Insights into Catalytic and tRNA Recognition Mechanism of the Dual-Specific tRNA Methyltransferase from Thermococcus kodakarensis


Abstract
1. Introduction
2. Materials and Methods
2.1. Mutagenesis and Purification of TkTrm10 Variants
2.2. In Vitro Transcription and Preparation of tRNA Radio-Labeled Substrates
2.3. Single Turnover Kinetics
2.4. pH-Rate Analysis
2.5. Metal Dependence Assays
2.6. Differential Scanning Fluorimetry
2.7. Filter Binding Assay
3. Results
3.1. pH-Rate Analysis to Assess the Mechanism of m1R9 Formation
3.2. Effect of Metals and tRNA Structural Stability on TkTrm10 Catalysis
3.3. Mutational Analysis of Conserved Residues
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Jackman, J.E.; Alfonzo, J.D. Transfer RNA modifications: Nature’s combinatorial chemistry playground. Wiley Interdiscip. Rev. RNA 2013, 4, 35–48. [Google Scholar] [CrossRef] [PubMed]
- Hori, H. Methylated nucleosides in tRNA and tRNA methyltransferases. Front. Genet. 2014, 5, 144. [Google Scholar] [CrossRef] [PubMed]
- Motorin, Y.; Helm, M. RNA nucleotide methylation. Wiley Interdiscip. Rev. RNA 2011, 2, 611–631. [Google Scholar] [CrossRef] [PubMed]
- Hou, Y.-M.; Perona, J.J. Stereochemical mechanisms of tRNA methyltransferases. FEBS Lett. 2010, 584, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Krishnamohan, A.; Jackman, J.E. A Family Divided: Distinct Structural and Mechanistic Features of the SpoU-TrmD (SPOUT) Methyltransferase Superfamily. Biochemistry 2018. [Google Scholar] [CrossRef] [PubMed]
- Jackman, J.E.; Montange, R.K.; Malik, H.S.; Phizicky, E.M. Identification of the yeast gene encoding the tRNA m1G methyltransferase responsible for modification at position 9. RNA 2003, 9, 574–585. [Google Scholar] [CrossRef] [PubMed]
- Shao, Z.; Yan, W.; Peng, J.; Zuo, X.; Zou, Y.; Li, F.; Gong, D.; Ma, R.; Wu, J.; Shi, Y.; et al. Crystal structure of tRNA m1G9 methyltransferase Trm10: insight into the catalytic mechanism and recognition of tRNA substrate. Nucleic Acids Res. 2014, 42, 509–525. [Google Scholar] [CrossRef]
- Van Laer, B.; Roovers, M.; Wauters, L.; Kasprzak, J.M.; Dyzma, M.; Deyaert, E.; Kumar Singh, R.; Feller, A.; Bujnicki, J.M.; Droogmans, L.; et al. Structural and functional insights into tRNA binding and adenosine N1-methylation by an archaeal Trm10 homologue. Nucleic Acids Res. 2016, 44, 940–953. [Google Scholar] [CrossRef]
- Singh, R.K.; Feller, A.; Roovers, M.; Elder, D.V.A.N.; Wauters, L.; Droogmans, L.; Versées, W. Structural and biochemical analysis of the dual-specificity Trm10 enzyme from Thermococcus kodakaraensis prompts reconsideration of its catalytic mechanism. RNA 2018, 24, 1080–1092. [Google Scholar] [CrossRef]
