Identification of Transcription Factors Involved in the Regulation of Flowering in Adonis Amurensis Through Combined RNA-seq Transcriptomics and iTRAQ Proteomics

 ,
,
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. RNA-seq and Transcriptome Analyses
2.3. Analysis of Differentially Expressed Genes (DEGs)
2.4. Protein Extraction and iTRAQ Proteome Analysis
2.5. Quantitative Real-Time PCR (qPCR) for Validation
3. Results
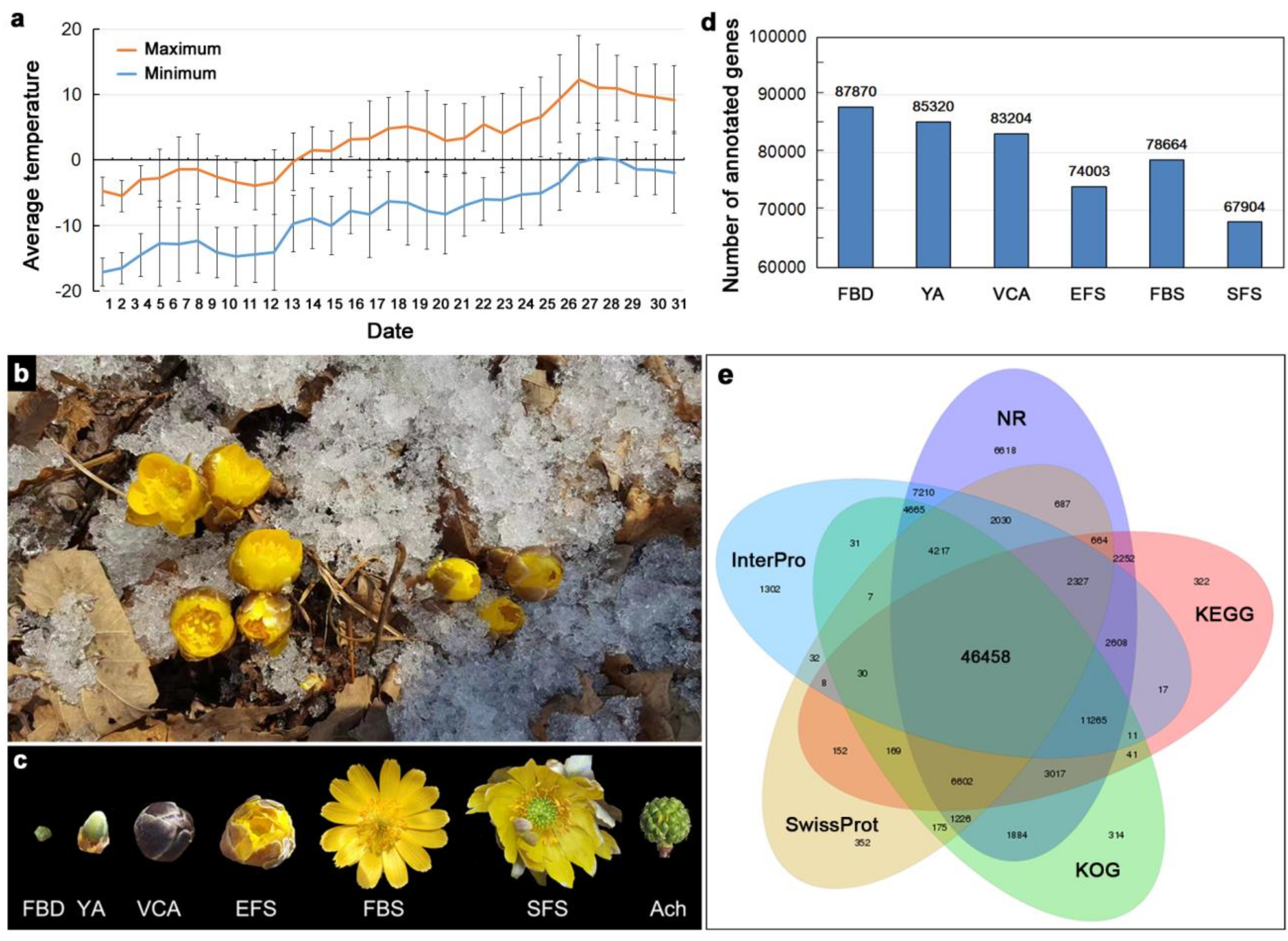
3.1. Transcriptome Sequencing of Floral Organs in A. amurensis
3.2. Identification of Differentially Expressed Genes (DEGs) During Flower Development
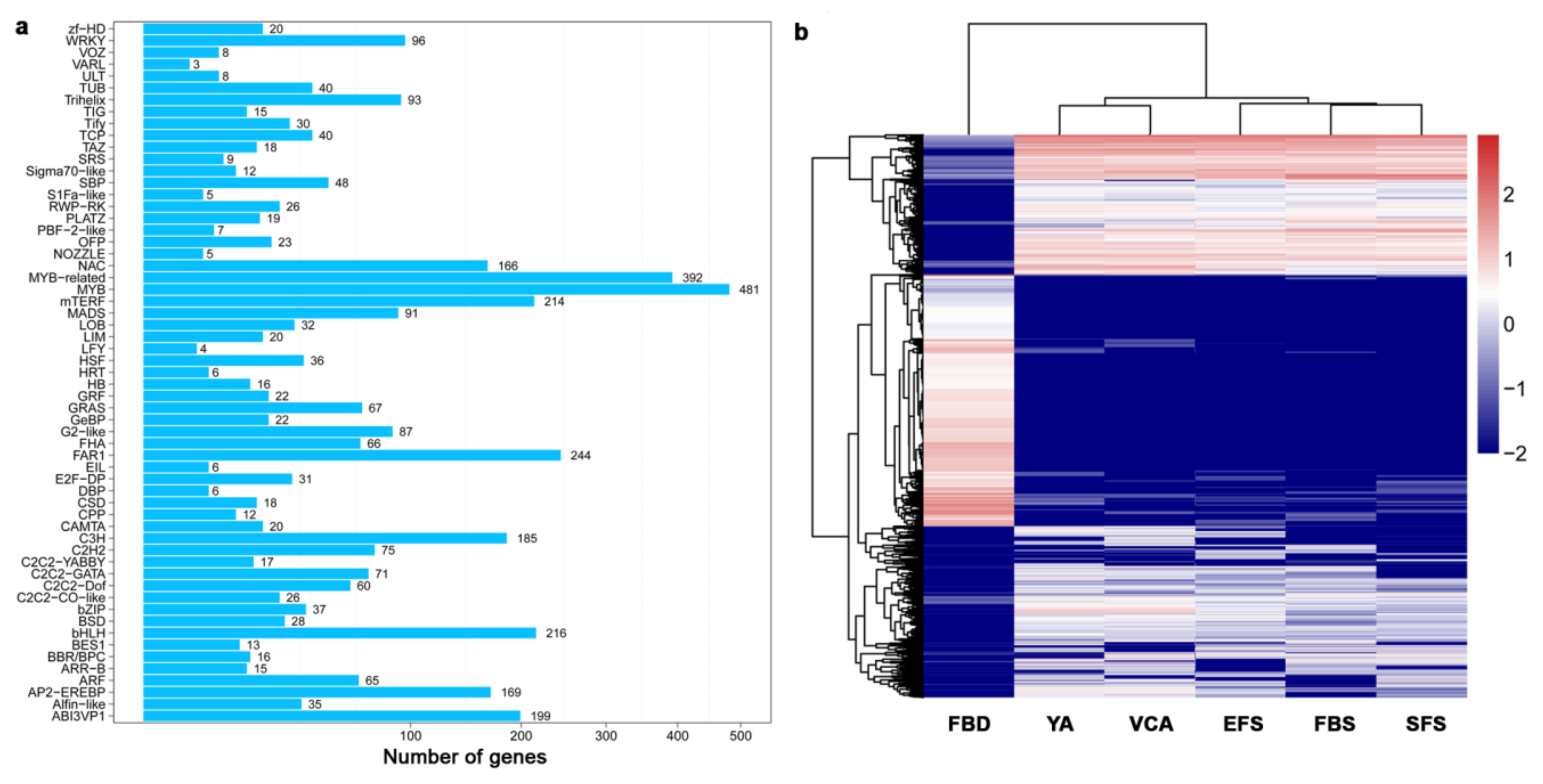
3.3. Identification and Classification of Transcription Factors (TF) During Flower Development
3.4. Proteome Sequencing of Floral Organs in A. amurensis
3.5. Identification of TFs Involved in Flowering by Combining Transcriptomic and Proteomic Data
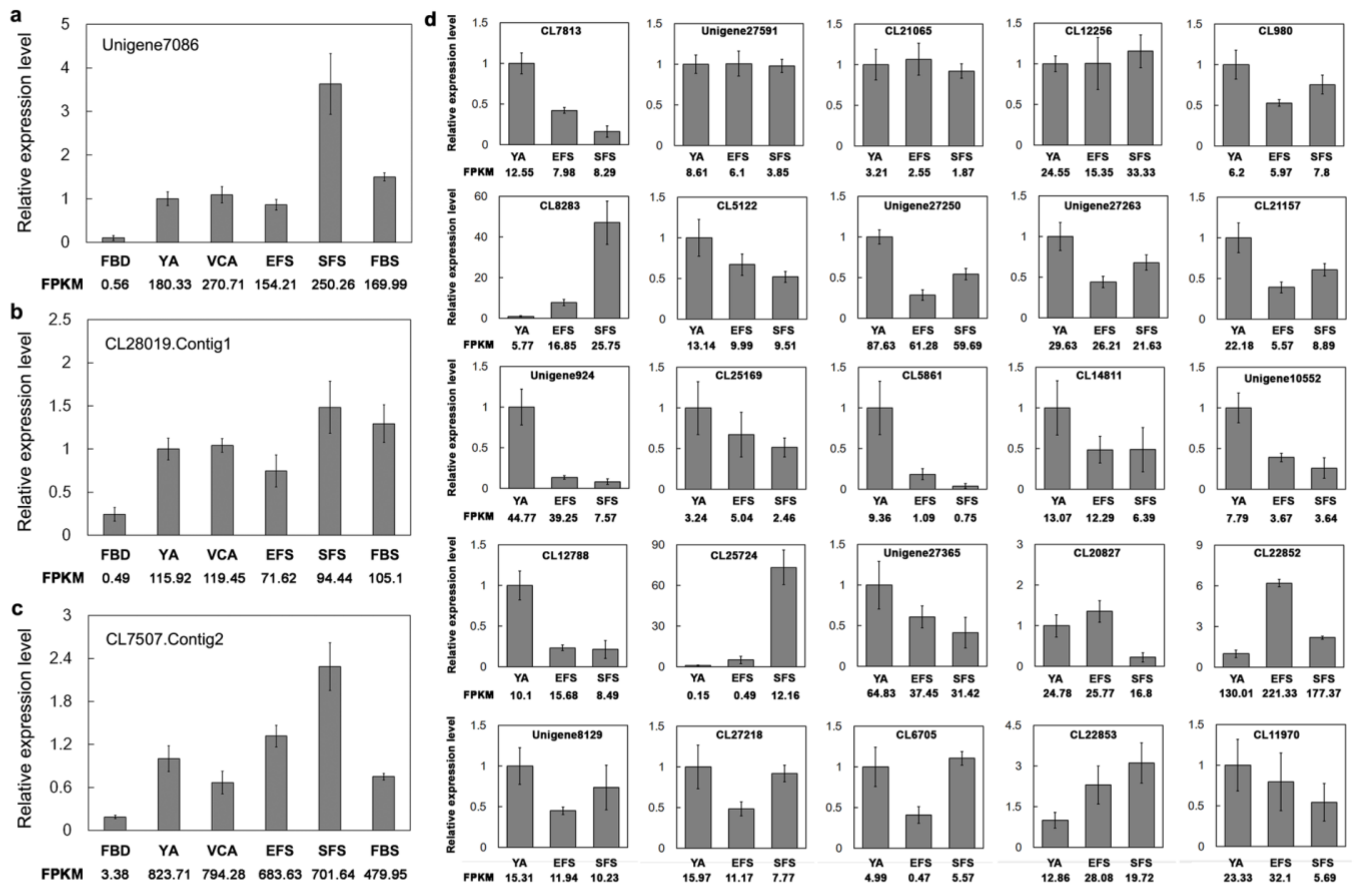
3.6. Verification of Gene Expression of TFs Involved in Flowering
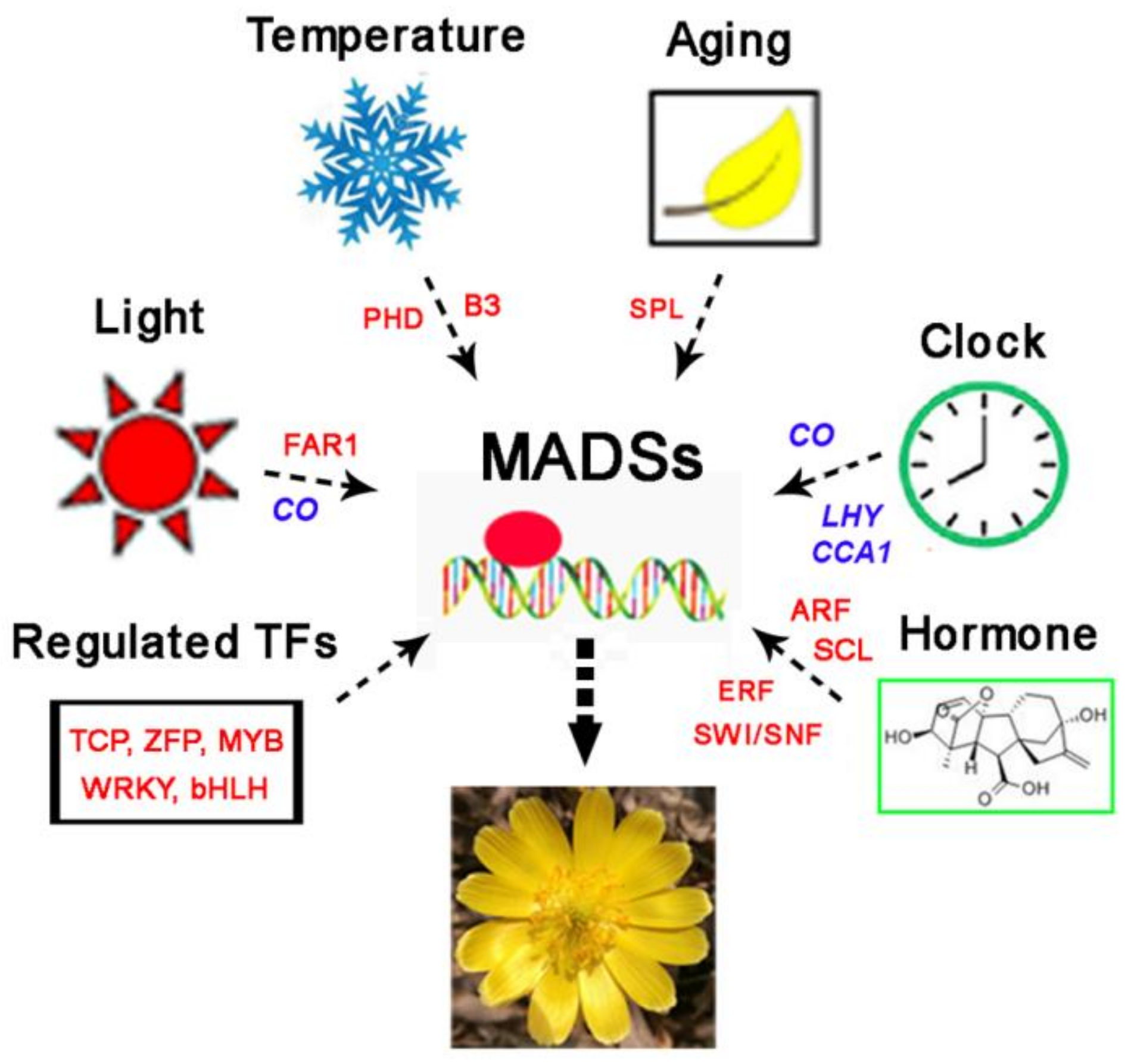
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| FAR1 | Far-red-impaired response 1 |
| PHY3 | Far-red elongated hypocotyls 3 |
| PHD | Plant homeodomain |
| ABI3VP1 | ABA-insensitive 3/viviparous 1 |
| SCL | Scarecrow-like |
| SWI/SNF | Switch/sucrose nonfermentable |
| ARF | Auxin response factor |
| ERF | Ethylene response factor |
| TCP | Teosinte branched 1/cycloidea/pcf |
| ZFP | Zinc finger protein |
| bHLH | Basic helix-loop-helix |
| MADS | Mcm 1/agamous/deficiens/srf |
| mTERF | Mitochondrial transcription termination factor |
| AP2-EREBPs | Apetala2-ethylene-responsive element binding proteins |
| CCA1 | Circadian clock associated 1 |
| LHY | Late elongated hypocotyl |
| TOC1 | Timing of cab expressin 1 |
| FT | Flowering locus T |
| TSF | Twin sister of FT |
| FLC | Flowering locus C |
| CO | Constans |
| SOC1 | Suppressor of overexpression of CO 1 |
