Functional SNPs in the Human Autoimmunity-Associated Locus 17q12-21

 , , and
, , and
Abstract
:1. Introduction
2. Materials and Methods
2.1. SNPs Selection
2.2. Publication Mining for Expression Quantitative Trait Loci (eQTL)
2.3. Enhancers Cloning
2.4. PCR Mutagenesis
2.5. Cells
2.6. Activation of U-937
2.7. Luciferase Reporter Assay
2.8. Statistical Analysis
3. Results
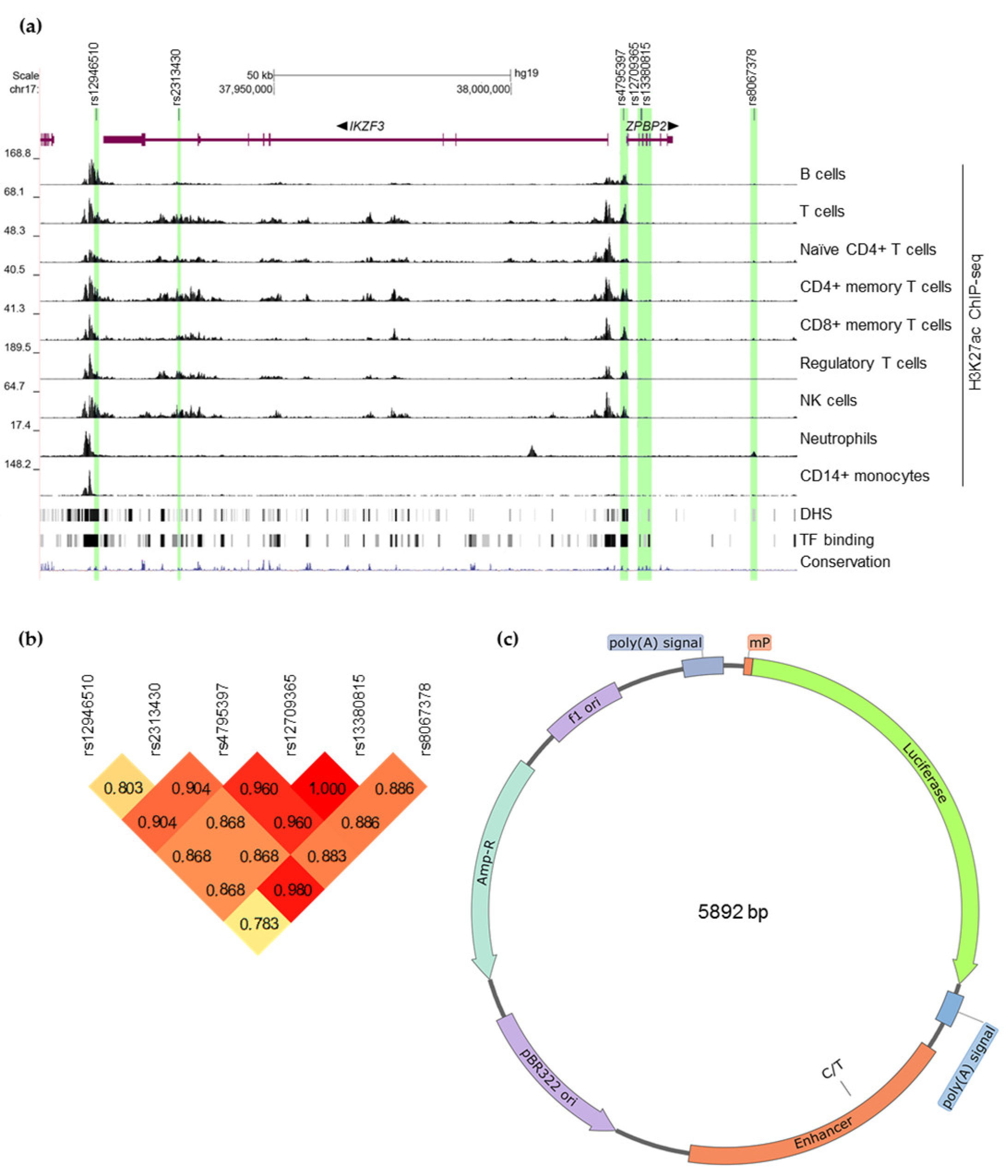
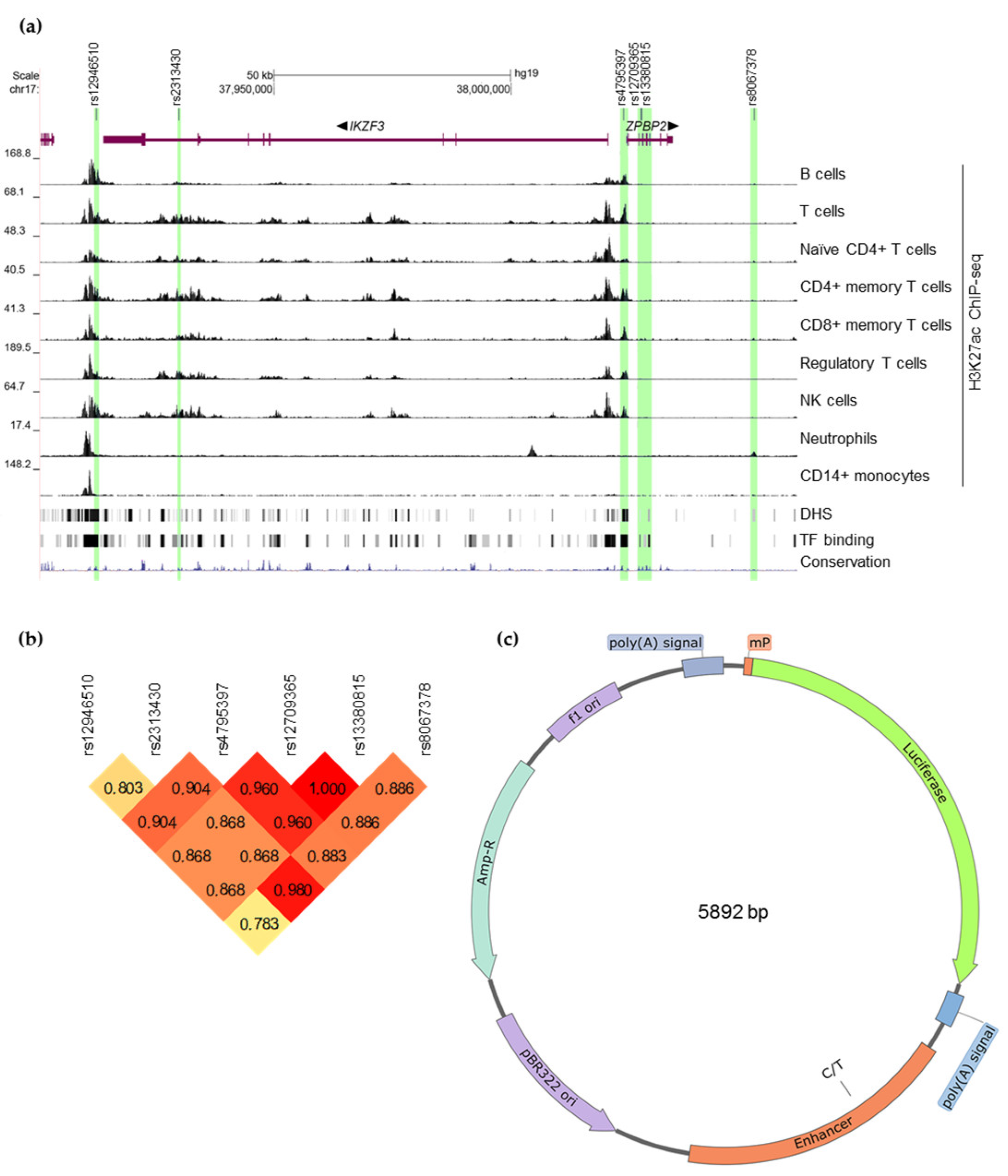
3.1. Selection of 17q12-21 SNPs for Experimental Validation
3.2. Possible Target Genes of the Selected Candidate SNPs
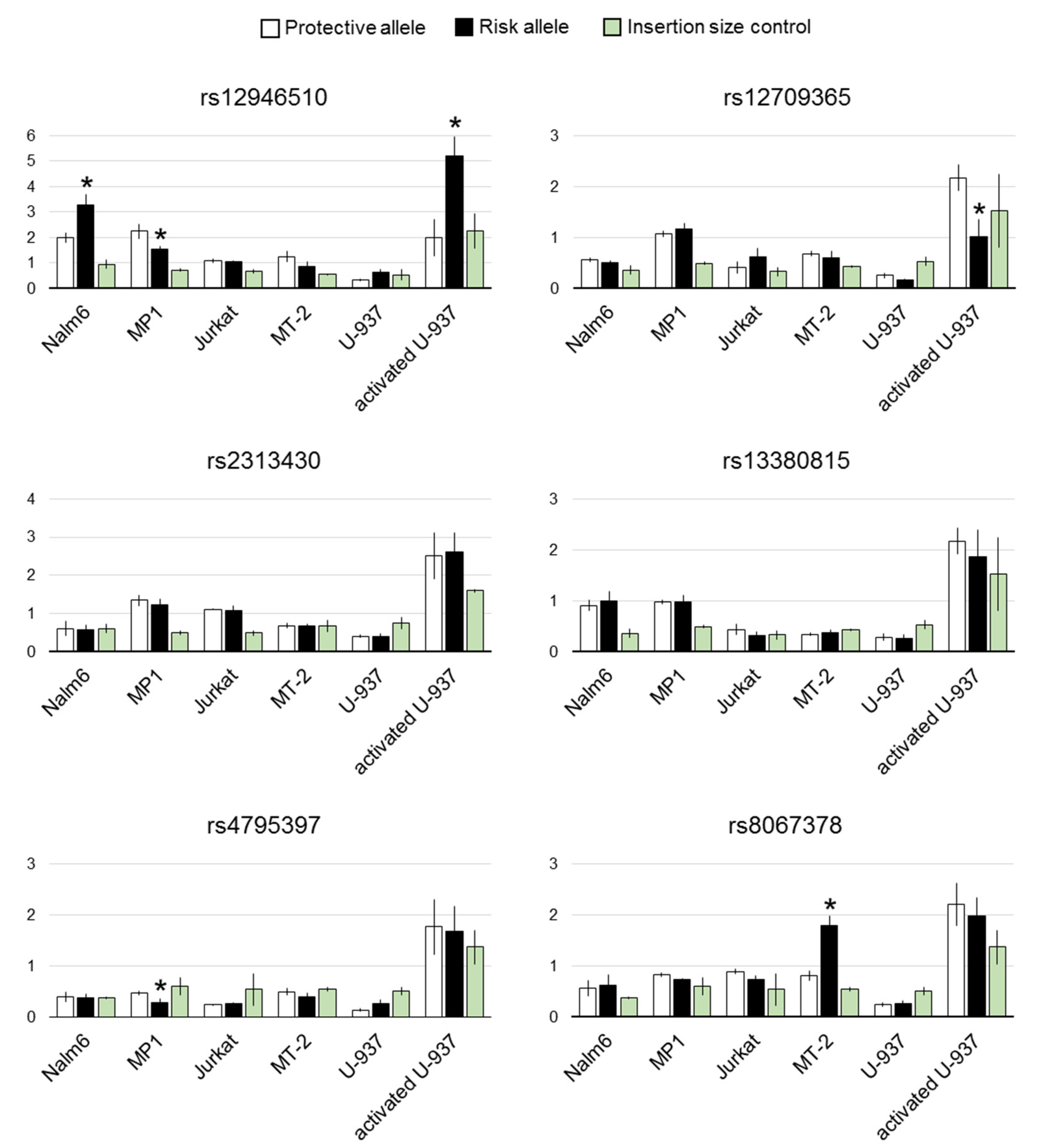
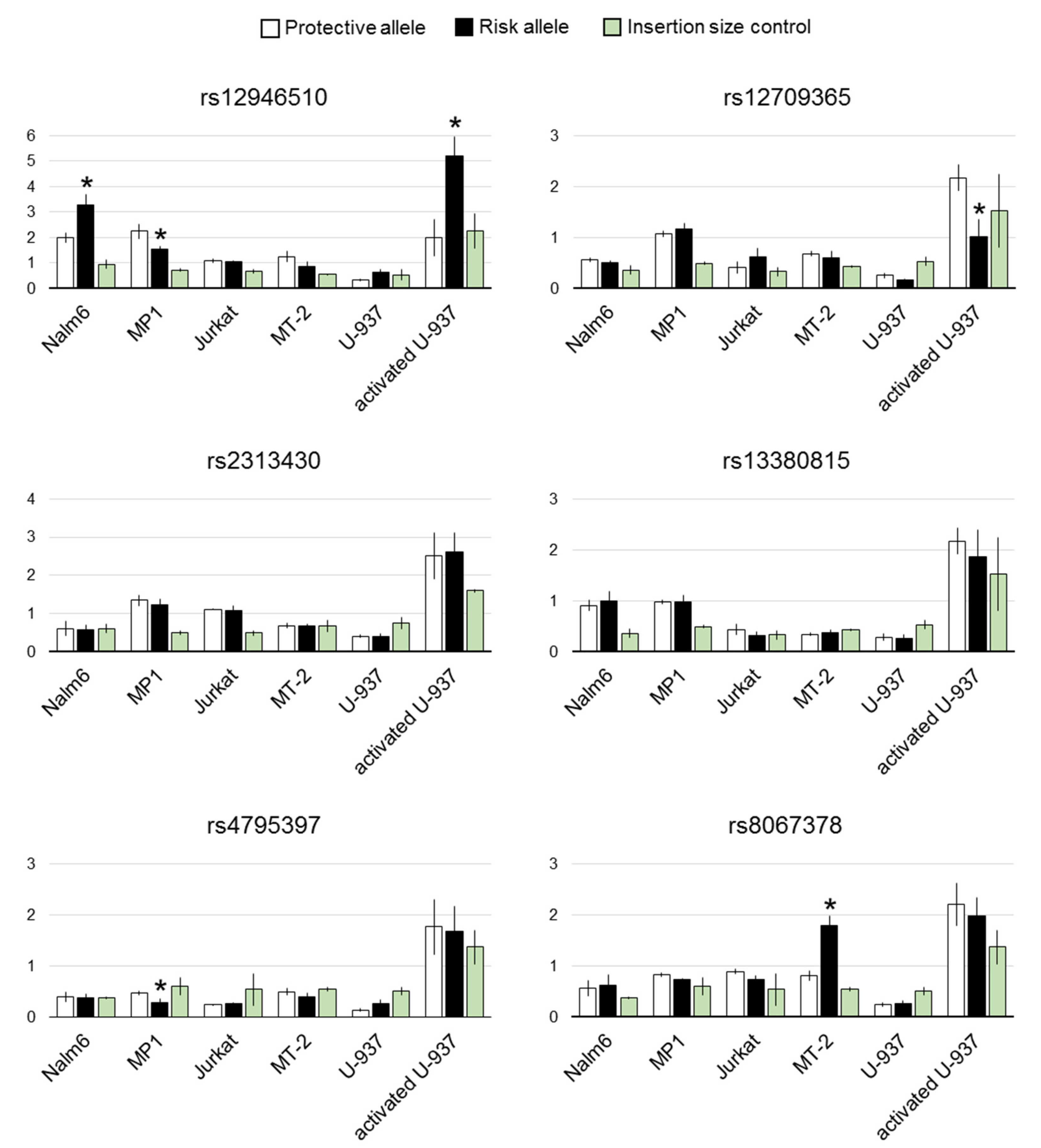
3.3. Effects of the Selected Variants on Transcription in Luciferase Reporter Assay
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| SNP | Single-Nucleotide Polymorphism |
| GWAS | Genome-Wide Association Study |
| PICS | Probabilistic Identification of Causal SNPs |
| PP | PICS Probability |
| eQTL | expression Quantitative Trait Loci |
| LD | Linkage Disequilibrium |
| LPS | Lipopolysaccharide |
| IFNγ | Interferon gamma |
| EBV | Epstein–Barr Virus |
| KGph3 | Phase 3 of the 1000 Genomes Project |
| RAF | Risk Allele Frequency |
| OR | Odds Ratio |
| DHS | DNase I hypersensitivity |
| CEU | Utah Residents with Northern and Western European Ancestry |
| YRI | Yoruba in Ibadan, Nigeria |
References
- Hall, J.M.; Lee, M.K.; Newman, B.; Morrow, J.E.; Anderson, L.A.; Huey, B.; King, M.C. Linkage of Early-Onset Familial Breast Cancer to Chromosome 17q21. Science 1990, 250, 1684–1689. [Google Scholar] [CrossRef] [PubMed]
- Dizier, M.H.; Besse-Schmittler, C.; Guilloud-Bataille, M.; Annesi-Maesano, I.; Boussaha, M.; Bousquet, J.; Charpin, D.; Degioanni, A.; Gormand, F.; Grimfeld, A.; et al. Genome Screen for Asthma and Related Phenotypes in the French EGEA Study. Am. J. Respir. Crit. Care Med. 2000, 162, 1812–1818. [Google Scholar] [CrossRef] [PubMed]
- Moffatt, M.F.; Kabesch, M.; Liang, L.; Dixon, A.L.; Strachan, D.; Heath, S.; Depner, M.; von Berg, A.; Bufe, A.; Rietschel, E.; et al. Genetic Variants Regulating ORMDL3 Expression Contribute to the Risk of Childhood Asthma. Nature 2007, 448, 470–473. [Google Scholar] [CrossRef] [PubMed]
