Phylostratigraphic Analysis Shows the Earliest Origination of the Abiotic Stress Associated Genes in A. thaliana



 and
and
Abstract
1. Introduction
2. Materials and Methods
2.1. Gene Sets Preparation
2.2. Network Reconstruction for Gene Sets
2.3. PAI/DI Calculation and Network Visualization
2.4. Significance of the Differences between PAI/DI Distributions in A. thaliana and Stress Gene Sets
2.5. Gene Ontology Annotation Enrichment for Stress Gene Sets
2.6. Relationship between Gene Network Structural and Evolutionary Characteristics
2.7. Evolutionary Changes of Gene Functions in Stress-Related Gene Networks
3. Results
3.1. GO Terms and Genes Associated with Abiotic Stress
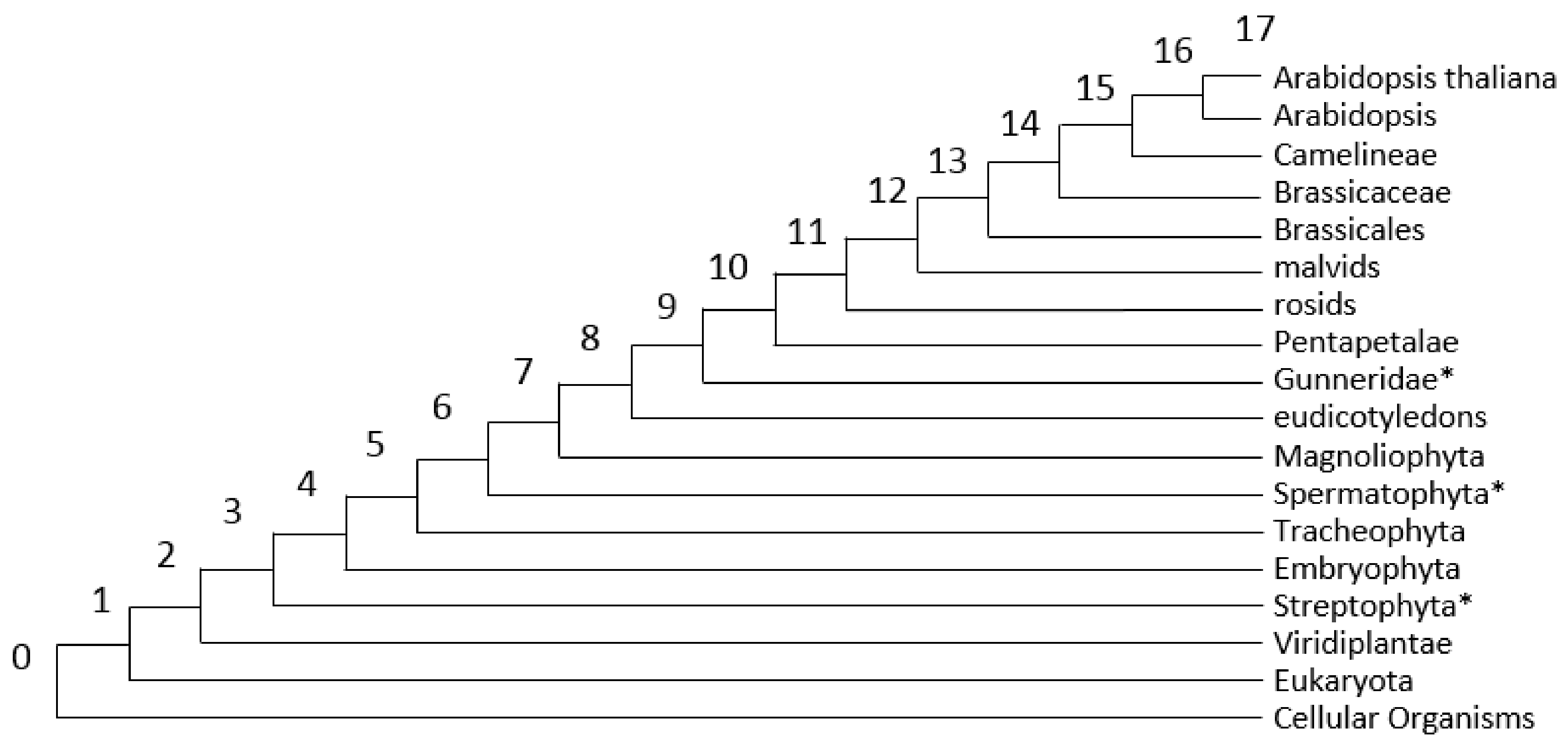
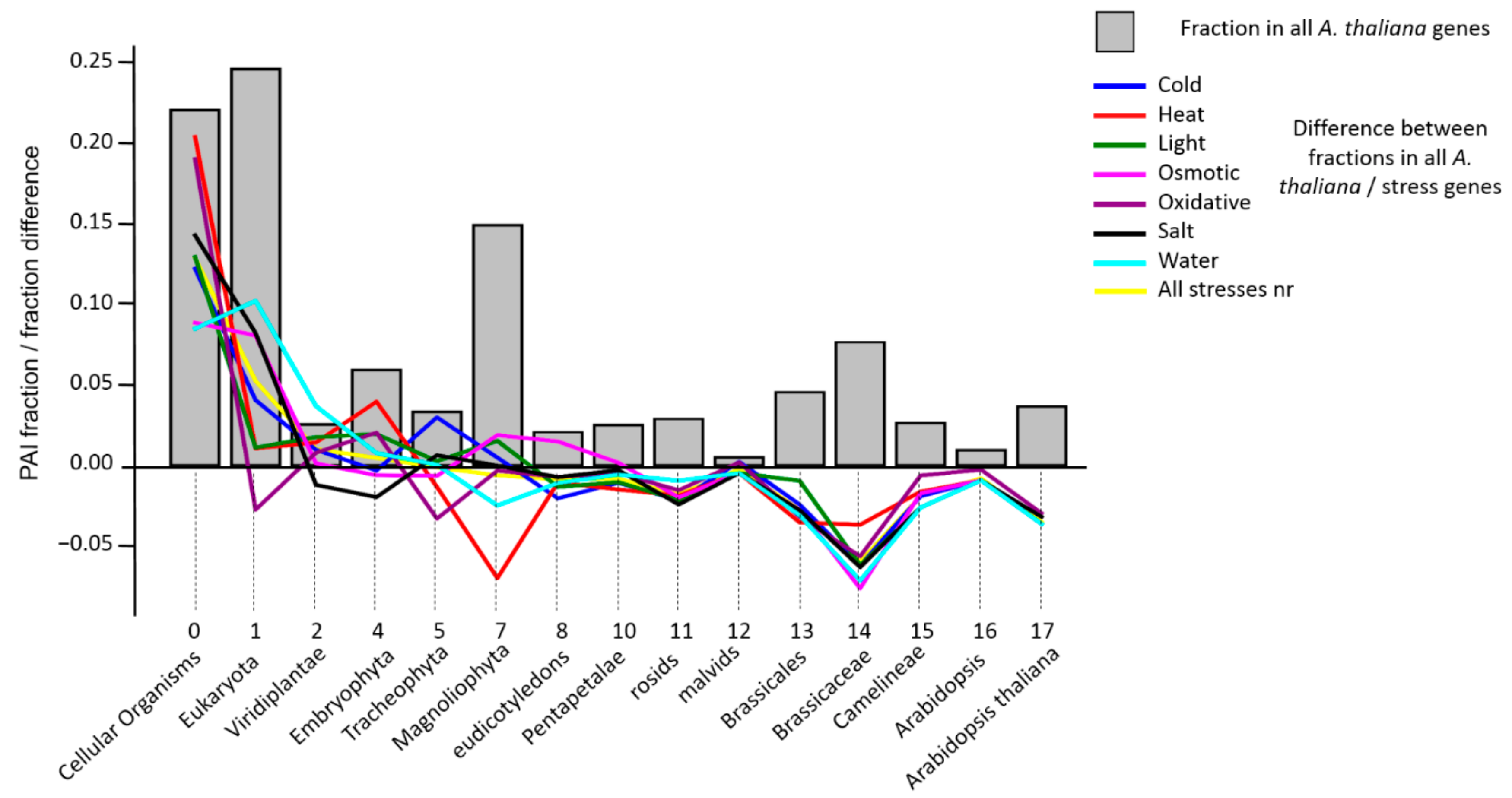
3.2. Analysis of PAI Distribution
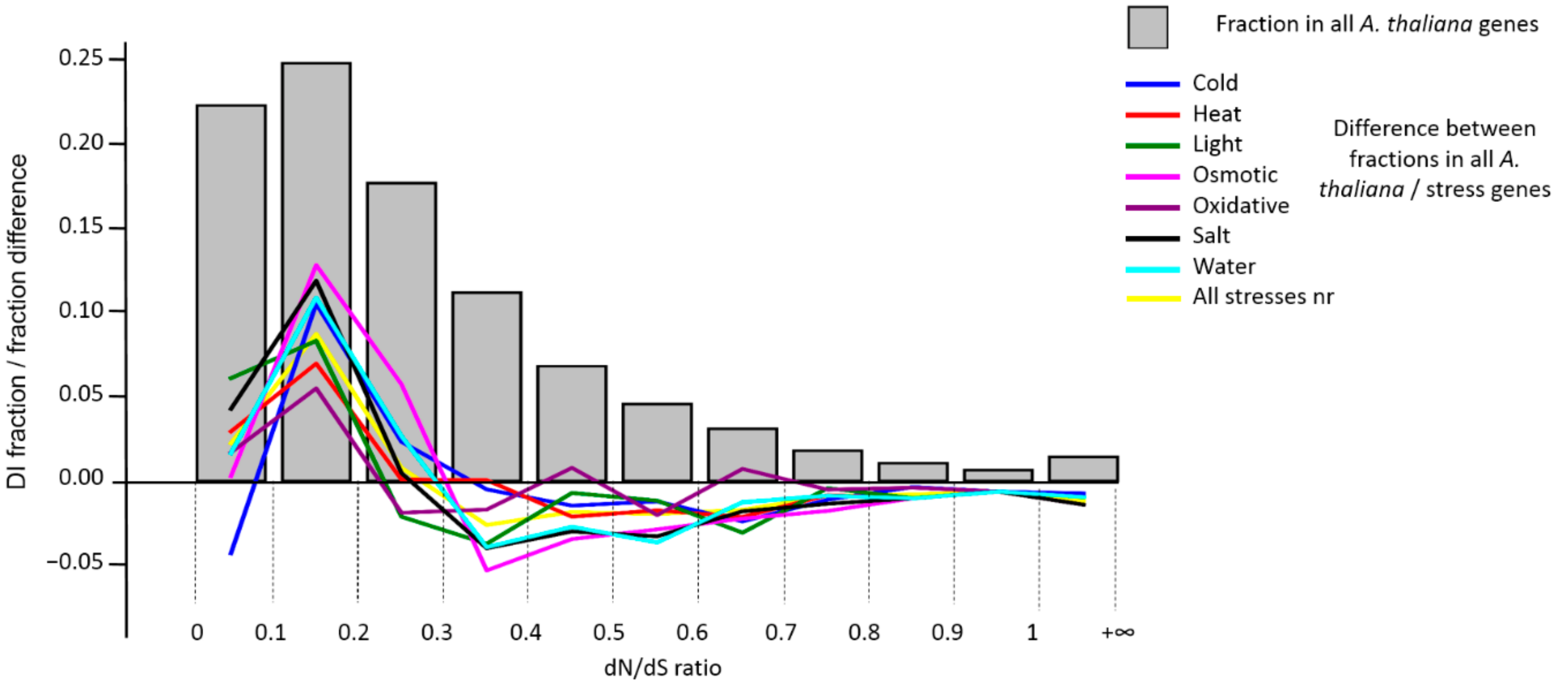
3.3. Analysis of DI Distribution
3.4. Network Clusters and Their Association with GO Terms
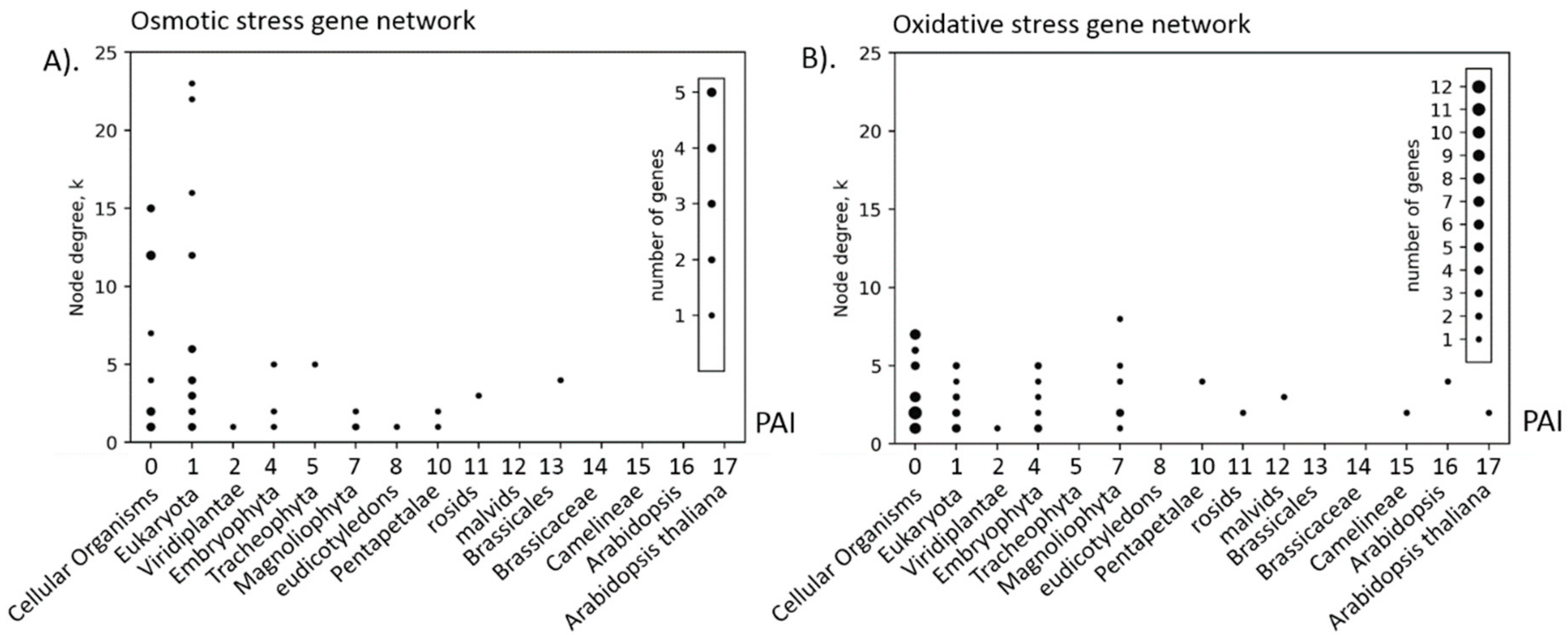
3.5. Relationship between Structural and Evolutionary Characteristics of Gene Networks
3.6. Evolution of Gene Network Function
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Matsukura, S.; Mizoi, J.; Yoshida, T.; Todaka, D.; Ito, Y.; Maruyama, K.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Comprehensive analysis of rice DREB2-type genes that encode transcription factors involved in the expression of abiotic stress-responsive genes. Mol. Genet. Genom. 2010, 283, 185–196. [Google Scholar] [CrossRef] [PubMed]
- Kumar, T.; Uzma; Khan, M.R.; Abbas, Z.; Ali, G.M. Genetic Improvement of Sugarcane for Drought and Salinity Stress Tolerance Using Arabidopsis Vacuolar Pyrophosphatase (AVP1) Gene. Mol. Biotechnol. 2014, 56, 199–209. [Google Scholar] [CrossRef] [PubMed]
- Cramer, G.R.; Urano, K.; Delrot, S.; Pezzotti, M.; Shinozaki, K. Effects of abiotic stress on plants: A systems biology perspective. BMC Plant Biol. 2011, 11, 163. [Google Scholar] [CrossRef] [PubMed]
- Haak, D.C.; Fukao, T.; Grene, R.; Hua, Z.; Ivanov, R.; Perrella, G.; Li, S. Multilevel Regulation of Abiotic Stress Responses in Plants. Front. Plant Sci. 2017, 8. [Google Scholar] [CrossRef]
- Kranner, I.; Minibayeva, F.V.; Beckett, R.P.; Seal, C.E. What is stress? Concepts, definitions and applications in seed science. New Phytol. 2010, 188, 655–673. [Google Scholar] [CrossRef]
- Kotak, S.; Larkindale, J.; Lee, U.; von Koskull-Döring, P.; Vierling, E.; Scharf, K.-D. Complexity of the heat stress response in plants. Curr. Opin. Plant Biol. 2007, 10, 310–316. [Google Scholar] [CrossRef]
- Ohama, N.; Sato, H.; Shinozaki, K.; Yamaguchi-Shinozaki, K. Transcriptional Regulatory Network of Plant Heat Stress Response. Trends Plant Sci. 2017, 22, 53–65. [Google Scholar] [CrossRef]
- Müller, M.; Munné-Bosch, S. Ethylene Response Factors: A Key Regulatory Hub in Hormone and Stress Signaling. Plant Physiol. 2015, 169, 32–41. [Google Scholar] [CrossRef]
- Ye, Y.; Ding, Y.; Jiang, Q.; Wang, F.; Sun, J.; Zhu, C. The role of receptor-like protein kinases (RLKs) in abiotic stress response in plants. Plant Cell Rep. 2017, 36, 235–242. [Google Scholar] [CrossRef]
- Shao, H.; Chu, L.; Zhao, C.; Guo, Q.; Liu, X.; Ribaut, J.-M. Plant gene regulatory network system under abiotic stress. Acta Biol. Szeged. 2006, 50, 1–9. [Google Scholar]
- Zhu, J.-K. Abiotic Stress Signaling and Responses in Plants. Cell 2016, 167, 313–324. [Google Scholar] [CrossRef] [PubMed]
- Hirayama, T.; Shinozaki, K. Research on plant abiotic stress responses in the post-genome era: Past, present and future. Plant J. 2010, 61, 1041–1052. [Google Scholar] [CrossRef] [PubMed]