| VRN2 | Vernalization 2 |
| VIN3 | Vernalization insensitive 3 |
| SVP | Short vegetative phase |
| GA | Gibberellin |
| GA20ox | Gibberellin 20 oxidase |
| SPL | Squamosa promoter binding protein-like |
| FUL | Fruitfull |
| PEP1 | Perpetual flowering 1 |
References
- Baurle, I.; Dean, C. The timing of developmental transitions in plants. Cell 2006, 125, 655–664. [Google Scholar] [CrossRef] [PubMed]
- Fornara, F.; de Montaigu, A.; Coupland, G. SnapShot: Control of flowering in Arabidopsis. Cell 2010, 141, 550. [Google Scholar] [CrossRef] [PubMed]
- Ausin, I.; Alonso-Blanco, C.; Martinez-Zapater, J.M. Environmental regulation of flowering. Int. J. Dev. Biol. 2005, 49, 689–705. [Google Scholar] [CrossRef] [PubMed]
- Mutasa-Gottgens, E.; Hedden, P. Gibberellin as a factor in floral regulatory networks. J. Exp. Bot. 2009, 60, 1979–1989. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.G. The autonomous pathway: Epigenetic and post-transcriptional gene regulation in the control of Arabidopsis flowering time. Curr. Opin. Plant Biol. 2004, 7, 570–574. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W.; Czech, B.; Weigel, D. miR156-regulated SPL transcription factors define an endogenous flowering pathway in Arabidopsis thaliana. Cell 2009, 138, 738–749. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Xi, W.; Shen, L.; Tan, C.; Yu, H. Regulation of floral patterning by flowering time genes. Dev. Cell 2009, 16, 711–722. [Google Scholar] [CrossRef] [PubMed]
- Imaizumi, T.; Kay, S.A. Photoperiodic control of flowering: Not only by coincidence. Trends Plant Sci. 2006, 11, 550–558. [Google Scholar] [CrossRef] [PubMed]
- Niwa, Y.; Ito, S.; Nakamichi, N.; Mizoguchi, T.; Niinuma, K.; Yamashino, T.; Mizuno, T. Genetic linkages of the circadian clock-associated genes, TOC1, CCA1 and LHY, in the photoperiodic control of flowering time in Arabidopsis thaliana. Plant Cell Physiol. 2007, 48, 925–937. [Google Scholar] [CrossRef] [PubMed]