- Stein, M.M.; Thompson, E.E.; Schoettler, N.; Helling, B.A.; Magnaye, K.M.; Stanhope, C.; Igartua, C.; Morin, A.; Washington, C.; Nicolae, D.; et al. A Decade of Research on the 17q12-21 Asthma Locus: Piecing Together the Puzzle. J. Allergy Clin. Immunol. 2018, 142, 749–764.e3. [Google Scholar] [CrossRef] [PubMed]
- Mells, G.F.; Floyd, J.A.B.; Morley, K.I.; Cordell, H.J.; Franklin, C.S.; Shin, S.-Y.; Heneghan, M.A.; Neuberger, J.M.; Donaldson, P.T.; Day, D.B.; et al. Genome-Wide Association Study Identifies 12 New Susceptibility Loci for Primary Biliary Cirrhosis. Nat. Genet. 2011, 43, 329–332. [Google Scholar] [CrossRef]
- Hirschfield, G.M.; Liu, X.; Han, Y.; Gorlov, I.P.; Lu, Y.; Xu, C.; Lu, Y.; Chen, W.; Juran, B.D.; Coltescu, C.; et al. Variants at IRF5-TNPO3, 17q12-21 and MMEL1 Are Associated with Primary Biliary Cirrhosis. Nat. Genet. 2010, 42, 655–657. [Google Scholar] [CrossRef]
- Stahl, E.A.; Raychaudhuri, S.; Remmers, E.F.; Xie, G.; Eyre, S.; Thomson, B.P.; Li, Y.; Kurreeman, F.A.S.; Zhernakova, A.; Hinks, A.; et al. Genome-Wide Association Study Meta-Analysis Identifies Seven New Rheumatoid Arthritis Risk Loci. Nat. Genet. 2010, 42, 508–514. [Google Scholar] [CrossRef]
- Saleh, N.M.; Raj, S.M.; Smyth, D.J.; Wallace, C.; Howson, J.M.M.; Bell, L.; Walker, N.M.; Stevens, H.E.; Todd, J.A. Genetic Association Analyses of Atopic Illness and Proinflammatory Cytokine Genes with Type 1 Diabetes. Diabetes Metab. Res. Rev. 2011, 27, 838–843. [Google Scholar] [CrossRef]
- Anderson, C.A.; Boucher, G.; Lees, C.W.; Franke, A.; D’Amato, M.; Taylor, K.D.; Lee, J.C.; Goyette, P.; Imielinski, M.; Latiano, A.; et al. Meta-Analysis Identifies 29 Additional Ulcerative Colitis Risk Loci, Increasing the Number of Confirmed Associations to 47. Nat. Genet. 2011, 43, 246–252. [Google Scholar] [CrossRef]
- Barrett, J.C.; Hansoul, S.; Nicolae, D.L.; Cho, J.H.; Duerr, R.H.; Rioux, J.D.; Brant, S.R.; Silverberg, M.S.; Taylor, K.D.; Barmada, M.M.; et al. Genome-Wide Association Defines More than 30 Distinct Susceptibility Loci for Crohn’s Disease. Nat. Genet. 2008, 40, 955–962. [Google Scholar] [CrossRef]