- Helm, M.; Giegé, R.; Florentz, C. A Watson-Crick base-pair-disrupting methyl group (m1A9) is sufficient for cloverleaf folding of human mitochondrial tRNALys. Biochemistry 1999, 38, 13338–13346. [Google Scholar] [CrossRef]
- Gillis, D.; Krishnamohan, A.; Yaacov, B.; Shaag, A.; Jackman, J.E.; Elpeleg, O. TRMT10A dysfunction is associated with abnormalities in glucose homeostasis, short stature and microcephaly. J. Med. Genet. 2014, 51, 581–586. [Google Scholar] [CrossRef] [PubMed]
- Zung, A.; Kori, M.; Burundukov, E.; Ben-Yosef, T.; Tatoor, Y.; Granot, E. Homozygous deletion of TRMT10A as part of a contiguous gene deletion in a syndrome of failure to thrive, delayed puberty, intellectual disability and diabetes mellitus. Am. J. Med. Genet. A 2015, 167, 3167–3173. [Google Scholar] [CrossRef] [PubMed]
- Igoillo-Esteve, M.; Genin, A.; Lambert, N.; Désir, J.; Pirson, I.; Abdulkarim, B.; Simonis, N.; Drielsma, A.; Marselli, L.; Marchetti, P.; et al. tRNA methyltransferase homolog gene TRMT10A mutation in young onset diabetes and primary microcephaly in humans. PLoS Genet. 2013, 9, e1003888. [Google Scholar] [CrossRef] [PubMed]
- Yew, T.W.; McCreight, L.; Colclough, K.; Ellard, S.; Pearson, E.R. tRNA methyltransferase homologue gene TRMT10A mutation in young adult-onset diabetes with intellectual disability, microcephaly and epilepsy. Diabet. Med. 2015. [Google Scholar] [CrossRef]
- Metodiev, M.D.; Thompson, K.; Alston, C.L.; Morris, A.A.M.; Bahi-buisson, N.; Pyle, A.; Griffin, H.; He, L.; Assouline, Z.; Siira, S.; et al. Recessive Mutations in TRMT10C Cause Defects in Mitochondrial RNA Processing and Multiple Respiratory Chain Deficiencies. Am. J. Hum. Genet. 2016, 993–1000. [Google Scholar] [CrossRef] [PubMed]
- Gustavsson, M.; Ronne, H. Evidence that tRNA modifying enzymes are important in vivo targets for 5-fluorouracil in yeast. RNA 2008, 14, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Kempenaers, M.; Roovers, M.; Oudjama, Y.; Tkaczuk, K.L.; Bujnicki, J.M.; Droogmans, L. New archaeal methyltransferases forming 1-methyladenosine or 1-methyladenosine and 1-methylguanosine at position 9 of tRNA. Nucleic Acids Res. 2010, 38, 6533–6543. [Google Scholar] [CrossRef]
- Vilardo, E.; Nachbagauer, C.; Buzet, A.; Taschner, A.; Holzmann, J.; Rossmanith, W. A subcomplex of human mitochondrial RNase P is a bifunctional methyltransferase–extensive moonlighting in mitochondrial tRNA biogenesis. Nucleic Acids Res. 2012, 40, 11583–11593. [Google Scholar] [CrossRef]
- Krishnamohan, A.; Jackman, J.E. Mechanistic features of the atypical tRNA m1G9 SPOUT methyltransferase, Trm10. Nucleic Acids Res. 2017, 45, 9019–9029. [Google Scholar] [CrossRef]
- Swinehart, W.E.; Henderson, J.C.; Jackman, J.E. Unexpected expansion of tRNA substrate recognition by the yeast m1G9 methyltransferase Trm10. RNA 2013, 19, 1137–1146. [Google Scholar] [CrossRef]