- Dietz, K.-J.; Jacquot, J.-P.; Harris, G. Hubs and bottlenecks in plant molecular signalling networks. New Phytol. 2010, 188, 919–938. [Google Scholar] [CrossRef] [PubMed]
- Vandereyken, K.; Van Leene, J.; De Coninck, B.; Cammue, B.P.A. Hub Protein Controversy: Taking a Closer Look at Plant Stress Response Hubs. Front. Plant Sci. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Jha, U.C.; Bohra, A.; Singh, N.P. Heat stress in crop plants: Its nature, impacts and integrated breeding strategies to improve heat tolerance. Plant Breed. 2014, 133, 679–701. [Google Scholar] [CrossRef]
- Guo, Y.-L. Gene family evolution in green plants with emphasis on the origination and evolution of Arabidopsis thaliana genes. Plant J. 2013, 73, 941–951. [Google Scholar] [CrossRef]
- Choudhary, M.K.; Basu, D.; Datta, A.; Chakraborty, N.; Chakraborty, S. Dehydration-responsive Nuclear Proteome of Rice (Oryza sativa L.) Illustrates Protein Network, Novel Regulators of Cellular Adaptation, and Evolutionary Perspective. Mol. Cell. Proteom. 2009, 8, 1579–1598. [Google Scholar] [CrossRef]
- Prasch, C.M.; Sonnewald, U. Signaling events in plants: Stress factors in combination change the picture. Environ. Exp. Bot. 2015, 114, 4–14. [Google Scholar] [CrossRef]
- Doroshkov, A.V.; Bobrovskikh, A.V. Using the methods of systems biology for predicting perspective target genes to select c3 and c4 cereals for oxidative stress resistance. Vavilov J. Genet. Breed. 2018, 22, 122–131. [Google Scholar] [CrossRef]
- Domazet-Loso, T.; Tautz, D. An Ancient Evolutionary Origin of Genes Associated with Human Genetic Diseases. Mol. Biol. Evol. 2008, 25, 2699–2707. [Google Scholar] [CrossRef]
- Domazet-Lošo, T.; Brajković, J.; Tautz, D. A phylostratigraphy approach to uncover the genomic history of major adaptations in metazoan lineages. Trends Genet. 2007, 23, 533–539. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Lošo, T.; Tautz, D. A phylogenetically based transcriptome age index mirrors ontogenetic divergence patterns. Nature 2010, 468, 815–818. [Google Scholar] [CrossRef] [PubMed]
- Quint, M.; Drost, H.-G.; Gabel, A.; Ullrich, K.K.; Bönn, M.; Grosse, I. A transcriptomic hourglass in plant embryogenesis. Nature 2012, 490, 98–101. [Google Scholar] [CrossRef] [PubMed]
- Durrant, M.; Boyer, J.; Zhou, W.; Baldwin, I.T.; Xu, S. Evidence of an evolutionary hourglass pattern in herbivory-induced transcriptomic responses. New Phytol. 2017, 215, 1264–1273. [Google Scholar] [CrossRef]
- Zhang, L.; Tan, Y.; Fan, S.; Zhang, X.; Zhang, Z. Phylostratigraphic analysis of gene co-expression network reveals the evolution of functional modules for ovarian cancer. Sci. Rep. 2019, 9, 2623. [Google Scholar] [CrossRef]
- Ruprecht, C.; Proost, S.; Hernandez-Coronado, M.; Ortiz-Ramirez, C.; Lang, D.; Rensing, S.A.; Becker, J.D.; Vandepoele, K.; Mutwil, M. Phylogenomic analysis of gene co-expression networks reveals the evolution of functional modules. Plant J. 2017, 90, 447–465. [Google Scholar] [CrossRef]
- Mustafin, Z.S.; Lashin, S.A.; Matushkin, Y.G.; Gunbin, K.V.; Afonnikov, D.A. Orthoscape: A cytoscape application for grouping and visualization KEGG based gene networks by taxonomy and homology principles. BMC Bioinform. 2017, 18, 1–9. [Google Scholar] [CrossRef][Green Version]
- Gene Ontology Consortium. Gene Ontology Consortium: Going forward. Nucleic Acids Res. 2015, 43, D1049–D1056. [Google Scholar] [CrossRef]
- Lamesch, P.; Berardini, T.Z.; Li, D.; Swarbreck, D.; Wilks, C.; Sasidharan, R.; Muller, R.; Dreher, K.; Alexander, D.L.; Garcia-Hernandez, M.; et al. The Arabidopsis Information Resource (TAIR): Improved gene annotation and new tools. Nucleic Acids Res. 2012, 40, D1202–D1210. [Google Scholar] [CrossRef]