- Kreiner, E.; Waage, J.; Standl, M.; Brix, S.; Pers, T.H.; Couto Alves, A.; Warrington, N.M.; Tiesler, C.M.T.; Fuertes, E.; Franke, L.; et al. Shared Genetic Variants Suggest Common Pathways in Allergy and Autoimmune Diseases. J. Allergy Clin. Immunol. 2017, 140, 771–781. [Google Scholar] [CrossRef] [PubMed]
- John, L.B.; Ward, A.C. The Ikaros Gene Family: Transcriptional Regulators of Hematopoiesis and Immunity. Mol. Immunol. 2011, 48, 1272–1278. [Google Scholar] [CrossRef] [PubMed]
- Theofilopoulos, A.N.; Kono, D.H.; Baccala, R. The Multiple Pathways to Autoimmunity. Nat. Immunol. 2017, 18, 716–724. [Google Scholar] [CrossRef] [PubMed]
- Gutierrez-Arcelus, M.; Rich, S.S.; Raychaudhuri, S. Autoimmune Diseases—Connecting Risk Alleles with Molecular Traits of the Immune System. Nat. Rev. Genet. 2016, 17, 160–174. [Google Scholar] [CrossRef] [PubMed]
- Spain, S.L.; Barrett, J.C. Strategies for Fine-Mapping Complex Traits. Hum. Mol. Genet. 2015, 24, R111–R119. [Google Scholar] [CrossRef] [PubMed]
- Farh, K.K.H.; Marson, A.; Zhu, J.; Kleinewietfeld, M.; Housley, W.J.; Beik, S.; Shoresh, N.; Whitton, H.; Ryan, R.J.H.; Shishkin, A.A.; et al. Genetic and Epigenetic Fine Mapping of Causal Autoimmune Disease Variants. Nature 2015, 518, 337–343. [Google Scholar] [CrossRef]
- Schmiedel, B.J.; Seumois, G.; Samaniego-Castruita, D.; Cayford, J.; Schulten, V.; Chavez, L.; Ay, F.; Sette, A.; Peters, B.; Vijayanand, P. 17q21 Asthma-Risk Variants Switch CTCF Binding and Regulate IL-2 Production by T Cells. Nat. Commun. 2016, 7, 13426. [Google Scholar] [CrossRef]
- Buckley, M.; Gjyshi, A.; Mendoza-Fandiño, G.; Baskin, R.; Carvalho, R.S.; Carvalho, M.A.; Woods, N.T.; Monteiro, A.N.A. Enhancer Scanning to Locate Regulatory Regions in Genomic Loci. Nat. Protoc. 2016, 11, 46–60. [Google Scholar] [CrossRef]
- Finemapping. Available online: https://pubs.broadinstitute.org/pubs/finemapping/dataportal.php (accessed on 14 November 2018).
