From Genotype to Phenotype: Through Chromatin


Abstract
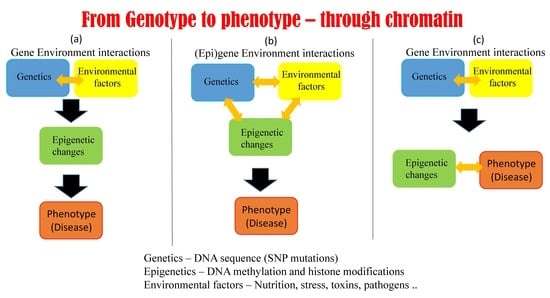
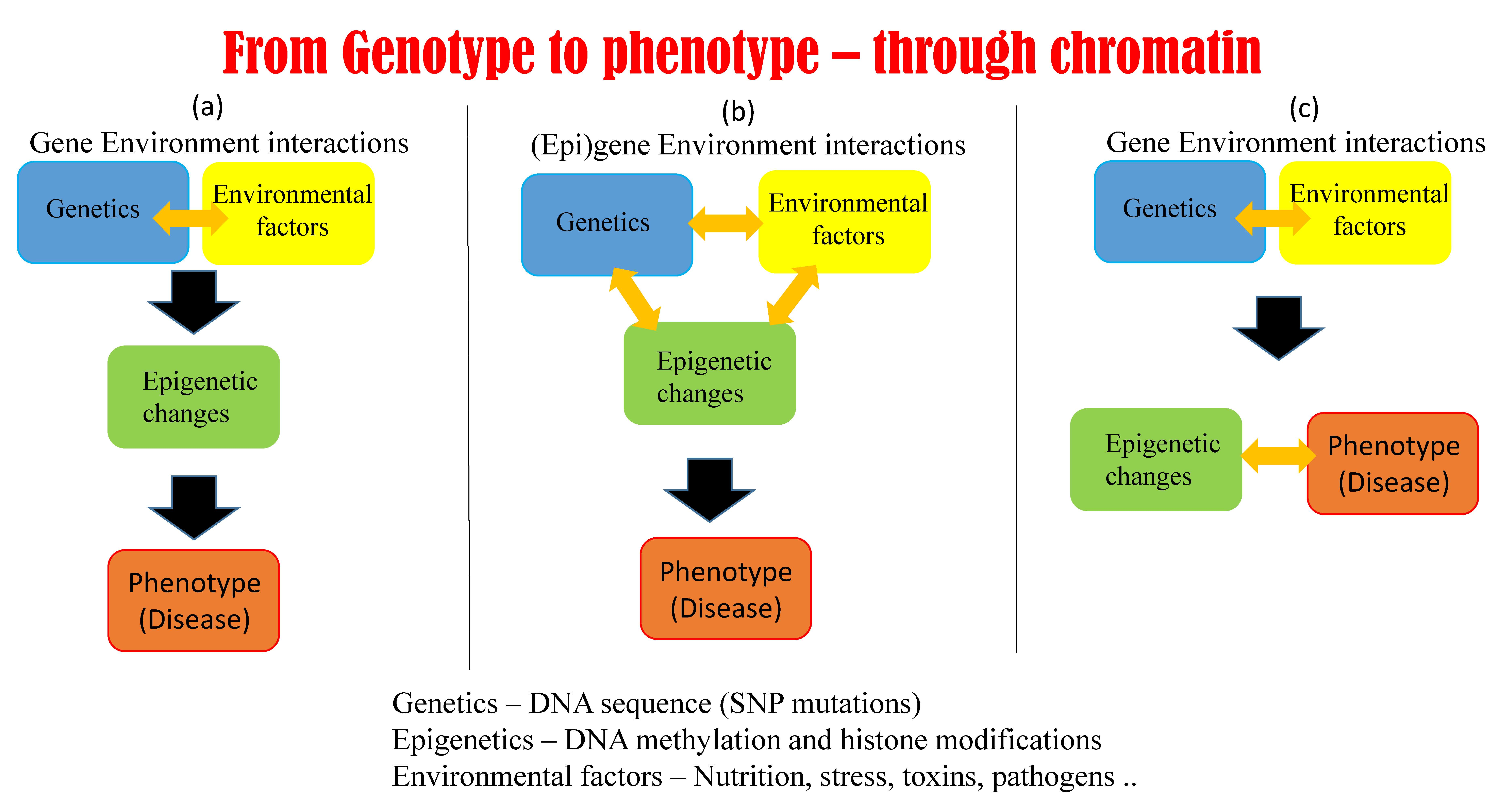
:
1. Introduction
1.1. Definition of Epigenetics
1.2. Broadening the Definition of Epigenetics
1.3. Epigenetic Mechanisms Regulate Gene Expression Using Environmental Cues
2. Chromatin Modifications and the Genome Organization
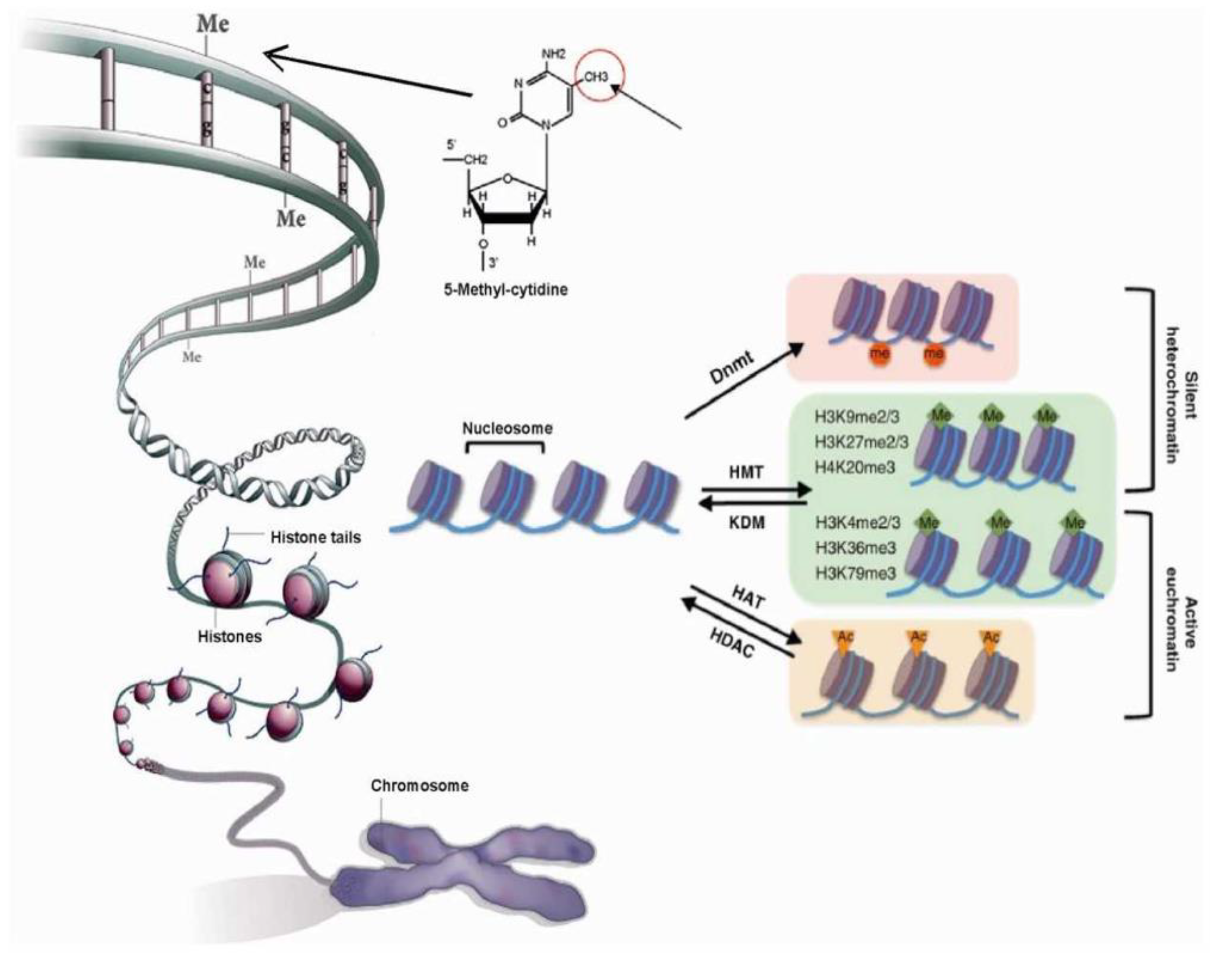
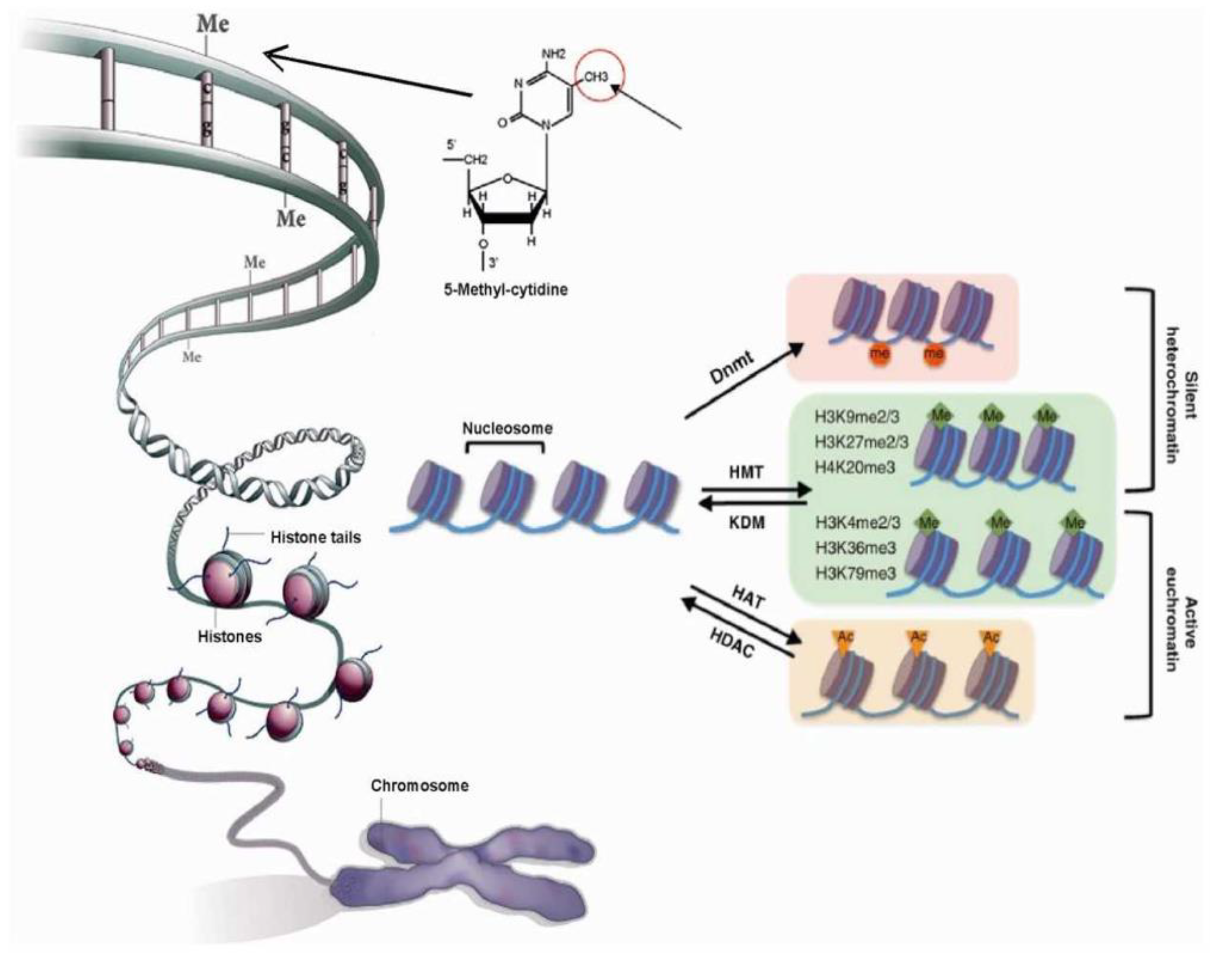
2.1. Chromatin’s Structure Defines Its Function
2.2. Chromatin Structure is Dynamic and Marked by Histone Modifications
3. Epigenetics in Disease Context
3.1. Genome-Wide Studies Are Not Enough
3.2. Largescale Epigenetic Studies in Cancer
3.2.1. Epigenetic Mechanisms Are Major Drivers in Cancer
3.2.2. Epigenetic Mechanisms in Hematopoietic Malignancies and Their Therapeutic Implications
3.2.3. Epigenetic Targets for Cancer Therapy
3.3. Largescale Epigenetic Studies in Other Diseases
4. Computational Approaches towards Epigenetic Data Analysis and Integration
4.1. Epigenetic Data Integration to Understand the “Epigenetic Code”
The Function of Epigenetic Modifications Still Remains Understudied
4.2. Linking Epigenetic Mechanisms to Phenotypes: Epigenetic Epidemiology
4.2.1. More Data Equals More Challenges
4.2.2. New Data Integration Opportunities
4.2.3. Epigenome-Wide Association Studies Analyses Are Informative Only about an Association and Not Causality
4.2.4. Causality Inference from Translational Studies
4.3. Combining Levels of Epigenetic Marks within Genomic Regions
5. Conclusions
5.1. Possible Scenarios Linking Epigenetics, Genetics, and Phenotype
5.2. New Approaches and Technologies Must Aim on Establishing a Causal Link between Epigenetics and Disease
5.3. Epigenetic Studies and Therapies Have an Important Role in Shaping the Future of Medicine
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Nicoglou, A. Waddington’s epigenetics or the pictorial meetings of development and genetics. Hist. Philos. Life Sci. 2018, 40, 61. [Google Scholar] [CrossRef] [PubMed]
- Bird, A.P.; Wolffe, A.P. Methylation-induced repression—Belts, braces, and chromatin. Cell 1999, 99, 451–454. [Google Scholar] [CrossRef]
- Mohn, F.; Weber, M.; Rebhan, M.; Roloff, T.C.; Richter, J.; Stadler, M.B.; Bibel, M.; Schübeler, D. Lineage-specific polycomb targets and de novo DNA methylation define restriction and potential of neuronal progenitors. Mol. Cell 2008, 30, 755–766. [Google Scholar] [CrossRef] [PubMed]
- Bird, A. DNA methylation patterns and epigenetic memory. Genes Dev. 2002, 16, 6–21. [Google Scholar] [CrossRef] [PubMed]
- Morgan, H.D.; Santos, F.; Green, K.; Dean, W.; Reik, W. Epigenetic reprogramming in mammals. Hum. Mol. Genet. 2005, 14, R47–R58. [Google Scholar] [CrossRef] [PubMed]
- Reik, W. Stability and flexibility of epigenetic gene regulation in mammalian development. Nature 2007, 447, 425–432. [Google Scholar] [CrossRef] [PubMed]
- Turner, B.M. Defining an epigenetic code. Nat. Cell Biol. 2007, 9, 2–6. [Google Scholar] [CrossRef] [PubMed]
- Bannister, A.J.; Kouzarides, T. Regulation of chromatin by histone modifications. Cell Res. 2011, 21, 381–395. [Google Scholar] [CrossRef]
- Jenuwein, T.; Allis, C.D. Translating the histone code. Science 2001, 293, 1074–1080. [Google Scholar] [CrossRef]
- Strahl, B.D.; Allis, C.D. The language of covalent histone modifications. Nature 2000, 403, 41–45. [Google Scholar] [CrossRef]
- Zhou, V.W.; Goren, A.; Bernstein, B.E. Charting histone modifications and the functional organization of mammalian genomes. Nat. Rev. Genet. 2011, 12, 7–18. [Google Scholar] [CrossRef] [PubMed]
