Pharmacogenomic and Pharmacotranscriptomic Profiling of Childhood Acute Lymphoblastic Leukemia: Paving the Way to Personalized Treatment

 , ,
, ,
Abstract
:1. Introduction
2. Childhood Acute Lymphoblastic Leukemia
3. Glucocorticoid Drugs
4. Vincristine
5. Asparaginase
6. Anthracyclines
7. Thiopurine Drugs
8. Methotrexate
9. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Geraghty, J.C. ‘Omics’ and ‘Omes’—The Future of Personalised Medicine. Available online: https://www.centogene.com/science/omics-and-omes-the-future-of-personalised-medicine.html (accessed on 2 April 2019).
- Georgitsi, M.; Zukic, B.; Pavlovic, S.; Patrinos, G.P. Transcriptional regulation and pharmacogenomics. Pharmacogenomics 2011, 12, 655–673. [Google Scholar] [CrossRef] [PubMed]
- Stojiljkovic, M.; P Patrinos, G.; Pavlovic, S. Clinical Applicability of Sequence Variations in Genes Related to Drug Metabolism. Curr. Drug Metab. 2011, 12, 445–454. [Google Scholar] [CrossRef] [PubMed]
- Amos, W.; Driscoll, E.; Hoffman, J.I. Candidate genes versus genome-wide associations: Which are better for detecting genetic susceptibility to infectious disease? Proc. Biol. Sci. 2011, 278, 1183–1188. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Robison, L.L.; Look, A.T. Acute lymphoblastic leukaemia. Lancet 2008, 371, 1030–1043. [Google Scholar] [CrossRef]
- Greaves, M.F.; Wiemels, J. Origins of chromosome translocations in childhood leukaemia. Nat. Rev. Cancer 2003, 3, 639–649. [Google Scholar] [CrossRef] [PubMed]
- Moricke, A.; Zimmermann, M.; Valsecchi, M.G.; Stanulla, M.; Biondi, A.; Mann, G.; Locatelli, F.; Cazzaniga, G.; Niggli, F.; Arico, M.; et al. Dexamethasone vs prednisone in induction treatment of pediatric ALL: Results of the randomized trial AIEOP-BFM ALL 2000. Blood 2016, 127, 2101–2112. [Google Scholar] [CrossRef] [PubMed]
- Hunger, S.P.; Mullighan, C.G. Acute Lymphoblastic Leukemia in Children. N. Engl. J. Med. 2015, 373, 1541–1552. [Google Scholar] [CrossRef] [PubMed]
- Pui, C.H.; Pei, D.; Sandlund, J.T.; Ribeiro, R.C.; Rubnitz, J.E.; Raimondi, S.C.; Onciu, M.; Campana, D.; Kun, L.E.; Jeha, S.; et al. Long-term results of St Jude Total Therapy Studies 11, 12, 13A, 13B, and 14 for childhood acute lymphoblastic leukemia. Leukemia 2010, 24, 371–382. [Google Scholar] [CrossRef] [PubMed]
- Schrappe, M.; Reiter, A.; Ludwig, W.-D.; Harbott, J.; Zimmermann, M.; Hiddemann, W.; Niemeyer, C.; Henze, G.; Feldges, A.; Zintl, F.; et al. Improved outcome in childhood acute lymphoblastic leukemia despite reduced use of anthracyclines and cranial radiotherapy: Results of trial ALL-BFM 90. Blood 2000, 95, 3310–3322. [Google Scholar] [PubMed]
- Gervasini, G.; Vagace, J.M. Impact of genetic polymorphisms on chemotherapy toxicity in childhood acute lymphoblastic leukemia. Front Genet 2012, 3, 249. [Google Scholar] [CrossRef] [PubMed]
- Mullighan, C.G.; Zhang, J.; Kasper, L.H.; Lerach, S.; Payne-Turner, D.; Phillips, L.A.; Heatley, S.L.; Holmfeldt, L.; Collins-Underwood, J.R.; Ma, J.; et al. CREBBP mutations in relapsed acute lymphoblastic leukaemia. Nature 2011, 471, 235–239. [Google Scholar] [CrossRef] [PubMed]
- Ross, C.J.; Visscher, H.; Rassekh, S.R.; Castro-Pastrana, L.I.; Shereck, E.; Carleton, B.; Hayden, M.R. Pharmacogenomics of serious adverse drug reactions in pediatric oncology. J. Popul. Ther. Clin. Pharmacol. 2011, 18, e134–e151. [Google Scholar] [PubMed]
- Thomas, A. How can we improve on the already impressive results in pediatric ALL? Hematol. Am. Soc. Hematol. Educ. Program 2015, 15, 414–419. [Google Scholar] [CrossRef] [PubMed]
- Mlakar, V.; Huezo-Diaz Curtis, P.; Satyanarayana Uppugunduri, C.R.; Krajinovic, M.; Ansari, M. Pharmacogenomics in Pediatric Oncology: Review of Gene-Drug Associations for Clinical Use. Int. J. Mol. Sci. 2016, 17, 1502. [Google Scholar] [CrossRef] [PubMed]
- Krajinovic, M.; Lemieux-Blanchard, E.; Chiasson, S.; Primeau, M.; Costea, I.; Moghrabi, A. Role of polymorphisms in MTHFR and MTHFD1 genes in the outcome of childhood acute lymphoblastic leukemia. Pharm. J. 2004, 4, 66–72. [Google Scholar] [CrossRef] [PubMed]
- Schmiegelow, K.; Forestier, E.; Kristinsson, J.; Soderhall, S.; Vettenranta, K.; Weinshilboum, R.; Wesenberg, F. Thiopurine methyltransferase activity is related to the risk of relapse of childhood acute lymphoblastic leukemia: Results from the NOPHO ALL-92 study. Leukemia 2009, 23, 557–564. [Google Scholar] [CrossRef] [PubMed]
- Inaba, H.; Pui, C.H. Glucocorticoid use in acute lymphoblastic leukaemia. Lancet Oncol. 2010, 11, 1096–1106. [Google Scholar] [CrossRef]
- Lin, K.T.; Wang, L.H. New dimension of glucocorticoids in cancer treatment. Steroids 2016, 111, 84–88. [Google Scholar] [CrossRef] [PubMed]
- Eipel, O.T.; Nemeth, K.; Torok, D.; Csordas, K.; Hegyi, M.; Ponyi, A.; Ferenczy, A.; Erdelyi, D.J.; Csoka, M.; Kovacs, G.T. The glucocorticoid receptor gene polymorphism N363S predisposes to more severe toxic side effects during pediatric acute lymphoblastic leukemia (ALL) therapy. Int. J. Hematol. 2013, 97, 216–222. [Google Scholar] [CrossRef] [PubMed]
- Tissing, W.J.; Meijerink, J.P.; den Boer, M.L.; Brinkhof, B.; van Rossum, E.F.; van Wering, E.R.; Koper, J.W.; Sonneveld, P.; Pieters, R. Genetic variations in the glucocorticoid receptor gene are not related to glucocorticoid resistance in childhood acute lymphoblastic leukemia. Clin. Cancer Res. 2005, 11, 6050–6056. [Google Scholar] [CrossRef] [PubMed]
- Xue, L.; Li, C.; Wang, Y.; Sun, W.; Ma, C.; He, Y.; Yu, Y.; Cai, L.; Wang, L. Single nucleotide polymorphisms in non-coding region of the glucocorticoid receptor gene and prednisone response in childhood acute lymphoblastic leukemia. Leuk. Lymphoma 2015, 56, 1704–1709. [Google Scholar] [CrossRef] [PubMed]
- Gasic, V.; Zukic, B.; Stankovic, B.; Janic, D.; Dokmanovic, L.; Lazic, J.; Krstovski, N.; Dolzan, V.; Jazbec, J.; Pavlovic, S.; et al. Pharmacogenomic markers of glucocorticoid response in the initial phase of remission induction therapy in childhood acute lymphoblastic leukemia. Radiol. Oncol. 2018, 52, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Zalewski, G.; Wasilewska, A.; Zoch-Zwierz, W.; Chyczewski, L. Response to prednisone in relation to NR3C1 intron B polymorphisms in childhood nephrotic syndrome. Pediatr. Nephrol. 2008, 23, 1073–1078. [Google Scholar] [CrossRef] [PubMed]
- Gregers, J.; Green, H.; Christensen, I.J.; Dalhoff, K.; Schroeder, H.; Carlsen, N.; Rosthoej, S.; Lausen, B.; Schmiegelow, K.; Peterson, C. Polymorphisms in the ABCB1 gene and effect on outcome and toxicity in childhood acute lymphoblastic leukemia. Pharm. J. 2015, 15, 372–379. [Google Scholar] [CrossRef] [PubMed]
- Jamroziak, K.; Mlynarski, W.; Balcerczak, E.; Mistygacz, M.; Trelinska, J.; Mirowski, M.; Bodalski, J.; Robak, T. Functional C3435T polymorphism of MDR1 gene: An impact on genetic susceptibility and clinical outcome of childhood acute lymphoblastic leukemia. Eur. J. Haematol. 2004, 72, 314–321. [Google Scholar] [CrossRef] [PubMed]
- Marino, S.; Verzegnassi, F.; Tamaro, P.; Stocco, G.; Bartoli, F.; Decorti, G.; Rabusin, M. Response to glucocorticoids and toxicity in childhood acute lymphoblastic leukemia: Role of polymorphisms of genes involved in glucocorticoid response. Pediatr. Blood Cancer 2009, 53, 984–991. [Google Scholar] [CrossRef] [PubMed]
- Anderer, G.; Schrappe, M.; Brechlin, A.M.; Lauten, M.; Muti, P.; Welte, K.; Stanulla, M. Polymorphisms within glutathione S-transferase genes and initial response to glucocorticoids in childhood acute lymphoblastic leukaemia. Pharmacogenetics 2000, 10, 715–726. [Google Scholar] [CrossRef] [PubMed]
- Meissner, B.; Stanulla, M.; Ludwig, W.D.; Harbott, J.; Moricke, A.; Welte, K.; Schrappe, M. The GSTT1 deletion polymorphism is associated with initial response to glucocorticoids in childhood acute lymphoblastic leukemia. Leukemia 2004, 18, 1920–1923. [Google Scholar] [CrossRef] [PubMed]
