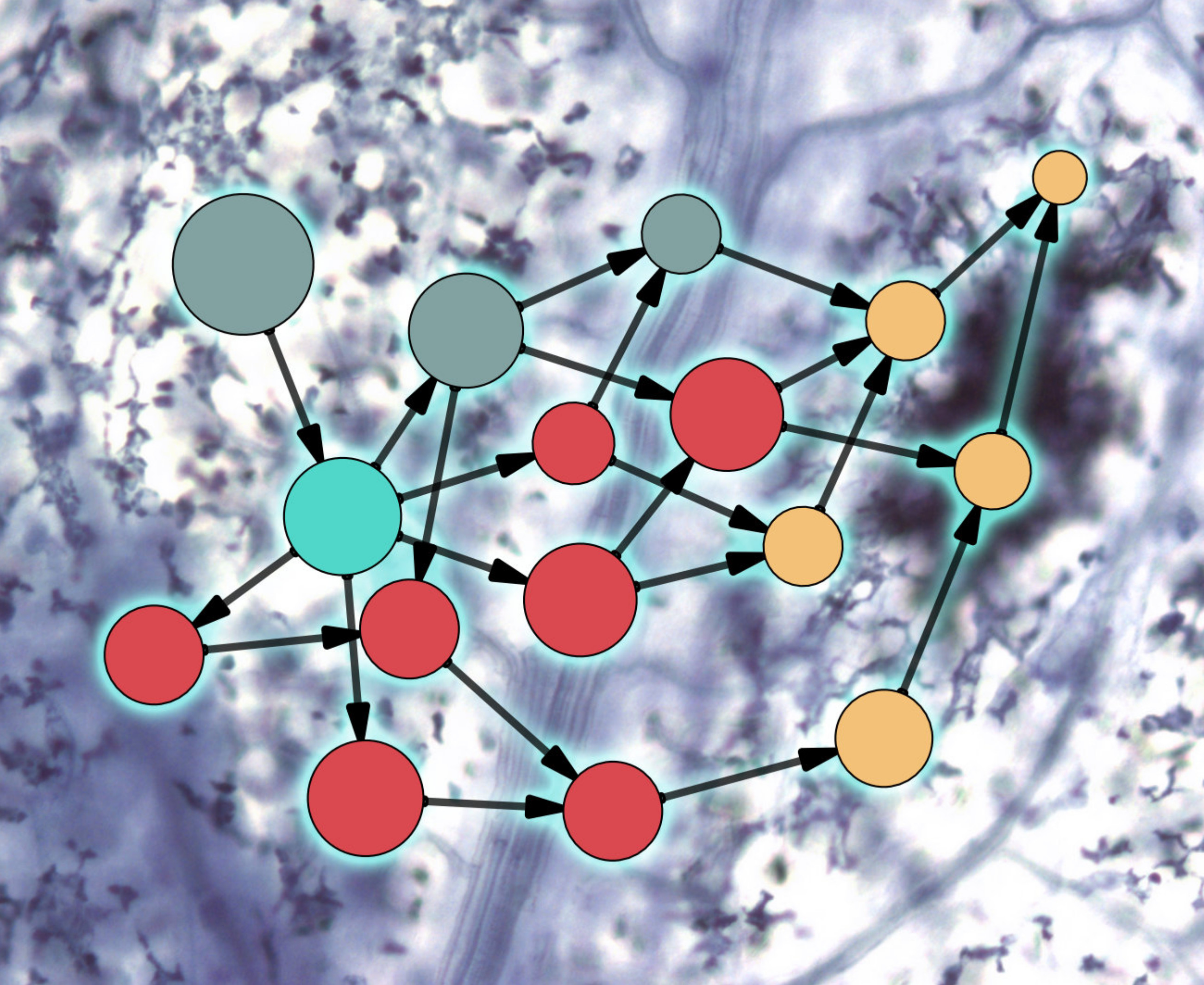{kind=link}
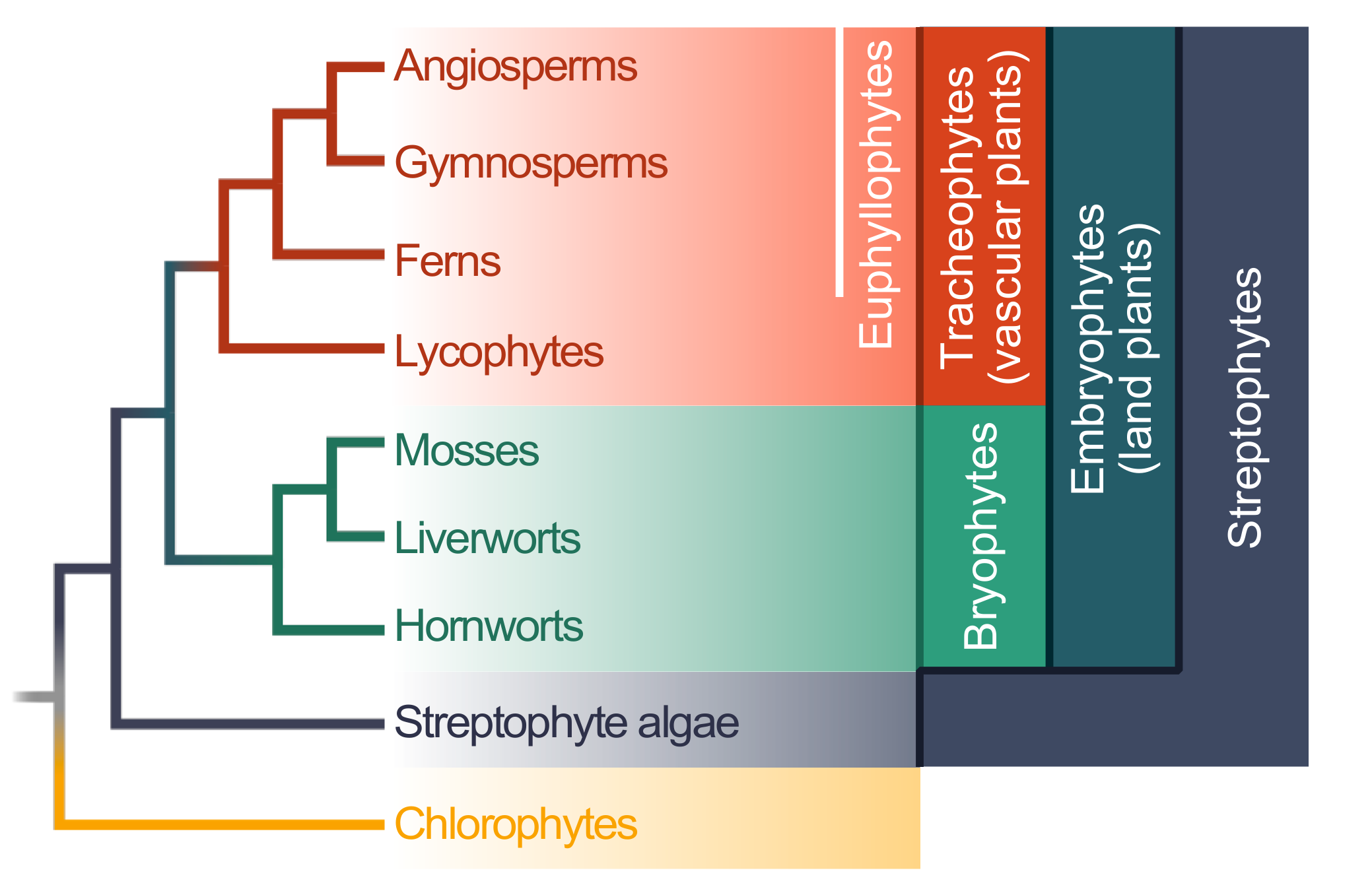{kind=link}
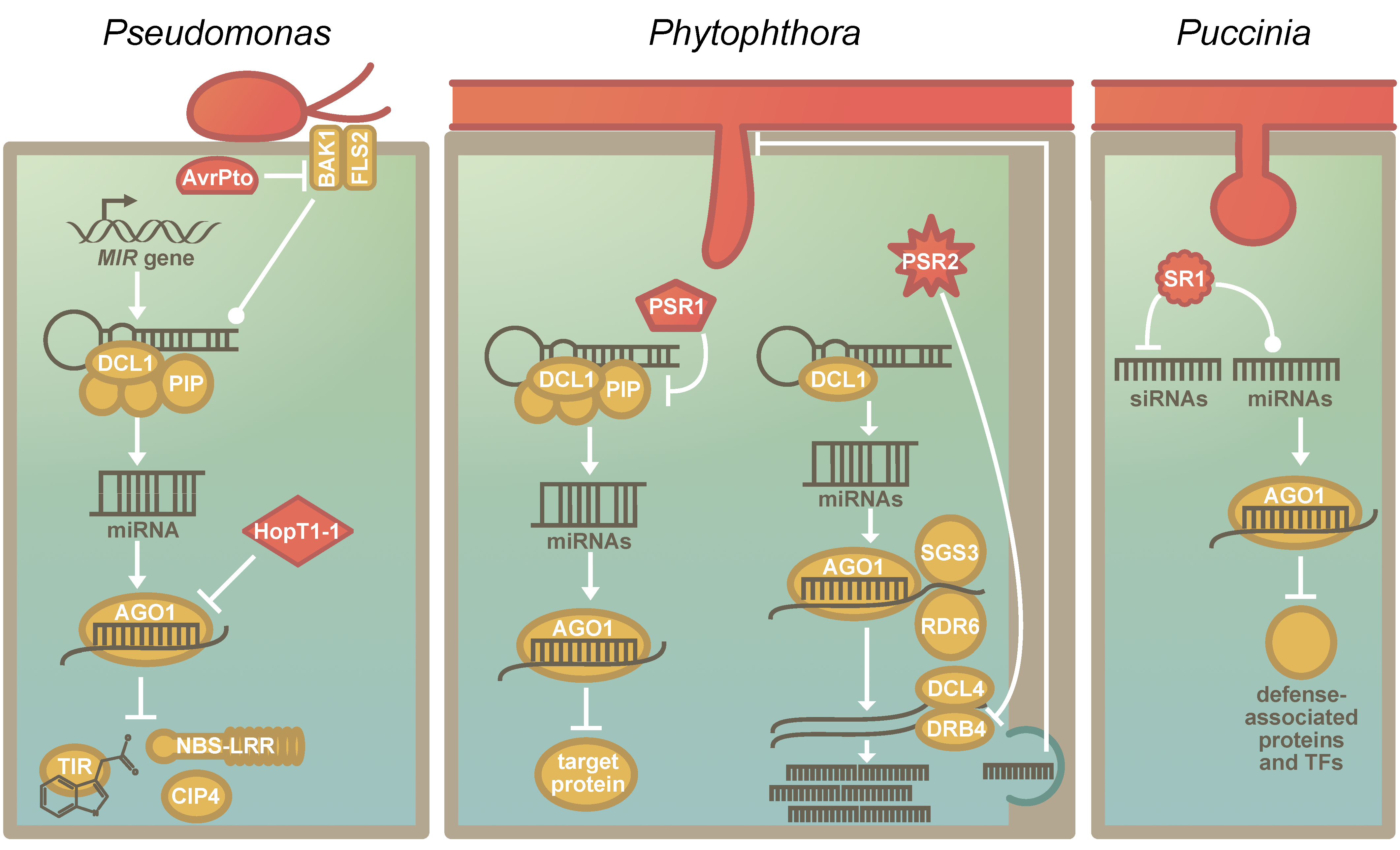{kind=link}
1. Introduction
2. Evolution of Plant–Microbe Interaction Networks
3. The Evolution of microRNA-Mediated Regulation of Resistance Genes
4. Additional microRNAs Regulating Plant Immunity
5. The RNA Silencing Machinery as a Common Target of Pathogens
6. Conclusions
Funding
Conflicts of Interest
References
- Rasmussen, S.; Barah, P.; Suarez-Rodriguez, M.C.; Bressendorff, S.; Friis, P.; Costantino, P.; Bones, A.M.; Nielsen, H.B.; Mundy, J. Transcript responses to combinations of stresses in Arabidopsis. Plant Phys. 2013, 161, 1783–1794. [Google Scholar] [CrossRef]
- Coolen, S.; Proietti, S.; Hickman, R.; Davila Olivas, N.H.; Huang, P.-P.; Van Verk, M.C.; Van Pelt, J.A.; Wittenberg, A.H.J.; De Vos, M.; Prins, M.; et al. Transcriptome dynamics of Arabidopsis during sequential biotic and abiotic stresses. Plant J. 2016, 86, 249–267. [Google Scholar] [CrossRef]
- Zuluaga, A.P.; Vega-Arreguín, J.C.; Fei, Z.; Matas, A.J.; Patev, S.; Fry, W.E.; Rose, J.K.C. Analysis of the tomato leaf transcriptome during successive hemibiotrophic stages of a compatible interaction with the oomycete pathogen Phytophthora infestans. Mol. Plant Pathol. 2016, 17, 42–54. [Google Scholar] [CrossRef] [PubMed]
- Tsuda, K.; Somssich, I.E. Transcriptional networks in plant immunity. New Phytol. 2015, 206, 932–947. [Google Scholar] [CrossRef]
- Song, L.; Huang, S.-S.C.; Wise, A.; Castanon, R.; Nery, J.R.; Chen, H.; Watanabe, M.; Thomas, J.; Bar-Joseph, Z.; Ecker, J.R. A transcription factor hierarchy defines an environmental stress response network. Science 2016, 354, aag1550. [Google Scholar] [CrossRef]
- Scheres, B.; van der Putten, W.H. The plant perceptron connects environment to development. Nature 2017, 543, 337–345. [Google Scholar] [CrossRef]
- Schornack, S.; Huitema, E.; Cano, L.M.; Bozkurt, T.O.; Oliva, R.; van Damme, M.; Schwizer, S.; Raffaele, S.; Chaparro-Garcia, A.; Farrer, R.; et al. Ten things to know about oomycete effectors. Mol. Plant Pathol. 2009, 10, 795–803. [Google Scholar] [CrossRef] [PubMed]
- Kong, L.; Qui, X.; Kang, J.; Wang, Y.; Chen, H.; Huang, J.; Qiu, M.; Zhao, Y.; Kong, G.; Ma, Z.; et al. A Phytophthora effector manipulates host histone acetylation and reprograms defense gene expression to promote infection. Curr. Biol. 2017, 27, 1–11. [Google Scholar] [CrossRef] [PubMed]
- Vijayapalani, P.; Hewezi, T.; Pontvianne, F.; Baum, T.J. An effector from the cyst nematode Heterodera schachtii derepresses host rRNA genes by altering histone acetylation. Plant Cell 2018, 30, 2795–2812. [Google Scholar] [CrossRef]
- Navarro, L.; Jay, F.; Nomura, K.; He, S.Y.; Voinnet, O. Suppression of the microRNA pathway by bacterial effector proteins. Science 2008, 321, 964–967. [Google Scholar] [CrossRef] [PubMed]
- Qiao, Y.; Liu, L.; Xiong, Q.; Flores, C.; Wong, J.; Shi, J.; Wang, X.; Liu, X.; Xiang, Q.; Jiang, S.; et al. Oomycete pathogens encode RNA silencing suppressors. Nat. Genet. 2013, 45, 330–333. [Google Scholar] [CrossRef]
- Yin, C.; Ramachandran, S.R.; Zhai, Y.; Bu, C.; Pappu, H.R.; Hulbert, S.H. A novel fungal effector from Puccinia graminis suppressing RNA silencing and plant defense responses. New Phytol. 2019, 222, 1561–1572. [Google Scholar] [CrossRef]