- Fairfax, B.P.; Humburg, P.; Makino, S.; Naranbhai, V.; Wong, D.; Lau, E.; Jostins, L.; Plant, K.; Andrews, R.; McGee, C.; et al. Innate Immune Activity Conditions the Effect of Regulatory Variants upon Monocyte Gene Expression. Science 2014, 343, 1246949. [Google Scholar] [CrossRef]
- Zeller, T.; Wild, P.; Szymczak, S.; Rotival, M.; Schillert, A.; Castagne, R.; Maouche, S.; Germain, M.; Lackner, K.; Rossmann, H.; et al. Genetics and Beyond—The Transcriptome of Human Monocytes and Disease Susceptibility. PLoS ONE 2010, 5, e10693. [Google Scholar] [CrossRef]
- Westra, H.-J.; Peters, M.J.; Esko, T.; Yaghootkar, H.; Schurmann, C.; Kettunen, J.; Christiansen, M.W.; Fairfax, B.P.; Schramm, K.; Powell, J.E.; et al. Systematic Identification of Trans EQTLs as Putative Drivers of Known Disease Associations. Nat. Genet. 2013, 45, 1238–1243. [Google Scholar] [CrossRef] [PubMed]
- Lloyd-Jones, L.R.; Holloway, A.; McRae, A.; Yang, J.; Small, K.; Zhao, J.; Zeng, B.; Bakshi, A.; Metspalu, A.; Dermitzakis, M.; et al. The Genetic Architecture of Gene Expression in Peripheral Blood. Am. J. Hum. Genet. 2017, 100, 228–237. [Google Scholar] [CrossRef] [PubMed]
- Wright, F.A.; Sullivan, P.F.; Brooks, A.I.; Zou, F.; Sun, W.; Xia, K.; Madar, V.; Jansen, R.; Chung, W.; Zhou, Y.-H.; et al. Heritability and Genomics of Gene Expression in Peripheral Blood. Nat. Genet. 2014, 46, 430–437. [Google Scholar] [CrossRef] [PubMed]
- Bryois, J.; Buil, A.; Evans, D.M.; Kemp, J.P.; Montgomery, S.B.; Conrad, D.F.; Ho, K.M.; Ring, S.; Hurles, M.; Deloukas, P.; et al. Cis and Trans Effects of Human Genomic Variants on Gene Expression. PLoS Genet. 2014, 10, e1004461. [Google Scholar] [CrossRef] [PubMed]
- Fehrmann, R.S.N.; Jansen, R.C.; Veldink, J.H.; Westra, H.-J.; Arends, D.; Bonder, M.J.; Fu, J.; Deelen, P.; Groen, H.J.M.; Smolonska, A.; et al. Trans-eQTLs Reveal That Independent Genetic Variants Associated with a Complex Phenotype Converge on Intermediate Genes, with a Major Role for the HLA. PLoS Genet. 2011, 7, e1002197. [Google Scholar] [CrossRef] [PubMed]
- Dimas, A.S.; Deutsch, S.; Stranger, B.E.; Montgomery, S.B.; Borel, C.; Attar-Cohen, H.; Ingle, C.; Beazley, C.; Gutierrez Arcelus, M.; Sekowska, M.; et al. Common Regulatory Variation Impacts Gene Expression in a Cell Type-Dependent Manner. Science 2009, 325, 1246–1250. [Google Scholar] [CrossRef] [PubMed]
- Murphy, A.; Chu, J.-H.; Xu, M.; Carey, V.J.; Lazarus, R.; Liu, A.; Szefler, S.J.; Strunk, R.; Demuth, K.; Castro, M.; et al. Mapping of Numerous Disease-Associated Expression Polymorphisms in Primary Peripheral Blood CD4+ Lymphocytes. Hum. Mol. Genet. 2010, 19, 4745–4757. [Google Scholar] [CrossRef]
- Westra, H.-J.; Arends, D.; Esko, T.; Peters, M.J.; Schurmann, C.; Schramm, K.; Kettunen, J.; Yaghootkar, H.; Fairfax, B.P.; Andiappan, A.K.; et al. Cell Specific eQTL Analysis without Sorting Cells. PLoS Genet. 2015, 11, e1005223. [Google Scholar] [CrossRef]
- Heap, G.A.; Trynka, G.; Jansen, R.C.; Bruinenberg, M.; Swertz, M.A.; Dinesen, L.C.; Hunt, K.A.; Wijmenga, C.; Vanheel, D.A.; Franke, L. Complex Nature of SNP Genotype Effects on Gene Expression in Primary Human Leucocytes. BMC Med. Genom. 2009, 2, 1. [Google Scholar] [CrossRef]
- Idaghdour, Y.; Czika, W.; Shianna, K.V.; Lee, S.H.; Visscher, P.M.; Martin, H.C.; Miclaus, K.; Jadallah, S.J.; Goldstein, D.B.; Wolfinger, R.D.; et al. Geographical Genomics of Human Leukocyte Gene Expression Variation in Southern Morocco. Nat. Genet. 2010, 42, 62–67. [Google Scholar] [CrossRef]
- Göring, H.H.H.; Curran, J.E.; Johnson, M.P.; Dyer, T.D.; Charlesworth, J.; Cole, S.A.; Jowett, J.B.M.; Abraham, L.J.; Rainwater, D.L.; Comuzzie, A.G.; et al. Discovery of Expression QTLs Using Large-Scale Transcriptional Profiling in Human Lymphocytes. Nat. Genet. 2007, 39, 1208–1216. [Google Scholar] [CrossRef] [PubMed]
- Heinzen, E.L.; Ge, D.; Cronin, K.D.; Maia, J.M.; Shianna, K.V.; Gabriel, W.N.; Welsh-Bohmer, K.A.; Hulette, C.M.; Denny, T.N.; Goldstein, D.B. Tissue-Specific Genetic Control of Splicing: Implications for the Study of Complex Traits. PLoS Biol. 2008, 6, e1. [Google Scholar] [CrossRef] [PubMed]
- Fairfax, B.P.; Makino, S.; Radhakrishnan, J.; Plant, K.; Leslie, S.; Dilthey, A.; Ellis, P.; Langford, C.; Vannberg, F.O.; Knight, J.C. Genetics of Gene Expression in Primary Immune Cells Identifies Cell Type-Specific Master Regulators and Roles of HLA Alleles. Nat. Genet. 2012, 44, 502–510. [Google Scholar] [CrossRef] [PubMed]
- Powell, J.E.; Henders, A.K.; McRae, A.F.; Caracella, A.; Smith, S.; Wright, M.J.; Whitfield, J.B.; Dermitzakis, E.T.; Martin, N.G.; Visscher, P.M.; et al. The Brisbane Systems Genetics Study: Genetical Genomics Meets Complex Trait Genetics. PLoS ONE 2012, 7, e35430. [Google Scholar] [CrossRef] [PubMed]
- SNAP Pairwise LD tool. Available online: http://archive.broadinstitute.org/mpg/snap/ldsearchpw.php (accessed on 14 November 2018).