- Stanulla, M.; Schrappe, M.; Brechlin, A.M.; Zimmermann, M.; Welte, K. Polymorphisms within glutathione S-transferase genes (GSTM1, GSTT1, GSTP1) and risk of relapse in childhood B-cell precursor acute lymphoblastic leukemia: A case-control study. Blood 2000, 95, 1222. [Google Scholar] [PubMed]
- Kamdem, L.K.; Hamilton, L.; Cheng, C.; Liu, W.; Yang, W.; Johnson, J.A.; Pui, C.H.; Relling, M.V. Genetic predictors of glucocorticoid-induced hypertension in children with acute lymphoblastic leukemia. Pharm. Genom. 2008, 18, 507–514. [Google Scholar] [CrossRef] [PubMed]
- Montano, A.; Forero-Castro, M.; Marchena-Mendoza, D.; Benito, R.; Hernandez-Rivas, J.M. New Challenges in Targeting Signaling Pathways in Acute Lymphoblastic Leukemia by NGS Approaches: An Update. Cancers 2018, 10, 110. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Cheng, C.; Devidas, M.; Cao, X.; Campana, D.; Yang, W.; Fan, Y.; Neale, G.; Cox, N.; Scheet, P.; et al. Genome-wide association study identifies germline polymorphisms associated with relapse of childhood acute lymphoblastic leukemia. Blood 2012, 120, 4197–4204. [Google Scholar] [CrossRef] [PubMed]
- Bolton, J.L.; Hayward, C.; Direk, N.; Lewis, J.G.; Hammond, G.L.; Hill, L.A.; Anderson, A.; Huffman, J.; Wilson, J.F.; Campbell, H.; et al. Genome wide association identifies common variants at the SERPINA6/SERPINA1 locus influencing plasma cortisol and corticosteroid binding globulin. PLoS Genet. 2014, 10, e1004474. [Google Scholar] [CrossRef] [PubMed]
- Karol, S.E.; Yang, W.; Van Driest, S.L.; Chang, T.Y.; Kaste, S.; Bowton, E.; Basford, M.; Bastarache, L.; Roden, D.M.; Denny, J.C.; et al. Genetics of glucocorticoid-associated osteonecrosis in children with acute lymphoblastic leukemia. Blood 2015, 126, 1770–1776. [Google Scholar] [CrossRef] [PubMed]
- Kawedia, J.D.; Kaste, S.C.; Pei, D.; Panetta, J.C.; Cai, X.; Cheng, C.; Neale, G.; Howard, S.C.; Evans, W.E.; Pui, C.H.; et al. Pharmacokinetic, pharmacodynamic, and pharmacogenetic determinants of osteonecrosis in children with acute lymphoblastic leukemia. Blood 2011, 117, 2340–2347; quiz 2556. [Google Scholar] [CrossRef] [PubMed]
- Bottini, N.; MacMurray, J.; Peters, W.; Rostamkhani, M.; Comings, D.E. Association of the acid phosphatase (ACP1) gene with triglyceride levels in obese women. Mol. Genet. Metab. 2002, 77, 226–229. [Google Scholar] [CrossRef]
- Gasic, V.; Stankovic, B.; Zukic, B.; Janic, D.; Dokmanovic, L.; Krstovski, N.; Lazic, J.; Milosevic, G.; Lucafò, M.; Stocco, G.; et al. Expression Pattern of Long Non-Coding RNA Growth Arrest-Specific 5 in the Remission Induction Therapy in Childhood Acute Lymphoblastic Leukemia. J. Med. Biochem. 2018. [Google Scholar] [CrossRef]
- Garabedian, M.J.; Logan, S.K. Glucocorticoid receptor DNA binding decoy is a gas. Sci. Signal 2010, 3, pe5. [Google Scholar] [CrossRef] [PubMed]
- Chen, H.; Zhang, D.; Zhang, G.; Li, X.; Liang, Y.; Kasukurthi, M.V.; Li, S.; Borchert, G.M.; Huang, J. A semantics-oriented computational approach to investigate microRNA regulation on glucocorticoid resistance in pediatric acute lymphoblastic leukemia. BMC Med Inform. Decis. Mak. 2018, 18, 57. [Google Scholar] [CrossRef] [PubMed]
- Aries, I.M.; Jerchel, I.S.; van den Dungen, R.E.; van den Berk, L.C.; Boer, J.M.; Horstmann, M.A.; Escherich, G.; Pieters, R.; den Boer, M.L. EMP1, a novel poor prognostic factor in pediatric leukemia regulates prednisolone resistance, cell proliferation, migration and adhesion. Leukemia 2014, 28, 1828–1837. [Google Scholar] [CrossRef] [PubMed]
- Morales, S.A.; Mareninov, S.; Coulam, P.; Wadehra, M.; Goodglick, L.; Braun, J.; Gordon, L.K. Functional consequences of interactions between FAK and epithelial membrane protein 2 (EMP2). Investig. Ophthalmol. Vis. Sci. 2009, 50, 4949–4956. [Google Scholar] [CrossRef] [PubMed]
- Gauld, S.B.; Cambier, J.C. Src-family kinases in B-cell development and signaling. Oncogene 2004, 23, 8001–8006. [Google Scholar] [CrossRef] [PubMed]
- Palacios, E.H.; Weiss, A. Function of the Src-family kinases, Lck and Fyn, in T-cell development and activation. Oncogene 2004, 23, 7990–8000. [Google Scholar] [CrossRef] [PubMed]
- Paugh, S.W.; Bonten, E.J.; Savic, D.; Ramsey, L.B.; Thierfelder, W.E.; Gurung, P.; Malireddi, R.K.; Actis, M.; Mayasundari, A.; Min, J.; et al. NALP3 inflammasome upregulation and CASP1 cleavage of the glucocorticoid receptor cause glucocorticoid resistance in leukemia cells. Nat. Genet. 2015, 47, 607–614. [Google Scholar] [CrossRef] [PubMed]
- Pottier, N.; Yang, W.; Assem, M.; Panetta, J.C.; Pei, D.; Paugh, S.W.; Cheng, C.; Den Boer, M.L.; Relling, M.V.; Pieters, R.; et al. The SWI/SNF chromatin-remodeling complex and glucocorticoid resistance in acute lymphoblastic leukemia. J. Natl. Cancer Inst. 2008, 100, 1792–1803. [Google Scholar] [CrossRef] [PubMed]
- Jordan, M.A.; Wilson, L. Microtubules as a target for anticancer drugs. Nat. Reviews. Cancer 2004, 4, 253–265. [Google Scholar] [CrossRef] [PubMed]
- Ceppi, F.; Langlois-Pelletier, C.; Gagne, V.; Rousseau, J.; Ciolino, C.; De Lorenzo, S.; Kevin, K.M.; Cijov, D.; Sallan, S.E.; Silverman, L.B.; et al. Polymorphisms of the vincristine pathway and response to treatment in children with childhood acute lymphoblastic leukemia. Pharmacogenomics 2014, 15, 1105–1116. [Google Scholar] [CrossRef] [PubMed]
- Egbelakin, A.; Ferguson, M.J.; MacGill, E.A.; Lehmann, A.S.; Topletz, A.R.; Quinney, S.K.; Li, L.; McCammack, K.C.; Hall, S.D.; Renbarger, J.L. Increased risk of vincristine neurotoxicity associated with low CYP3A5 expression genotype in children with acute lymphoblastic leukemia. Pediatr. Blood Cancer 2011, 56, 361–367. [Google Scholar] [CrossRef] [PubMed]
- Hartman, A.; van Schaik, R.H.; van der Heiden, I.P.; Broekhuis, M.J.; Meier, M.; den Boer, M.L.; Pieters, R. Polymorphisms in genes involved in vincristine pharmacokinetics or pharmacodynamics are not related to impaired motor performance in children with leukemia. Leuk. Res. 2010, 34, 154–159. [Google Scholar] [CrossRef] [PubMed]
- Moore, A.S.; Norris, R.; Price, G.; Nguyen, T.; Ni, M.; George, R.; van Breda, K.; Duley, J.; Charles, B.; Pinkerton, R. Vincristine pharmacodynamics and pharmacogenetics in children with cancer: A limited-sampling, population modelling approach. J. Paediatr. Child Health 2011, 47, 875–882. [Google Scholar] [CrossRef] [PubMed]
- Renbarger, J.L.; McCammack, K.C.; Rouse, C.E.; Hall, S.D. Effect of race on vincristine-associated neurotoxicity in pediatric acute lymphoblastic leukemia patients. Pediatr. Blood Cancer 2008, 50, 769–771. [Google Scholar] [CrossRef] [PubMed]
- Aplenc, R.; Glatfelter, W.; Han, P.; Rappaport, E.; La, M.; Cnaan, A.; Blackwood, M.A.; Lange, B.; Rebbeck, T. CYP3A genotypes and treatment response in paediatric acute lymphoblastic leukaemia. Br. J. Haematol. 2003, 122, 240–244. [Google Scholar] [CrossRef] [PubMed]
- Kishi, S.; Cheng, C.; French, D.; Pei, D.; Das, S.; Cook, E.H.; Hijiya, N.; Rizzari, C.; Rosner, G.L.; Frudakis, T.; et al. Ancestry and pharmacogenetics of antileukemic drug toxicity. Blood 2007, 109, 4151–4157. [Google Scholar] [CrossRef] [PubMed]
- Plasschaert, S.L.; Groninger, E.; Boezen, M.; Kema, I.; de Vries, E.G.; Uges, D.; Veerman, A.J.; Kamps, W.A.; Vellenga, E.; de Graaf, S.S.; et al. Influence of functional polymorphisms of the MDR1 gene on vincristine pharmacokinetics in childhood acute lymphoblastic leukemia. Clin. Pharm. 2004, 76, 220–229. [Google Scholar] [CrossRef]
- Dennison, J.B.; Kulanthaivel, P.; Barbuch, R.J.; Renbarger, J.L.; Ehlhardt, W.J.; Hall, S.D. Selective metabolism of vincristine in vitro by CYP3A5. Drug Metab. Dispos. Biol. Fate Chem. 2006, 34, 1317–1327. [Google Scholar] [CrossRef] [PubMed]