- Shim, J.S.; Kubota, A.; Imaizumi, T. Circadian clock and photoperiodic flowering in Arabidopsis: CONSTANS is a Hub for signal integration. Plant Physiol. 2017, 173, 5–15. [Google Scholar] [CrossRef] [PubMed]
- Suarez-Lopez, P.; Wheatley, K.; Robson, F.; Onouchi, H.; Valverde, F.; Coupland, G. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 2001, 410, 1116–1120. [Google Scholar] [CrossRef] [PubMed]
- Searle, I.; He, Y.; Turck, F.; Vincent, C.; Fornara, F.; Krober, S.; Amasino, R.A.; Coupland, G. The transcription factor FLC confers a flowering response to vernalization by repressing meristem competence and systemic signaling in Arabidopsis. Genes Dev. 2006, 20, 898–912. [Google Scholar] [CrossRef]
- Berry, S.; Dean, C. Environmental perception and epigenetic memory: Mechanistic insight through FLC. Plant J. 2015, 83, 133–148. [Google Scholar] [CrossRef] [PubMed]
- Bastow, R.; Mylne, J.S.; Lister, C.; Lippman, Z.; Martienssen, R.A.; Dean, C. Vernalization requires epigenetic silencing of FLC by histone methylation. Nature 2004, 427, 164–167. [Google Scholar] [CrossRef] [PubMed]
- Wood, C.C.; Robertson, M.; Tanner, G.; Peacock, W.J.; Dennis, E.S.; Helliwell, C.A. The Arabidopsis thaliana vernalization response requires a polycomb-like protein complex that also includes VERNALIZATION INSENSITIVE 3. Proc. Natl. Acad. Sci. USA 2006, 103, 14631–14636. [Google Scholar] [CrossRef]
- Sung, S.; Amasino, R.M. Vernalization in Arabidopsis thaliana is mediated by the PHD finger protein VIN3. Nature 2004, 427, 159–164. [Google Scholar] [CrossRef]
- Lee, J.H.; Yoo, S.J.; Park, S.H.; Hwang, I.; Lee, J.S.; Ahn, J.H. Role of SVP in the control of flowering time by ambient temperature in Arabidopsis. Genes Dev. 2007, 21, 397–402. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.W. Regulation of flowering time by the miR156-mediated age pathway. J. Exp. Bot. 2014, 65, 4723–4730. [Google Scholar] [CrossRef] [PubMed]
