Diagnostic and Therapeutic Strategies for Fluoropyrimidine Treatment of Patients Carrying Multiple DPYD Variants

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients
2.2. Dihydropyrimidine Dehydrogenase Enzyme Activity Measurements
2.3. Molecular Methods for Estimation of Phasing
2.4. Frequencies of Compound Heterozygous DPYD Carriers
Exome Aggregation Consortium and Genome Aggregation Database
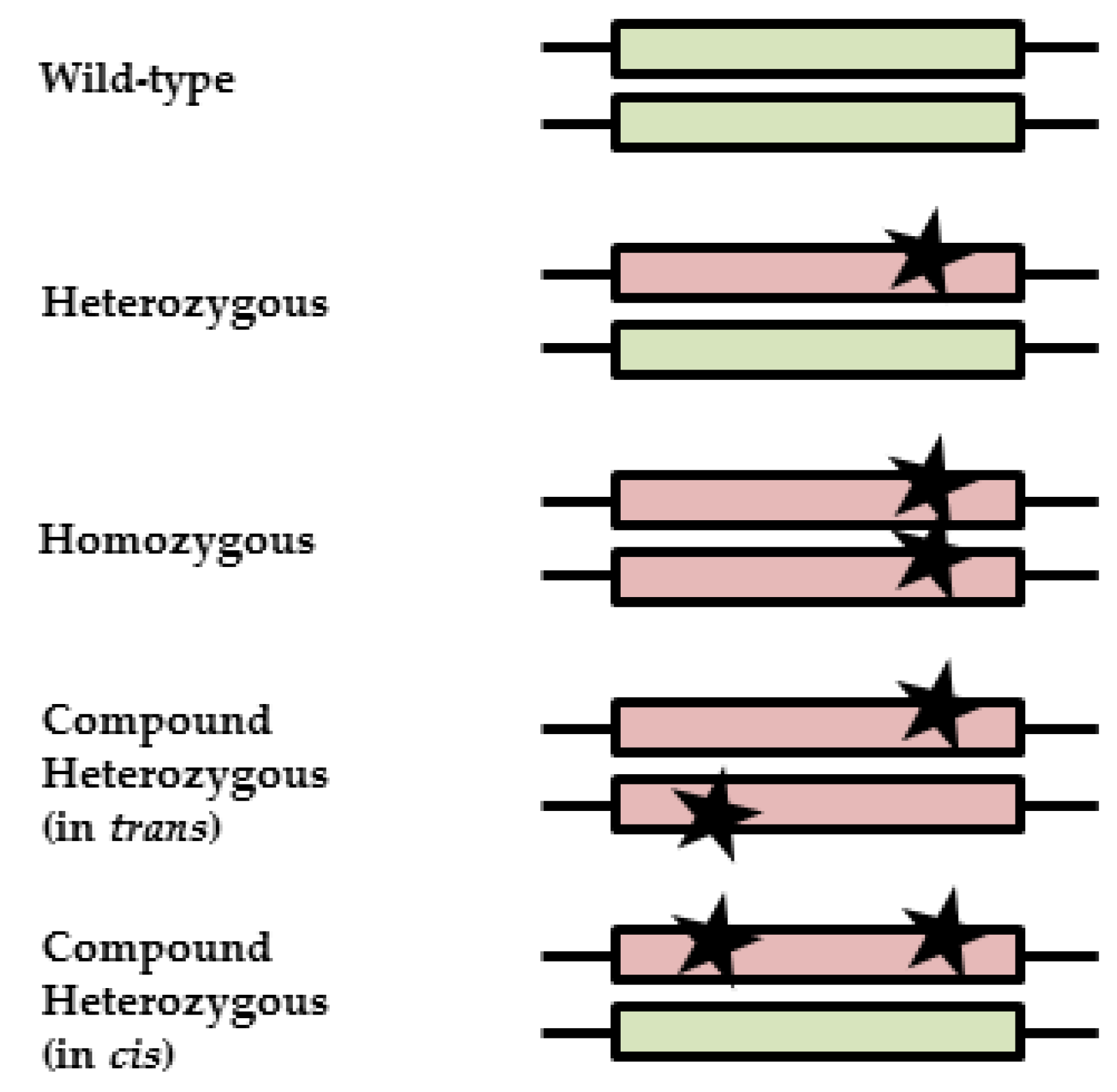
2.5. Phasing in Compound Heterozygous DPYD Carriers
2.5.1. Genome of the Netherlands Datasets
2.5.2. 1000 Genomes Database
2.5.3. Exome Trios Leiden University Medical Centre Database
3. Results
3.1. Patient Cases and Clinical Implications
3.2. Preventing Toxicity
3.3. Frequencies of Compound Heterozygous DPYD Carriers without Phasing Information
3.4. Frequencies of Compound Heterozygous DPYD Carriers with Phasing Information
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Scrip’s Cancer Chemotherapy Report; Scrip world pharmaceutical news; PJB Publications Ltd.: London, UK, 2002.