- Korneev, K.V.; Sviriaeva, E.N.; Mitkin, N.A.; Gorbacheva, A.M.; Uvarova, A.N.; Ustiugova, A.S.; Afanasyeva, M.A.; Kulakovskiy, I.V.; Schwartz, A.M.; Kuprash, D.V. Minor C Allele of the SNP Rs7873784 Creates a Strong PU.1 Binding Site and Promotes Enhancer Activity of 3’-Untranslated Region of TLR4 Gene in U937 Monocytes. Manuscript in preparation.
- McGovern, D.P.B.; Gardet, A.; Törkvist, L.; Goyette, P.; Essers, J.; Taylor, K.D.; Neale, B.M.; Ong, R.T.H.; Lagacé, C.; Li, C.; et al. Genome-Wide Association Identifies Multiple Ulcerative Colitis Susceptibility Loci. Nat. Genet. 2010, 42, 332–337. [Google Scholar] [CrossRef] [PubMed]
- Beecham, A.H.; Patsopoulos, N.A.; Xifara, D.K.; Davis, M.F.; Kemppinen, A.; Cotsapas, C.; Shah, T.S.; Spencer, C.; Booth, D.; Goris, A.; et al. Analysis of Immune-Related Loci Identifies 48 New Susceptibility Variants for Multiple Sclerosis. Nat. Genet. 2013, 45, 1353–1362. [Google Scholar] [CrossRef] [PubMed]
- Franke, A.; McGovern, D.P.B.; Barrett, J.C.; Wang, K.; Radford-Smith, G.L.; Ahmad, T.; Lees, C.W.; Balschun, T.; Lee, J.; Roberts, R.; et al. Genome-Wide Meta-Analysis Increases to 71 the Number of Confirmed Crohn’s Disease Susceptibility Loci. Nat. Genet. 2010, 42, 1118–1125. [Google Scholar] [CrossRef]
- Plagnol, V.; Howson, J.M.M.; Smyth, D.J.; Walker, N.; Hafler, J.P.; Wallace, C.; Stevens, H.; Jackson, L.; Simmonds, M.J.; Bingley, P.J.; et al. Genome-Wide Association Analysis of Autoantibody Positivity in Type 1 Diabetes Cases. PLoS Genet. 2011, 7, e1002216. [Google Scholar] [CrossRef]
- Eyre, S.; Bowes, J.; Diogo, D.; Lee, A.; Barton, A.; Martin, P.; Zhernakova, A.; Stahl, E.; Viatte, S.; McAllister, K.; et al. High-Density Genetic Mapping Identifies New Susceptibility Loci for Rheumatoid Arthritis. Nat. Genet. 2012, 44, 1336–1340. [Google Scholar] [CrossRef]
- Liu, J.Z.; Almarri, M.A.; Gaffney, D.J.; Mells, G.F.; Jostins, L.; Cordell, H.J.; Ducker, S.J.; Day, D.B.; Heneghan, M.A.; Neuberger, J.M.; et al. Dense Fine-Mapping Study Identifies New Susceptibility Loci for Primary Biliary Cirrhosis. Nat. Genet. 2012, 44, 1137–1141. [Google Scholar] [CrossRef]
- 1000 Genomes|A Deep Catalog of Human Genetic Variation. Available online: http://www.internationalgenome.org/ (accessed on 14 November 2018).
- Deplancke, B.; Alpern, D.; Gardeux, V. The Genetics of Transcription Factor DNA Binding Variation. Cell 2016, 166, 538–554. [Google Scholar] [CrossRef] [PubMed]
- Brodie, A.; Azaria, J.R.; Ofran, Y. How Far from the SNP May the Causative Genes Be? Nucleic Acids Res. 2016, 44, 6046–6054. [Google Scholar] [CrossRef] [PubMed]
- Brown, A.A.; Viñuela, A.; Delaneau, O.; Spector, T.D.; Small, K.S.; Dermitzakis, E.T. Predicting Causal Variants Affecting Expression by Using Whole-Genome Sequencing and RNA-Seq from Multiple Human Tissues. Nat. Genet. 2017, 49, 1747–1751. [Google Scholar] [CrossRef] [PubMed]
- Emilsson, V.; Thorleifsson, G.; Zhang, B.; Leonardson, A.S.; Zink, F.; Zhu, J.; Carlson, S.; Helgason, A.; Walters, G.B.; Gunnarsdottir, S.; et al. Genetics of Gene Expression and Its Effect on Disease. Nature 2008, 452, 423–428. [Google Scholar] [CrossRef] [PubMed]
- Aguet, F.; Brown, A.A.; Castel, S.E.; Davis, J.R.; He, Y.; Jo, B.; Mohammadi, P.; Park, Y.Y.; Parsana, P.; Segrè, A.V.; et al. Genetic Effects on Gene Expression across Human Tissues. Nature 2017, 550, 204–213. [Google Scholar] [CrossRef] [PubMed]
- Lonsdale, J.; Thomas, J.; Salvatore, M.; Phillips, R.; Lo, E.; Shad, S.; Hasz, R.; Walters, G.; Garcia, F.; Young, N.; et al. The Genotype-Tissue Expression (GTEx) Project. Nat. Genet. 2013, 45, 580–585. [Google Scholar] [CrossRef] [PubMed]
- UCSC Genome Browser. Available online: https://genome.ucsc.edu/ (accessed on 14 November 2018).
- Kent, W.J.; Sugnet, C.W.; Furey, T.S.; Roskin, K.M.; Pringle, T.H.; Zahler, A.M.; Haussler, D. The Human Genome Browser at UCSC. Genome Res. 2002, 12, 996–1006. [Google Scholar] [CrossRef]
- ENCODE: Encyclopedia of DNA Elements. Available online: https://www.encodeproject.org/ (accessed on 14 November 2018).