- McLellan, H.; Boevink, P.C.; Armstrong, M.R.; Pritchard, L.; Gomez, S.; Morales, J.; Whisson, S.C.; Beynon, J.L.; Birch, P.R. An RxLR from Phytophthora infestans prevents re-localisation of two plant NAC transcription factors from the endoplasmic reticulum to the nucleus. PLoS Pathog. 2013, 9, e1003670. [Google Scholar] [CrossRef]
- Kay, S.; Hahn, S.; Marois, E.; Hause, G.; Bonas, U. A bacterial effector acts as a plant transcription factor and induces a cell size regulator. Science 2007, 318, 648–651. [Google Scholar] [CrossRef]
- Boch, J.; Scholze, H.; Schornack, S.; Landgraf, A.; Hahn, S.; Kay, S.; Lahaye, T.; Nickstadt, A.; Bonas, U. Breaking the code of DNA binding specificity of TAL-type III effectors. Science 2009, 326, 1509–1512. [Google Scholar] [CrossRef]
- Bogdanove, A.J.; Schornack, S.; Lahaye, T. TAL effectors: Finding plant genes for disease and defense. Curr. Opin. Plant Biol. 2010, 13, 394–401. [Google Scholar] [CrossRef]
- Weiberg, A.; Wang, M.; Lin, F.M.; Zhao, H.; Zhang, Z.; Kaloshian, I.; Huang, H.D.; Jin, H. Fungal small RNAs suppress plant immunity by hijacking host RNA interference pathways. Science 2013, 342, 118–123. [Google Scholar] [CrossRef]
- Wang, M.; Weiberg, A.; Dellota, E., Jr.; Yamane, D.; Jin, H. Botrytis small RNA Bc-siR37 suppresses plant defense genes by cross-kingdom RNAi. RNA Biol. 2017, 13, 421–428. [Google Scholar] [CrossRef]
- Mukhtar, M.S.; Carvunis, A.-R.; Dreze, M.; Epple, P.; Steinbrenner, J.; Moore, J.; Tasan, M.; Galli, M.; Hao, T.; Nishimura, M.T.; et al. Independently evolved virulence effectors converge onto hubs in a plant immune system network. Science 2011, 333, 596–601. [Google Scholar] [CrossRef]
- Weßling, R.; Epple, P.; Altmann, S.; He, Y.; Yang, L.; Henz, S.R.; McDonald, N.; Wiley, K.; Bader, K.C.; Gläßer, C.; et al. Convergent targeting of a common host protein-network by pathogen effectors from three kingdoms of life. Cell Host Microbe 2014, 16, 364–375. [Google Scholar] [CrossRef]
- Carella, P.; Evangelisti, E.; Schornack, S. Sticking to it: Phytopathogen effector molecules may converge on evolutionarily conserved host targets in green plants. Curr. Opin. Plant Biol. 2018, 44, 174–180. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.L.; Puttick, M.N.; Clark, J.W.; Edwards, D.; Kenrick, P.; Pressel, S.; Wellman, C.H.; Yang, Z.; Schneider, H.; Donoghue, P.C.J. The timescale of early land plant evolution. Proc. Natl. Acad. Sci. USA 2018, 115, E2274–E2283. [Google Scholar] [CrossRef] [PubMed]
- Puttick, M.N.; Morris, J.L.; Williams, T.A.; Cox, C.J.; Edwards, D.; Kenrick, P.; Pressel, S.; Wellman, C.H.; Schneider, H.; Pisani, D.; et al. The interrelationships of land plants and the nature of the ancestral embryophyte. Curr. Biol. 2018, 28, 733–745. [Google Scholar] [CrossRef]
- De Sousa, F.; Foster, P.G.; Donoghue, P.C.J.; Cox, C.J. Nuclear protein phylogenies support the monophyly of the three bryophyte groups (Bryophyta Schimp.). New Phytol. 2018, 222, 565–575. [Google Scholar] [CrossRef]
- Rensing, S.A. Plant evolution: Phylogenetic relationships between the earliest land plants. Curr. Biol. 2018, 28, R210–R213. [Google Scholar] [CrossRef]