- Zentner, G.E.; Henikoff, S. Regulation of nucleosome dynamics by histone modifications. Nat. Struct. Mol. Biol. 2013, 20, 259–266. [Google Scholar] [CrossRef]
- Allis, C.D.; Jenuwein, T. The molecular hallmarks of epigenetic control. Nat. Rev. Genet. 2016, 17, 487–500. [Google Scholar] [CrossRef] [PubMed]
- Maleszewska, M.; Kaminska, B. Is Glioblastoma an Epigenetic Malignancy? Cancers 2013, 5, 1120–1139. [Google Scholar] [CrossRef] [PubMed]
- Frías-Lasserre, D.; Villagra, C.A. The Importance of ncRNAs as Epigenetic Mechanisms in Phenotypic Variation and Organic Evolution. Front. Microbiol. 2017, 8, 2483. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.T. Lessons from X-chromosome inactivation: Long ncRNA as guides and tethers to the epigenome. Genes Dev. 2009, 23, 1831–1842. [Google Scholar] [CrossRef] [PubMed]
- Jirtle, R.L.; Skinner, M.K. Environmental epigenomics and disease susceptibility. Nat. Rev. Genet. 2007, 8, 253–262. [Google Scholar] [CrossRef]
- Okano, M.; Bell, D.W.; Haber, D.A.; Li, E. DNA methyltransferases Dnmt3a and Dnmt3b are essential for de novo methylation and mammalian development. Cell 1999, 99, 247–257. [Google Scholar] [CrossRef]
- Philibert, R.A.; Beach, S.R.H.; Brody, G.H. The DNA methylation signature of smoking: An archetype for the identification of biomarkers for behavioral illness. Neb. Symp. Motiv. 2014, 61, 109–127. [Google Scholar]
- Joubert, B.R.; Felix, J.F.; Yousefi, P.; Bakulski, K.M.; Just, A.C.; Breton, C.; Reese, S.E.; Markunas, C.A.; Richmond, R.C.; Xu, C.-J.; et al. DNA Methylation in Newborns and Maternal Smoking in Pregnancy: Genome-wide Consortium Meta-analysis. Am. J. Hum. Genet. 2016, 98, 680–696. [Google Scholar] [CrossRef]
- Keleher, M.R.; Zaidi, R.; Shah, S.; Oakley, M.E.; Pavlatos, C.; El Idrissi, S.; Xing, X.; Li, D.; Wang, T.; Cheverud, J.M. Maternal high-fat diet associated with altered gene expression, DNA methylation, and obesity risk in mouse offspring. PLoS ONE 2018, 13, e0192606. [Google Scholar] [CrossRef] [PubMed]
- Li, E. Chromatin modification and epigenetic reprogramming in mammalian development. Nat. Rev. Genet. 2002, 3, 662–673. [Google Scholar] [CrossRef] [PubMed]
- Trojer, P.; Reinberg, D. Facultative heterochromatin: Is there a distinctive molecular signature? Mol. Cell 2007, 28, 1–13. [Google Scholar] [CrossRef] [PubMed]
- Filion, G.J.; van Bemmel, J.G.; Braunschweig, U.; Talhout, W.; Kind, J.; Ward, L.D.; Brugman, W.; de Castro, I.J.; Kerkhoven, R.M.; Bussemaker, H.J.; et al. Systematic Protein Location Mapping Reveals Five Principal Chromatin Types in Drosophila Cells. Cell 2010, 143, 212–224. [Google Scholar] [CrossRef]
- Barski, A.; Cuddapah, S.; Cui, K.; Roh, T.-Y.; Schones, D.E.; Wang, Z.; Wei, G.; Chepelev, I.; Zhao, K. High-resolution profiling of histone methylations in the human genome. Cell 2007, 129, 823–837. [Google Scholar] [CrossRef]
- Dixon, J.R.; Selvaraj, S.; Yue, F.; Kim, A.; Li, Y.; Shen, Y.; Hu, M.; Liu, J.S.; Ren, B. Topological domains in mammalian genomes identified by analysis of chromatin interactions. Nature 2012, 485, 376–380. [Google Scholar] [CrossRef]
- Filippova, D.; Patro, R.; Duggal, G.; Kingsford, C. Identification of alternative topological domains in chromatin. Algorithms Mol. Biol. AMB 2014, 9, 14. [Google Scholar] [CrossRef]
- Hnisz, D.; Weintraub, A.S.; Day, D.S.; Valton, A.-L.; Bak, R.O.; Li, C.H.; Goldmann, J.; Lajoie, B.R.; Fan, Z.P.; Sigova, A.A.; et al. Activation of proto-oncogenes by disruption of chromosome neighborhoods. Science 2016, 351, 1454–1458. [Google Scholar] [CrossRef]