- ENCODE Project Consortium. An Integrated Encyclopedia of DNA Elements in the Human Genome. Nature 2012, 489, 57–74. [Google Scholar] [CrossRef]
- Gibbs, R.A.; Boerwinkle, E.; Doddapaneni, H.; Han, Y.; Korchina, V.; Kovar, C.; Lee, S.; Muzny, D.; Reid, J.G.; Zhu, Y.; et al. A Global Reference for Human Genetic Variation. Nature 2015, 526, 68–74. [Google Scholar] [CrossRef]
- Hurwitz, R.; Hozier, J.; LeBien, T.; Minowada, J.; Gajl-Peczalska, K.; Kubonishi, I.; Kersey, J. Characterization of a Leukemic Cell Line of the Pre-B Phenotype. Int. J. Cancer 1979, 23, 174–180. [Google Scholar] [CrossRef]
- Pistillo, M.P.; Tanigaki, N.; Mazzoleni, O.; Ciccone, E.; Hämmerling, U.; Park, M.; Terasaki, P.I.; Ferrara, G.B. Human Lymphoblastoid Cell Lines Secreting Antibodies with Restricted HLA Specificity. Immunogenetics 1987, 25, 145–151. [Google Scholar] [CrossRef] [PubMed]
- Schneider, U.; Schwenk, H.-U.H.-U.U.; Bornkamm, G. Characterization of EBV-genome Negative “Null” and “T” Cell Lines Derived from Children with Acute Lymphoblastic Leukemia and Leukemic Transformed Non-Hodgkin Lymphoma. Int. J. Cancer 1977, 19, 621–626. [Google Scholar] [CrossRef] [PubMed]
- Fujita, T.; Shibuya, H.; Ohashi, T.; Yamanishi, K.; Taniguchi, T. Regulation of Human Interleukin-2 Gene: Functional DNA Sequences in the 5′ Flanking Region for the Gene Expression in Activated T Lymphocytes. Cell 1986, 46, 401–407. [Google Scholar] [CrossRef]
- Roose, J.P.; Diehn, M.; Tomlinson, M.G.; Lin, J.; Alizadeh, A.A.; Botstein, D.; Brown, P.O.; Weiss, A. T Cell Receptor-Independent Basal Signaling via Erk and Abl Kinases Suppresses RAG Gene Expression. PLoS Biol. 2003, 1, e53. [Google Scholar] [CrossRef] [PubMed]
- Hamano, R.; Wu, X.; Wang, Y.; Oppenheim, J.J.; Chen, X. Characterization of MT-2 Cells as a Human Regulatory T Cell-like Cell Line. Cell. Mol. Immunol. 2015, 12, 780–782. [Google Scholar] [CrossRef] [PubMed]
- Sundström, C.; Nilsson, K. Establishment and Characterization of a Human Histiocytic Lymphoma Cell Line (U-937). Int. J. Cancer 1976, 17, 565–577. [Google Scholar] [CrossRef] [PubMed]
- PERFECTOS-APE: PrEdicting Regulatory Functional Effect by Approximate P-value Estimation. Available online: http://opera.autosome.ru/perfectosape/ (accessed on 14 November 2018).
- Vorontsov, I.E.; Kulakovskiy, I.V.; Khimulya, G.; Nikolaeva, D.D.; Makeev, V.J. PERFECTOS-APE: PrEdicting Regulatory Functional Effect by Approximate P-value Estimation. In Proceedings of the International Conference on Bioinformatics Models, Methods and Algorithms, Lisbon, Portugal, 12–15 January 2015; Volume 1, pp. 102–108. [Google Scholar]
- JASPAR 2018. Available online: http://jaspar.genereg.net/ (accessed on 14 November 2018).
- Schwartz, A.M.; Putlyaeva, L.V.; Covich, M.; Klepikova, A.V.; Akulich, K.A.; Vorontsov, I.E.; Korneev, K.V.; Dmitriev, S.E.; Polanovsky, O.L.; Sidorenko, S.P.; et al. Early B-Cell Factor 1 (EBF1) Is Critical for Transcriptional Control of SLAMF1 Gene in Human B Cells. Biochim. Biophys. Acta Gene Regul. Mech. 2016, 1859, 1259–1268. [Google Scholar] [CrossRef]
- MacArthur, J.; Bowler, E.; Cerezo, M.; Gil, L.; Hall, P.; Hastings, E.; Junkins, H.; McMahon, A.; Milano, A.; Morales, J.; et al. The New NHGRI-EBI Catalog of Published Genome-Wide Association Studies (GWAS Catalog). Nucleic Acids Res. 2017, 45, D896–D901. [Google Scholar] [CrossRef]
- Schaid, D.J.; Chen, W.; Larson, N.B. From Genome-Wide Associations to Candidate Causal Variants by Statistical Fine-Mapping. Nat. Rev. Genet. 2018, 19, 491–504. [Google Scholar] [CrossRef]
- Wang, J.H.; Avitahl, N.; Cariappa, A.; Friedrich, C.; Ikeda, T.; Renold, A.; Andrikopoulos, K.; Liang, L.; Pillai, S.; Morgan, B.A.; et al. Aiolos Regulates B Cell Activation and Maturation to Effector State. Immunity 1998, 9, 543–553. [Google Scholar] [CrossRef]
- He, B.; Chen, C.; Teng, L.; Tan, K. Global View of Enhancer-Promoter Interactome in Human Cells. Proc. Natl. Acad. Sci. USA 2014, 111, E2191–E2199. [Google Scholar] [CrossRef] [PubMed]
- Swanson, B.J.; Jäck, H.-M.; Lyons, G.E. Characterization of Myocyte Enhancer Factor 2 (MEF2) Expression in B and T Cells: MEF2C Is a B Cell-Restricted Transcription Factor in Lymphocytes. Mol. Immunol. 1998, 35, 445–458. [Google Scholar] [CrossRef]