- Ochiai, A. Zoogeographic studies on the soleoid fishes found in Japan and its neighbouring regions. Bull. Jpn. Soc. Sci. Fish. 1957, 22, 526–530. [Google Scholar] [CrossRef]
- Szklarczyk, D.; Franceschini, A.; Wyder, S.; Forslund, K.; Heller, D.; Huerta-Cepas, J.; Simonovic, M.; Roth, A.; Santos, A.; Tsafou, K.P.; et al. STRING v10: Protein–protein interaction networks, integrated over the tree of life. Nucleic Acids Res. 2015, 43, D447–D452. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software Environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef]
- Kanehisa, M.; Furumichi, M.; Tanabe, M.; Sato, Y.; Morishima, K. KEGG: New perspectives on genomes, pathways, diseases and drugs. Nucleic Acids Res. 2017, 45, D353–D361. [Google Scholar] [CrossRef] [PubMed]
- Yang, Z. PAML 4: Phylogenetic analysis by maximum likelihood. Mol. Biol. Evol. 2007, 24, 1586–1591. [Google Scholar] [CrossRef] [PubMed]
- Cui, X.; Lv, Y.; Chen, M.; Nikoloski, Z.; Twell, D.; Zhang, D. Young genes out of the male: An insight from evolutionary age analysis of the pollen transcriptome. Mol. Plant. 2015, 8, 935–945. [Google Scholar] [CrossRef] [PubMed]
- Cheng, X.; Hui, J.H.L.; Lee, Y.Y.; Wan Law, P.T.; Kwan, H.S. A “developmental hourglass” in fungi. Mol. Biol. Evol. 2015, 32, 1556–1566. [Google Scholar] [CrossRef]
- Lei, L.; Steffen, J.G.; Osborne, E.J.; Toomajian, C. Plant organ evolution revealed by phylotranscriptomics in Arabidopsis thaliana. Sci. Rep. 2017, 7, 7567. [Google Scholar] [CrossRef]
- Drost, H.G.; Gabel, A.; Grosse, I.; Quint, M. Evidence for active maintenance of phylotranscriptomic hourglass patterns in animal and plant embryogenesis. Mol. Biol. Evol. 2015, 32, 1221–1231. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44–57. [Google Scholar] [CrossRef]
- Ferreira, J.A.; Zwinderman, A.H. On the Benjamini–Hochberg method. Ann. Stat. 2006, 34, 1827–1849. [Google Scholar] [CrossRef]
- Schaeffer, S.E. Graph clustering. Comput. Sci. Rev. 2007, 1, 27–64. [Google Scholar] [CrossRef]
- Xu, J.; Li, Y. Discovering disease-genes by topological features in human protein–protein interaction network. Bioinformatics 2006, 22, 2800–2805. [Google Scholar] [CrossRef] [PubMed]
- Hagberg, A.A.; Schult, D.A.; Swart, P.J. Exploring network structure, dynamics, and function using networkx. In Proceedings of the 7th Python in Science Conference (SciPy2008), Pasadena, CA, USA, 19–24 August 2008; Varoquaux, G., Vaught, T., Millman, J., Eds.; 2008; pp. 11–15. [Google Scholar]
- Newman, M.E.J. Mixing patterns in networks. Phys. Rev. E 2003, 67, 026126. [Google Scholar] [CrossRef] [PubMed]
- van Zanten, M.; Snoek, L.B.; Proveniers, M.C.G.; Peeters, A.J.M. The many functions of ERECTA. Trends Plant Sci. 2009, 14, 214–218. [Google Scholar] [CrossRef] [PubMed]
- Shen, H.; Zhong, X.; Zhao, F.; Wang, Y.; Yan, B.; Li, Q.; Chen, G.; Mao, B.; Wang, J.; Li, Y.; et al. Overexpression of receptor-like kinase ERECTA improves thermotolerance in rice and tomato. Nat. Biotechnol. 2015, 33, 996–1003. [Google Scholar] [CrossRef] [PubMed]
- Kurepa, J.; Walker, J.M.; Smalle, J.; Gosink, M.M.; Davis, S.J.; Durham, T.L.; Sung, D.-Y.; Vierstra, R.D. The small ubiquitin-like modifier (SUMO) protein modification system in Arabidopsis. Accumulation of SUMO1 and -2 conjugates is increased by stress. J. Biol. Chem. 2003, 278, 6862–6872. [Google Scholar] [CrossRef]
- Datta, M.; Kaushik, S.; Jyoti, A.; Mathur, N.; Kothari, S.L.; Jain, A. SIZ1-mediated SUMOylation during phosphate homeostasis in plants: Looking beyond the tip of the iceberg. Semin. Cell Dev. Biol. 2018, 74, 123–132. [Google Scholar] [CrossRef]