- Silvers, R.; Keller, H.; Schwalbe, H.; Hengesbach, M. Differential Scanning Fluorimetry for Monitoring RNA Stability. Chembiochem 2015, 1109–1114. [Google Scholar] [CrossRef] [PubMed]
- Sakaguchi, R.; Lahoud, G.; Christian, T.; Gamper, H.; Hou, Y.-M. A divalent metal ion-dependent N1-methyl transfer to G37-tRNA. Chem. Biol. 2014, 21, 1351–1360. [Google Scholar] [CrossRef] [PubMed]
- Sievers, F.; Wilm, A.; Dineen, D.; Gibson, T.J.; Karplus, K.; Li, W.; Lopez, R.; Mcwilliam, H.; Remmert, M.; Sö Ding, J.; et al. Fast, scalable generation of high-quality protein multiple sequence alignments using Clustal Omega. Mol. Syst. Biol. 2011, 7, 539. [Google Scholar] [CrossRef]
- Swinehart, W.E.; Jackman, J.E. Diversity in mechanism and function of tRNA methyltransferases. RNA Biol. 2015, 12, 398–411. [Google Scholar] [CrossRef] [PubMed]
- Dégut, C.; Ponchon, L.; Folly-Klan, M.; Barraud, P.; Tisné, C. The m1A58 modification in eubacterial tRNA: An overview of tRNA recognition and mechanism of catalysis by TrmI. Biophys. Chem. 2016, 210, 27–34. [Google Scholar] [CrossRef] [PubMed]
- Stewart, R.; Harris, M.G. Amino group acidity in nucleotide bases. Can. J. Chem. 1977, 55, 3807–3814. [Google Scholar] [CrossRef]
- Kettani, A.; Guéron, M.; Leroy, J.-L. Amino Proton Exchange Processes in Mononucleosides. J. Am. Chem. Soc. 1997, 119, 1108–1115. [Google Scholar] [CrossRef]
- Schubert, H.L.; Blumenthal, R.M.; Cheng, X. Many paths to methyltransfer: A chronicle of convergence. Trends Biochem. Sci. 2003, 28, 329–335. [Google Scholar] [CrossRef]
- Rydberg, B.; Lindahl, T. Nonenzymatic methylation of DNA by the intracellular methyl group donor S-adenosyl-L-methionine is a potentially mutagenic reaction. EMBO J. 1982, 1, 211–216. [Google Scholar] [CrossRef]
- Barraud, P.; Golinelli-Pimpaneau, B.; Atmanene, C.; Sanglier, S.; Van Dorsselaer, A.; Droogmans, L.; Dardel, F.; Tisné, C. Crystal Structure of Thermus thermophilus tRNA m1A58 Methyltransferase and Biophysical Characterization of Its Interaction with tRNA. J. Mol. Biol. 2008. [Google Scholar] [CrossRef]
- Oerum, S.; Dégut, C.; Barraud, P.; Tisné, C. m1A Post-Transcriptional Modification in tRNAs. Biomolecules 2017, 7, 20. [Google Scholar] [CrossRef] [PubMed]
- Tkaczuk, K.L.; Dunin-Horkawicz, S.; Purta, E.; Bujnicki, J.M. Structural and evolutionary bioinformatics of the SPOUT superfamily of methyltransferases. BMC Bioinform. 2007, 8, 73. [Google Scholar] [CrossRef] [PubMed]
- Christian, T.; Lahoud, G.; Liu, C.; Hoffmann, K.; Perona, J.J.; Hou, Y.-M. Mechanism of N-methylation by the tRNA m1G37 methyltransferase Trm5. RNA 2010, 16, 2484–2492. [Google Scholar] [CrossRef] [PubMed]