- Walko, C.M.; Lindley, C. Capecitabine: A review. Clin. Ther. 2005, 27, 23–44. [Google Scholar] [CrossRef] [PubMed]
- Malet-Martino, M.; Martino, R. Clinical studies of three oral prodrugs of 5-fluorouracil (capecitabine, UFT, S-1): A review. Oncologist 2002, 7, 288–323. [Google Scholar] [CrossRef] [PubMed]
- Rosmarin, D.; Palles, C.; Pagnamenta, A.; Kaur, K.; Pita, G.; Martin, M.; Domingo, E.; Jones, A.; Howarth, K.; Freeman-Mills, L.; et al. A candidate gene study of capecitabine-related toxicity in colorectal cancer identifies new toxicity variants at DPYD and a putative role for ENOSF1 rather than TYMS. Gut 2015, 64, 111–120. [Google Scholar] [CrossRef]
- Terrazzino, S.; Cargnin, S.; Del Re, M.; Danesi, R.; Canonico, P.L.; Genazzani, A.A. DPYD IVS14+1G>A and 2846A>T genotyping for the prediction of severe fluoropyrimidine-related toxicity: A meta-analysis. Pharmacogenomics 2013, 14, 1255–1272. [Google Scholar] [CrossRef]
- Saltz, L.B.; Niedzwiecki, D.; Hollis, D.; Goldberg, R.M.; Hantel, A.; Thomas, J.P.; Fields, A.L.; Mayer, R.J. Irinotecan fluorouracil plus leucovorin is not superior to fluorouracil plus leucovorin alone as adjuvant treatment for stage III colon cancer: Results of CALGB 89803. J. Clin. Oncol. 2007, 25, 3456–3461. [Google Scholar] [CrossRef] [PubMed]
- Van Kuilenburg, A.B. Dihydropyrimidine dehydrogenase and the efficacy and toxicity of 5-fluorouracil. Eur. J. Cancer 2004, 40, 939–950. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, F.J.; Fernandez-Salguero, P. Diagnostic analysis, clinical importance and molecular basis of dihydropyrimidine dehydrogenase deficiency. Trends Pharmacol. Sci. 1995, 16, 325–327. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.; Meijer, J.; Maurer, D.; Dobritzsch, D.; Meinsma, R.; Los, M.; Knegt, L.C.; Zoetekouw, L.; Jansen, R.L.; Dezentje, V.; et al. Severe fluoropyrimidine toxicity due to novel and rare DPYD missense mutations, deletion and genomic amplification affecting DPD activity and mRNA splicing. Biochim. Biophys. Acta 2017, 1863, 721–730. [Google Scholar] [CrossRef]
- Meulendijks, D.; Henricks, L.M.; van Kuilenburg, A.B.; Jacobs, B.A.; Aliev, A.; Rozeman, L.; Meijer, J.; Beijnen, J.H.; de Graaf, H.; Cats, A.; et al. Patients homozygous for DPYD c.1129-5923C>G/haplotype B3 have partial DPD deficiency and require a dose reduction when treated with fluoropyrimidines. Cancer Chemother. Pharmacol. 2016, 78, 875–880. [Google Scholar] [CrossRef]
- Offer, S.M.; Fossum, C.C.; Wegner, N.J.; Stuflesser, A.J.; Butterfield, G.L.; Diasio, R.B. Comparative functional analysis of DPYD variants of potential clinical relevance to dihydropyrimidine dehydrogenase activity. Cancer Res. 2014, 74, 2545–2554. [Google Scholar] [CrossRef]
- Henricks, L.M.; Lunenburg, C.A.; Meulendijks, D.; Gelderblom, H.; Cats, A.; Swen, J.J.; Schellens, J.H.; Guchelaar, H.J. Translating DPYD genotype into DPD phenotype: Using the DPYD gene activity score. Pharmacogenomics 2015, 16, 1277–1286. [Google Scholar] [CrossRef] [PubMed]