- Bilichak, A.; Yao, Y.; Titov, V.; Golubov, A.; Kovalchuk, I. Genome stability in the uvh6 mutant of Arabidopsis thaliana. Plant Cell Rep. 2014, 33, 979–991. [Google Scholar] [CrossRef]
- Liu, Z.; Hall, J.D.; Mount, D.W. Arabidopsis UVH3 gene is a homolog of the Saccharomyces cerevisiae RAD2 and human XPG DNA repair genes. Plant J. 2001, 26, 329–338. [Google Scholar] [CrossRef]
- Virdi, K.S.; Wamboldt, Y.; Kundariya, H.; Laurie, J.D.; Keren, I.; Kumar, K.R.S.; Block, A.; Basset, G.; Luebker, S.; Elowsky, C.; et al. MSH1 Is a Plant Organellar DNA Binding and Thylakoid Protein under Precise Spatial Regulation to Alter Development. Mol. Plant 2016, 9, 245–260. [Google Scholar] [CrossRef]
- Shedge, V.; Davila, J.; Arrieta-Montiel, M.P.; Mohammed, S.; Mackenzie, S.A. Extensive Rearrangement of the Arabidopsis Mitochondrial Genome Elicits Cellular Conditions for Thermotolerance. Plant Physiol. 2010, 152, 1960–1970. [Google Scholar] [CrossRef] [PubMed]
- Zhao, S.; Liu, L. IUCr Expression and crystallographic studies of the Arabidopsis thaliana GDP- D -mannose pyrophosphorylase VTC1. Acta Crystallogr. Sect. F Struct. Biol. Commun. 2016, 72, 795–798. [Google Scholar] [CrossRef] [PubMed]
- Laing, W.A.; Martínez-Sánchez, M.; Wright, M.A.; Bulley, S.M.; Brewster, D.; Dare, A.P.; Rassam, M.; Wang, D.; Storey, R.; Macknight, R.C.; et al. An Upstream Open Reading Frame Is Essential for Feedback Regulation of Ascorbate Biosynthesis in Arabidopsis. Plant Cell 2015, 27, 772–786. [Google Scholar] [CrossRef] [PubMed]
- Gong, Z.; Dong, C.; Lee, H.; Zhu, J.; Xiong, L.; Gong, D.; Stevenson, B.; Zhu, J.-K. A DEAD Box RNA Helicase Is Essential for mRNA Export and Important for Development and Stress Responses in Arabidopsis. Plant Cell 2005, 17, 256–267. [Google Scholar] [CrossRef] [PubMed]
- Wu, S.-J.; Wang, L.-C.; Yeh, C.-H.; Lu, C.-A.; Wu, S.-J. Isolation and characterization of the Arabidopsis heat-intolerant 2 (hit2) mutant reveal the essential role of the nuclear export receptor EXPORTIN1A (XPO1A) in plant heat tolerance. New Phytol. 2010, 186, 833–842. [Google Scholar] [CrossRef] [PubMed]
- Cho, S.H.; von Schwartzenberg, K.; Quatrano, R. The Role of Abscisic Acid in Stress Tolerance. In Annual Plant Reviews Online; John Wiley & Sons, Ltd.: Chichester, UK, 2018; pp. 282–297. [Google Scholar]
- Basu, S.; Rabara, R. Abscisic acid—An enigma in the abiotic stress tolerance of crop plants. Plant Gene 2017, 11, 90–98. [Google Scholar] [CrossRef]
- Kazan, K. Diverse roles of jasmonates and ethylene in abiotic stress tolerance. Trends Plant Sci. 2015, 20, 219–229. [Google Scholar] [CrossRef]
- Kulik, A.; Wawer, I.; Krzywińska, E.; Bucholc, M.; Dobrowolska, G. SnRK2 Protein Kinases—Key Regulators of Plant Response to Abiotic Stresses. OMICS J. Integr. Biol. 2011, 15, 859–872. [Google Scholar] [CrossRef]
- Kim, M.-H.; Sasaki, K.; Imai, R. Cold shock domain protein 3 regulates freezing tolerance in Arabidopsis thaliana. J. Biol. Chem. 2009, 284, 23454–23460. [Google Scholar] [CrossRef]
- Chi, W.-T.; Fung, R.W.M.; Liu, H.-C.; Hsu, C.-C.; Charng, Y.-Y. Temperature-induced lipocalin is required for basal and acquired thermotolerance in Arabidopsis. Plant. Cell Environ. 2009, 32, 917–927. [Google Scholar] [CrossRef]
- Frenette Charron, J.-B.; Ouellet, F.; Pelletier, M.; Danyluk, J.; Chauve, C.; Sarhan, F. Identification, Expression, and Evolutionary Analyses of Plant Lipocalins. Plant Physiol. 2005, 139, 2017–2028. [Google Scholar] [CrossRef] [PubMed]