- Kaiser, V.B.; Taylor, M.S.; Semple, C.A. Mutational Biases Drive Elevated Rates of Substitution at Regulatory Sites across Cancer Types. PLoS Genet. 2016, 12, e1006207. [Google Scholar] [CrossRef]
- Mifsud, B.; Tavares-Cadete, F.; Young, A.N.; Sugar, R.; Schoenfelder, S.; Ferreira, L.; Wingett, S.W.; Andrews, S.; Grey, W.; Ewels, P.A.; et al. Mapping long-range promoter contacts in human cells with high-resolution capture Hi-C. Nat. Genet. 2015, 47, 598–606. [Google Scholar] [CrossRef]
- Corces, M.R.; Corces, V.G. The three-dimensional cancer genome. Curr. Opin. Genet. Dev. 2016, 36, 1–7. [Google Scholar] [CrossRef] [PubMed]
- Achinger-Kawecka, J.; Clark, S.J. Disruption of the 3D cancer genome blueprint. Epigenomics 2017, 9, 47–55. [Google Scholar] [CrossRef] [PubMed]
- Kouzarides, T. SnapShot: Histone-modifying enzymes. Cell 2007, 131, 822. [Google Scholar] [CrossRef] [PubMed]
- Margueron, R.; Trojer, P.; Reinberg, D. The key to development: Interpreting the histone code? Curr. Opin. Genet. Dev. 2005, 15, 163–176. [Google Scholar] [CrossRef] [PubMed]
- Mantsoki, A.; Devailly, G.; Joshi, A. CpG island erosion, polycomb occupancy and sequence motif enrichment at bivalent promoters in mammalian embryonic stem cells. Sci. Rep. 2015, 5, 16791. [Google Scholar] [CrossRef] [PubMed]
- Bartke, T.; Vermeulen, M.; Xhemalce, B.; Robson, S.C.; Mann, M.; Kouzarides, T. Nucleosome-interacting proteins regulated by DNA and histone methylation. Cell 2010, 143, 470–484. [Google Scholar] [CrossRef] [PubMed]
- Tabolacci, E.; Chiurazzi, P. Epigenetics, fragile X syndrome and transcriptional therapy. Am. J. Med. Genet. A. 2013, 161A, 2797–2808. [Google Scholar] [CrossRef]
- Welter, D.; MacArthur, J.; Morales, J.; Burdett, T.; Hall, P.; Junkins, H.; Klemm, A.; Flicek, P.; Manolio, T.; Hindorff, L.; et al. The NHGRI GWAS Catalog, a curated resource of SNP-trait associations. Nucleic Acids Res. 2014, 42, D1001–D1006. [Google Scholar] [CrossRef]
- Visscher, P.M.; Brown, M.A.; McCarthy, M.I.; Yang, J. Five years of GWAS discovery. Am. J. Hum. Genet. 2012, 90, 7–24. [Google Scholar] [CrossRef]
- Northcott, P.A.; Nakahara, Y.; Wu, X.; Feuk, L.; Ellison, D.W.; Croul, S.; Mack, S.; Kongkham, P.N.; Peacock, J.; Dubuc, A.; et al. Multiple recurrent genetic events converge on control of histone lysine methylation in medulloblastoma. Nat. Genet. 2009, 41, 465–472. [Google Scholar] [CrossRef]
- Roadmap Epigenomics Consortium; Kundaje, A.; Meuleman, W.; Ernst, J.; Bilenky, M.; Yen, A.; Heravi-Moussavi, A.; Kheradpour, P.; Zhang, Z.; Wang, J.; et al. Integrative analysis of 111 reference human epigenomes. Nature 2015, 518, 317–330. [Google Scholar] [CrossRef] [PubMed]
- Corradin, O.; Scacheri, P.C. Enhancer variants: Evaluating functions in common disease. Genome Med. 2014, 6, 85. [Google Scholar] [CrossRef] [PubMed]
- Corces, M.R.; Granja, J.M.; Shams, S.; Louie, B.H.; Seoane, J.A.; Zhou, W.; Silva, T.C.; Groeneveld, C.; Wong, C.K.; Cho, S.W.; et al. The chromatin accessibility landscape of primary human cancers. Science 2018, 362, eaav1898. [Google Scholar] [CrossRef] [PubMed]
- Severson, T.M.; Kim, Y.; Joosten, S.E.P.; Schuurman, K.; van der Groep, P.; Moelans, C.B.; Ter Hoeve, N.D.; Manson, Q.F.; Martens, J.W.; van Deurzen, C.H.M.; et al. Characterizing steroid hormone receptor chromatin binding landscapes in male and female breast cancer. Nat. Commun. 2018, 9, 482. [Google Scholar] [CrossRef] [PubMed]