- Wang, R.; Farrona, S.; Vincent, C.; Joecker, A.; Schoof, H.; Turck, F.; Alonso-Blanco, C.; Coupland, G.; Albani, M.C. PEP1 regulates perennial flowering in Arabis alpina. Nature 2009, 459, 423–427. [Google Scholar] [CrossRef]
- Hyun, Y.; Vincent, C.; Tilmes, V.; Bergonzi, S.; Kiefer, C.; Richter, R.; Martinez-Gallegos, R.; Severing, E.; Coupland, G. A regulatory circuit conferring varied flowering response to cold in annual and perennial plants. Science 2019, 363, 409–412. [Google Scholar] [CrossRef]
- Wang, H.; Chang, X.; Lin, J.; Chang, Y.; Chen, J.C.; Reid, M.S.; Jiang, C.Z. Transcriptome profiling reveals regulatory mechanisms underlying corolla senescence in petunia. Hortic. Res. 2018, 5, 16. [Google Scholar] [CrossRef] [PubMed]
- Liu, J.; Fu, X.; Dong, Y.; Lu, J.; Ren, M.; Zhou, N.; Wang, C. MIKC(C)-type MADS-box genes in Rosa chinensis: The remarkable expansion of ABCDE model genes and their roles in floral organogenesis. Hortic. Res. 2018, 5, 25. [Google Scholar] [CrossRef]
- Guo, X.; Yu, C.; Luo, L.; Wan, H.; Zhen, N.; Li, Y.; Cheng, T.; Wang, J.; Pan, H.; Zhang, Q. Developmental transcriptome analysis of floral transition in Rosa odorata var. gigantea. Plant Mol. Biol. 2018, 97, 113–130. [Google Scholar] [CrossRef] [PubMed]
- Zhou, A.; Ma, H.; Liu, E.; Jiang, T.; Feng, S.; Gong, S.; Wang, J. Transcriptome sequencing of Dianthus spiculifolius and analysis of the genes involved in responses to combined cold and drought stress. Int. J. Mol. Sci. 2017, 18, 849. [Google Scholar] [CrossRef]
- Li, B.; Dewey, C.N. RSEM: Accurate transcript quantification from RNA-Seq data with or without a reference genome. BMC Bioinform. 2011, 12, 323. [Google Scholar] [CrossRef]
- Mortazavi, A.; Williams, B.A.; McCue, K.; Schaeffer, L.; Wold, B. Mapping and quantifying mammalian transcriptomes by RNA-Seq. Nat. Methods 2008, 5, 621–628. [Google Scholar] [CrossRef]