- Deenen, M.J.; Meulendijks, D.; Cats, A.; Sechterberger, M.K.; Severens, J.L.; Boot, H.; Smits, P.H.; Rosing, H.; Mandigers, C.M.; Soesan, M.; et al. Upfront genotyping of DPYD*2A to individualize fluoropyrimidine therapy: A safety and cost analysis. J. Clin. Oncol. 2016, 34, 227–234. [Google Scholar] [CrossRef] [PubMed]
- Lunenburg, C.A.; van Staveren, M.C.; Gelderblom, H.; Guchelaar, H.J.; Swen, J.J. Evaluation of clinical implementation of prospective DPYD genotyping in 5-fluorouracil- or capecitabine-treated patients. Pharmacogenomics 2016, 17, 721–729. [Google Scholar] [CrossRef] [PubMed]
- Henricks, L.M.; Lunenburg, C.; de Man, F.M.; Meulendijks, D.; Frederix, G.W.J.; Kienhuis, E.; Creemers, G.J.; Baars, A.; Dezentje, V.O.; Imholz, A.L.T.; et al. DPYD genotype-guided dose individualisation of fluoropyrimidine therapy in patients with cancer: A prospective safety analysis. Lancet Oncol. 2018, 19, 1459–1467. [Google Scholar] [CrossRef]
- Lunenburg, C.A.; Henricks, L.M.; Guchelaar, H.J.; Swen, J.J.; Deenen, M.J.; Schellens, J.H.; Gelderblom, H. Prospective DPYD genotyping to reduce the risk of fluoropyrimidine-induced severe toxicity: Ready for prime time. Eur. J. Cancer 2016, 54, 40–48. [Google Scholar] [CrossRef] [PubMed]
- Meulendijks, D.; Henricks, L.M.; Sonke, G.S.; Deenen, M.J.; Froehlich, T.K.; Amstutz, U.; Largiader, C.R.; Jennings, B.A.; Marinaki, A.M.; Sanderson, J.D.; et al. Clinical relevance of DPYD variants c.1679T>G, c.1236G>A/HapB3, and c.1601G>A as predictors of severe fluoropyrimidine-associated toxicity: A systematic review and meta-analysis of individual patient data. Lancet Oncol. 2015, 16, 1639–1650. [Google Scholar] [CrossRef]
- Result Survey Screening for DPD Deficiency. Medische Oncologie. October 2016. Available online: https://www.nvmo.org/magazine/ (accessed on 1 November 2016).
- KNMP. Royal Dutch Society for the Advancement of Pharmacy. Fluorouracil/Capecitabine DPD Gene Activity Score and Guidelines. Available online: https://kennisbank.knmp.nl/article/farmacogenetica/2552-4893-4894.html (accessed on 5 May 2017).
- Amstutz, U.; Henricks, L.M.; Offer, S.M.; Barbarino, J.; Schellens, J.H.M.; Swen, J.J.; Klein, T.E.; McLeod, H.L.; Caudle, K.E.; Diasio, R.B.; et al. Clinical pharmacogenetics implementation consortium (CPIC) guideline for dihydropyrimidine dehydrogenase genotype and fluoropyrimidine dosing: 2017 update. Clin. Pharmacol. Ther. 2018, 103, 210–216. [Google Scholar] [CrossRef]
- Henricks, L.M.; Kienhuis, E.; de Man, F.M.; van der Veldt, A.A.M.; Hamberg, P.; van Kuilenburg, A.B.P.; van Schaik, R.H.N.; Lunenburg, C.A.T.C.; Guchelaar, H.J.; Schellens, J.H.M.; et al. Treatment algorithm for homozygous or compound heterozygous DPYD variant allele carriers with low dose capecitabine. JCO Precis. Oncol. 2017, 1, 1–10, Published online October 6. [Google Scholar] [CrossRef]
- Federa. Available online: federa.org (accessed on 9 September 2017).