- Ooi, W.F.; Xing, M.; Xu, C.; Yao, X.; Ramlee, M.K.; Lim, M.C.; Cao, F.; Lim, K.; Babu, D.; Poon, L.-F.; et al. Epigenomic profiling of primary gastric adenocarcinoma reveals super-enhancer heterogeneity. Nat. Commun. 2016, 7, 12983. [Google Scholar] [CrossRef] [PubMed]
- Müller-Tidow, C.; Klein, H.-U.; Hascher, A.; Isken, F.; Tickenbrock, L.; Thoennissen, N.; Agrawal-Singh, S.; Tschanter, P.; Disselhoff, C.; Wang, Y.; et al. Profiling of histone H3 lysine 9 trimethylation levels predicts transcription factor activity and survival in acute myeloid leukemia. Blood 2010, 116, 3564–3571. [Google Scholar] [CrossRef] [PubMed]
- Turcan, S.; Makarov, V.; Taranda, J.; Wang, Y.; Fabius, A.W.M.; Wu, W.; Zheng, Y.; El-Amine, N.; Haddock, S.; Nanjangud, G.; et al. Mutant-IDH1-dependent chromatin state reprogramming, reversibility, and persistence. Nat. Genet. 2018, 50, 62–72. [Google Scholar] [CrossRef]
- Agrawal-Singh, S.; Isken, F.; Agelopoulos, K.; Klein, H.-U.; Thoennissen, N.H.; Koehler, G.; Hascher, A.; Bäumer, N.; Berdel, W.E.; Thiede, C.; et al. Genome-wide analysis of histone H3 acetylation patterns in AML identifies PRDX2 as an epigenetically silenced tumor suppressor gene. Blood 2012, 119, 2346–2357. [Google Scholar] [CrossRef]
- Hoemme, C.; Peerzada, A.; Behre, G.; Wang, Y.; McClelland, M.; Nieselt, K.; Zschunke, M.; Disselhoff, C.; Agrawal, S.; Isken, F.; et al. Chromatin modifications induced by PML-RARalpha repress critical targets in leukemogenesis as analyzed by ChIP-Chip. Blood 2008, 111, 2887–2895. [Google Scholar] [CrossRef]
- Salzberg, A.C.; Harris-Becker, A.; Popova, E.Y.; Keasey, N.; Loughran, T.P.; Claxton, D.F.; Grigoryev, S.A. Genome-wide mapping of histone H3K9me2 in acute myeloid leukemia reveals large chromosomal domains associated with massive gene silencing and sites of genome instability. PLoS ONE 2017, 12, e0173723. [Google Scholar] [CrossRef]
- Hlady, R.A.; Sathyanarayan, A.; Thompson, J.J.; Zhou, D.; Wu, Q.; Pham, K.; Lee, J.H.; Liu, C.; Robertson, K.D. Integrating the Epigenome to Identify Novel Drivers of Hepatocellular Carcinoma. Hepatol. Baltim. Md 2018. [Google Scholar]
- Kelso, T.W.R.; Porter, D.K.; Amaral, M.L.; Shokhirev, M.N.; Benner, C.; Hargreaves, D.C. Chromatin accessibility underlies synthetic lethality of SWI/SNF subunits in ARID1A-mutant cancers. eLife 2017, 6, e30506. [Google Scholar] [CrossRef] [PubMed]
- Coetzee, S.G.; Shen, H.C.; Hazelett, D.J.; Lawrenson, K.; Kuchenbaecker, K.; Tyrer, J.; Rhie, S.K.; Levanon, K.; Karst, A.; Drapkin, R.; et al. Cell-type-specific enrichment of risk-associated regulatory elements at ovarian cancer susceptibility loci. Hum. Mol. Genet. 2015, 24, 3595–3607. [Google Scholar] [CrossRef] [PubMed]
- Valencia, A.M.; Kadoch, C. Chromatin regulatory mechanisms and therapeutic opportunities in cancer. Nat. Cell Biol. 2019. [Google Scholar] [CrossRef] [PubMed]
- Kulis, M.; Esteller, M. DNA Methylation and Cancer. Adv. Genet. 2010. [Google Scholar] [CrossRef]
- Bernhart, S.H.; Kretzmer, H.; Holdt, L.M.; Jühling, F.; Ammerpohl, O.; Bergmann, A.K.; Northoff, B.H.; Doose, G.; Siebert, R.; Stadler, P.F.; et al. Changes of bivalent chromatin coincide with increased expression of developmental genes in cancer. Sci. Rep. 2016, 6, 37393. [Google Scholar] [CrossRef] [PubMed]