- Li, F.-W.; Brouwer, P.; Carretero-Paulet, L.; Cheng, S.; de Vries, J.; Delaux, P.-M.; Eily, A.; Koppers, N.; Li-Yaung, K.; Li, Z.; et al. Fern genomes elucidate land plant evolution and cyanobacterial symbioses. Nat. Plants 2018, 4, 460–472. [Google Scholar] [CrossRef] [PubMed]
- Provart, N.J.; Alonso, J.; Assmann, S.M.; Bergmann, D.; Brady, S.M.; Brkljacic, J.; Browse, J.; Chapple, C.; Colot, V.; Cutler, S.; et al. 50 years of Arabidopsis research: Highlights and future directions. New Phytol. 2016, 209, 921–944. [Google Scholar] [CrossRef] [PubMed]
- Rensing, S.A. Why we need more non-seed plant models. New Phytol. 2017, 216, 355–360. [Google Scholar] [CrossRef]
- Chang, C.; Bowman, J.L.; Meyerowitz, E.M. Field guide to plant model systems. Cell 2016, 167, 325–339. [Google Scholar] [CrossRef]
- Rensing, S.A.; Lang, D.; Zimmer, A.D.; Terry, A.; Salamov, A.; Shapiro, H.; Nishiyama, T.; Perroud, P.F.; Lindquist, E.A.; Kamisugi, Y.; et al. The Physcomitrella genome reveals evolutionary insights into the conquest of land by plants. Science 2008, 319, 64–69. [Google Scholar] [CrossRef]
- Lang, D.; Ullrich, K.K.; Murat, F.; Fuchs, J.; Jenkins, J.; Haas, F.B.; Piednoel, M.; Gundlach, H.; Van Bel, M.; Meyberg, R.; et al. The Physcomitrella patens chromosome-scale assembly reveals moss genome structure and evolution. Plant J. 2018, 93, 515–533. [Google Scholar] [CrossRef]
- Perroud, P.-F.; Haas, F.B.; Hiss, M.; Ullrich, K.K.; Alboresi, A.; Amirebrahimi, M.; Barry, K.; Bassi, R.; Bonhomme, S.; Chen, H.; et al. The Physcomitrella patens gene atlas project: Large-scale RNA-seq based expression data. Plant J. 2018, 95, 168–182. [Google Scholar] [CrossRef]
- Bowman, J.L.; Kohchi, T.; Yamato, K.T.; Jenkins, J.; Shu, S.; Ishizaki, K.; Yamaoka, S.; Nishihama, R.; Nakamura, Y.; Berger, F.; et al. Insights into land plant evolution garnered from the Marchantia polymorpha genome. Cell 2017, 171, 287–304. [Google Scholar] [CrossRef] [PubMed]
- Catarino, B.; Hetherington, A.J.; Emms, D.M.; Kelly, S.; Dolan, L. The stepwise increase in the number of transcription factor families in the precambrian predated the diversification of plants on land. Mol. Biol. Evol. 2016, 33, 2815–2819. [Google Scholar] [CrossRef]
- Wilhelmsson, P.K.I.; Mühlich, C.; Ullrich, K.K.; Rensing, S.A. Comprehensive genome-wide classification reveals that many plant-specific transcription factors evolved in streptophyte algae. Genome Biol. Evol. 2017, 9, 3384–3397. [Google Scholar] [CrossRef] [PubMed]
- Delwiche, C.F.; Cooper, E.D. The evolutionary origin of a terrestrial flora. Curr. Biol. 2015, 25, R899–R910. [Google Scholar] [CrossRef]
- De Vries, J.; Archibald, J.M. Plant evolution: Landmarks on the path to terrestrial life. New Phytol. 2018, 217, 1428–1434. [Google Scholar] [CrossRef] [PubMed]
- Bowman, J.L.; Briginshaw, L.N.; Fisher, T.J.; Flores-Sandoval, E. Something ancient and something neofunctionalized—Evolution of land plant hormone signaling pathways. Curr. Opin. Plant Biol. 2019, 47, 64–77. [Google Scholar] [CrossRef]
- Umezawa, T.; Nakashima, K.; Miyakawa, T.; Kuromori, T.; Tanokura, M.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Molecular basis of the core regulatory network in ABA responses: Sensing, signaling and transport. Plant Cell Physiol. 2010, 51, 1821–1839. [Google Scholar] [CrossRef] [PubMed]