- Reiner, A.; Yekutieli, D.; Benjamini, Y. Identifying differentially expressed genes using false discovery rate controlling procedures. Bioinformatics 2003, 19, 368–375. [Google Scholar] [CrossRef]
- Xie, Y.; Xu, L.; Wang, Y.; Fan, L.; Chen, Y.; Tang, M.; Luo, X.; Liu, L. Comparative proteomic analysis provides insight into a complex regulatory network of taproot formation in radish (Raphanus sativus L.). Hortic. Res. 2018, 5, 51. [Google Scholar] [CrossRef]
- Wang, H.; Deng, X.W. Arabidopsis FHY3 defines a key phytochrome A signaling component directly interacting with its homologous partner FAR1. EMBO J. 2002, 21, 1339–1349. [Google Scholar] [CrossRef]
- King, G.J.; Chanson, A.H.; McCallum, E.J.; Ohme-Takagi, M.; Byriel, K.; Hill, J.M.; Martin, J.L.; Mylne, J.S. The Arabidopsis B3 domain protein VERNALIZATION1 (VRN1) is involved in processes essential for development, with structural and mutational studies revealing its DNA-binding surface. J. Biol. Chem. 2013, 288, 3198–3207. [Google Scholar] [CrossRef]
- Zhang, Z.L.; Ogawa, M.; Fleet, C.M.; Zentella, R.; Hu, J.; Heo, J.O.; Lim, J.; Kamiya, Y.; Yamaguchi, S.; Sun, T.P. Scarecrow-like 3 promotes gibberellin signaling by antagonizing master growth repressor DELLA in Arabidopsis. Proc. Natl. Acad. Sci. USA 2011, 108, 2160–2165. [Google Scholar] [CrossRef] [PubMed]
- Sarnowska, E.A.; Rolicka, A.T.; Bucior, E.; Cwiek, P.; Tohge, T.; Fernie, A.R.; Jikumaru, Y.; Kamiya, Y.; Franzen, R.; Schmelzer, E.; et al. DELLA-interacting SWI3C core subunit of switch/sucrose nonfermenting chromatin remodeling complex modulates gibberellin responses and hormonal cross talk in Arabidopsis. Plant Physiol. 2013, 163, 305–317. [Google Scholar] [CrossRef]
- Santner, A.; Calderon-Villalobos, L.I.; Estelle, M. Plant hormones are versatile chemical regulators of plant growth. Nat. Chem. Biol. 2009, 5, 301–307. [Google Scholar] [CrossRef] [PubMed]