- Deng, J.; Shoemaker, R.; Xie, B.; Gore, A.; LeProust, E.M.; Antosiewicz-Bourget, J.; Egli, D.; Maherali, N.; Park, I.-H.; Yu, J.; et al. Targeted bisulfite sequencing reveals changes in DNA methylation associated with nuclear reprogramming. Nat. Biotechnol. 2009, 27, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Easwaran, H.; Johnstone, S.E.; Van Neste, L.; Ohm, J.; Mosbruger, T.; Wang, Q.; Aryee, M.J.; Joyce, P.; Ahuja, N.; Weisenberger, D.; et al. A DNA hypermethylation module for the stem/progenitor cell signature of cancer. Genome Res. 2012, 22, 837–849. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Zhao, Z.; Ozark, P.A.; Fantini, D.; Marshall, S.A.; Rendleman, E.J.; Cozzolino, K.A.; Louis, N.; He, X.; Morgan, M.A.; et al. Resetting the epigenetic balance of Polycomb and COMPASS function at enhancers for cancer therapy. Nat. Med. 2018, 24, 758–769. [Google Scholar] [CrossRef] [PubMed]
- Mikeska, T.; Craig, J.M. DNA methylation biomarkers: Cancer and beyond. Genes 2014, 5, 821–864. [Google Scholar] [CrossRef]
- Da Costa, E.M.; McInnes, G.; Beaudry, A.; Raynal, N.J.-M. DNA Methylation–Targeted Drugs. Cancer J. 2017, 23, 270–276. [Google Scholar] [CrossRef] [PubMed]
- Soto-Ramírez, N.; Arshad, S.H.; Holloway, J.W.; Zhang, H.; Schauberger, E.; Ewart, S.; Patil, V.; Karmaus, W. The interaction of genetic variants and DNA methylation of the interleukin-4 receptor gene increase the risk of asthma at age 18 years. Clin. Epigenet. 2013, 5, 1. [Google Scholar] [CrossRef] [PubMed]
- James, S.J.; Melnyk, S.; Jernigan, S.; Pavliv, O.; Trusty, T.; Lehman, S.; Seidel, L.; Gaylor, D.W.; Cleves, M.A. A functional polymorphism in the reduced folate carrier gene and DNA hypomethylation in mothers of children with autism. Am. J. Med. Genet. Part B Neuropsychiatr. Genet. 2010, 153B, 1209–1220. [Google Scholar] [CrossRef] [PubMed]
- Teijido, O.; Cacabelos, R. Pharmacoepigenomic Interventions as Novel Potential Treatments for Alzheimer’s and Parkinson’s Diseases. Int. J. Mol. Sci. 2018, 19, 3199. [Google Scholar] [CrossRef] [PubMed]
- Soragni, E.; Miao, W.; Iudicello, M.; Jacoby, D.; De Mercanti, S.; Clerico, M.; Longo, F.; Piga, A.; Ku, S.; Campau, E.; et al. Epigenetic therapy for Friedreich ataxia. Ann. Neurol. 2014, 76, 489–508. [Google Scholar] [CrossRef] [PubMed]
- Polak, P.; Karlic, R.; Koren, A.; Thurman, R.; Sandstrom, R.; Lawrence, M.S.; Reynolds, A.; Rynes, E.; Vlahovicek, K.; Stamatoyannopoulos, J.A.; et al. Cell-of-origin chromatin organization shapes the mutational landscape of cancer. Nature 2015, 518, 360–364. [Google Scholar] [CrossRef] [PubMed]
- Hou, X.; He, X.; Wang, K.; Hou, N.; Fu, J.; Jia, G.; Zuo, X.; Xiong, H.; Pang, M. Genome-Wide Network-Based Analysis of Colorectal Cancer Identifies Novel Prognostic Factors and an Integrative Prognostic Index. Cell. Physiol. Biochem. 2018, 49, 1703–1716. [Google Scholar] [CrossRef] [PubMed]
- Zhu, B.; Song, N.; Shen, R.; Arora, A.; Machiela, M.J.; Song, L.; Landi, M.T.; Ghosh, D.; Chatterjee, N.; Baladandayuthapani, V.; et al. Integrating Clinical and Multiple Omics Data for Prognostic Assessment across Human Cancers. Sci. Rep. 2017, 7, 16954. [Google Scholar] [CrossRef] [PubMed]
- Sekhon, A.; Singh, R.; Qi, Y. DeepDiff: DEEP-learning for predicting DIFFerential gene expression from histone modifications. Bioinformatics 2018, 34, i891–i900. [Google Scholar] [CrossRef] [PubMed]
- Wu, J.; Xu, J.; Liu, B.; Yao, G.; Wang, P.; Lin, Z.; Huang, B.; Wang, X.; Li, T.; Shi, S.; et al. Chromatin analysis in human early development reveals epigenetic transition during ZGA. Nature 2018, 557, 256–260. [Google Scholar] [CrossRef] [PubMed]
- Hon, G.C.; Hawkins, R.D.; Ren, B. Predictive chromatin signatures in the mammalian genome. Hum. Mol. Genet. 2009, 18, R195–R201. [Google Scholar] [CrossRef]