- Uemura, M.; Tominaga, Y.; Nakagawara, C.; Shigematsu, S.; Minami, A.; Kawamura, Y. Responses of the plasma membrane to low temperatures. Physiol. Plant. 2006, 126, 81–89. [Google Scholar] [CrossRef]
- Abo-Ogiala, A.; Carsjens, C.; Diekmann, H.; Fayyaz, P.; Herrfurth, C.; Feussner, I.; Polle, A. Temperature-induced lipocalin (TIL) is translocated under salt stress and protects chloroplasts from ion toxicity. J. Plant Physiol. 2014, 171, 250–259. [Google Scholar] [CrossRef] [PubMed]
- Rymarquis, L.A.; Souret, F.F.; Green, P.J. Evidence that XRN4, an Arabidopsis homolog of exoribonuclease XRN1, preferentially impacts transcripts with certain sequences or in particular functional categories. RNA 2011, 17, 501–511. [Google Scholar] [CrossRef] [PubMed]
- Potuschak, T.; Vansiri, A.; Binder, B.M.; Lechner, E.; Vierstra, R.D.; Genschik, P. The Exoribonuclease XRN4 Is a Component of the Ethylene Response Pathway in Arabidopsis. Plant Cell 2006, 18, 3047–3057. [Google Scholar] [CrossRef]
- Nguyen, A.H.; Matsui, A.; Tanaka, M.; Mizunashi, K.; Nakaminami, K.; Hayashi, M.; Iida, K.; Toyoda, T.; Nguyen, D.; Van Seki, M. Loss of Arabidopsis 5′–3′ Exoribonuclease AtXRN4 Function Enhances Heat Stress Tolerance of Plants Subjected to Severe Heat Stress. Plant Cell Physiol. 2015, 56, 1762–1772. [Google Scholar] [CrossRef]
- Merret, R.; Descombin, J.; Juan, Y.; Favory, J.-J.; Carpentier, M.-C.; Chaparro, C.; Charng, Y.; Deragon, J.-M.; Bousquet-Antonelli, C. XRN4 and LARP1 Are Required for a Heat-Triggered mRNA Decay Pathway Involved in Plant Acclimation and Survival during Thermal Stress. Cell Rep. 2013, 5, 1279–1293. [Google Scholar] [CrossRef] [PubMed]
- Etchells, J.P.; Provost, C.M.; Turner, S.R. Plant Vascular Cell Division Is Maintained by an Interaction between PXY and Ethylene Signalling. PLoS Genet. 2012, 8, e1002997. [Google Scholar] [CrossRef]
- Bapteste, E.; Huneman, P. Towards a Dynamic Interaction Network of Life to unify and expand the evolutionary theory. BMC Biol. 2018, 16, 56. [Google Scholar] [CrossRef]
- Ahuja, I.; de Vos, R.C.H.; Bones, A.M.; Hall, R.D. Plant molecular stress responses face climate change. Trends Plant Sci. 2010, 15, 664–674. [Google Scholar] [CrossRef]
- Hansen, B.O.; Vaid, N.; Musialak-Lange, M.; Janowski, M.; Mutwil, M. Elucidating gene function and function evolution through comparison of co-expression networks of plants. Front. Plant Sci. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Domazet-Lošo, T.; Tautz, D. Phylostratigraphic tracking of cancer genes suggests a link to the emergence of multicellularity in metazoa. BMC Biol. 2010, 8, 66. [Google Scholar] [CrossRef] [PubMed]
- Wolf, Y.I.; Novichkov, P.S.; Karev, G.P.; Koonin, E.V.; Lipman, D.J. The universal distribution of evolutionary rates of genes and distinct characteristics of eukaryotic genes of different apparent ages. Proc. Natl. Acad. Sci. USA 2009, 106, 7273–7280. [Google Scholar] [CrossRef] [PubMed]
- Albà, M.M.; Castresana, J. Inverse Relationship Between Evolutionary Rate and Age of Mammalian Genes. Mol. Biol. Evol. 2005, 22, 598–606. [Google Scholar] [CrossRef] [PubMed]
- Krylov, D.M. Gene Loss, Protein Sequence Divergence, Gene Dispensability, Expression Level, and Interactivity Are Correlated in Eukaryotic Evolution. Genome Res. 2003, 13, 2229–2235. [Google Scholar] [CrossRef] [PubMed]
- Dickerson, J.E.; Zhu, A.; Robertson, D.L.; Hentges, K.E. Defining the Role of Essential Genes in Human Disease. PLoS ONE 2011, 6, e27368. [Google Scholar] [CrossRef]
- Chae, L.; Lee, I.; Shin, J.; Rhee, S.Y. Towards understanding how molecular networks evolve in plants. Curr. Opin. Plant Biol. 2012, 15, 177–184. [Google Scholar] [CrossRef]