- National Cancer Institute: Common Terminology Criteria for Adverse Events v4.03. Available online: https://evs.nci.nih.gov/ftp1/CTCAE/CTCAE_4.03_2010-06-14_QuickReference_8.5x11.pdf (accessed on 9 September 2017).
- Meulendijks, D.; Cats, A.; Beijnen, J.H.; Schellens, J.H. Improving safety of fluoropyrimidine chemotherapy by individualizing treatment based on dihydropyrimidine dehydrogenase activity—Ready for clinical practice? Cancer Treat. Rev. 2016, 50, 23–34. [Google Scholar] [CrossRef]
- Van Staveren, M.C.; van Kuilenburg, A.B.; Guchelaar, H.J.; Meijer, J.; Punt, C.J.; de Jong, R.S.; Gelderblom, H.; Maring, J.G. Evaluation of an oral uracil loading test to identify DPD-deficient patients using a limited sampling strategy. Br. J. Clin. Pharmacol. 2016, 81, 553–561. [Google Scholar] [CrossRef]
- Henricks, L.M.; Siemerink, E.J.M.; Rosing, H.; Meijer, J.; Goorden, S.M.I.; Polstra, A.M.; Zoetekouw, L.; Cats, A.; Schellens, J.H.M.; van Kuilenburg, A.B.P. Capecitabine-based treatment of a patient with a novel DPYD genotype and complete dihydropyrimidine dehydrogenase deficiency. Int. J. Cancer 2018, 142, 424–430. [Google Scholar] [CrossRef]
- Van Kuilenburg, A.B.; Van Lenthe, H.; Tromp, A.; Veltman, P.C.; Van Gennip, A.H. Pitfalls in the diagnosis of patients with a partial dihydropyrimidine dehydrogenase deficiency. Clin. Chem. 2000, 46, 9–17. [Google Scholar] [PubMed]
- Van Kuilenburg, A.B.; Meinsma, R.; Zoetekouw, L.; Van Gennip, A.H. Increased risk of grade IV neutropenia after administration of 5-fluorouracil due to a dihydropyrimidine dehydrogenase deficiency: High prevalence of the IVS14+1g>a mutation. Int. J. Cancer 2002, 101, 253–258. [Google Scholar] [CrossRef]
- Regan, J.F.; Kamitaki, N.; Legler, T.; Cooper, S.; Klitgord, N.; Karlin-Neumann, G.; Wong, C.; Hodges, S.; Koehler, R.; Tzonev, S.; et al. A rapid molecular approach for chromosomal phasing. PLoS ONE 2015, 10, e0118270. [Google Scholar] [CrossRef] [PubMed]
- Buermans, H.P.; Vossen, R.H.; Anvar, S.Y.; Allard, W.G.; Guchelaar, H.J.; White, S.J.; den Dunnen, J.T.; Swen, J.J.; van der Straaten, T. flexible and scalable full-length CYP2D6 long amplicon PacBio sequencing. Hum. Mutat. 2017, 38, 310–316. [Google Scholar] [CrossRef]
- Van der Straaten, T.; Swen, J.; Baak-Pablo, R.; Guchelaar, H.J. Use of plasmid-derived external quality control samples in pharmacogenetic testing. Pharmacogenomics 2008, 9, 1261–1266. [Google Scholar] [CrossRef] [PubMed]
- ExAC. Exome Aggregation Consortium. ExAC Browser (Beta). Available online: http://exac.broadinstitute.org/ (accessed on 13 December 2017).
- gnomAD. genome Aggregation Database. gnomAD browser (Beta). Available online: http://gnomad.broadinstitute.org/ (accessed on 14 July 2017).
- Francioli, L.; Menelaou, A.; Pulit, S.; van Dijk, F.; Palamara, P.; Elbers, C.; Neerincx, P.; Ye, K.; Guryev, V.; Kloosterman, W.; et al. Genome of The Netherlands Consortium. Whole-genome sequence variation, population structure and demographic history of the Dutch population. Nat. Genet. 2014, 46, 818–825. [Google Scholar] [CrossRef] [PubMed]
- Python. Python Software Foundation©. Available online: https://www.python.org/ (accessed on 9 September 2017).