- Rada-Iglesias, A. Is H3K4me1 at enhancers correlative or causative? Nat. Genet. 2018, 50, 4–5. [Google Scholar] [CrossRef]
- Laird, P.W. Principles and challenges of genomewide DNA methylation analysis. Nat. Rev. Genet. 2010, 11, 191–203. [Google Scholar] [CrossRef] [PubMed]
- Rakyan, V.K.; Down, T.A.; Balding, D.J.; Beck, S. Epigenome-wide association studies for common human diseases. Nat. Rev. Genet. 2012, 12, 529–541. [Google Scholar] [CrossRef] [PubMed]
- Jiang, S.; Mortazavi, A. Integrating ChIP-seq with other functional genomics data. Brief. Funct. Genom. 2018, 17, 104–115. [Google Scholar] [CrossRef]
- Lappalainen, T.; Greally, J.M. Associating cellular epigenetic models with human phenotypes. Nat. Rev. Genet. 2017, 18, 441–451. [Google Scholar] [CrossRef]
- Ritchie, M.D.; Holzinger, E.R.; Li, R.; Pendergrass, S.A.; Kim, D. Methods of integrating data to uncover genotype-phenotype interactions. Nat. Rev. Genet. 2015, 16, 85–97. [Google Scholar] [CrossRef] [PubMed]
- Auerbach, J.; Howey, R.; Jiang, L.; Justice, A.; Li, L.; Oualkacha, K.; Sayols-Baixeras, S.; Aslibekyan, S.W. Causal modeling in a multi-omic setting: Insights from GAW20. BMC Genet. 2018, 19, 74. [Google Scholar] [CrossRef]
- Buenrostro, J.D.; Giresi, P.G.; Zaba, L.C.; Chang, H.Y.; Greenleaf, W.J. Transposition of native chromatin for fast and sensitive epigenomic profiling of open chromatin, DNA-binding proteins and nucleosome position. Nat. Methods 2013, 10, 1213–1218. [Google Scholar] [CrossRef] [PubMed]
- Devailly, G.; Mantsoki, A.; Joshi, A. Heat*seq: An interactive web tool for high-throughput sequencing experiment comparison with public data. Bioinforma. Oxf. Engl. 2016, 32, 3354–3356. [Google Scholar] [CrossRef] [PubMed]
- Romanescu, R.G.; Espin-Garcia, O.; Ma, J.; Bull, S.B. Integrating epigenetic, genetic, and phenotypic data to uncover gene-region associations with triglycerides in the GOLDN study 06 Biological Sciences 0604 Genetics. BMC Proc. 2018, 12, 57. [Google Scholar] [CrossRef] [PubMed]
- Liang, L.; Willis-Owen, S.A.G.; Laprise, C.; Wong, K.C.C.; Davies, G.A.; Hudson, T.J.; Binia, A.; Hopkin, J.M.; Yang, I.V.; Grundberg, E.; et al. An epigenome-wide association study of total serum immunoglobulin e concentration. Nature 2015, 520, 670–674. [Google Scholar] [CrossRef] [PubMed]
- Wang, B.; DeStefano, A.L.; Lin, H. Integrative methylation score to identify epigenetic modifications associated with lipid changes resulting from fenofibrate treatment in families. BMC Proc. 2018, 12, 28. [Google Scholar] [CrossRef] [PubMed]
- Shah, S.; Bonder, M.J.; Marioni, R.E.; Zhu, Z.; McRae, A.F.; Zhernakova, A.; Harris, S.E.; Liewald, D.; Henders, A.K.; Mendelson, M.M.; et al. Improving Phenotypic Prediction by Combining Genetic and Epigenetic Associations. Am. J. Hum. Genet. 2015, 97, 75–85. [Google Scholar] [CrossRef] [PubMed]
- Zheng, R.; Wan, C.; Mei, S.; Qin, Q.; Wu, Q.; Sun, H.; Chen, C.-H.; Brown, M.; Zhang, X.; Meyer, C.A.; et al. Cistrome Data Browser: Expanded datasets and new tools for gene regulatory analysis. Nucleic Acids Res. 2018, 1–7. [Google Scholar] [CrossRef]
- Ward, L.D.; Kellis, M. HaploReg v4: Systematic mining of putative causal variants, cell types, regulators and target genes for human complex traits and disease. Nucleic Acids Res. 2016, 44, D877–D881. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein-protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef]