- Vitkup, D.; Kharchenko, P.; Wagner, A. Influence of metabolic network structure and function on enzyme evolution. Genome Biol. 2006, 7, R39. [Google Scholar] [CrossRef]
- Alvarez-Ponce, D.; Fares, M.A. Evolutionary rate and duplicability in the Arabidopsis thaliana protein–protein interaction network. Genome Biol. Evol. 2012, 4, 1263–1274. [Google Scholar] [CrossRef]
- Masalia, R.R.; Bewick, A.J.; Burke, J.M. Connectivity in gene coexpression networks negatively correlates with rates of molecular evolution in flowering plants. PLoS ONE 2017, 12, e0182289. [Google Scholar] [CrossRef]
- Alvarez-Ponce, D.; Feyertag, F.; Chakraborty, S. Position matters: Network centrality considerably impacts rates of protein evolution in the human protein–protein interaction network. Genome Biol. Evol. 2017, 9, 1742–1756. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Crawford, R.M.M. Biochemical and ecological similarities in marsh plants and diving animals. Naturwissenschaften 1978, 65, 194–201. [Google Scholar] [CrossRef]
- Nurnberger, T.; Brunner, F.; Kemmerling, B.; Piater, L. Innate immunity in plants and animals: Striking similarities and obvious differences. Immunol. Rev. 2004, 198, 249–266. [Google Scholar] [CrossRef] [PubMed]
- Apel, K.; Hirt, H. REACTIVE OXYGEN SPECIES: Metabolism, Oxidative Stress, and Signal Transduction. Annu. Rev. Plant Biol. 2004, 55, 373–399. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Bhatla, S.C.; Demidchik, V. Plants and human beings engage similar molecular crosstalk with nitric oxide under stress conditions. Funct. Plant Biol. 2019, 46, 695–701. [Google Scholar] [CrossRef]
- Giri, J.; Dansana, P.K.; Kothari, K.S.; Sharma, G.; Vij, S.; Tyagi, A.K. SAPs as novel regulators of abiotic stress response in plants. BioEssays 2013, 35, 639–648. [Google Scholar] [CrossRef]
- Soltis, P.S.; Soltis, D.E. Ancient WGD events as drivers of key innovations in angiosperms. Curr. Opin. Plant Biol. 2016, 30, 159–165. [Google Scholar] [CrossRef]
- Blanc, G.; Hokamp, K.; Wolfe, K.H. A recent polyploidy superimposed on older large-scale duplications in the Arabidopsis genome. Genome Res. 2003, 13, 137–144. [Google Scholar] [CrossRef]
- Bowers, J.E.; Chapman, B.A.; Rong, J.; Paterson, A.H. Unravelling angiosperm genome evolution by phylogenetic analysis of chromosomal duplication events. Nature 2003, 422, 433–438. [Google Scholar] [CrossRef]
- Lysak, M.A. Brassicales: An update on chromosomal evolution and ancient polyploidy. Plant Syst. Evol. 2018, 304, 757–762. [Google Scholar] [CrossRef]
- Jiao, Y.; Wickett, N.J.; Ayyampalayam, S.; Chanderbali, A.S.; Landherr, L.; Ralph, P.E.; Tomsho, L.P.; Hu, Y.; Liang, H.; Soltis, P.S.; et al. Ancestral polyploidy in seed plants and angiosperms. Nature 2011, 473, 97–100. [Google Scholar] [CrossRef] [PubMed]
- Panchy, N.; Lehti-Shiu, M.; Shiu, S.H. Evolution of gene duplication in plants. Plant Physiol. 2016, 171, 2294–2316. [Google Scholar] [CrossRef] [PubMed]
- Schranz, M.E.; Mohammadin, S.; Edger, P.P. Ancient whole genome duplications, novelty and diversification: The WGD Radiation Lag-Time Model. Curr. Opin. Plant Biol. 2012, 15, 147–153. [Google Scholar] [CrossRef] [PubMed]
- Bohnert, H.J.; Sheveleva, E. Plant stress adaptations—Making metabolism move. Curr. Opin. Plant Biol. 1998, 1, 267–274. [Google Scholar] [CrossRef]
- Wahid, A.; Gelani, S.; Ashraf, M.; Foolad, M.R. Heat tolerance in plants: An overview. Environ. Exp. Bot. 2007, 61, 199–223. [Google Scholar] [CrossRef]
- Shinozaki, K.; Yamaguchi-Shinozaki, K. Gene networks involved in drought stress response and tolerance. J. Exp. Bot. 2006, 58, 221–227. [Google Scholar] [CrossRef]