- Licausi, F.; Ohme-Takagi, M.; Perata, P. APETALA2/ethylene responsive factor (AP2/ERF) transcription factors: Mediators of stress responses and developmental programs. New Phytol. 2013, 199, 639–649. [Google Scholar] [CrossRef]
- Kubota, A.; Ito, S.; Shim, J.S.; Johnson, R.S.; Song, Y.H.; Breton, G.; Goralogia, G.S.; Kwon, M.S.; Laboy Cintron, D.; Koyama, T.; et al. TCP4-dependent induction of CONSTANS transcription requires GIGANTEA in photoperiodic flowering in Arabidopsis. PLoS Genet. 2017, 13, e1006856. [Google Scholar] [CrossRef] [PubMed]
- Lucero, L.E.; Manavella, P.A.; Gras, D.E.; Ariel, F.D.; Gonzalez, D.H. Class I and class II TCP transcription factors modulate SOC1-dependent flowering at multiple levels. Mol. Plant 2017, 10, 1571–1574. [Google Scholar] [CrossRef] [PubMed]
- Kim, S.; Choi, K.; Park, C.; Hwang, H.J.; Lee, I. SUPPRESSOR OF FRIGIDA4, encoding a C2H2-Type zinc finger protein, represses flowering by transcriptional activation of Arabidopsis FLOWERING LOCUS C. Plant Cell 2006, 18, 2985–2998. [Google Scholar] [CrossRef]
- Yan, Y.; Shen, L.; Chen, Y.; Bao, S.; Thong, Z.; Yu, H. A MYB-domain protein EFM mediates flowering responses to environmental cues in Arabidopsis. Dev. Cell 2014, 30, 437–448. [Google Scholar] [CrossRef] [PubMed]
- Yu, Y.; Liu, Z.; Wang, L.; Kim, S.G.; Seo, P.J.; Qiao, M.; Wang, N.; Li, S.; Cao, X.; Park, C.M.; et al. WRKY71 accelerates flowering via the direct activation of FLOWERING LOCUS T and LEAFY in Arabidopsis thaliana. Plant J. 2016, 85, 96–106. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Wang, H.; Li, X.; Liang, G.; Yu, D. Two DELLA-interacting proteins bHLH48 and bHLH60 regulate flowering under long-day conditions in Arabidopsis thaliana. J. Exp. Bot. 2017, 68, 2757–2767. [Google Scholar] [CrossRef]