- Fabregat, A.; Jupe, S.; Matthews, L.; Sidiropoulos, K.; Gillespie, M.; Garapati, P.; Haw, R.; Jassal, B.; Korninger, F.; May, B.; et al. The Reactome Pathway Knowledgebase. Nucleic Acids Res. 2018, 46, D649–D655. [Google Scholar] [CrossRef]
- Himmelstein, D.S.; Lizee, A.; Hessler, C.; Brueggeman, L.; Chen, S.L.; Hadley, D.; Green, A.; Khankhanian, P.; Baranzini, S.E. Systematic integration of biomedical knowledge prioritizes drugs for repurposing. eLife 2017, 6, e26726. [Google Scholar] [CrossRef]
- Haaland, Ø.A.; Lie, R.T.; Romanowska, J.; Gjerdevik, M.; Gjessing, H.K.; Jugessur, A. A Genome-Wide Search for Gene-Environment Effects in Isolated Cleft Lip with or without Cleft Palate Triads Points to an Interaction between Maternal Periconceptional Vitamin Use and Variants in ESRRG. Front. Genet. 2018, 9, 1–16. [Google Scholar] [CrossRef]
- Lawlor, D.A.; Harbord, R.M.; Sterne, J.A.C.; Timpson, N.; Davey Smith, G. Mendelian randomization: Using genes as instruments for making causal inferences in epidemiology. Stat. Med. 2008, 27, 1133–1163. [Google Scholar] [CrossRef] [PubMed]
- Relton, C.L.; Davey Smith, G. Two-step epigenetic mendelian randomization: A strategy for establishing the causal role of epigenetic processes in pathways to disease. Int. J. Epidemiol. 2012, 41, 161–176. [Google Scholar] [CrossRef] [PubMed]
- Latvala, A.; Ollikainen, M. Mendelian randomization in (epi)genetic epidemiology: An effective tool to be handled with care. Genome Biol. 2016, 17, 156. [Google Scholar] [CrossRef] [PubMed]
- Dekkers, K.F.; van Iterson, M.; Slieker, R.C.; Moed, M.H.; Bonder, M.J.; van Galen, M.; Mei, H.; Zhernakova, D.V.; van den Berg, L.H.; Deelen, J.; et al. Blood lipids influence DNA methylation in circulating cells. Genome Biol. 2016, 17, 138. [Google Scholar] [CrossRef] [PubMed]
- Cecil, C.A.M.; Walton, E.; Pingault, J.-B.; Provençal, N.; Pappa, I.; Vitaro, F.; Côté, S.; Szyf, M.; Tremblay, R.E.; Tiemeier, H.; et al. DRD4 methylation as a potential biomarker for physical aggression: An epigenome-wide, cross-tissue investigation. Am. J. Med. Genet. B Neuropsychiatr. Genet. 2018, 177, 746–764. [Google Scholar] [CrossRef] [PubMed]
- Howey, R.A.J.; Cordell, H.J. Application of Bayesian networks to GAW20 genetic and blood lipid data. BMC Proc. 2018, 12, 19. [Google Scholar] [CrossRef] [PubMed]
- Justice, A.E.; Howard, A.G.; Fernández-Rhodes, L.; Graff, M.; Tao, R.; North, K.E. Direct and indirect genetic effects on triglycerides through omics and correlated phenotypes. BMC Proc. 2018, 12, 22. [Google Scholar] [CrossRef] [PubMed]
- Yu, P.; Xiao, S.; Xin, X.; Song, C.-X.; Huang, W.; McDee, D.; Tanaka, T.; Wang, T.; He, C.; Zhong, S. Spatiotemporal clustering of the epigenome reveals rules of dynamic gene regulation. Genome Res. 2013, 23, 352–364. [Google Scholar] [CrossRef]
- Fiziev, P.; Ernst, J. ChromTime: Modeling spatio-temporal dynamics of chromatin marks. Genome Biol. 2018, 19, 109. [Google Scholar] [CrossRef]
- Guo, S.; Diep, D.; Plongthongkum, N.; Fung, H.L.; Zhang, K.; Zhang, K. Identification of methylation haplotype blocks AIDS in deconvolution of heterogeneous tissue samples and tumor tissue-of-origin mapping from plasma DNA. Nat. Genet. 2017, 49, 635–642. [Google Scholar] [CrossRef]
- Romanowska, J.; Haaland, Ø.A.; Jugessur, A.; Gjerdevik, M.; Xu, Z.; Taylor, J.; Wilcox, A.J.; Jonassen, I.; Lie, R.T.; Håkon, K. Gjessing Integrating genome-wide methylation and genotype data to elucidate how region-wise methylation level might influence allele-defined relative risks. Submitted. 2019. [Google Scholar]