- Hauser, F.; Waadt, R.; Schroeder, J.I. Evolution of abscisic acid synthesis and signalling mechanisms. Curr. Biol. 2011, 21, R346–R355. [Google Scholar] [CrossRef]
- Eklund, D.M.; Kanei, M.; Flores-Sandoval, E.; Ishizaki, K.; Nishihama, R.; Kohchi, T.; Lagercrantz, U.; Bhalerao, R.P.; Sakata, Y.; Bowman, J.L. An evolutionarily conserved abscisic acid signaling pathway regulates dormancy in the liverwort Marchantia polymorpha. Curr. Biol. 2019, 28, 3691–3699. [Google Scholar] [CrossRef]
- De Vries, J.; Curtis, B.A.; Gould, S.B.; Archibald, J.M. Embryophyte stress signaling evolved in the algal progenitors of land plants. Proc. Natl. Acad. Sci. USA 2018, 115, E3471–E3480. [Google Scholar] [CrossRef]
- De Vries, S.; de Vries, J.; von Dahlen, J.; Gould, S.B.; Archibald, J.M.; Rose, L.E.; Slamovits, C.H. On plant defense signaling networks and early land plant evolution. Commun. Integr. Biol. 2018, 11, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Peng, Y.; Sun, T.; Zhang, Y. Perception of salicylic acid in Physcomitrella patens. Front. Plant Sci. 2017, 8, 2145. [Google Scholar] [CrossRef] [PubMed]
- Hori, K.; Maruyama, F.; Fujisawa, T.; Togashi, T.; Yamamoto, N.; Seo, M.; Sato, S.; Yamada, T.; Mori, H.; Tajima, N.; et al. Klebsormidium flaccidum genome reveals primary factors for plant terrestrial adaptation. Nat. Commun. 2014, 5, 3978. [Google Scholar] [CrossRef] [PubMed]
- Ju, C.; Van de Poel, B.; Cooper, E.D.; Thierer, J.H.; Gibbons, T.R.; Delwiche, C.F.l.; Chang, C. Conservation of ethylene as a plant hormone over 450 million years of evolution. Nat. Plants. 2015, 1, 14004. [Google Scholar] [CrossRef] [PubMed]
- Ohtaka, K.; Hori, K.; Kanno, Y.; Seo, M.; Ohta, H. Primitive auxin response without TIR1 and Aux/IAA in the charophyte alga Klebsormidium nitens. Plant Physiol. 2017, 174, 1621–1632. [Google Scholar] [CrossRef]
- Nishiyama, T.; Sakayama, H.; de Vries, J.; Saint-Marcoux, D.; Ullrich, K.K.; Haas, F.B.; Vanderstraeten, L.; Becker, D.; Lang, D.; Vosolsobě, S.; et al. The Chara genome: Secondary complexity and implications for plant terrestrialization. Cell 2018, 174, 448–464. [Google Scholar] [CrossRef]
- De Vries, J.; de Vries, S.; Slamovits, C.H.; Rose, L.E.; Archibald, J.M. How embryophytic is the biosynthesis of phenylpropanoids and their derivatives in streptophyte algae? Plant Cell Physiol. 2017, 58, 934–945. [Google Scholar] [CrossRef] [PubMed]
- Monte, I.; Ishida, S.; Zamarreño, A.M.; Hamber, M.; Franco-Zorilla, J.M.; García-Casado, G.; Gouhier-Darimont, C.; Reymond, P.; Takahashi, K.; García-Mina, J.M.; et al. Ligand-receptor co-evolution shaped the jasmonate pathway in land plants. Nat. Chem. Biol. 2018, 14, 480–488. [Google Scholar] [CrossRef]
- Monte, I.; Franco-Zorilla, J.M.; García-Casado, G.; Zamarreño, A.M.; García-Mina, J.M.; Nishihama, R.; Kohchi, T.; Solano, R. A single JAZ repressor controls the jasmonate pathway in Marchantia polymorpha. Mol. Plant. 2019, 12, 185–198. [Google Scholar] [CrossRef]
- Delaux, P.-M.; Radhakrishnan, G.V.; Jayaraman, D.; Cheema, J.; Malbreil, M.; Volkening, J.D.; Sekimoto, H.; Nishiyama, T.; Melkonian, M.; Pokorny, L.; et al. Algal ancestor of land plants was preadapted for symbiosis. Proc. Natl. Acad. Sci. USA 2015, 112, 13390–13395. [Google Scholar] [CrossRef]
- Zipfel, C.; Oldroyd, G.E.D. Plant signalling in symbiosis and immunity. Nature 2017, 543, 328–336. [Google Scholar] [CrossRef]