- Christian, T.; Gamper, H.; Hou, Y. Conservation of structure and mechanism by Trm5 enzymes. RNA 2013, 1192–1199. [Google Scholar] [CrossRef] [PubMed]
- Goto-Ito, S.; Ito, T.; Kuratani, M.; Bessho, Y.; Yokoyama, S. Tertiary structure checkpoint at anticodon loop modification in tRNA functional maturation. Nat. Struct. Mol. Biol. 2009, 16, 1109–1115. [Google Scholar] [CrossRef] [PubMed]






{kind=link}
{kind=link}
{kind=link}
{kind=link}
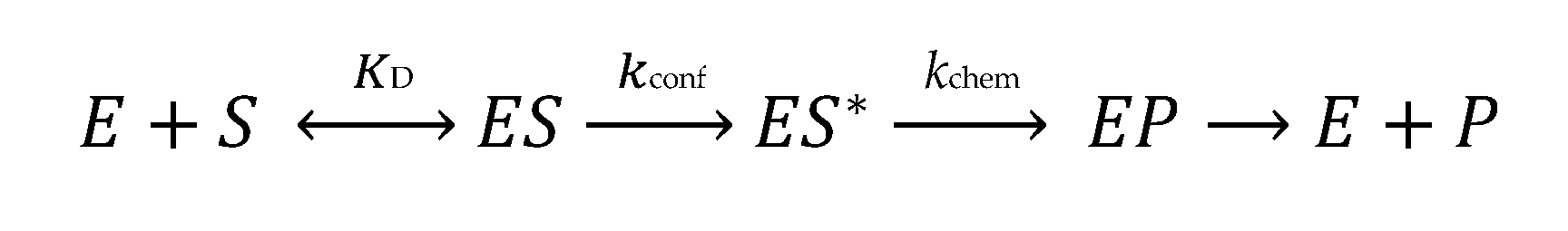{kind=link}
| Substrate | kobs min−1 (pH 8.0) | Assay Temperature |
|---|---|---|
| m1G9 | ||
| SctRNAPhe-G9 | 1.39 ± 0.13 | 40 °C |
| SctRNAGly | 0.15 ± 0.04 | 40 °C |
| TktRNAArg | ≤0.012 a | 50 °C |
| m1A9 | ||
| SctRNAPhe | 0.56 ± 0.11 | 40 °C |
| TktRNAThr | 0.12 ± 0.02 | 50 °C |
| TkTrm10 Variant | m1A9 kobs (min−1) | Theoretical kobs (min−1) c | m1A9 FD a | m1G9 kobs (min−1) | Theoretical kobs (min−1)c | m1G9 FD a |
|---|---|---|---|---|---|---|
| WT | 0.56 ± 0.11 | - | - | 1.4 ± 0.13 | - | - |
| D104N | 0.49 ± 0.08 | - | 1.2 | 1.74 ± 0.20 | - | 0.8 |
| E115Q | 0.38 ± 0.06 | - | 1.5 | 0.42 ± 0.07 | - | 3.3 |
| D206N | 0.17 ± 0.04 | - | 3.4 | 0.57 ± 0.10 | - | 2.4 |
| D245N | 0.40 ± 0.06 | - | 1.4 | 0.34 ± 0.01 | - | 4.0 |
| D206N+D245N | 0.14 ± 0.03 | 0.12 | 4.0 | 0.23 ± 0.01 | 0.14 | 6.0 |
| D104N+D206N+D245N (3D) | 0.09 ± 0.01 | 0.10 | 6.0 | 0.32 ± 0.06 | 0.17 | 4.4 |
| D104N+D206N+D245N+E115Q (3D+E) | 0.013 ± 0.007 | 0.07 | 56 | 0.13 ± 0.01 | 0.05 | 11 |
| D104A | 0.035 ± 0.007 | - | 16 | 0.071 ± 0.003 | - | 20 |
| D245A | 0.021 ± 0.008 | - | 26 | 0.021 ± 0.008 | - | 68 |
| D104A+E115Q+D245A (DED) | <0.0006 b | 0.0009 | 1555 | <0.0006 b | 0.0004 | 3487 |
| Q122A | 0.14 ± 0.02 | - | 4.0 | 0.03 ± 0.01 | - | 42 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Krishnamohan, A.; Dodbele, S.; Jackman, J.E. Insights into Catalytic and tRNA Recognition Mechanism of the Dual-Specific tRNA Methyltransferase from Thermococcus kodakarensis. Genes 2019, 10, 100. https://doi.org/10.3390/genes10020100
Krishnamohan A, Dodbele S, Jackman JE. Insights into Catalytic and tRNA Recognition Mechanism of the Dual-Specific tRNA Methyltransferase from Thermococcus kodakarensis. Genes. 2019; 10(2):100. https://doi.org/10.3390/genes10020100
Chicago/Turabian StyleKrishnamohan, Aiswarya, Samantha Dodbele, and Jane E. Jackman. 2019. "Insights into Catalytic and tRNA Recognition Mechanism of the Dual-Specific tRNA Methyltransferase from Thermococcus kodakarensis" Genes 10, no. 2: 100. https://doi.org/10.3390/genes10020100
APA StyleKrishnamohan, A., Dodbele, S., & Jackman, J. E. (2019). Insights into Catalytic and tRNA Recognition Mechanism of the Dual-Specific tRNA Methyltransferase from Thermococcus kodakarensis. Genes, 10(2), 100. https://doi.org/10.3390/genes10020100