- Kuehl, P.; Zhang, J.; Lin, Y.; Lamba, J.; Assem, M.; Schuetz, J.; Watkins, P.B.; Daly, A.; Wrighton, S.A.; Hall, S.D.; et al. Sequence diversity in CYP3A promoters and characterization of the genetic basis of polymorphic CYP3A5 expression. Nat. Genet. 2001, 27, 383–391. [Google Scholar] [CrossRef] [PubMed]
- Diouf, B.; Crews, K.R.; Lew, G.; Pei, D.; Cheng, C.; Bao, J.; Zheng, J.J.; Yang, W.; Fan, Y.; Wheeler, H.E.; et al. Association of an inherited genetic variant with vincristine-related peripheral neuropathy in children with acute lymphoblastic leukemia. JAMA 2015, 313, 815–823. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Camino, A.; Martin-Guerrero, I.; Lopez-Lopez, E.; Echebarria-Barona, A.; Zabalza, I.; Ruiz, I.; Guerra-Merino, I.; Garcia-Orad, A. Lack of association of the CEP72 rs924607 TT genotype with vincristine-related peripheral neuropathy during the early phase of pediatric acute lymphoblastic leukemia treatment in a Spanish population. Pharm. Genom. 2016, 26, 100–102. [Google Scholar] [CrossRef] [PubMed]
- Diouf, B.; Crews, K.R.; Evans, W.E. Vincristine pharmacogenomics: ‘winner’s curse’ or a different phenotype? Pharm. Genom. 2016, 26, 51–52. [Google Scholar] [CrossRef] [PubMed]
- Dennison, J.B.; Jones, D.R.; Renbarger, J.L.; Hall, S.D. Effect of CYP3A5 expression on vincristine metabolism with human liver microsomes. J. Pharmacol. Exp. Ther. 2007, 321, 553–563. [Google Scholar] [CrossRef] [PubMed]
- Berg, S.L.; Parsons, D.W. The Pharmacogenomics of Vincristine-Induced Neuropathy: On Pins and Needles. JAMA Oncol. 2015, 1, 975–976. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, E.; Gutierrez-Camino, A.; Astigarraga, I.; Navajas, A.; Echebarria-Barona, A.; Garcia-Miguel, P.; Garcia de Andoin, N.; Lobo, C.; Guerra-Merino, I.; Martin-Guerrero, I.; et al. Vincristine pharmacokinetics pathway and neurotoxicity during early phases of treatment in pediatric acute lymphoblastic leukemia. Pharmacogenomics 2016, 17, 731–741. [Google Scholar] [CrossRef] [PubMed]
- Abaji, R.; Ceppi, F.; Patel, S.; Gagne, V.; Xu, C.J.; Spinella, J.F.; Colombini, A.; Parasole, R.; Buldini, B.; Basso, G.; et al. Genetic risk factors for VIPN in childhood acute lymphoblastic leukemia patients identified using whole-exome sequencing. Pharmacogenomics 2018, 19, 1181–1193. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Lei, K.; Yuan, X.; Wu, X.; Zhuang, Y.; Xu, T.; Xu, R.; Han, M. SUN1/2 and Syne/Nesprin-1/2 complexes connect centrosome to the nucleus during neurogenesis and neuronal migration in mice. Neuron 2009, 64, 173–187. [Google Scholar] [CrossRef] [PubMed]
- Kenmochi, N.; Suzuki, T.; Uechi, T.; Magoori, M.; Kuniba, M.; Higa, S.; Watanabe, K.; Tanaka, T. The human mitochondrial ribosomal protein genes: Mapping of 54 genes to the chromosomes and implications for human disorders. Genomics 2001, 77, 65–70. [Google Scholar] [CrossRef] [PubMed]
- Zhu, H.; Wan, X.; Li, J.; Han, L.; Bo, X.; Chen, W.; Lu, C.; Shen, Z.; Xu, C.; Chen, L.; et al. Computational Prediction and Validation of BAHD1 as a Novel Molecule for Ulcerative Colitis. Sci. Rep. 2015, 5, 12227. [Google Scholar] [CrossRef] [PubMed]
- Sun, Z.; Wu, Y.; Ordog, T.; Baheti, S.; Nie, J.; Duan, X.; Hojo, K.; Kocher, J.P.; Dyck, P.J.; Klein, C.J. Aberrant signature methylome by DNMT1 hot spot mutation in hereditary sensory and autonomic neuropathy 1E. Epigenetics 2014, 9, 1184–1193. [Google Scholar] [CrossRef] [PubMed]
- Li, L.; Sajdyk, T.; Smith, E.M.L.; Chang, C.W.; Li, C.; Ho, R.H.; Hutchinson, R.; Wells, E.; Skiles, J.L.; Winick, N.; et al. Genetic Variants Associated With Vincristine-Induced Peripheral Neuropathy in Two Populations of Children With Acute Lymphoblastic Leukemia. Clin. Pharm. 2018. [Google Scholar] [CrossRef] [PubMed]
- Robertson, N.G.; Cremers, C.W.; Huygen, P.L.; Ikezono, T.; Krastins, B.; Kremer, H.; Kuo, S.F.; Liberman, M.C.; Merchant, S.N.; Miller, C.E.; et al. Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum. Mol. Genet. 2006, 15, 1071–1085. [Google Scholar] [CrossRef] [PubMed]
- Wittamer, V.; Franssen, J.D.; Vulcano, M.; Mirjolet, J.F.; Le Poul, E.; Migeotte, I.; Brezillon, S.; Tyldesley, R.; Blanpain, C.; Detheux, M.; et al. Specific recruitment of antigen-presenting cells by chemerin, a novel processed ligand from human inflammatory fluids. J. Exp. Med. 2003, 198, 977–985. [Google Scholar] [CrossRef] [PubMed]
- Schotte, D.; De Menezes, R.X.; Akbari Moqadam, F.; Khankahdani, L.M.; Lange-Turenhout, E.; Chen, C.; Pieters, R.; Den Boer, M.L. MicroRNA characterize genetic diversity and drug resistance in pediatric acute lymphoblastic leukemia. Haematologica 2011, 96, 703–711. [Google Scholar] [CrossRef] [PubMed]
- Akbari Moqadam, F.; Lange-Turenhout, E.A.; Aries, I.M.; Pieters, R.; den Boer, M.L. MiR-125b, miR-100 and miR-99a co-regulate vincristine resistance in childhood acute lymphoblastic leukemia. Leuk. Res. 2013, 37, 1315–1321. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, E.; Gutierrez-Camino, A.; Pinan, M.A.; Sanchez-Toledo, J.; Uriz, J.J.; Ballesteros, J.; Garcia-Miguel, P.; Navajas, A.; Garcia-Orad, A. Pharmacogenetics of microRNAs and microRNAs biogenesis machinery in pediatric acute lymphoblastic leukemia. PLoS ONE 2014, 9, e91261. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Camino, A.; Umerez, M.; Martin-Guerrero, I.; Garcia de Andoin, N.; Santos, B.; Sastre, A.; Echebarria-Barona, A.; Astigarraga, I.; Navajas, A.; Garcia-Orad, A. Mir-pharmacogenetics of Vincristine and peripheral neurotoxicity in childhood B-cell acute lymphoblastic leukemia. Pharm. J. 2018, 18, 704–712. [Google Scholar] [CrossRef] [PubMed]
- Franca, R.; Rebora, P.; Bertorello, N.; Fagioli, F.; Conter, V.; Biondi, A.; Colombini, A.; Micalizzi, C.; Zecca, M.; Parasole, R.; et al. Pharmacogenetics and induction/consolidation therapy toxicities in acute lymphoblastic leukemia patients treated with AIEOP-BFM ALL 2000 protocol. Pharm. J. 2017, 17, 4–10. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.A.; Smith, C.; Yang, W.; Mullighan, C.G.; Qu, C.; Larsen, E.; Bowman, W.P.; Liu, C.; Ramsey, L.B.; Chang, T.; et al. Genome-wide analysis links NFATC2 with asparaginase hypersensitivity. Blood 2015, 126, 69–75. [Google Scholar] [CrossRef] [PubMed]
- Duval, M. Comparison of Escherichia coli-asparaginase with Erwinia-asparaginase in the treatment of childhood lymphoid malignancies: Results of a randomized European Organisation for Research and Treatment of Cancer—Children’s Leukemia Group phase 3 trial. Blood 2002, 99, 2734–2739. [Google Scholar] [CrossRef] [PubMed]
- Dinndorf, P.A.; Gootenberg, J.; Cohen, M.H.; Keegan, P.; Pazdur, R. FDA Drug Approval Summary: Pegaspargase (Oncaspar®) for the First-Line Treatment of Children with Acute Lymphoblastic Leukemia (ALL). oncologist 2007, 12, 991–998. [Google Scholar] [CrossRef] [PubMed]
- Rousseau, J.; Gagne, V.; Labuda, M.; Beaubois, C.; Sinnett, D.; Laverdiere, C.; Moghrabi, A.; Sallan, S.E.; Silverman, L.B.; Neuberg, D.; et al. ATF5 polymorphisms influence ATF function and response to treatment in children with childhood acute lymphoblastic leukemia. Blood 2011, 118, 5883–5890. [Google Scholar] [CrossRef] [PubMed]
- Ben Tanfous, M.; Sharif-Askari, B.; Ceppi, F.; Laaribi, H.; Gagne, V.; Rousseau, J.; Labuda, M.; Silverman, L.B.; Sallan, S.E.; Neuberg, D.; et al. Polymorphisms of asparaginase pathway and asparaginase-related complications in children with acute lymphoblastic leukemia. Clin. Cancer Res. 2015, 21, 329–334. [Google Scholar] [CrossRef] [PubMed]
- Abaji, R.; Gagne, V.; Xu, C.J.; Spinella, J.F.; Ceppi, F.; Laverdiere, C.; Leclerc, J.M.; Sallan, S.E.; Neuberg, D.; Kutok, J.L.; et al. Whole-exome sequencing identified genetic risk factors for asparaginase-related complications in childhood ALL patients. Oncotarget 2017, 8, 43752–43767. [Google Scholar] [CrossRef] [PubMed]