- Honma, T.; Goto, K. Complexes of MADS-box proteins are sufficient to convert leaves into floral organs. Nature 2001, 409, 525–529. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xiao, X.; Wang, X.; Zeng, J.; Kang, H.; Fan, X.; Sha, L.; Zhang, H.; Zhou, Y. RNA-Seq and iTRAQ reveal the dwarfing mechanism of dwarf polish wheat (Triticum polonicum L.). Int. J. Biol. Sci. 2016, 12, 653–666. [Google Scholar] [CrossRef][Green Version]
- Ye, X.; Wang, H.; Chen, P.; Fu, B.; Zhang, M.; Li, J.; Zheng, X.; Tan, B.; Feng, J. Combination of iTRAQ proteomics and RNA-seq transcriptomics reveals multiple levels of regulation in phytoplasma-infected Ziziphus jujuba Mill. Hortic. Res. 2017, 4, 17080. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wang, Y.; Zhang, Y.; Wang, W.; Irish, V.F.; Huang, T. RABBIT EARS regulates the transcription of TCP4 during petal development in Arabidopsis. J. Exp. Bot. 2016, 67, 6473–6480. [Google Scholar] [CrossRef] [PubMed]
- Van Es, S.W.; Silveira, S.R.; Rocha, D.I.; Bimbo, A.; Martinelli, A.P.; Dornelas, M.C.; Angenent, G.C.; Immink, R.G.H. Novel functions of the Arabidopsis transcription factor TCP5 in petal development and ethylene biosynthesis. Plant J. 2018, 94, 867–879. [Google Scholar] [CrossRef]
- Gao, H.; Zheng, X.M.; Fei, G.; Chen, J.; Jin, M.; Ren, Y.; Wu, W.; Zhou, K.; Sheng, P.; Zhou, F.; et al. Ehd4 encodes a novel and Oryza-genus-specific regulator of photoperiodic flowering in rice. PLoS Genet. 2013, 9, e1003281. [Google Scholar] [CrossRef] [PubMed]
- Chao, Y.H.; Zhang, T.J.; Yang, Q.C.; Kang, J.M.; Sun, Y.; Gruber, M.Y.; Qin, Z.H. Expression of the alfalfa CCCH-type zinc finger protein gene MsZFN delays flowering time in transgenic Arabidopsis thaliana. Plant Sci. 2014, 215, 92–99. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Wang, H.; Yu, D. Arabidopsis WRKY transcription factors WRKY12 and WRKY13 oppositely regulate flowering under short-day conditions. Mol. Plant 2016, 9, 1492–1503. [Google Scholar] [CrossRef]