- IGSR. The International Genome Sample Resource. Available online: http://www.internationalgenome.org/ (accessed on 29 June 2017).
- Li, H. Tabix: Fast retrieval of sequence features from generic TAB-delimited files. Bioinformatics 2011, 27, 718–719. [Google Scholar] [CrossRef] [PubMed]
- R Core Team. R: A Language and Environment for Statistical Computing 2018. Available online: https://www.R-project.org. (accessed on 9 September 2017).
- Toffoli, G.; Giodini, L.; Buonadonna, A.; Berretta, M.; De, P.A.; Scalone, S.; Miolo, G.; Mini, E.; Nobili, S.; Lonardi, S.; et al. Clinical validity of a DPYD-based pharmacogenetic test to predict severe toxicity to fluoropyrimidines. Int. J. Cancer 2015, 137, 2971–2980. [Google Scholar] [CrossRef] [PubMed]
- Johnson, M.R.; Wang, K.; Diasio, R.B. Profound dihydropyrimidine dehydrogenase deficiency resulting from a novel compound heterozygote genotype. Clin. Cancer Res. 2002, 8, 768–774. [Google Scholar]
- Jacobs, B.A.; Deenen, M.J.; Pluim, D.; van Hasselt, J.G.; Krahenbuhl, M.D.; van Geel, R.M.; de Vries, N.; Rosing, H.; Meulendijks, D.; Burylo, A.M.; et al. Pronounced between-subject and circadian variability in thymidylate synthase and dihydropyrimidine dehydrogenase enzyme activity in human volunteers. Br. J. Clin. Pharmacol. 2016, 82, 706–716. [Google Scholar] [CrossRef] [PubMed]
- Meulendijks, D.; Henricks, L.M.; Amstutz, U.; Froehlich, T.K.; Largiader, C.R.; Beijnen, J.H.; de Boer, A.; Deenen, M.J.; Cats, A.; Schellens, J.H. Rs895819 in MIR27A improves the predictive value of DPYD variants to identify patients at risk of severe fluoropyrimidine-associated toxicity. Int. J. Cancer 2016, 138, 2752–2761. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Patient # | Primary Tumor | Treatment | Capecitabine Dose | Executed Assays |
|---|---|---|---|---|
| 1 | BC | CAP | 1000 mg/m2/bid | Genotyping (R), DPD activity (R), in-house technique (R), droplet digital PCR (R) |
| 2 | BC | CAP | 800 mg bid (50%) | Genotyping (P), DPD activity (R), in-house technique (R) |
| 3 | CRC | CAP + OX | 900 mg bid (50%) 1 | Genotyping (P), DPD activity (P), PacBio (R) |
| 4 | BC | CAP | 1500 mg bid | Genotyping (R), DPD activity (R 2) |
| 5 | CRC | CAP + RT | 800 mg bid (50%) | Genotyping (P + R 3), DPD activity (R 4), PacBio (R) |
| 6 | CRC | CAP + OX | 1000 mg/m2/bid | Genotyping (R), DPD activity (R) |
| 7 | CRC | CAP + OX + BEV | 1000 mg/m2/bid | Genotyping (R), DPD activity (R) |
| Patient # | DPYD Variants | Phasing | GAS [12] | DPWG Dose Advice (% of Regular Dose) | DPD Activity (nmol/(mg×h)) | Percentage of DPD Activity 1 |
|---|---|---|---|---|---|---|
| 1 | DPYD*2A + c.1236G>A | in trans | 0.5 | 25% | 0.9 | 9% |
| 2 | DPYD*2A + c.2846A>T | in trans | 0.5 | 25% | 6.0 | 60% |
| 3 | c.1236G>A + c.2846A>T | in trans | 1 | 50% | 4.5 | 45% |
| 4 | DPYD*2A + c.2846A>T | unknown | X | X | 0.11 | 1% |
| 5 | DPYD*2A + c.2846A>T | in cis | 1 | 50% | 7.2 | 72% |