- Mori, S.; Bernardi, R.; Laurent, A.; Resnati, M.; Crippa, A.; Gabrieli, A.; Keough, R.; Gonda, T.J.; Blasi, F. Myb-binding protein 1A (MYBBP1A) is essential for early embryonic development, controls cell cycle and mitosis, and acts as a tumor suppressor. PLoS ONE 2012, 7, e39723. [Google Scholar] [CrossRef] [PubMed]
- Owen, H.R.; Elser, M.; Cheung, E.; Gersbach, M.; Kraus, W.L.; Hottiger, M.O. MYBBP1a is a novel repressor of NF-kappaB. J. Mol. Biol. 2007, 366, 725–736. [Google Scholar] [CrossRef] [PubMed]
- Luo, S.X.; Li, S.; Zhang, X.H.; Zhang, J.J.; Long, G.H.; Dong, G.F.; Su, W.; Deng, Y.; Liu, Y.; Zhao, J.M.; et al. Genetic polymorphisms of interleukin-16 and risk of knee osteoarthritis. PLoS ONE 2015, 10, e0123442. [Google Scholar] [CrossRef] [PubMed]
- Lehti, M.S.; Henriksson, H.; Rummukainen, P.; Wang, F.; Uusitalo-Kylmala, L.; Kiviranta, R.; Heino, T.J.; Kotaja, N.; Sironen, A. Cilia-related protein SPEF2 regulates osteoblast differentiation. Sci. Rep. 2018, 8, 859. [Google Scholar] [CrossRef] [PubMed]
- Kang, S.M.; Rosales, J.L.; Meier-Stephenson, V.; Kim, S.; Lee, K.Y.; Narendran, A. Genome-wide loss-of-function genetic screening identifies opioid receptor mu1 as a key regulator of L-asparaginase resistance in pediatric acute lymphoblastic leukemia. Oncogene 2017, 36, 5910–5913. [Google Scholar] [CrossRef] [PubMed]
- Friesen, C.; Roscher, M.; Hormann, I.; Fichtner, I.; Alt, A.; Hilger, R.A.; Debatin, K.M.; Miltner, E. Cell death sensitization of leukemia cells by opioid receptor activation. Oncotarget 2013, 4, 677–690. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Fernandez, C.A.; Smith, C.; Yang, W.; Cheng, C.; Panetta, J.C.; Kornegay, N.; Liu, C.; Ramsey, L.B.; Karol, S.E.; et al. Genome-Wide Study Links PNPLA3 Variant with Elevated Hepatic Transaminase After Acute Lymphoblastic Leukemia Therapy. Clin. Pharm. 2017, 102, 131–140. [Google Scholar] [CrossRef] [PubMed]
- Kienesberger, P.C.; Oberer, M.; Lass, A.; Zechner, R. Mammalian patatin domain containing proteins: A family with diverse lipolytic activities involved in multiple biological functions. J. Lipid Res. 2009, 50, S63–S68. [Google Scholar] [CrossRef] [PubMed]
- Chen, S.H.; Pei, D.; Yang, W.; Cheng, C.; Jeha, S.; Cox, N.J.; Evans, W.E.; Pui, C.H.; Relling, M.V. Genetic variations in GRIA1 on chromosome 5q33 related to asparaginase hypersensitivity. Clin. Pharm. 2010, 88, 191–196. [Google Scholar] [CrossRef] [PubMed]
- Blumenthal, M.N.; Langefeld, C.D.; Beaty, T.H.; Bleecker, E.R.; Ober, C.; Lester, L.; Lange, E.; Barnes, K.C.; Wolf, R.; King, R.A.; et al. A genome-wide search for allergic response (atopy) genes in three ethnic groups: Collaborative Study on the Genetics of Asthma. Hum. Genet. 2004, 114, 157–164. [Google Scholar] [CrossRef] [PubMed]
- Kerner, B.; Jasinska, A.J.; DeYoung, J.; Almonte, M.; Choi, O.W.; Freimer, N.B. Polymorphisms in the GRIA1 gene region in psychotic bipolar disorder. Am. J. Med Genet. B Neuropsychiatr. Genet. 2009, 150B, 24–32. [Google Scholar] [CrossRef] [PubMed]
- Kutszegi, N.; Semsei, A.F.; Gezsi, A.; Sagi, J.C.; Nagy, V.; Csordas, K.; Jakab, Z.; Lautner-Csorba, O.; Gabor, K.M.; Kovacs, G.T.; et al. Subgroups of Paediatric Acute Lymphoblastic Leukaemia Might Differ Significantly in Genetic Predisposition to Asparaginase Hypersensitivity. PLoS ONE 2015, 10, e0140136. [Google Scholar] [CrossRef] [PubMed]
- Rajic, V.; Debeljak, M.; Goricar, K.; Jazbec, J. Polymorphisms in GRIA1 gene are a risk factor for asparaginase hypersensitivity during the treatment of childhood acute lymphoblastic leukemia. Leuk. Lymphoma 2015, 56, 3103–3108. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, C.A.; Smith, C.; Yang, W.; Date, M.; Bashford, D.; Larsen, E.; Bowman, W.P.; Liu, C.; Ramsey, L.B.; Chang, T.; et al. HLA-DRB1*07:01 is associated with a higher risk of asparaginase allergies. Blood 2014, 124, 1266–1276. [Google Scholar] [CrossRef] [PubMed]
- Kutszegi, N.; Yang, X.; Gezsi, A.; Schermann, G.; Erdelyi, D.J.; Semsei, A.F.; Gabor, K.M.; Sagi, J.C.; Kovacs, G.T.; Falus, A.; et al. HLA-DRB1*07:01-HLA-DQA1*02:01-HLA-DQB1*02:02 haplotype is associated with a high risk of asparaginase hypersensitivity in acute lymphoblastic leukemia. Haematologica 2017, 102, 1578–1586. [Google Scholar] [CrossRef] [PubMed]
- Rao, A.; Luo, C.; Hogan, P.G. Transcription factors of the NFAT family: Regulation and function. Annu. Rev. Immunol. 1997, 15, 707–747. [Google Scholar] [CrossRef] [PubMed]
- Xanthoudakis, S.; Viola, J.P.; Shaw, K.T.; Luo, C.; Wallace, J.D.; Bozza, P.T.; Luk, D.C.; Curran, T.; Rao, A. An enhanced immune response in mice lacking the transcription factor NFAT1. Science 1996, 272, 892–895. [Google Scholar] [CrossRef] [PubMed]
- Wolthers, B.O.; Frandsen, T.L.; Patel, C.J.; Abaji, R.; Attarbaschi, A.; Barzilai, S.; Colombini, A.; Escherich, G.; Grosjean, M.; Krajinovic, M.; et al. Trypsin encoding PRSS1-PRSS2 variation influence the risk of asparaginase-associated pancreatitis in children with acute lymphoblastic leukemia: A Ponte di Legno toxicity working group report. Haematologica 2018. [Google Scholar] [CrossRef] [PubMed]
- Hojfeldt, S.G.; Wolthers, B.O.; Tulstrup, M.; Abrahamsson, J.; Gupta, R.; Harila-Saari, A.; Heyman, M.; Henriksen, L.T.; Jonsson, O.G.; Lahteenmaki, P.M.; et al. Genetic predisposition to PEG-asparaginase hypersensitivity in children treated according to NOPHO ALL2008. Br. J. Haematol. 2019, 184, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Gil, A.; Ritter, O.; Saul, V.V.; Wilhelm, J.; Yang, C.Y.; Grosschedl, R.; Imai, Y.; Kuba, K.; Kracht, M.; Schmitz, M.L. The CCR4-NOT complex contributes to repression of Major Histocompatibility Complex class II transcription. Sci. Rep. 2017, 7, 3547. [Google Scholar] [CrossRef] [PubMed]
- Steinke, J.W.; Borish, L.; Rosenwasser, L.J. 5. Genetics of hypersensitivity. J. Allergy Clin. Immunol. 2003, 111, S495–S501. [Google Scholar] [CrossRef] [PubMed]
- Di Marco, A.; Cassinelli, G.; Arcamone, F. The discovery of daunorubicin. Cancer Treat. Rep. 1981, 65 (Suppl. 4), 3–8. [Google Scholar]
- Champoux, J.J. DNA topoisomerases: Structure, function, and mechanism. Annu. Rev. Biochem. 2001, 70, 369–413. [Google Scholar] [CrossRef] [PubMed]
- Gewirtz, D.A. A critical evaluation of the mechanisms of action proposed for the antitumor effects of the anthracycline antibiotics adriamycin and daunorubicin. Biochem. Pharmacol. 1999, 57, 727–741. [Google Scholar] [CrossRef]
- Armenian, S.; Bhatia, S. Predicting and Preventing Anthracycline-Related Cardiotoxicity. Am. Soc. Clin. Oncol. Educ. Book. Am. Soc. Clin. Oncology. Annu. Meet. 2018. [Google Scholar] [CrossRef] [PubMed]
- Blanco, J.G.; Leisenring, W.M.; Gonzalez-Covarrubias, V.M.; Kawashima, T.I.; Davies, S.M.; Relling, M.V.; Robison, L.L.; Sklar, C.A.; Stovall, M.; Bhatia, S. Genetic polymorphisms in the carbonyl reductase 3 gene CBR3 and the NAD(P)H:quinone oxidoreductase 1 gene NQO1 in patients who developed anthracycline-related congestive heart failure after childhood cancer. Cancer 2008, 112, 2789–2795. [Google Scholar] [CrossRef] [PubMed]
- Visscher, H.; Ross, C.J.; Rassekh, S.R.; Barhdadi, A.; Dube, M.P.; Al-Saloos, H.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; et al. Pharmacogenomic prediction of anthracycline-induced cardiotoxicity in children. J. Clin. Oncol. 2012, 30, 1422–1428. [Google Scholar] [CrossRef] [PubMed]
- Nagasawa, K.; Nagai, K.; Ohnishi, N.; Yokoyama, T.; Fujimoto, S. Contribution of Specific Transport Systems to Anthracycline Transport in Tumor and Normal Cells. Curr. Drug Metab. 2001, 2, 355–366. [Google Scholar] [CrossRef] [PubMed]