- Koo, S.C.; Bracko, O.; Park, M.S.; Schwab, R.; Chun, H.J.; Park, K.M.; Seo, J.S.; Grbic, V.; Balasubramanian, S.; Schmid, M.; et al. Control of lateral organ development and flowering time by the Arabidopsis thaliana MADS-box gene AGAMOUS-LIKE6. Plant J. 2010, 62, 807–816. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Values | Nr | InterPro | KOG | KEGG | Swiss-Prot | Nt | GO | Overall |
|---|---|---|---|---|---|---|---|---|
| Number | 103,730 | 82,218 | 80,112 | 75,943 | 65,136 | 63,978 | 39,219 | 113,899 |
| Percentage | 39.39% | 31.22% | 30.42% | 28.84% | 24.73% | 24.29% | 14.89% | 43.25% |
| Total Spectra | Spectra | Unique Spectra | Peptide | Unique Peptide | Protein |
|---|---|---|---|---|---|
| 368,236 | 83,842 | 68,563 | 41,938 | 36,250 | 8427 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zhou, A.; Sun, H.; Dai, S.; Feng, S.; Zhang, J.; Gong, S.; Wang, J. Identification of Transcription Factors Involved in the Regulation of Flowering in Adonis Amurensis Through Combined RNA-seq Transcriptomics and iTRAQ Proteomics. Genes 2019, 10, 305. https://doi.org/10.3390/genes10040305
Zhou A, Sun H, Dai S, Feng S, Zhang J, Gong S, Wang J. Identification of Transcription Factors Involved in the Regulation of Flowering in Adonis Amurensis Through Combined RNA-seq Transcriptomics and iTRAQ Proteomics. Genes. 2019; 10(4):305. https://doi.org/10.3390/genes10040305
Chicago/Turabian StyleZhou, Aimin, Hongwei Sun, Shengyue Dai, Shuang Feng, Jinzhu Zhang, Shufang Gong, and Jingang Wang. 2019. "Identification of Transcription Factors Involved in the Regulation of Flowering in Adonis Amurensis Through Combined RNA-seq Transcriptomics and iTRAQ Proteomics" Genes 10, no. 4: 305. https://doi.org/10.3390/genes10040305
APA StyleZhou, A., Sun, H., Dai, S., Feng, S., Zhang, J., Gong, S., & Wang, J. (2019). Identification of Transcription Factors Involved in the Regulation of Flowering in Adonis Amurensis Through Combined RNA-seq Transcriptomics and iTRAQ Proteomics. Genes, 10(4), 305. https://doi.org/10.3390/genes10040305