- Parida, A.K.; Das, A.B. Salt tolerance and salinity effects on plants: A review. Ecotoxicol. Environ. Saf. 2005, 60, 324–349. [Google Scholar] [CrossRef]
- Hazman, M.; Hause, B.; Eiche, E.; Riemann, M.; Nick, P. Different forms of osmotic stress evoke qualitatively different responses in rice. J. Plant Physiol. 2016, 202, 45–56. [Google Scholar] [CrossRef]
- Shao, H.-B.; Guo, Q.-J.; Chu, L.-Y.; Zhao, X.-N.; Su, Z.-L.; Hu, Y.-C.; Cheng, J.-F. Understanding molecular mechanism of higher plant plasticity under abiotic stress. Colloids Surf. B Biointerfaces 2007, 54, 37–45. [Google Scholar] [CrossRef]
- Pandey, P.; Ramegowda, V.; Senthil-Kumar, M. Shared and unique responses of plants to multiple individual stresses and stress combinations: Physiological and molecular mechanisms. Front. Plant Sci. 2015, 6. [Google Scholar] [CrossRef]
- He, X.; Zhang, J. Toward a Molecular Understanding of Pleiotropy. Genetics 2006, 173, 1885–1891. [Google Scholar] [CrossRef] [PubMed]
- Pritykin, Y.; Ghersi, D.; Singh, M. Genome-Wide Detection and Analysis of Multifunctional Genes. PLoS Comput. Biol. 2015, 11, e1004467. [Google Scholar] [CrossRef] [PubMed]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
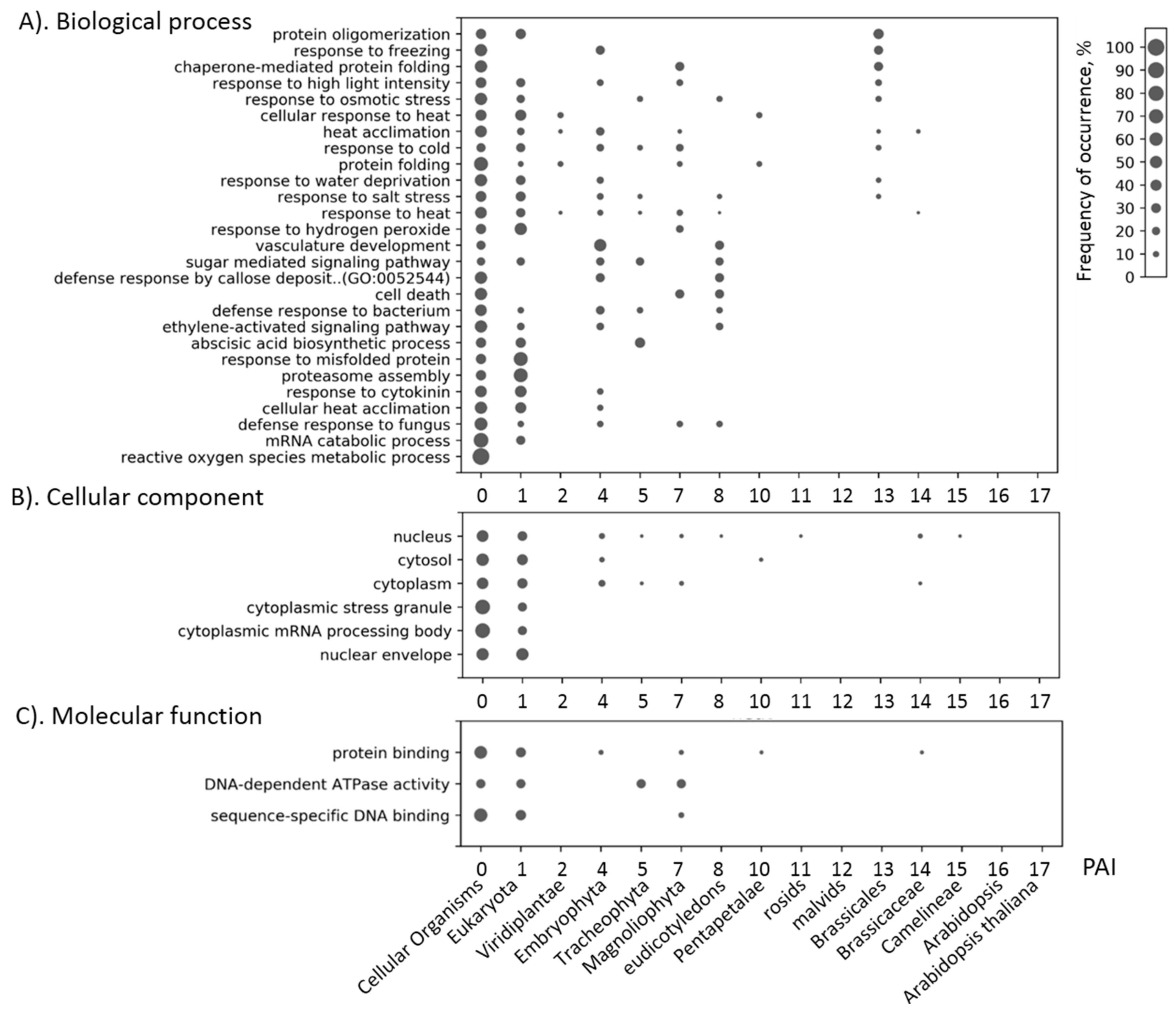{kind=link}
| Stress Type | Number of GO Terms | Number of Genes | KEGG Number of Genes |
|---|---|---|---|
| Cold | 4 | 150 | 144 |
| Heat | 14 | 102 | 102 |
| Light | 48 | 155 | 141 |
| Osmotic | 23 | 116 | 114 |
| Oxidative | 28 | 154 | 152 |
| Salt | 17 | 231 | 230 |
| Water | 27 | 215 | 211 |
| Salt | Heat | Light | Water | Cold | Osmotic | Oxidative | Unique Genes | |
|---|---|---|---|---|---|---|---|---|
| Salt | 232 | 13 | 7 (0.03) | 41 * | 18 | 47 | 18 | 126 |
| −0.06 | (0.18) | −0.08 | −0.2 | −0.08 | −0.54 | |||
| Heat | 13 | 102 | 8 | 11 | 8 | 8 | 6 | 72 |
| −0.13 | −0.08 | −0.11 | −0.08 | −0.08 | −0.06 | −0.71 | ||
| Light | 7 | 8 | 155 | 12 | 9 | 3 | 6 | 120 |
| −0.05 | −0.05 | −0.08 | −0.06 | −0.02 | −0.04 | −0.77 | ||
| Water | 41 | 11 | 12 | 216 | 18 | 29 | 11 | 124 |
| −0.2 | −0.05 | −0.06 | −0.09 | −0.14 | −0.05 | −0.59 | ||
| Cold | 18 | 8 | 9 (0.06) | 18 | 150 | 17 | 6 | 93 |
| −0.12 | −0.05 | −0.12 | −0.12 | −0.04 | −0.64 | |||
| Osmotic | 47 | 8 | 3 | 29 | 17 | 117 | 12 | 35 |
| −0.41 | −0.07 | −0.03 | −0.25 | −0.15 | −0.1 | −0.3 | ||
| Oxidative | 18 | 6 | 6 | 11 | 6 | 12 | 154 | 118 |
| −0.12 | −0.04 | −0.04 | −0.07 | −0.04 | −0.08 | −0.77 |
| Stress | Cold | Heat | Llight | Osmotic | Oxidative | Salt | Water | All Stress nr |
|---|---|---|---|---|---|---|---|---|
| p(PAIstress < PAIrand) | 100,000 * | 100,000 | 100,000 | 100,000 | 100,000 | 100,000 | 100,000 | 100,000 |
| p(ChiSqstress < ChiSqrand) | 163 | 153 | 303 | 2132 | 20 | 1 | 1 | 0 ** |
| p (dfPAIi stress < dfPAIi rand) for specific phylostratum | ||||||||
| 00_Cellular organisms | 28 | 0 ** | 11 | 1051 | 0 ** | 0 ** | 153 | 0 ** |
| 01_Eukaryota | 11,218 | 35,449 | 34,137 | 2009 | 75,372 | 194 | 34 | 78 |
| 02_Viridiplantae | 15,729 | 11,571 | 6603 | 32,387 | 18,710 | 83,664 | 96 | 6394 |
| 04_Embryophyta | 47,489 | 3778 | 12,708 | 51,099 | 11,320 | 87,444 | 26,046 | 25,790 |
| 05_Tracheophyta | 2181 | 65,732 | 32,104 | 51,865 | 99,386 | 22,936 | 39,481 | 53,618 |
| 07_Magnoliophyta | 38,170 | 97,432 | 26,294 | 23,881 | 49,013 | 45,825 | 82,207 | 100,000 |
| 08_eudicotyledons | 94,830 | 62,021 | 78,520 | 8542 | 60,328 | 69,323 | 80,882 | 100,000 |
| 10_Pentapetalae | 69,619 | 72,175 | 68,242 | 31,311 | 52,357 | 50,365 | 60,197 | 97,992 |