- Rossi, D.; Rasi, S.; Franceschetti, S.; Capello, D.; Castelli, A.; De Paoli, L.; Ramponi, A.; Chiappella, A.; Pogliani, E.M.; Vitolo, U.; et al. Analysis of the host pharmacogenetic background for prediction of outcome and toxicity in diffuse large B-cell lymphoma treated with R-CHOP21. Leukemia 2009, 23, 1118–1126. [Google Scholar] [CrossRef] [PubMed]
- Wojnowski, L.; Kulle, B.; Schirmer, M.; Schluter, G.; Schmidt, A.; Rosenberger, A.; Vonhof, S.; Bickeboller, H.; Toliat, M.R.; Suk, E.K.; et al. NAD(P)H oxidase and multidrug resistance protein genetic polymorphisms are associated with doxorubicin-induced cardiotoxicity. Circulation 2005, 112, 3754–3762. [Google Scholar] [CrossRef] [PubMed]
- Rajic, V.; Aplenc, R.; Debeljak, M.; Prestor, V.V.; Karas-Kuzelicki, N.; Mlinaric-Rascan, I.; Jazbec, J. Influence of the polymorphism in candidate genes on late cardiac damage in patients treated due to acute leukemia in childhood. Leuk. Lymphoma 2009, 50, 1693–1698. [Google Scholar] [CrossRef] [PubMed]
- Visscher, H.; Ross, C.J.; Rassekh, S.R.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; van der Pal, H.J.; Rogers, P.C.; Rieder, M.J.; et al. Validation of variants in SLC28A3 and UGT1A6 as genetic markers predictive of anthracycline-induced cardiotoxicity in children. Pediatr. Blood Cancer 2013, 60, 1375–1381. [Google Scholar] [CrossRef] [PubMed]
- Visscher, H.; Rassekh, S.R.; Sandor, G.S.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; van der Pal, H.J.; Rogers, P.C.; Rieder, M.J.; Carleton, B.C.; et al. Genetic variants in SLC22A17 and SLC22A7 are associated with anthracycline-induced cardiotoxicity in children. Pharmacogenomics 2015, 16, 1065–1076. [Google Scholar] [CrossRef] [PubMed]
- Aminkeng, F.; Bhavsar, A.P.; Visscher, H.; Rassekh, S.R.; Li, Y.; Lee, J.W.; Brunham, L.R.; Caron, H.N.; van Dalen, E.C.; Kremer, L.C.; et al. A coding variant in RARG confers susceptibility to anthracycline-induced cardiotoxicity in childhood cancer. Nat. Genet. 2015, 47, 1079–1084. [Google Scholar] [CrossRef] [PubMed]
- Bilbija, D.; Haugen, F.; Sagave, J.; Baysa, A.; Bastani, N.; Levy, F.O.; Sirsjo, A.; Blomhoff, R.; Valen, G. Retinoic acid signalling is activated in the postischemic heart and may influence remodelling. PLoS ONE 2012, 7, e44740. [Google Scholar] [CrossRef] [PubMed]
- Delacroix, L.; Moutier, E.; Altobelli, G.; Legras, S.; Poch, O.; Choukrallah, M.A.; Bertin, I.; Jost, B.; Davidson, I. Cell-specific interaction of retinoic acid receptors with target genes in mouse embryonic fibroblasts and embryonic stem cells. Mol. Cell Biol. 2010, 30, 231–244. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Liu, W.; Sun, C.L.; Armenian, S.H.; Hakonarson, H.; Hageman, L.; Ding, Y.; Landier, W.; Blanco, J.G.; Chen, L.; et al. Hyaluronan synthase 3 variant and anthracycline-related cardiomyopathy: A report from the children’s oncology group. J. Clin. Oncol. 2014, 32, 647–653. [Google Scholar] [CrossRef] [PubMed]
- Perik, P.J.; de Vries, E.G.; Gietema, J.A.; van der Graaf, W.T.; Sleijfer, D.T.; Suurmeijer, A.J.; van Veldhuisen, D.J. The dilemma of the strive for apoptosis in oncology: Mind the heart. Crit. Rev. Oncol. Hematol. 2005, 53, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Weinshilboum, R.M.; Sladek, S.L. Mercaptopurine pharmacogenetics: Monogenic inheritance of erythrocyte thiopurine methyltransferase activity. Am. J. Hum. Genet. 1980, 32, 651–662. [Google Scholar] [PubMed]
- Relling, M.V.; Gardner, E.E.; Sandborn, W.J.; Schmiegelow, K.; Pui, C.H.; Yee, S.W.; Stein, C.M.; Carrillo, M.; Evans, W.E.; Klein, T.E.; et al. Clinical Pharmacogenetics Implementation Consortium guidelines for thiopurine methyltransferase genotype and thiopurine dosing. Clin. Pharm. 2011, 89, 387–391. [Google Scholar] [CrossRef] [PubMed]
- Karas-Kuzelicki, N.; Jazbec, J.; Milek, M.; Mlinaric-Rascan, I. Heterozygosity at the TPMT gene locus, augmented by mutated MTHFR gene, predisposes to 6-MP related toxicities in childhood ALL patients. Leukemia 2008, 23, 971. [Google Scholar] [CrossRef] [PubMed]
- Karas-Kuzelicki, N.; Milek, M.; Mlinaric-Rascan, I. MTHFR and TYMS genotypes influence TPMT activity and its differential modulation in males and females. Clin. Biochem. 2010, 43, 37–42. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Yang, W.; Pei, D.; Cheng, C.; Smith, C.; Landier, W.; Hageman, L.; Chen, Y.; Yang, J.J.; Crews, K.R.; et al. Genomewide Approach Validates Thiopurine Methyltransferase Activity Is a Monogenic Pharmacogenomic Trait. Clin. Pharm. 2017, 101, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Tamm, R.; Magi, R.; Tremmel, R.; Winter, S.; Mihailov, E.; Smid, A.; Moricke, A.; Klein, K.; Schrappe, M.; Stanulla, M.; et al. Polymorphic variation in TPMT is the principal determinant of TPMT phenotype: A meta-analysis of three genome-wide association studies. Clin. Pharm. 2017, 101, 684–695. [Google Scholar] [CrossRef] [PubMed]
- Gerbek, T.; Ebbesen, M.; Nersting, J.; Frandsen, T.L.; Appell, M.L.; Schmiegelow, K. Role of TPMT and ITPA variants in mercaptopurine disposition. Cancer Chemother. Pharm. 2018, 81, 579–586. [Google Scholar] [CrossRef] [PubMed]
- Liu, C.; Janke, L.J.; Yang, J.J.; Evans, W.E.; Schuetz, J.D.; Relling, M.V. Differential effects of thiopurine methyltransferase (TPMT) and multidrug resistance-associated protein gene 4 (MRP4) on mercaptopurine toxicity. Cancer Chemother. Pharm. 2017, 80, 287–293. [Google Scholar] [CrossRef] [PubMed]
- Tanaka, Y.; Manabe, A.; Fukushima, H.; Suzuki, R.; Nakadate, H.; Kondoh, K.; Nakamura, K.; Koh, K.; Fukushima, T.; Tsuchida, M.; et al. Multidrug resistance protein 4 (MRP4) polymorphisms impact the 6-mercaptopurine dose tolerance during maintenance therapy in Japanese childhood acute lymphoblastic leukemia. Pharm. J. 2015, 15, 380–384. [Google Scholar] [CrossRef] [PubMed]
- Milosevic, G.; Kotur, N.; Krstovski, N.; Lazic, J.; Zukic, B.; Stankovic, B.; Janic, D.; Katsila, T.; Patrinos, G.P.; Pavlovic, S.; et al. Variants in TPMT, ITPA, ABCC4 and ABCB1 Genes as Predictors of 6-mercaptopurine Induced Toxicity in Children with Acute Lymphoblastic Leukemia. J. Med. Biochem. 2018, 37, 320–327. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Li, L.; Yang, P.; Yang, L.; Zheng, J.E.; Zhou, Y.; Han, Y. Optimal predictor for 6-mercaptopurine intolerance in Chinese children with acute lymphoblastic leukemia: NUDT15, TPMT, or ITPA genetic variants? BMC Cancer 2018, 18, 516. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Landier, W.; Yang, W.; Liu, C.; Hageman, L.; Cheng, C.; Pei, D.; Chen, Y.; Crews, K.R.; Kornegay, N.; et al. Inherited NUDT15 variant is a genetic determinant of mercaptopurine intolerance in children with acute lymphoblastic leukemia. J. Clin. Oncol. 2015, 33, 1235–1242. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Nishii, R.; Perez-Andreu, V.; Yang, W.; Klussmann, F.A.; Zhao, X.; Lin, T.N.; Hoshitsuki, K.; Nersting, J.; Kihira, K.; et al. NUDT15 polymorphisms alter thiopurine metabolism and hematopoietic toxicity. Nat. Genet. 2016, 48, 367–373. [Google Scholar] [CrossRef] [PubMed]
- Liang, D.C.; Yang, C.P.; Liu, H.C.; Jaing, T.H.; Chen, S.H.; Hung, I.J.; Yeh, T.C.; Lin, T.H.; Lai, C.L.; Lai, C.Y.; et al. NUDT15 gene polymorphism related to mercaptopurine intolerance in Taiwan Chinese children with acute lymphoblastic leukemia. Pharm. J. 2016, 16, 536–539. [Google Scholar] [CrossRef] [PubMed]
- Moriyama, T.; Yang, Y.L.; Nishii, R.; Ariffin, H.; Liu, C.; Lin, T.N.; Yang, W.; Lin, D.T.; Yu, C.H.; Kham, S.; et al. Novel variants in NUDT15 and thiopurine intolerance in children with acute lymphoblastic leukemia from diverse ancestry. Blood 2017, 130, 1209–1212. [Google Scholar] [CrossRef] [PubMed]
- Relling, M.V.; Schwab, M.; Whirl-Carrillo, M.; Suarez-Kurtz, G.; Pui, C.H.; Stein, C.M.; Moyer, A.M.; Evans, W.E.; Klein, T.E.; Antillon-Klussmann, F.G.; et al. Clinical Pharmacogenetics Implementation Consortium Guideline for Thiopurine Dosing Based on TPMT and NUDT15 Genotypes: 2018 Update. Clin. Pharm. 2018. [Google Scholar] [CrossRef] [PubMed]