- Belkhadir, Y.; Subramaniam, R.; Dangle, J.L. Plant disease resistance protein signaling: NBS-LRR proteins and their partners. Curr. Opin. Plant Biol. 2004, 7, 391–399. [Google Scholar] [CrossRef]
- Yue, J.-X.; Meyers, B.C.; Chen, J.-Q.; Tian, D.; Yang, S. Trancing the origin and evolutionary history of plant nucleotide-binding site-leucine-rich repeat (NBS-LRR) genes. New Phytol. 2012, 193, 1049–1063. [Google Scholar] [CrossRef]
- Gao, Y.; Wang, W.; Zhang, T.; Gong, Z.; Zhao, H.; Han, G.-Z. Out of the water: The origin and early diversification of plant R-genes. Plant Physiol. 2018, 177, 82–89. [Google Scholar] [CrossRef]
- Shao, Z.-Q.; Xue, J.-Y.; Wang, Q.; Wang, B.; Chen, J.-Q. Revisiting the origin of plant NBS-LRR genes. Trends Plant Sci. 2019, 24, 9–12. [Google Scholar] [CrossRef]
- Han, G.Z. Origin and evolution of the plant immune system. New Phytol. 2019, 222, 70–83. [Google Scholar] [CrossRef]
- DeYoung, B.J.; Innes, R.W. Plant NBS-LRR proteins in pathogen sensing and host defense. Nat. Immunol. 2006, 7, 1243–1249. [Google Scholar] [CrossRef]
- Zhai, J.; Jeong, D.-H.; De Paoli, E.; Park, S.; Rosen, B.D.; Li, Y.; González, A.J.; Yan, Z.; Kitto, S.L.; Grusak, M.A.; et al. MicroRNAs as master regulators of the plant NB-LRR defense gene family via the production of phased, trans-acting siRNAs. Genes Dev. 2011, 25, 2540–2553. [Google Scholar] [CrossRef]
- Shivaprasad, P.V.; Chen, H.M.; Patel, K.; Bond, D.M.; Santos, B.A.; Baulcombe, D.C. A microRNA superfamily regulates nucleotide bindingsite-leucine-rich repeats and other mRNAs. Plant Cell 2012, 24, 859–874. [Google Scholar] [CrossRef] [PubMed]
- Kurihara, Y.; Watanabe, Y. Arabidopsis micro-RNA biogenesis through Dicer-like 1 protein functions. Proc. Natl. Acad. Sci. USA 2004, 101, 12753–12758. [Google Scholar] [CrossRef]
- Nagano, H.; Fukudome, A.; Hiraguri, A.; Moriyama, H.; Fukuhara, T. Distinct substrate specificities of Arabidopsis DCL3 and DCL4. Nucleic Acids Res. 2014, 42, 1845–1856. [Google Scholar] [CrossRef] [PubMed]
- Rogers, K.; Chen, X. Biogenesis, turnover, and of action of plant microRNAs. Plant Cell 2013, 25, 2383–2399. [Google Scholar] [CrossRef]
- Fagard, M.; Boutet, S.; Morel, J.-B.; Bellini, C.; Vaucheret, H. AGO1, QDE-2, and RDE-1 are related proteins required for post-transcriptional gene silencing in plants, quelling in fungi, and RNA interference in animals. Proc. Natl. Acad. Sci. USA 2000, 97, 11650–11654. [Google Scholar] [CrossRef]
- De Vries, S.; Kukuk, A.; von Dahlen, J.K.; Schnake, A.; Kloesges, T.; Rose, L.E. Expression profiling across wild and cultivated tomatoes supports the relevance of early miR482/2118 suppression for Phytophthora resistance. Proc. Biol. Sci. 2018, 285, 20172560. [Google Scholar] [CrossRef]
- Ouyang, S.; Park, G.; Atamian, H.S.; Han, C.S.; Stajich, J.E.; Kaloshian, I.; Borkovich, K.A. MicroRNAs suppress NB domain genes in tomato that confer resistance to Fusarium oxysporum. PLoS Pathog. 2014, 10, e1004464. [Google Scholar] [CrossRef]
- Allen, E.; Xie, Z.; Gustafson, A.M.; Carrington, J.C. microRNA-directed phasing during trans-acting siRNA biogenesis in plants. Cell 2005, 121, 207–221. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Peragine, A.; Park, M.Y.; Poethig, R.S. A pathway for the biogenesis of trans-acting siRNAs in Arabidopsis. Genes Dev. 2005, 19, 2164–2175. [Google Scholar] [CrossRef]