| 11_rosids | 91,840 | 79,304 | 91,447 | 83,906 | 81,256 | 99,084 | 72,339 | 99,935 |
| 12_malvids | 15,610 | 39,144 | 49,460 | 42,420 | 16,775 | 67,429 | 63,996 | 78,703 |
| 13_Brassicales | 89,388 | 94,844 | 61,746 | 89,433 | 96,971 | 97,955 | 98,746 | 100,000 |
| 14_Brassicaceae | 99,913 | 89,314 | 99,872 | 99,981 | 99,765 | 99,998 | 100,000 | 100,000 |
| 15_Camelineae | 89,271 | 74,850 | 97,699 | 80,248 | 56,525 | 99,796 | 99,627 | 100,000 |
| 16_Arabidopsis | 74,084 | 61,724 | 73,454 | 65,695 | 41,687 | 88,476 | 86,494 | 99,736 |
| 17_A. thaliana | 99,520 | 97,657 | 99,446 | 98,525 | 97,458 | 99,811 | 99,948 | 100,000 |
| Stress | Cold | Heat | Light | Osmotic | Oxidative | Salt | Water | All Stresses nr |
|---|---|---|---|---|---|---|---|---|
| p(DIstress < DIrand) | 96,272 * | 99,700 | 99,993 | 99,993 | 98,140 | 100,000 | 99,999 | 100,000 |
| p(ChiSqstress < ChiSqrand) | 7010 | 43,398 | 4707 | 589 | 52,740 | 10 | 77 | 0 ** |
| p(NDI stress < NDI rand) for specific DI bin | ||||||||
| [0, 0.1] | 93,375 | 28,064 | 6137 | 53,230 | 38,498 | 10,701 | 38,527 | 3109 |
| (0.1, 0.2] | 67 | 2567 | 440 | 25 | 2633 | 0 ** | 3 | 0 ** |
| (0.2, 0.3] | 15,832 | 38,676 | 66,383 | 3174 | 64,490 | 32,404 | 9731 | 18,452 |
| (0.3, 0.4] | 49,562 | 40,493 | 90,569 | 96,058 | 69,027 | 97,027 | 96,333 | 99,395 |
| (0.4, 0.5] | 71,783 | 75,719 | 58,004 | 92,193 | 30,113 | 96,979 | 94,603 | 99,451 |
| (0.5, 0.6] | 70,023 | 73,560 | 68,603 | 92,023 | 86,198 | 99,567 | 99,808 | 99,973 |
| (0.6, 0.7] | 95,009 | 84,956 | 99,071 | 88,993 | 22,528 | 93,999 | 82,333 | 99,957 |
| (0.7, 0.8] | 76,327 | 58,418 | 51,318 | 88,964 | 55,802 | 93,241 | 77,350 | 98,435 |
| (0.8, 0.9] | 47,813 | 67,878 | 79,538 | 72,465 | 50,554 | 92,166 | 90,674 | 99,617 |
| (0.9, 1] | 61,938 | 49,902 | 61,515 | 53,707 | 64,025 | 78,219 | 75,853 | 99,712 |
| (1, +∞) | 62,703 | 78,089 | 87,708 | 81,801 | 89,300 | 96,544 | 82,213 | 99,985 |
| Stress | r(k, PAI) | p-Value |
|---|---|---|
| Сold | 0.004 | 0.974 |
| Heat | −0.361 * | 0.003 |
| Light | −0.125 | 0.248 |
| Osmotic | −0.379 | 0.006 |
| Oxidative | 0.019 | 0.875 |
| Salt | −0.266 | 0.006 |
| Water | −0.061 | 0.524 |
| Stress | Np | ra | σ(ra) |
|---|---|---|---|
| Сold | 148 | 0.251 | 0.345 |
| Heat | 196 | 0.126 | 0.294 |
| Light | 178 | 0.026 | 0.348 |
| Osmotic | 148 | 0.192 | 0.327 |
| Oxidative | 111 | 0.567 | 0.344 |
| Salt | 203 | 0.143 | 0.293 |
| Water | 213 | 0.031 | 0.307 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Mustafin, Z.S.; Zamyatin, V.I.; Konstantinov, D.K.; Doroshkov, A.V.; Lashin, S.A.; Afonnikov, D.A. Phylostratigraphic Analysis Shows the Earliest Origination of the Abiotic Stress Associated Genes in A. thaliana. Genes 2019, 10, 963. https://doi.org/10.3390/genes10120963
Mustafin ZS, Zamyatin VI, Konstantinov DK, Doroshkov AV, Lashin SA, Afonnikov DA. Phylostratigraphic Analysis Shows the Earliest Origination of the Abiotic Stress Associated Genes in A. thaliana. Genes. 2019; 10(12):963. https://doi.org/10.3390/genes10120963
Chicago/Turabian StyleMustafin, Zakhar S., Vladimir I. Zamyatin, Dmitrii K. Konstantinov, Aleksej V. Doroshkov, Sergey A. Lashin, and Dmitry A. Afonnikov. 2019. "Phylostratigraphic Analysis Shows the Earliest Origination of the Abiotic Stress Associated Genes in A. thaliana" Genes 10, no. 12: 963. https://doi.org/10.3390/genes10120963
APA StyleMustafin, Z. S., Zamyatin, V. I., Konstantinov, D. K., Doroshkov, A. V., Lashin, S. A., & Afonnikov, D. A. (2019). Phylostratigraphic Analysis Shows the Earliest Origination of the Abiotic Stress Associated Genes in A. thaliana. Genes, 10(12), 963. https://doi.org/10.3390/genes10120963