- Kong, N.R.; Davis, M.; Chai, L.; Winoto, A.; Tjian, R. MEF2C and EBF1 Co-Regulate B Cell-Specific Transcription. PLoS Genet. 2016, 12, e1005845. [Google Scholar] [CrossRef] [PubMed]
- Mansson, R.; Welinder, E.; Åhsberg, J.; Lin, Y.C.; Benner, C.; Glass, C.K.; Lucas, J.S.; Sigvardsson, M.; Murre, C. Positive Intergenic Feedback Circuitry, Involving EBF1 and FOXO1, Orchestrates B-Cell Fate. Proc. Natl. Acad. Sci. USA 2012, 109, 21028–21033. [Google Scholar] [CrossRef] [PubMed]
- Verlaan, D.J.; Berlivet, S.; Hunninghake, G.M.; Madore, A.-M.; Larivière, M.; Moussette, S.; Grundberg, E.; Kwan, T.; Ouimet, M.; Ge, B.; et al. Allele-Specific Chromatin Remodeling in the ZPBP2/GSDMB/ORMDL3 Locus Associated with the Risk of Asthma and Autoimmune Disease. Am. J. Hum. Genet. 2009, 85, 377–393. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
{kind=link}
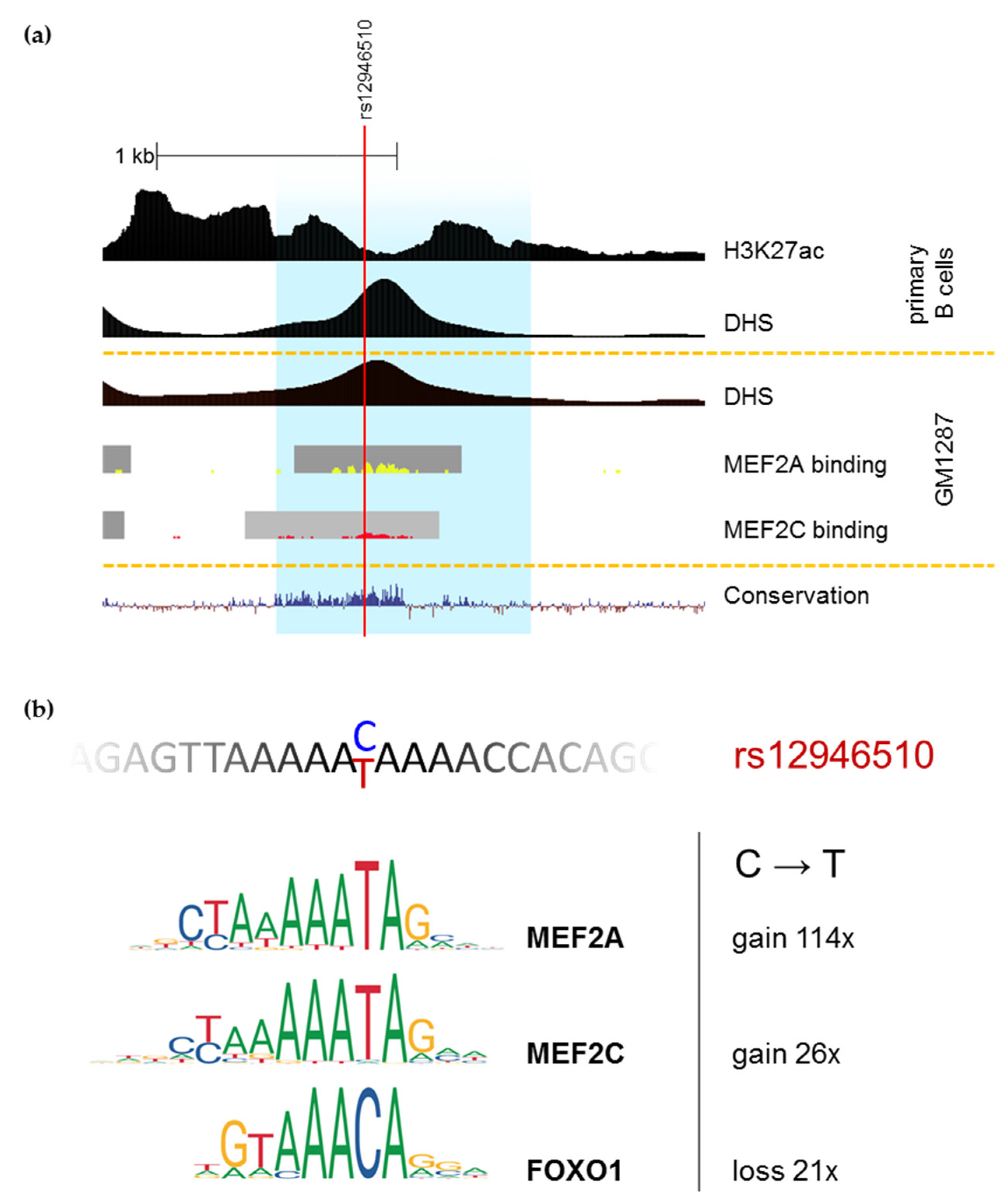{kind=link}
| SNP | Enhancer (Chr17 Hg19 Coordinates) | Length, bp | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|---|---|
| rs12946510 | 37912040- 37913027 | 1057 | TTTggatccAATGCAATTCCAGTGGGGGT | TTTgtcgacGCCCCTCAGTAGCTGGTTTT |
| rs2313430 | 37929651- 37930102 | 718 | TTTggatccGGACATCAGGCCTTTGGGAA | TTAgtcgacTGAACTGGGGAAGAGGGACA |
| rs4795397 | 38023183- 38024680 | 1705 | AAAggatccTGGTTAAGTCTCCTCTCATAGGATT | AAAgtcgacTAAAACGCGGGCATTGGACT |
| rs12709365/rs13380815 | 38026822- 38029614 | 3029 | TTTagatctAAGGACTTCAGACGAGCGTT | TTTgtcgacTACTCCAGCTCTCTTTTGAGAA |
| rs8067378 | 38050770- 38051866 | 1448 | TTTggatccCACCTGCTCCTGTCTGATGC | TTTgtcgacGAGCCCATTGCAAGCAGTCT |
| Control For | Hg19 Coordinates | Length, bp | Forward Primer (5′-3′) | Reverse Primer (5′-3′) |
|---|---|---|---|---|
| rs2313430 | chr17:38087769- 38088481 | 713 | TTTggatccATACAGTGATTGCATTTGCTTCG | TTTgtcgacTGATCATCGCCATCTTCATTTACTT |
| rs12946510 | chr10:6101364- 6102272 * | 909 | AAggatccGCTGTACCCAGTGCGTAG | TATgtcgacTACTGCAAAGTGGCTATGAAG |
| rs4795397/rs806737 | chr17:38087831- 38089396 | 1566 | AAAggatccGAGCCATGAGGTGATAATTATGGAA | AAAgtcgacATGAAAAAGATCACCCTAAATCCCT |
| rs12709365/rs13380815 | chr18:69576417- 69579429 | 3013 | ATTTggatccTGGAAGTTCAGTGAGTGTGTC | TATTgtcgacTCCTCATGCTTCCGGTTGTC |
| SNP | Forward primer (5′-3′) | Reverse primer (5′-3′) |
|---|---|---|
| rs12946510 | GAGTTAAAAATAAAACCACAGCAA | CTGTGGTTTTATTTTTAACTCTGT |
| rs2313430 | GAGATCTTTTTTTCATGTTCTTTTC | GAACATGAAAAAAAGATCTCACTCA |
| rs4795397 | GAAAAGGCCAGTCGGGCTCCATC | TGGAGCCCGACTGGCCTTTTCTG |
| rs12709365 | CCTTGGAACATAGGTATTATTAATTA | ATTAATAATACCTATGTTCCAAGGCA |
| rs13380815 | ATGACAGAATTGAGATTTTGTGGGA | CCACAAAATCTCAATTCTGTCATAT |
| rs8067378 | CGTTATAAATGGGGAAAAACGTT | TTTTTCCCCATTTATAACGTTACA |
| Cell Line | Pulse Voltage, V | Pulse Width, ms | Pulse Number |
|---|---|---|---|
| MP1 | 1300 | 30 | 1 |
| Jurkat | 1350 | 10 | 3 |
| Nalm6 | 1300 | 30 | 1 |
| MT-2 | 1400 | 30 | 1 |
| U-937 | 1400 | 30 | 1 |
| Fine-Mapping Study | Candidate SNP | Risk Allele | RAF in European Population (KGph3) | Ancestral/Alternative Allele | Index SNP | GWAS (Ref. #) | Associated Disease | Association p-Value | OR |
|---|---|---|---|---|---|---|---|---|---|
| Farh et al., 2015; Schmiedel et al., 2016 | rs12946510 (intergenic) | T | 0,47 | C/T | rs12946510 | [39] | Multiple sclerosis | 2.90 × 10−9 | 1.07 |
| Schmiedel et al., 2016 | rs2313430 (intronic) | T | 0,52 | T/C | rs2305480 | [38] | Ulcerative colitis | 3.01 × 10−8 | 1.15 |
| Schmiedel et al., 2016 | rs4795397 (intergenic) | G | 0,48 | A/G | rs2305480 | [38] | Ulcerative colitis | 3.01 × 10−8 | 1.15 |
| Farh et al., 2015 | rs12709365 (intronic) | G | 0,47 | A/G | rs2872507 | [9] | Ulcerative colitis | 5 × 10−11 | 1.15 |
| [10] | Crohn’s disease | 5 × 10−9 | 1.12 | ||||||
| [40] | Crohn’s disease | 2 × 10−9 | 1.14 | ||||||
| [41] | Type 1 diabetes autoantibodies | 2 × 10−6 | 1.10 | ||||||
| [7] | Rheumatoid arthritis | 9 × 10−7 | 1.10 | ||||||
| rs12936409 | [42] | Rheumatoid arthritis | 2.8 × 10−9 | 1.10 | |||||
| Farh et al., 2015 | rs13380815 (intronic) | G | 0,47 | A/G | See above | ||||
| Farh et al., 2015 | rs8067378 (intergenic) | G | 0,51 | G/A | rs8067378 | [38] | Ulcerative colitis | 9.74 × 10−8 | 1.12 |