- Axtell, M.J.; Jan, C.; Rajagopalan, R.; Bartel, D.P. A two-hit trigger for siRNA biogenesis in plants. Cell 2006, 127, 565–577. [Google Scholar] [CrossRef]
- Chen, H.-M.; Chen, L.-T.; Patel, K.; Li, Y.-H.; Baulcombe, D.C.; Wu, S.-H. 22-nucleotide RNAs trigger secondary siRNA biogenesis in plants. Proc. Natl. Acad. Sci. USA 2010, 107, 15269–15274. [Google Scholar] [CrossRef]
- Cuperus, J.T.; Carbonell, A.; Fahlgren, N.; Garcia-Ruiz, H.; Burke, R.T.; Takeda, A.; Sullivan, C.M.; Montgomery, T.A.; Carrington, J.C. Unique functionality of 22-nt miRNAs in triggering RDR6-dependent siRNA biogenesis from target transcripts in Arabidopsis. Nat. Struct. Mol. Biol. 2010, 17, 997–1003. [Google Scholar] [CrossRef]
- De Vries, S.; Kloesges, T.; Rose, L.E. Evolutionarily dynamic, but robust, targeting of resistance genes by the miR482/2118 gene family in the Solanaceae. Genome Biol. Evol. 2015, 19, 3307–3321. [Google Scholar] [CrossRef]
- Nozawa, M.; Miura, S.; Nei, M. Origins and evolution of microRNA genes in plant species. Genome Biol. Evol. 2012, 4, 230–239. [Google Scholar] [CrossRef]
- Xia, R.; Xu, J.; Arikit, S.; Meyers, B.C. Extensive families of miRNAs and PHAS loci Norway spruce demonstrate the complex phasiRNA networks in seed plants. Mol. Biol. Evol. 2015, 32, 2905–2918. [Google Scholar] [CrossRef]
- Zhai, J.; Zhang, H.; Arikit, S.; Huang, K.; Nan, G.-L.; Walbot, V.; Meyers, B.C. Spatiotemporally dynamic, cell-type–dependent premeiotic and meiotic phasiRNAs in maize anthers. Proc. Natl. Acd. Sci. USA 2015, 112, 3146–3151. [Google Scholar] [CrossRef]
- Boccara, M.; Sarazin, A.; Thiébeauld, O.; Jay, F.; Voinnet, O.; Navarro, L.; Colot, V. The Arabidopsis miR482-RDR6 silencing pathway modulates PAMP- and effector-triggered immunity through the post-transcriptional control of disease resistance genes. PLoS Pathog. 2014, 10, e1003883. [Google Scholar] [CrossRef]
- Canto-Pastor, A.; Santos, B.A.M.C.; Valli, A.A.; Summers, W.; Schornack, S.; Baulcombe, D.C. Enhanced resistance to bacterial and oomycete pathogens by short tandem target mimic RNAs in tomato. Proc. Natl. Acad. Sci. USA 2019, 116, 2755–2760. [Google Scholar] [CrossRef]
- Zhang, Y.; Xia, R.; Kuang, H.; Meyers, B.C. The diversification of plant NBS-LRR defense genes directs the evolution of microRNAs that target them. Mol. Biol. Evol. 2016, 33, 2692–2705. [Google Scholar] [CrossRef]
- Fahlgren, N.; Jogdeo, S.; Kasschau, K.D.; Sullivan, C.M.; Chapman, E.J.; Laubringer, S.; Smith, L.M.; Dasenko, M.; Givan, S.A.; Weigel, D.; et al. MicroRNA gene evolution in Arabidopsis lyrata and Arabidopsis thaliana. Plant Cell 2010, 22, 1074–1089. [Google Scholar] [CrossRef]
- Rose, L.E.; Bittner-Eddy, P.D.; Langley, C.H.; Holub, E.B.; Michelmore, R.W.; Beynon, J.L. The maintenance of extreme amino acid diversity at the disease resistance gene, RPP13, in Arabidopsis thaliana. Genetics 2004, 166, 1517–1527. [Google Scholar] [CrossRef]
- Clark, R.M.; Schweikert, G.; Toomajian, C.; Ossowski, S.; Zeller, G.; Shinn, P.; Warthmann, N.; Hu, T.T.; Fu, G.; Hinds, D.A.; et al. Common sequence polymorphisms shaping genetic diversity in Arabidopsis thaliana. Science 2007, 317, 338–342. [Google Scholar] [CrossRef]