- Stocco, G.; Yang, W.; Crews, K.R.; Thierfelder, W.E.; Decorti, G.; Londero, M.; Franca, R.; Rabusin, M.; Valsecchi, M.G.; Pei, D.; et al. PACSIN2 polymorphism influences TPMT activity and mercaptopurine-related gastrointestinal toxicity. Hum. Mol. Genet. 2012, 21, 4793–4804. [Google Scholar] [CrossRef] [PubMed]
- Stocco, G.; Franca, R.; Favretto, D.; Giurici, N.; Ventura, A.; Decorti, G.; Vinti, L.; Colombini, A.; Brivio, E.; Bettini, L.R.; et al. PACSIN2 rs2413739 Polymorphism and Thiopurine Pharmacokinetics: Validation Studies in Pediatric Patients. Blood 2017, 130, 4999. [Google Scholar]
- Smid, A.; Karas-Kuzelicki, N.; Jazbec, J.; Mlinaric-Rascan, I. PACSIN2 polymorphism is associated with thiopurine-induced hematological toxicity in children with acute lymphoblastic leukaemia undergoing maintenance therapy. Sci. Rep. 2016, 6, 30244. [Google Scholar] [CrossRef] [PubMed]
- Roberts, R.L.; Wallace, M.C.; Seinen, M.L.; Krishnaprasad, K.; Chew, A.; Lawrance, I.; Prosser, R.; Bampton, P.; Grafton, R.; Simms, L.; et al. PACSIN2 does not influence thiopurine-related toxicity in patients with inflammatory bowel disease. Am. J. Gastroenterol. 2014, 109, 925–927. [Google Scholar] [CrossRef] [PubMed]
- Meyer, J.A.; Wang, J.; Hogan, L.E.; Yang, J.J.; Dandekar, S.; Patel, J.P.; Tang, Z.; Zumbo, P.; Li, S.; Zavadil, J.; et al. Relapse-specific mutations in NT5C2 in childhood acute lymphoblastic leukemia. Nat. Genet. 2013, 45, 290–294. [Google Scholar] [CrossRef] [PubMed]
- Tzoneva, G.; Perez-Garcia, A.; Carpenter, Z.; Khiabanian, H.; Tosello, V.; Allegretta, M.; Paietta, E.; Racevskis, J.; Rowe, J.M.; Tallman, M.S.; et al. Activating mutations in the NT5C2 nucleotidase gene drive chemotherapy resistance in relapsed ALL. Nat. Med. 2013, 19, 368–371. [Google Scholar] [CrossRef] [PubMed]
- Lindqvist, C.M.; Lundmark, A.; Nordlund, J.; Freyhult, E.; Ekman, D.; Carlsson Almlof, J.; Raine, A.; Overnas, E.; Abrahamsson, J.; Frost, B.M.; et al. Deep targeted sequencing in pediatric acute lymphoblastic leukemia unveils distinct mutational patterns between genetic subtypes and novel relapse-associated genes. Oncotarget 2016, 7, 64071–64088. [Google Scholar] [CrossRef] [PubMed]
- Kotur, N.; Stankovic, B.; Kassela, K.; Georgitsi, M.; Vicha, A.; Leontari, I.; Dokmanovic, L.; Janic, D.; Krstovski, N.; Klaassen, K.; et al. 6-mercaptopurine influences TPMT gene transcription in a TPMT gene promoter variable number of tandem repeats-dependent manner. Pharmacogenomics 2012, 13, 283–295. [Google Scholar] [CrossRef] [PubMed]
- Zukic, B.; Radmilovic, M.; Stojiljkovic, M.; Tosic, N.; Pourfarzad, F.; Dokmanovic, L.; Janic, D.; Colovic, N.; Philipsen, S.; Patrinos, G.P.; et al. Functional analysis of the role of the TPMT gene promoter VNTR polymorphism in TPMT gene transcription. Pharmacogenomics 2010, 11, 547–557. [Google Scholar] [CrossRef] [PubMed]
- Kotur, N.; Dokmanovic, L.; Janic, D.; Stankovic, B.; Krstovski, N.; Tosic, N.; Katsila, T.; Patrinos, G.P.; Zukic, B.; Pavlovic, S. TPMT gene expression is increased during maintenance therapy in childhood acute lymphoblastic leukemia patients in a TPMT gene promoter variable number of tandem repeat-dependent manner. Pharmacogenomics 2015, 16, 1701–1712. [Google Scholar] [CrossRef] [PubMed]
- Beesley, A.H.; Cummings, A.J.; Freitas, J.R.; Hoffmann, K.; Firth, M.J.; Ford, J.; de Klerk, N.H.; Kees, U.R. The gene expression signature of relapse in paediatric acute lymphoblastic leukaemia: Implications for mechanisms of therapy failure. Br. J. Haematol. 2005, 131, 447–456. [Google Scholar] [CrossRef] [PubMed]
- Bhojwani, D.; Kang, H.; Moskowitz, N.P.; Min, D.J.; Lee, H.; Potter, J.W.; Davidson, G.; Willman, C.L.; Borowitz, M.J.; Belitskaya-Levy, I.; et al. Biologic pathways associated with relapse in childhood acute lymphoblastic leukemia: A Children’s Oncology Group study. Blood 2006, 108, 711–717. [Google Scholar] [CrossRef] [PubMed]
- Hogan, L.E.; Meyer, J.A.; Yang, J.; Wang, J.; Wong, N.; Yang, W.; Condos, G.; Hunger, S.P.; Raetz, E.; Saffery, R.; et al. Integrated genomic analysis of relapsed childhood acute lymphoblastic leukemia reveals therapeutic strategies. Blood 2011, 118, 5218–5226. [Google Scholar] [CrossRef] [PubMed]
- Staal, F.J.T.; de Ridder, D.; Szczepanski, T.; Schonewille, T.; van der Linden, E.C.E.; van Wering, E.R.; van der Velden, V.H.J.; van Dongen, J.J.M. Genome-wide expression analysis of paired diagnosis–relapse samples in ALL indicates involvement of pathways related to DNA replication, cell cycle and DNA repair, independent of immune phenotype. Leukemia 2010, 24, 491. [Google Scholar] [CrossRef] [PubMed]
- Yang, J.J.; Bhojwani, D.; Yang, W.; Cai, X.; Stocco, G.; Crews, K.; Wang, J.; Morrison, D.; Devidas, M.; Hunger, S.P.; et al. Genome-wide copy number profiling reveals molecular evolution from diagnosis to relapse in childhood acute lymphoblastic leukemia. Blood 2008, 112, 4178–4183. [Google Scholar] [CrossRef] [PubMed]
- Zaza, G.; Cheok, M.; Yang, W.; Panetta, J.C.; Pui, C.H.; Relling, M.V.; Evans, W.E. Gene expression and thioguanine nucleotide disposition in acute lymphoblastic leukemia after in vivo mercaptopurine treatment. Blood 2005, 106, 1778–1785. [Google Scholar] [CrossRef] [PubMed]
- Ansari, A.; Aslam, Z.; De Sica, A.; Smith, M.; Gilshenan, K.; Fairbanks, L.; Marinaki, A.; Sanderson, J.; Duley, J. Influence of xanthine oxidase on thiopurine metabolism in Crohn’s disease. Aliment. Pharmacol. Ther. 2008, 28, 749–757. [Google Scholar] [CrossRef] [PubMed]
- Huang, Y.; Anderle, P.; Bussey, K.J.; Barbacioru, C.; Shankavaram, U.; Dai, Z.; Reinhold, W.C.; Papp, A.; Weinstein, J.N.; Sadee, W. Membrane transporters and channels: Role of the transportome in cancer chemosensitivity and chemoresistance. Cancer Res. 2004, 64, 4294–4301. [Google Scholar] [CrossRef] [PubMed]
- Beesley, A.H.; Firth, M.J.; Anderson, D.; Samuels, A.L.; Ford, J.; Kees, U.R. Drug-gene modeling in pediatric T-cell acute lymphoblastic leukemia highlights importance of 6-mercaptopurine for outcome. Cancer Res. 2013, 73, 2749–2759. [Google Scholar] [CrossRef] [PubMed]
- Organista-Nava, J.; Gomez-Gomez, Y.; Illades-Aguiar, B.; Rivera-Ramirez, A.B.; Saavedra-Herrera, M.V.; Leyva-Vazquez, M.A. Overexpression of dihydrofolate reductase is a factor of poor survival in acute lymphoblastic leukemia. Oncol. Lett. 2018, 15, 8405–8411. [Google Scholar] [CrossRef] [PubMed]
- Wojtuszkiewicz, A.; Peters, G.J.; van Woerden, N.L.; Dubbelman, B.; Escherich, G.; Schmiegelow, K.; Sonneveld, E.; Pieters, R.; van de Ven, P.M.; Jansen, G.; et al. Methotrexate resistance in relation to treatment outcome in childhood acute lymphoblastic leukemia. J. Hematol. Oncol. 2015, 8, 61. [Google Scholar] [CrossRef] [PubMed]
- Kaluzna, E.; Strauss, E.; Zajac-Spychala, O.; Gowin, E.; Swiatek-Koscielna, B.; Nowak, J.; Fichna, M.; Mankowski, P.; Januszkiewicz-Lewandowska, D. Functional variants of gene encoding folate metabolizing enzyme and methotrexate-related toxicity in children with acute lymphoblastic leukemia. Eur. J. Pharm. 2015, 769, 93–99. [Google Scholar] [CrossRef] [PubMed]
- Roy Moulik, N.; Kumar, A.; Agrawal, S.; Awasthi, S.; Mahdi, A.A.; Kumar, A. Role of folate status and methylenetetrahydrofolate reductase genotype on the toxicity and outcome of induction chemotherapy in children with acute lymphoblastic leukemia. Leuk. Lymphoma 2015, 56, 1379–1384. [Google Scholar] [CrossRef] [PubMed]
- Araoz, H.V.; D’Aloi, K.; Foncuberta, M.E.; Sanchez La Rosa, C.G.; Alonso, C.N.; Chertkoff, L.; Felice, M. Pharmacogenetic studies in children with acute lymphoblastic leukemia in Argentina. Leuk. Lymphoma 2015, 56, 1370–1378. [Google Scholar] [CrossRef] [PubMed]