| 6 | DPYD*2A + c.1236G>A | unknown | X | X | 3.8 | 38% |
| 7 | DPYD*2A + c.1236G>A | unknown | X | X | 1.6 | 16% |
| Patient # | DPYD variants | Dose (% of Regular Dose) | Toxicity (Maximal CTC Grade) |
|---|---|---|---|
| 1 | DPYD*2A + c.1236G>A | 100% | 4 |
| 2 | DPYD*2A + c.2846A>T | 50% | 1–2 |
| 3 | c.1236G>A + c.2846A>T | 50% → 70% | 0 (on 50% dose) → 3 (on 70% dose) |
| 4 | DPYD*2A + c.2846A>T | 100% | 5 |
| 5 | DPYD*2A + c.2846A>T | 50% | 0 |
| 6 | DPYD*2A + c.1236G>A | 100% | 4 |
| 7 | DPYD*2A + c.1236G>A | 100% | 3 |
| Variants | DPYD*2A (rs3918290) | DPYD*13 (rs55886062) | c.1236G>A (rs56038477) | c.2846A>T (rs67376798) | |||||
|---|---|---|---|---|---|---|---|---|---|
| Databases | HW/HE/HM | MAF | HW/HE/HM | MAF | HW/HE/HM | MAF | HW/HE/HM | MAF | |
| GoNL | 489/7/0 | 0.7% | 494/2/0 | 0.2% | 475/21/0 | 2.1% | 490/6/0 | 0.6% | |
| 1000 Genomes | 405/2/0 | 0.2% | 406/1/0 | 0.1% | 389/18/0 | 2.2% | 403/4/0 | 0.5% | |
| Exome Trios LUMC | 946/15/0 | 0.8% | 946/0/0 | 0.00% | 946/46/0 | 2.3% | 946/2/0 | 0.1% | |
| ExAC | 60,627/624/5 | 0.5% | 60,320/42/0 | 0.03% | 60,652/1808/27 | 1.5% | 60,687/317/1 | 0.3% | |
| gnomAD | 138,489/1586/10 | 0.6% | 138,166/83/0 | 0.03% | 138,407/3841/39 | 1.4% | 138,478/792/1 | 0.3% | |
| Combination of DPYD Variants | Calculated Frequency |
|---|---|
| DPYD*2A + DPYD*13 | 0.0002% |
| DPYD*2A + c.1236G>A | 0.008% |
| DPYD*2A + c.2846A>T | 0.001% |
| DPYD*13 + c.1236G>A | 0.0005% |
| DPYD*13 + c.2846A>T | 0.0001% |
| c.1236G>A + c.2846A>T | 0.004% |
© 2018 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Lunenburg, C.A.T.C.; Henricks, L.M.; Van Kuilenburg, A.B.P.; Mathijssen, R.H.J.; Schellens, J.H.M.; Gelderblom, H.; Guchelaar, H.-J.; Swen, J.J. Diagnostic and Therapeutic Strategies for Fluoropyrimidine Treatment of Patients Carrying Multiple DPYD Variants. Genes 2018, 9, 585. https://doi.org/10.3390/genes9120585
Lunenburg CATC, Henricks LM, Van Kuilenburg ABP, Mathijssen RHJ, Schellens JHM, Gelderblom H, Guchelaar H-J, Swen JJ. Diagnostic and Therapeutic Strategies for Fluoropyrimidine Treatment of Patients Carrying Multiple DPYD Variants. Genes. 2018; 9(12):585. https://doi.org/10.3390/genes9120585
Chicago/Turabian StyleLunenburg, Carin A. T. C., Linda M. Henricks, André B. P. Van Kuilenburg, Ron H. J. Mathijssen, Jan H. M. Schellens, Hans Gelderblom, Henk-Jan Guchelaar, and Jesse J. Swen. 2018. "Diagnostic and Therapeutic Strategies for Fluoropyrimidine Treatment of Patients Carrying Multiple DPYD Variants" Genes 9, no. 12: 585. https://doi.org/10.3390/genes9120585
APA StyleLunenburg, C. A. T. C., Henricks, L. M., Van Kuilenburg, A. B. P., Mathijssen, R. H. J., Schellens, J. H. M., Gelderblom, H., Guchelaar, H.-J., & Swen, J. J. (2018). Diagnostic and Therapeutic Strategies for Fluoropyrimidine Treatment of Patients Carrying Multiple DPYD Variants. Genes, 9(12), 585. https://doi.org/10.3390/genes9120585