- Dolinoy, D.C.; Weidman, J.R.; Waterland, R.A.; Jirtle, R.L. Maternal genistein alters coat color and protects Avy mouse offspring from obesity by modifying the fetal epigenome. Environ. Health Perspect. 2006, 114, 567–572. [Google Scholar] [CrossRef] [PubMed]
- Briffa, J.F.; Wlodek, M.E.; Moritz, K.M. Transgenerational programming of nephron deficits and hypertension. Semin. Cell Dev. Biol. 2018. [Google Scholar] [CrossRef]
- Dekker, J.; Rippe, K.; Dekker, M.; Kleckner, N. Capturing chromosome conformation. Science 2002, 295, 1306–1311. [Google Scholar] [CrossRef] [PubMed]
- Lieberman-Aiden, E.; van Berkum, N.L.; Williams, L.; Imakaev, M.; Ragoczy, T.; Telling, A.; Amit, I.; Lajoie, B.R.; Sabo, P.J.; Dorschner, M.O.; et al. Comprehensive mapping of long-range interactions reveals folding principles of the human genome. Science 2009, 326, 289–293. [Google Scholar] [CrossRef]
- Fraser, J.; Williamson, I.; Bickmore, W.A.; Dostie, J. An Overview of Genome Organization and How We Got There: From FISH to Hi-C. Microbiol. Mol. Biol. Rev. MMBR 2015, 79, 347–372. [Google Scholar] [CrossRef]
- Wu, C.; Pan, W. Integration of Enhancer-Promoter Interactions with GWAS Summary Results Identifies Novel Schizophrenia-Associated Genes and Pathways. Genetics 2018, 209, 699–709. [Google Scholar] [CrossRef] [PubMed]
- Thurman, R.E.; Rynes, E.; Humbert, R.; Vierstra, J.; Maurano, M.T.; Haugen, E.; Sheffield, N.C.; Stergachis, A.B.; Wang, H.; Vernot, B.; et al. The accessible chromatin landscape of the human genome. Nature 2012, 489, 75–82. [Google Scholar] [CrossRef] [PubMed]
- Vipin, D.; Wang, L.; Devailly, G.; Michoel, T.; Joshi, A. Causal Transcription Regulatory Network Inference Using Enhancer Activity as a Causal Anchor. Int. J. Mol. Sci. 2018, 19, 3609. [Google Scholar] [CrossRef] [PubMed]
- Radkiewicz, C.; Johansson, A.L.V.; Dickman, P.W.; Lambe, M.; Edgren, G. Sex differences in cancer risk and survival: A Swedish cohort study. Eur. J. Cancer 2017, 84, 130–140. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
{kind=link}
| Num. | Data Type | Disease | Available data | # of Samples | Reference |
|---|---|---|---|---|---|
| 1 | ATAC-seq | 23 cancer types | Genotype, ATAC-seq, RNA-seq | 410 | [43] |
| 2 | ChIP-seq | Prostate cancer | H3K27ac, H3K4me3, H3K27me3 | 100 | GSE120738 |
| 3 | ChIP-seq | Breast cancer | H3K4me1, TFs | - | [44] |
| 4 | ChIP-seq | Adenocarcinoma | H3K27ac, H3K4me3, H3K4me1 | 94 | [45] |
| 5 | ChIP-seq | Acute myeloid leukemia | H3K9me3 | 108 | [46] |
| 6 | ChIP-seq | Glioma | Multiple | - | [47] |
| 7 | ChIP-on-chip | Acute myeloid leukemia | H3 | 73 | [48] |
| 8 | ChIP-on-chip | Acute promyelocytic leukemia | H3, H3K9me3, H3K4me3 | 372 | [49] |
| 9 | ChIP-seq | Acute myeloid leukemia | H3K9me2 | 16 | [50] |
| 10 | ChIP-seq | Hepatocarcinoma | Multiple | 5 | [51] |
| 11 | ATAC-seq, ChIP-seq | Colorectal cancer | Multiple | 4 | [52] |
| 12 | FAIRE-seq, ChIP-seq | Ovarian cancer | H3K27ac, H3K4me1 | 5 | [53] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Romanowska, J.; Joshi, A. From Genotype to Phenotype: Through Chromatin. Genes 2019, 10, 76. https://doi.org/10.3390/genes10020076
Romanowska J, Joshi A. From Genotype to Phenotype: Through Chromatin. Genes. 2019; 10(2):76. https://doi.org/10.3390/genes10020076
Chicago/Turabian StyleRomanowska, Julia, and Anagha Joshi. 2019. "From Genotype to Phenotype: Through Chromatin" Genes 10, no. 2: 76. https://doi.org/10.3390/genes10020076
APA StyleRomanowska, J., & Joshi, A. (2019). From Genotype to Phenotype: Through Chromatin. Genes, 10(2), 76. https://doi.org/10.3390/genes10020076