- Finkelstein, Y.; Blonquist, T.M.; Vijayanathan, V.; Stevenson, K.E.; Neuberg, D.S.; Silverman, L.B.; Vrooman, L.M.; Sallan, S.E.; Cole, P.D. A thymidylate synthase polymorphism is associated with increased risk for bone toxicity among children treated for acute lymphoblastic leukemia. Pediatr. Blood Cancer 2017, 64, e26393. [Google Scholar] [CrossRef] [PubMed]
- Yazicioglu, B.; Kaya, Z.; Guntekin Ergun, S.; Percin, F.; Kocak, U.; Yenicesu, I.; Gursel, T. Influence of Folate-Related Gene Polymorphisms on High-Dose Methotrexate-Related Toxicity and Prognosis in Turkish Children with Acute Lymphoblastic Leukemia. Turk. J. Haematol. 2017, 34, 143–150. [Google Scholar] [CrossRef] [PubMed]
- Lazic, J.; Kotur, N.; Krstovski, N.; Dokmanovic, L.; Zukic, B.; Predojevic-Samardzic, J.; Zivotic, M.; Milosevic, G.; Djoric, M.; Janic, D.; et al. Importance of pharmacogenetic markers in the methylenetetrahydrofolate reductase gene during methotrexate treatment in pediatric patients with acute lymphoblastic leukemia. Arch. Biol. Sci. 2017, 69, 239–246. [Google Scholar] [CrossRef]
- Yao, P.; He, X.; Zhang, R.; Tong, R.; Xiao, H. The influence of MTHFR genetic polymorphisms on adverse reactions after methotrexate in patients with hematological malignancies: A meta-analysis. Hematology 2019, 24, 10–19. [Google Scholar] [CrossRef] [PubMed]
- Zhu, C.; Liu, Y.W.; Wang, S.Z.; Li, X.L.; Nie, X.L.; Yu, X.T.; Zhao, L.B.; Wang, X.L. Associations between the C677T and A1298C polymorphisms of MTHFR and the toxicity of methotrexate in childhood malignancies: A meta-analysis. Pharm. J. 2018, 18, 450–459. [Google Scholar] [CrossRef] [PubMed]
- Giletti, A.; Esperon, P. Genetic markers in methotrexate treatments. Pharm. J. 2018, 18, 689–703. [Google Scholar] [CrossRef] [PubMed]
- Trevino, L.R.; Shimasaki, N.; Yang, W.; Panetta, J.C.; Cheng, C.; Pei, D.; Chan, D.; Sparreboom, A.; Giacomini, K.M.; Pui, C.H.; et al. Germline genetic variation in an organic anion transporter polypeptide associated with methotrexate pharmacokinetics and clinical effects. J. Clin. Oncol. 2009, 27, 5972–5978. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Panetta, J.C.; Smith, C.; Yang, W.; Fan, Y.; Winick, N.J.; Martin, P.L.; Cheng, C.; Devidas, M.; Pui, C.H.; et al. Genome-wide study of methotrexate clearance replicates SLCO1B1. Blood 2013, 121, 898–904. [Google Scholar] [CrossRef] [PubMed]
- Ramsey, L.B.; Bruun, G.H.; Yang, W.; Trevino, L.R.; Vattathil, S.; Scheet, P.; Cheng, C.; Rosner, G.L.; Giacomini, K.M.; Fan, Y.; et al. Rare versus common variants in pharmacogenetics: SLCO1B1 variation and methotrexate disposition. Genome Res. 2012, 22, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, E.; Martin-Guerrero, I.; Ballesteros, J.; Pinan, M.A.; Garcia-Miguel, P.; Navajas, A.; Garcia-Orad, A. Polymorphisms of the SLCO1B1 gene predict methotrexate-related toxicity in childhood acute lymphoblastic leukemia. Pediatr. Blood Cancer 2011, 57, 612–619. [Google Scholar] [CrossRef] [PubMed]
- Radtke, S.; Zolk, O.; Renner, B.; Paulides, M.; Zimmermann, M.; Moricke, A.; Stanulla, M.; Schrappe, M.; Langer, T. Germline genetic variations in methotrexate candidate genes are associated with pharmacokinetics, toxicity, and outcome in childhood acute lymphoblastic leukemia. Blood 2013, 121, 5145–5153. [Google Scholar] [CrossRef] [PubMed]
- Zhang, H.N.; He, X.L.; Wang, C.; Wang, Y.; Chen, Y.J.; Li, J.X.; Niu, C.H.; Gao, P. Impact of SLCO1B1 521T > C variant on leucovorin rescue and risk of relapse in childhood acute lymphoblastic leukemia treated with high-dose methotrexate. Pediatr. Blood Cancer 2014, 61, 2203–2207. [Google Scholar] [CrossRef] [PubMed]
- Liu, S.G.; Gao, C.; Zhang, R.D.; Zhao, X.X.; Cui, L.; Li, W.J.; Chen, Z.P.; Yue, Z.X.; Zhang, Y.Y.; Wu, M.Y.; et al. Polymorphisms in methotrexate transporters and their relationship to plasma methotrexate levels, toxicity of high-dose methotrexate, and outcome of pediatric acute lymphoblastic leukemia. Oncotarget 2017, 8, 37761–37772. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Lopez, E.; Ballesteros, J.; Pinan, M.A.; Sanchez de Toledo, J.; Garcia de Andoin, N.; Garcia-Miguel, P.; Navajas, A.; Garcia-Orad, A. Polymorphisms in the methotrexate transport pathway: A new tool for MTX plasma level prediction in pediatric acute lymphoblastic leukemia. Pharm. Genom. 2013, 23, 53–61. [Google Scholar] [CrossRef] [PubMed]
- Assaraf, Y.G. Molecular basis of antifolate resistance. Cancer Metastasis Rev. 2007, 26, 153–181. [Google Scholar] [CrossRef] [PubMed]
- Cheok, M.H.; Evans, W.E. Acute lymphoblastic leukaemia: A model for the pharmacogenomics of cancer therapy. Nat. Rev. Cancer 2006, 6, 117–129. [Google Scholar] [CrossRef] [PubMed]
- Wijdeven, R.H.; Pang, B.; Assaraf, Y.G.; Neefjes, J. Old drugs, novel ways out: Drug resistance toward cytotoxic chemotherapeutics. Drug Resist. Updates Rev. Comment. Antimicrob. Anticancer Chemother. 2016, 28, 65–81. [Google Scholar] [CrossRef] [PubMed]
- Belkov, V.M.; Krynetski, E.Y.; Schuetz, J.D.; Yanishevski, Y.; Masson, E.; Mathew, S.; Raimondi, S.; Pui, C.-H.; Relling, M.V.; Evans, W.E. Reduced Folate Carrier Expression in Acute Lymphoblastic Leukemia: A Mechanism for Ploidy but not Lineage Differences in Methotrexate Accumulation. Blood 1999, 93, 1643. [Google Scholar] [PubMed]
- Gorlick, R.; Goker, E.; Trippett, T.; Steinherz, P.; Elisseyeff, Y.; Mazumdar, M.; Flintoff, W.F.; Bertino, J.R. Defective Transport Is a Common Mechanism of Acquired Methotrexate Resistance in Acute Lymphocytic Leukemia and Is Associated With Decreased Reduced Folate Carrier Expression. Blood 1997, 89, 1013. [Google Scholar] [PubMed]
- Rots, M.G.; Willey, J.C.; Jansen, G.; Van Zantwijk, C.H.; Noordhuis, P.; DeMuth, J.P.; Kuiper, E.; Veerman, A.J.P.; Pieters, R.; Peters, G.J. mRNA expression levels of methotrexate resistance-related proteins in childhood leukemia as determined by a standardized competitive template-based RT-PCR method. Leukemia 2000, 14, 2166–2175. [Google Scholar] [CrossRef] [PubMed]
- Levy, A.S.; Sather, H.N.; Steinherz, P.G.; Sowers, R.; La, M.; Moscow, J.A.; Gaynon, P.S.; Uckun, F.M.; Bertino, J.R.; Gorlick, R. Reduced Folate Carrier and Dihydrofolate Reductase Expression in Acute Lymphocytic Leukemia May Predict Outcome: A Children’s Cancer Group Study. J. Pediatr. Hematol. Oncol. 2003, 25, 688–695. [Google Scholar] [CrossRef] [PubMed]
- Galpin, A.J.; Schuetz, J.D.; Masson, E.; Yanishevski, Y.; Synold, T.W.; Barredo, J.C.; Pui, C.H.; Relling, M.V.; Evans, W.E. Differences in folylpolyglutamate synthetase and dihydrofolate reductase expression in human B-lineage versus T-lineage leukemic lymphoblasts: Mechanisms for lineage differences in methotrexate polyglutamylation and cytotoxicity. Mol. Pharm. 1997, 52, 155–163. [Google Scholar] [CrossRef]
- Rots, M.G.; Pieters, R.; Kaspers, G.-J.L.; van Zantwijk, C.H.; Noordhuis, P.; Mauritz, R.; Veerman, A.J.P.; Jansen, G.; Peters, G.J. Differential Methotrexate Resistance in Childhood T- Versus Common/PreB-Acute Lymphoblastic Leukemia Can Be Measured by an In Situ Thymidylate Synthase Inhibition Assay, But Not by the MTT Assay. Blood 1999, 93, 1067. [Google Scholar] [PubMed]
- Kager, L.; Cheok, M.; Yang, W.; Zaza, G.; Cheng, Q.; Panetta, J.C.; Pui, C.H.; Downing, J.R.; Relling, M.V.; Evans, W.E. Folate pathway gene expression differs in subtypes of acute lymphoblastic leukemia and influences methotrexate pharmacodynamics. J. Clin. Invest. 2005, 115, 110–117. [Google Scholar] [CrossRef] [PubMed]
- Sorich, M.J.; Pottier, N.; Pei, D.; Yang, W.; Kager, L.; Stocco, G.; Cheng, C.; Panetta, J.C.; Pui, C.H.; Relling, M.V.; et al. In vivo response to methotrexate forecasts outcome of acute lymphoblastic leukemia and has a distinct gene expression profile. PLoS Med. 2008, 5, e83. [Google Scholar] [CrossRef] [PubMed]