- Rose, L.E.; Michelmore, R.W.; Langley, C.H. Natural variation in the Pto disease resistance gene within species of wild tomato (Lycopersicon) II. Population genetics of Pto. Genetics 2007, 175, 1307–1319. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Jiang, N.; Cui, J.; Yang, G.; He, X.; Meng, J.; Luan, Y. Comparative transcriptome analysis shows the defense response networks regulated by miR482b. Plant Cell Rep. 2019, 38, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Islam, W.; Islam, S. u.; Qasim, M.; Wang, L. Host-pathogen interactions modulated by small RNAs. RNA Biol. 2017, 14, 891–904. [Google Scholar] [CrossRef]
- Rose, L.E.; Overdijk, E.J.R.; van Damme, M. Small RNA molecules and their role in plant disease. Eur. J. Plant Pathol. 2019, 153, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Chávez Montes, R.A.; de Fátima Rosas-Cárdenas, F.; De Paoli, E.; Accerbi, M.; Rymarquis, L.A.; Mahalingam, G.; Marsch-Martínez, N.; Meyers, B.C.; Green, P.J.; de Folter, F. Sample sequencing of vascular plants demonstrates widespread conservation and divergence of microRNAs. Nat. Commun. 2014, 5, 3722. [Google Scholar] [CrossRef]
- Zhang, T.; Zhao, Y.-L.; Zhao, J.-H.; Wang, S.; Jin, Y.; Chen, Z.-Q.; Fang, Y.-Y.; Hua, C.-L.; Ding, S.-W.; Guo, H.-S. Cotton plants export microRNAs to inhibit virulence gene expression in a fungal pathogen. Nat. Plants 2016, 2, 16153. [Google Scholar] [CrossRef] [PubMed]
- Juan, Y.; Cui, J.; Li, J.; Jiang, N.; Liu, P.; Meng, J. Effective enhancement of resistance to Phytophthora infestans by overexpression of miR172a and b in Solanum lycopersicum. Planta 2018, 247, 127–138. [Google Scholar]
- Voinnet, O. Induction and suppression of RNA silencing: Insights from viral infections. Nat. Rev. Genet. 2005, 6, 206–220. [Google Scholar] [CrossRef]
- Vetukuri, R.R.; Whisson, S.C.; Grenville-Briggs, L.J. Phytophthora infestans effector Pi14054 is a novel candidate suppressor of host silencing mechanisms. Eur. J. Plant Pathol. 2017, 149, 771–777. [Google Scholar] [CrossRef]
- Qiao, Y.; Shi, J.; Zhai, Y.; Hou, Y.; Ma, W. Phytophthora effector targets a novel component of small RNA pathway in plants to promote infection. Proc. Natl. Acad. Sci. USA 2015, 112, 5850–5855. [Google Scholar] [CrossRef]
- Fei, Q.; Xia, R.; Meyers, B.C. Phased, secondary, small interfering RNAs in posttranscriptional regulatory networks. Plant Cell 2013, 25, 2400–2415. [Google Scholar] [CrossRef] [PubMed]
- Xiong, Q.; Ye, W.; Choi, D.; Wong, J.; Qiao, Y.; Tao, K.; Wang, Y.; Ma, W. Phytophthora suppressor of RNA silencing 2 is a conserved RxLR effector that promotes infection in soybean and Arabidopsis thaliana. Mol. Plant Microbe Interact. 2014, 27, 1379–1389. [Google Scholar] [CrossRef] [PubMed]
- De Vries, S.; von Dahlen, J.K.; Uhlmann, C.; Schnake, A.; Kloesges, T.; Rose, L.E. Signatures of selection and host-adapted gene expression of the Phytophthora infestans RNA silencing suppressor PSR2. Mol. Plant Pathol. 2017, 18, 110–124. [Google Scholar] [CrossRef]
- Ye, W.; Ma, W. Filamentous pathogen effectors interfering with small RNA silencing in plant hosts. Curr. Opin. Microbiol. 2016, 32, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Boutemy, L.S.; King, S.R.F.; Win, J.; Hughes, R.K.; Clarke, T.A.; Blumenschein, T.M.A.; Kamoun, S.; Banfield, M.J. Structures of Phytophthora RxLR effector proteins a conserved but adaptable fold underpins functional diversity. J. Biol. Chem. 2011, 286, 35834–35842. [Google Scholar] [CrossRef]