| rs8067378 | [43] | Primary biliary cirrhosis | 6.05 × 10−14 | 1.26 | |||||
| Gene | Candidate SNP | Cell Type | Evidence Type * | Reference |
|---|---|---|---|---|
| STARD3 | rs13380815/ rs12709365 | primary monocytes (24 h LPS-stimulated; IFNγ-stimulated; naïve) | by LD (r2 = 1) | [20] |
| PGAP3 | rs2313430/ rs8067378 | primary monocytes (2 h LPS-stimulated) | by LD (r2 = 1) | [20] |
| rs12709365/ rs13380815 | primary monocytes (24 h LPS-stimulated) | by LD (r2 = 1) | [20] | |
| rs2313430/ rs8067378 | primary monocytes (IFNγ-stimulated) | by LD (r2 = 1) | [20] | |
| rs12709365/ rs13380815 | naïve primary monocytes | by LD (r2 = 1) | [20] | |
| IKZF3 | rs2313430 | naïve primary monocytes | by LD (r2 = 1) | [21] |
| rs2313430/ rs8067378 | whole blood (meta-analysis) | by LD (r2 = 1) | [22] | |
| rs13380815/ rs12709365 | whole blood | by LD (r2 = 0.967) | [23] | |
| rs8067378/ rs2313430 | whole blood | by LD (r2 = 0.875) | [23] | |
| rs12946510 | whole blood | direct data | [24] | |
| ZPBP2 | rs2313430/ rs8067378 | lymphoblastoid cell lines | by LD (r2 = 0.967) | [25] |
| GSDMB | all | whole blood, spleen, EBV-immortalized B cells (except rs12946510) | direct data | GTEx V7 [50] |
| rs8067378 | whole blood | direct data | [26] | |
| rs12709365/ rs13380815 | whole blood | by LD (r2 = 1) | [26] | |
| rs8067378 | EBV-immortalized B cells | direct data | [27] | |
| rs2313430/ rs8067378 | primary peripheral blood CD4+ lymphocytes | by LD (r2 = 1) | [28] | |
| rs8067378/ rs2313430 | whole blood, meta-analysis | by LD (r2 = 1) | [22] | |
| rs8067378 | lymphocytes (inferred) | direct data | [29] | |
| rs2313430/ rs8067378 | whole blood | by LD (r2 = 1) | [23] | |
| ORMDL3 | all | whole blood, spleen, EBV-immortalized B cells | direct data | GTEx V7 [50] |
| rs13380815/ rs12709365 | whole blood | by LD (r2 = 1) | [30] | |
| rs8067378 | EBV-transformed B cell lines | direct data | [30] | |
| rs13380815/ rs12709365 | primary peripheral blood CD4+ lymphocytes | by LD (r2 = 0.874) | [28] | |
| rs8067378/ rs2313430 | primary monocytes (24 h LPS-stimulated) | by LD (r2 = 0.091) | [20] | |
| rs8067378/ rs2313430 | naïve primary monocytes | by LD (r2 = 1) | [20] | |
| rs4795397 | whole blood, meta-analysis | direct data | [22] | |
| rs8067378 | lymphocytes (inferred) | direct data | [29] | |
| rs2313430/ rs8067378 | whole blood | by LD (r2 = 1) | [23] | |
| rs2313430/ rs8067378 | lymphoblastoid cell lines | by LD (r2 = 1) | [25] | |
| GSDMA | rs8067378/ rs2313430 | primary monocytes (2 h and 24 h LPS-stimulated; IFNγ-stimulated) | by LD (r2 = 1) | [20] |
| PSMD3 | rs12709365/ rs13380815 | primary monocytes (24 h LPS-stimulated) | by LD (r2 = 0.874) | [20] |
| Name | Origin | Phenotype | References |
|---|---|---|---|
| Nalm6 | Pre-B cells cultured from the blood of a patient with non-T, non-B acute lymphoblastic leukemia | B cell precursors | [56] |
| MP1 | EBV-transformed peripheral B lymphocytes | Mature IgM-producing B cells | [57] |
| Jurkat | Cells derived from the peripheral blood of a patient with acute lymphoblastic leukemia | T helpers | [58,59,60] |
| MT-2 | Human T cell leukemia virus type 1 (HTLV-1) infected leukocytes from cord blood | Regulatory T cells | [61] |
| U-937 | Monocytic cell line derived from diffuse histiocytic lymphoma | Monocytes | [62] |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Ustiugova, A.S.; Korneev, K.V.; Kuprash, D.V.; Afanasyeva, M.A. Functional SNPs in the Human Autoimmunity-Associated Locus 17q12-21. Genes 2019, 10, 77. https://doi.org/10.3390/genes10020077
Ustiugova AS, Korneev KV, Kuprash DV, Afanasyeva MA. Functional SNPs in the Human Autoimmunity-Associated Locus 17q12-21. Genes. 2019; 10(2):77. https://doi.org/10.3390/genes10020077
Chicago/Turabian StyleUstiugova, Alina S., Kirill V. Korneev, Dmitry V. Kuprash, and Marina A. Afanasyeva. 2019. "Functional SNPs in the Human Autoimmunity-Associated Locus 17q12-21" Genes 10, no. 2: 77. https://doi.org/10.3390/genes10020077
APA StyleUstiugova, A. S., Korneev, K. V., Kuprash, D. V., & Afanasyeva, M. A. (2019). Functional SNPs in the Human Autoimmunity-Associated Locus 17q12-21. Genes, 10(2), 77. https://doi.org/10.3390/genes10020077