- Dulucq, S.; St-Onge, G.; Gagne, V.; Ansari, M.; Sinnett, D.; Labuda, D.; Moghrabi, A.; Krajinovic, M. DNA variants in the dihydrofolate reductase gene and outcome in childhood ALL. Blood 2008, 111, 3692–3700. [Google Scholar] [CrossRef] [PubMed]
- Rocha, J.C.; Cheng, C.; Liu, W.; Kishi, S.; Das, S.; Cook, E.H.; Sandlund, J.T.; Rubnitz, J.; Ribeiro, R.; Campana, D.; et al. Pharmacogenetics of outcome in children with acute lymphoblastic leukemia. Blood 2005, 105, 4752–4758. [Google Scholar] [CrossRef] [PubMed]
- Al-Mahayri, Z.N.; Patrinos, G.P.; Ali, B.R. Pharmacogenomics in pediatric acute lymphoblastic leukemia: Promises and limitations. Pharmacogenomics 2017, 18, 687–699. [Google Scholar] [CrossRef] [PubMed]
- Viennas, E.; Komianou, A.; Mizzi, C.; Stojiljkovic, M.; Mitropoulou, C.; Muilu, J.; Vihinen, M.; Grypioti, P.; Papadaki, S.; Pavlidis, C.; et al. Expanded national database collection and data coverage in the FINDbase worldwide database for clinically relevant genomic variation allele frequencies. Nucleic Acids Res. 2017, 45, D846–D853. [Google Scholar] [CrossRef] [PubMed]
- Mizzi, C.; Dalabira, E.; Kumuthini, J.; Dzimiri, N.; Balogh, I.; Basak, N.; Bohm, R.; Borg, J.; Borgiani, P.; Bozina, N.; et al. A European Spectrum of Pharmacogenomic Biomarkers: Implications for Clinical Pharmacogenomics. PLoS ONE 2016, 11, e0162866. [Google Scholar] [CrossRef] [PubMed]
- Hong, H.; Xu, L.; Su, Z.; Liu, J.; Ge, W.; Shen, J.; Fang, H.; Perkins, R.; Shi, L.; Tong, W. Pitfall of genome-wide association studies: Sources of inconsistency in genotypes and their effects. J. Biomed. Sci. Eng. 2012, 5, 557–573. [Google Scholar] [CrossRef]
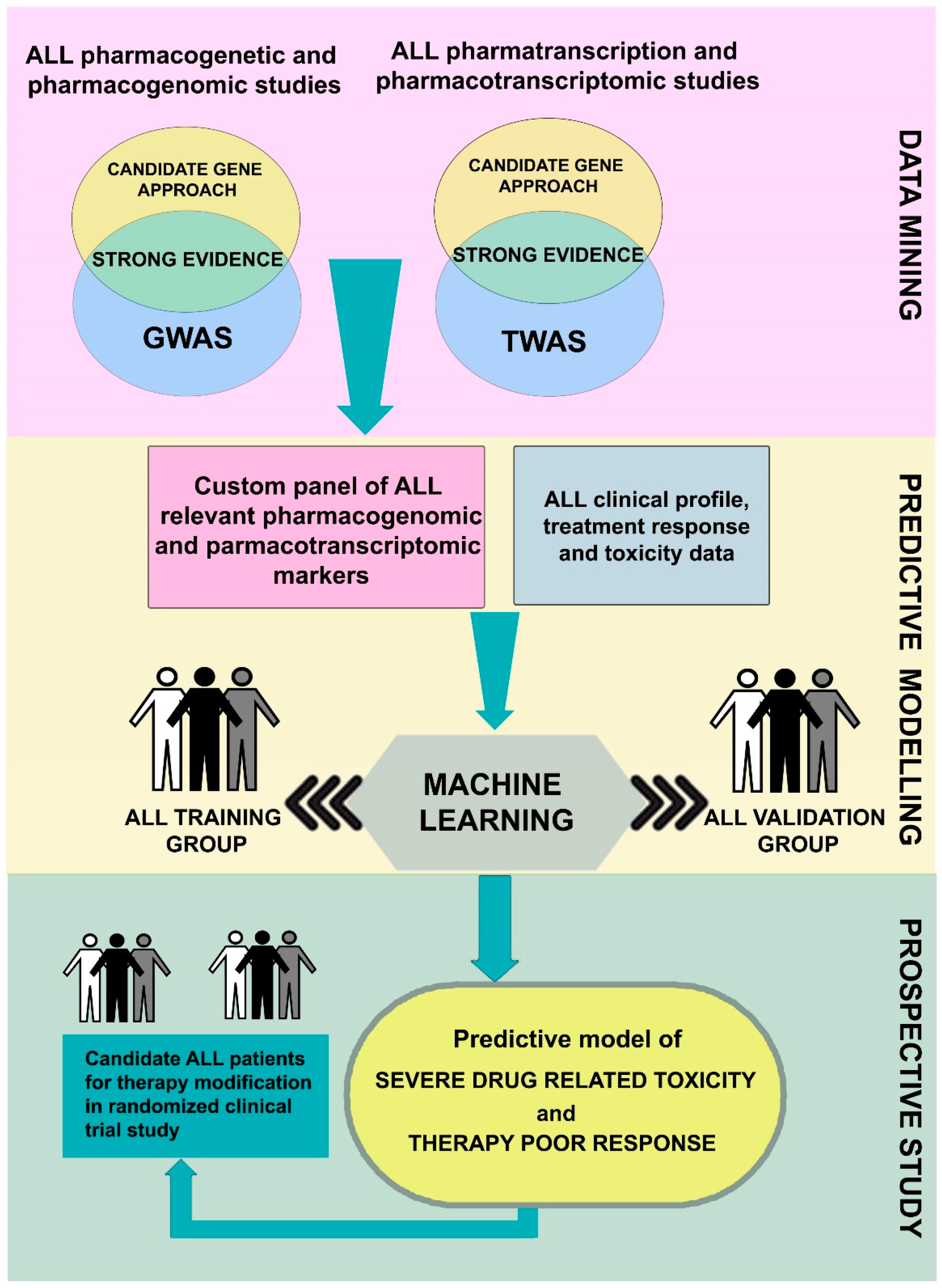

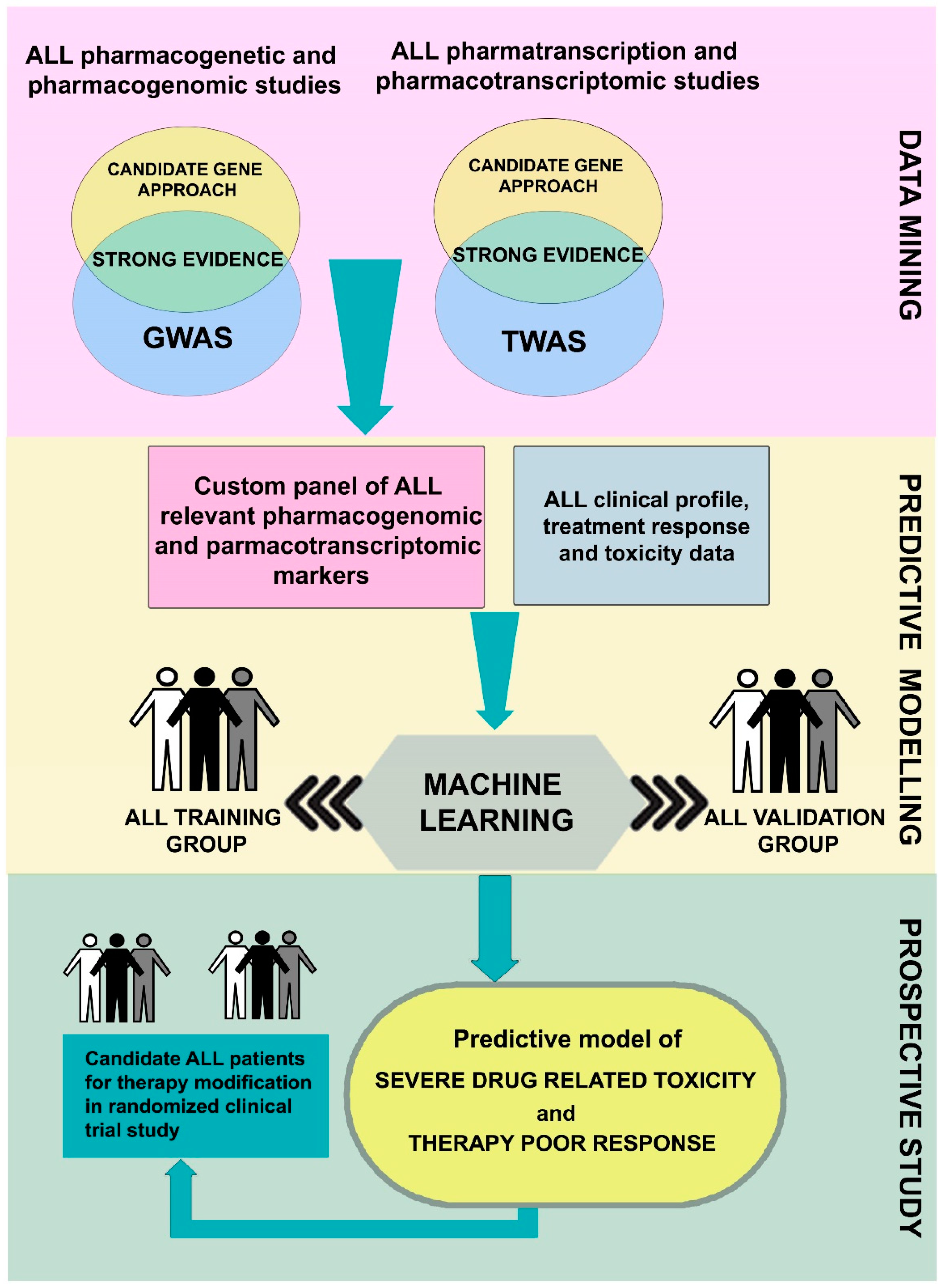{kind=link}
| Pharmacogene | Variant or RNA | Effect | Methodology | References |
|---|---|---|---|---|
| Glucocorticoid drugs | ||||
| ABCB1, WT 1-AS | rs6007758, rs41488548, rs10264856, rs4728709 | Higher clearance of DEX | Microarray | [33] |
| SERPINA6 | rs12589136 | Higher plasma cortisol levels | WES | [34] |
| Intergenic variant | rs10989692 | Increased risk of osteonecrosis | WES | [35] |
| ACP-1 | Multiple SNPs | Increased risk of osteonecrosis | Microarray | [36] |
| hsa-miR-142-3p, hsa-miR-17-5p | miRNA | High correlation with GC resistance | Omni-Search | [40] |
| EMP1 | mRNA | Higher expression in prednisone poor responders | Microarray | [41] |
| CASP1, NLRP3 | mRNA | High expression and subsequent high GC resistance | Microarray | [45] |
| SMARCA4, ARID1A, SMARCB1 | mRNA | Decreased expression is associated with GC resistance | Microarray | [46] |
| CREBBP | somatic mutations | Presence of damaging mutations leads to GC resistance | Microarray | [12] |
| Vincristine | ||||
| CEP72 | rs924607 | vincristine-related peripheral neuropathy | Microarray | [58] |
| ABCC2 | rs374006 rs12826 | vincristine-related peripheral neuropathy | Targeted DNA sequencing | [63] |
| SYNE2, MRPL47, BAHD1 * | rs2781377 rs10513762 rs3803357 * | vincristine-related peripheral neuropathy | WES | [64] |
| COCH | rs1045644 rs7963521 | vincristine-related peripheral neuropathy | Microarray | [69] |
| miR-125b, miR-99a, miR-100 | microRNA | resistance to vincristine | microRNA expression study | [72] |
| miR-300, DROSHA | rs12894467 rs639174 | vincristine-related peripheral neuropathy, vomits | Microarray | [74] |
| miR-3117-3p, miR-4481 | rs12402181 rs7896283 | vincristine-related peripheral neuropathy | Microarray | [75] |
| Asparaginase | ||||
| MYBBP1A, IL16, SPEF2 | rs3809849 rs11556218 rs34708521 | allergy, pancreatitis and thrombosis related to asparaginase, EFS, OS | WES | [82] |
| OPRM1 | microRNA | resistance to asparaginase | genome-wide RNAi screening | [87] |
| PNPLA3 | rs738409 | elevated alanine transaminase (ALT) levels leading to hepatotoxicity | Microarray | [89] |
| GRIA1 | rs4958351 | asparaginase hypersensitivity | Microarray | [91] |
| HLA-DRB1 | HLA-DRB1*07:01 | asparaginase hypersensitivity | Microarray | [95] |
| NFATC2, HLA-DRB1, GRIA1 | rs6021191 HLA-DRB1*07:01 rs4958351 | asparaginase hypersensitivity | Microarray | [77] |
| HLADRB1, HLADQ1 | HLADRB1*07:01 HLA-DQB1*02:02 | asparaginase hypersensitivity | targeted DNA sequencing | [97] |
| PRSS1-PRSS2 locus NFATC2 | rs4726576 rs10273639 rs62228256 | asparaginase hypersensitivity, pancreatitis | Microarray | [100] |
| CNOT3, HLADQA1, TAP2 | rs73062673 rs9272131 | asparaginase hypersensitivity | Microarray | [101] |
| Anthracyclines | ||||
| SLC28A3 | rs7853758 | anthracycline-induced cardiotoxicity | Microarray | [109] |
| UGT1A6, SLC28A3 | rs17863783 rs885004 rs7853758 * | anthracycline-induced cardiotoxicity | Microarray | [114] |
| SLC22A17, SLC22A7 | rs4982753 rs4149178 | anthracycline-induced cardiotoxicity | Microarray | [115] |
| RARG | rs2229774 | anthracycline-induced cardiotoxicity | Microarray | [116] |
| HAS3 | rs2232228 | anthracycline-induced cardiotoxicity | Microarray | [119] |
| Thiopurine drugs | ||||
| TPMT, NUDT15 | rs1142345, rs116855232 | 6-MP dose intensity | WGS | [132] |
| TPMT | rs1142345 | TPMT activity | WGS | [125,126] |
| PACSIN2 | rs2413739, mRNA | TPMT activity | WGS, RNA seq | [137] |
| NT5C2 | somatic mutations | Relapse | WES, RNA seq | [141,142] |
| XDH, SLC29A1, ADA, CASP7, TOPBP1, ANAPC5, CCT4 | mRNA | Level of TGN after initial MP treatment | Microarray | [152] |
| SLC25A3, ATP50, COX5B, COX7A2L; RPS19, RPL18, RPS25, RPL23; EEF1G, EIF3S5, eIF3k | mRNA | Level of TGN after initial 6-MP+MTX treatment | Microarray | [152] |
| PAICS, TYMS, CAD, ATIC, GART, MSH6 | mRNA | Late relapse, probably related to 6-MP and MTX | Microarray | [149] |
| SUOX, ABCC1 | mRNA | Thiopurine resistance | Microarray | [155] |
| Methotrexate | ||||
| SLCO1B1 | rs4149081, rs11045879, rs11045821, rs4149056 | MTX clearance | WGS | [167,168] |
| ABCC2, ABCC4 | rs3740065, rs9516519 | MTX plasma level | Targeted DNA sequencing | [174] |
| DHFR, TYMS, CTPS; BCL3, CDC20, CENPF, FAIM3; POLD3, RPA3, RNASEH2A, RPM1, 2AFX | mRNA | Reduction of circulating leukemia cells after initial treatment | Microarray | [185] |
| DHFR, TYMS | mRNA | 5-year disease free survival | Microarray | [185] |
| TYMS, MTHFD1, RUVBL2 | mRNA | MTX-PG accumulation after high dose MTX treatment in nonhyperdipoid B-ALL | Microarray | [184] |
| MTHFD2, PPAT, RUVBL2 | mRNA | MTX-PG accumulation after high dose MTX treatment in T-ALL | Microarray | [184] |
| ABCG2, ABCC4, TYMS | mRNA | MTX cytotoxic effect in nonhyperdipoid B-ALL, as measured by the reduction of circulating ALL cells | Microarray | [184] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pavlovic, S.; Kotur, N.; Stankovic, B.; Zukic, B.; Gasic, V.; Dokmanovic, L. Pharmacogenomic and Pharmacotranscriptomic Profiling of Childhood Acute Lymphoblastic Leukemia: Paving the Way to Personalized Treatment. Genes 2019, 10, 191. https://doi.org/10.3390/genes10030191
Pavlovic S, Kotur N, Stankovic B, Zukic B, Gasic V, Dokmanovic L. Pharmacogenomic and Pharmacotranscriptomic Profiling of Childhood Acute Lymphoblastic Leukemia: Paving the Way to Personalized Treatment. Genes. 2019; 10(3):191. https://doi.org/10.3390/genes10030191
Chicago/Turabian StylePavlovic, Sonja, Nikola Kotur, Biljana Stankovic, Branka Zukic, Vladimir Gasic, and Lidija Dokmanovic. 2019. "Pharmacogenomic and Pharmacotranscriptomic Profiling of Childhood Acute Lymphoblastic Leukemia: Paving the Way to Personalized Treatment" Genes 10, no. 3: 191. https://doi.org/10.3390/genes10030191
APA StylePavlovic, S., Kotur, N., Stankovic, B., Zukic, B., Gasic, V., & Dokmanovic, L. (2019). Pharmacogenomic and Pharmacotranscriptomic Profiling of Childhood Acute Lymphoblastic Leukemia: Paving the Way to Personalized Treatment. Genes, 10(3), 191. https://doi.org/10.3390/genes10030191