- Hou, Y.; Zhai, Y.; Feng, L.; Karimi, H.Z.; Rutter, B.D.; Zeng, L.; Choi, D.S.; Zhang, B.; Gu, W.; Chen, X.; et al. A Phytophthora effector suppresses trans-kingdom RNAi to promote disease susceptibility. Cell Host Microbe 2019, 25, 153–165. [Google Scholar] [CrossRef]
- Hiraguri, A.; Itoh, R.; Kondo, N.; Nomura, Y.; Aizawa, D.; Murai, Y.; Koiwa, H.; Seki, M.; Shinozaki, K.; Fukuhara, T. Specific interactions between Dicer-like proteins and HYL1/DRB-family dsRNA-binding proteins in Arabidopsis thaliana. Plant Mol. Biol. 2005, 57, 173–188. [Google Scholar] [CrossRef] [PubMed]
- Adenot, X.; Elmayan, T.; Lauressergues, D.; Boutet, S.; Bouché, N.; Gasciolli, V.; Vaucheret, H. DRB4-dependent TAS3 trans-actting siRNAs control leaf morphology through AGO7. Curr. Biol. 2006, 16, 927–932. [Google Scholar] [CrossRef]
- Nakazawa, Y.; Hiraguri, A.; Moriyama, H.; Fukuhara, T. The dsRNA-binding protein DRB4 interacts with the Dicer-like protein DCL4 in vivo and functions in the trans-acting siRNA pathway. Plant Mol. Biol. 2007, 63, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Carella, P.; Gogleva, A.; Tomaselli, M.; Alfs, C.; Schornack, S. Phytophthora palmivora establishes tissue-specific intracellular infection structures in the earliest divergent land plant lineage. Proc. Natl. Acad. Sci. USA 2018, 115, E3846–E3855. [Google Scholar] [CrossRef]
- Cho, S.H.; Coruh, C.; Axtell, M.J. miR156 and miR390 regulate tasiRNA accumulation and developmental timing in Physcomitrella patens. Plant Cell 2012, 24, 4837–4849. [Google Scholar] [CrossRef] [PubMed]
- Tsuzuki, A.; Nishihama, R.; Ishizaki, K.; Kurihara, Y.; Matsui, M.; Bowman, J.L.; Kohchi, T.; Hamada, T.; Watanabe, Y. Profiling and characterization of small RNAs in the liverwort, Marchantia polymorpha, belonging to the first diverged land plants. Plant Cell Physiol. 2016, 57, 359–372. [Google Scholar] [CrossRef] [PubMed]
- Clavel, M.; Pélissier, T.; Montavon, T.; Tschopp, M.-A.; Pouch-Pélissier, M.-N.; Descombin, J.; Jean, V.; Dunoyer, P.; Bousquet-Antonelli, C.; Deragon, J.-M. Evolutionary history of double-stranded RNA binding proteins in plants: Identification of new cofactors involved in easiRNA biogenesis. Plant Mol. Biol. 2016, 91, 131–147. [Google Scholar] [CrossRef]


© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
de Vries, S.; de Vries, J.; Rose, L.E. The Elaboration of miRNA Regulation and Gene Regulatory Networks in Plant–Microbe Interactions. Genes 2019, 10, 310. https://doi.org/10.3390/genes10040310
de Vries S, de Vries J, Rose LE. The Elaboration of miRNA Regulation and Gene Regulatory Networks in Plant–Microbe Interactions. Genes. 2019; 10(4):310. https://doi.org/10.3390/genes10040310
Chicago/Turabian Stylede Vries, Sophie, Jan de Vries, and Laura E. Rose. 2019. "The Elaboration of miRNA Regulation and Gene Regulatory Networks in Plant–Microbe Interactions" Genes 10, no. 4: 310. https://doi.org/10.3390/genes10040310
APA Stylede Vries, S., de Vries, J., & Rose, L. E. (2019). The Elaboration of miRNA Regulation and Gene Regulatory Networks in Plant–Microbe Interactions. Genes, 10(4), 310. https://doi.org/10.3390/genes10040310