Computational Inference of Gene Co-Expression Networks for the identification of Lung Carcinoma Biomarkers: An Ensemble Approach

 ,
,  ,
,  ,
,  and
and 
Abstract
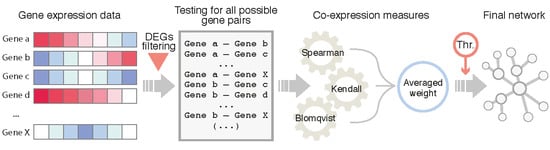
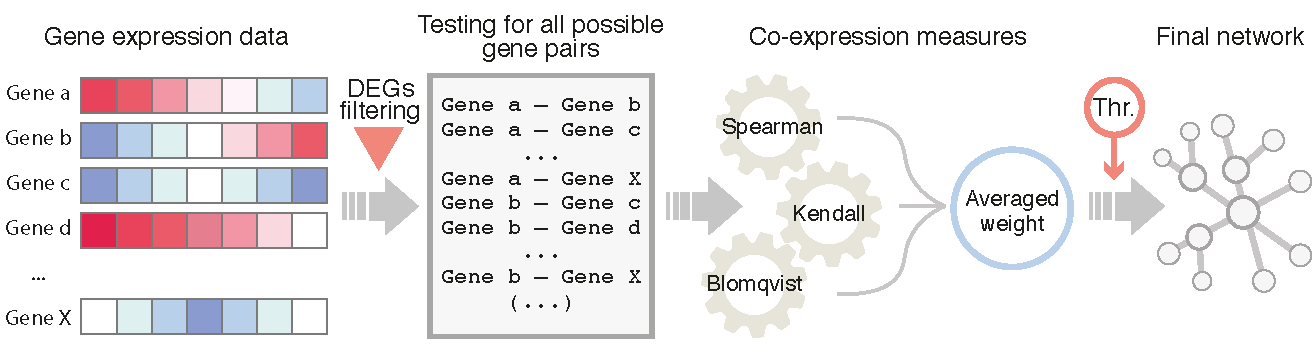
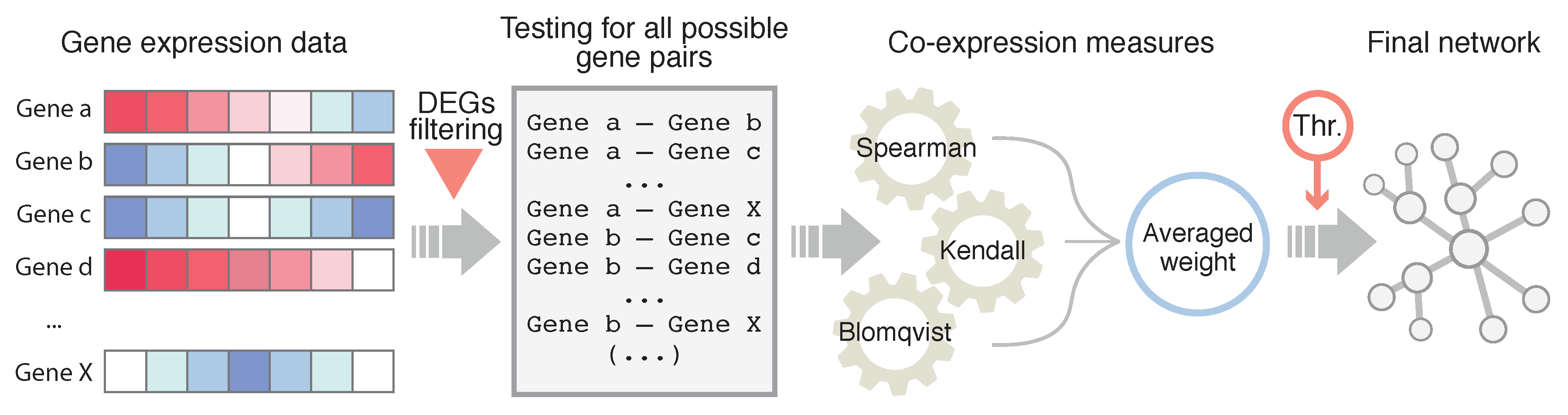
1. Introduction
1.1. Related Works
2. Materials and Methods
2.1. Lung Cancer Dataset
2.2. Data Preprocessing
2.3. Differential Expression Analysis
2.4. Network Inference
2.5. GN Analysis: Topology and Enrichment Analyses
3. Results
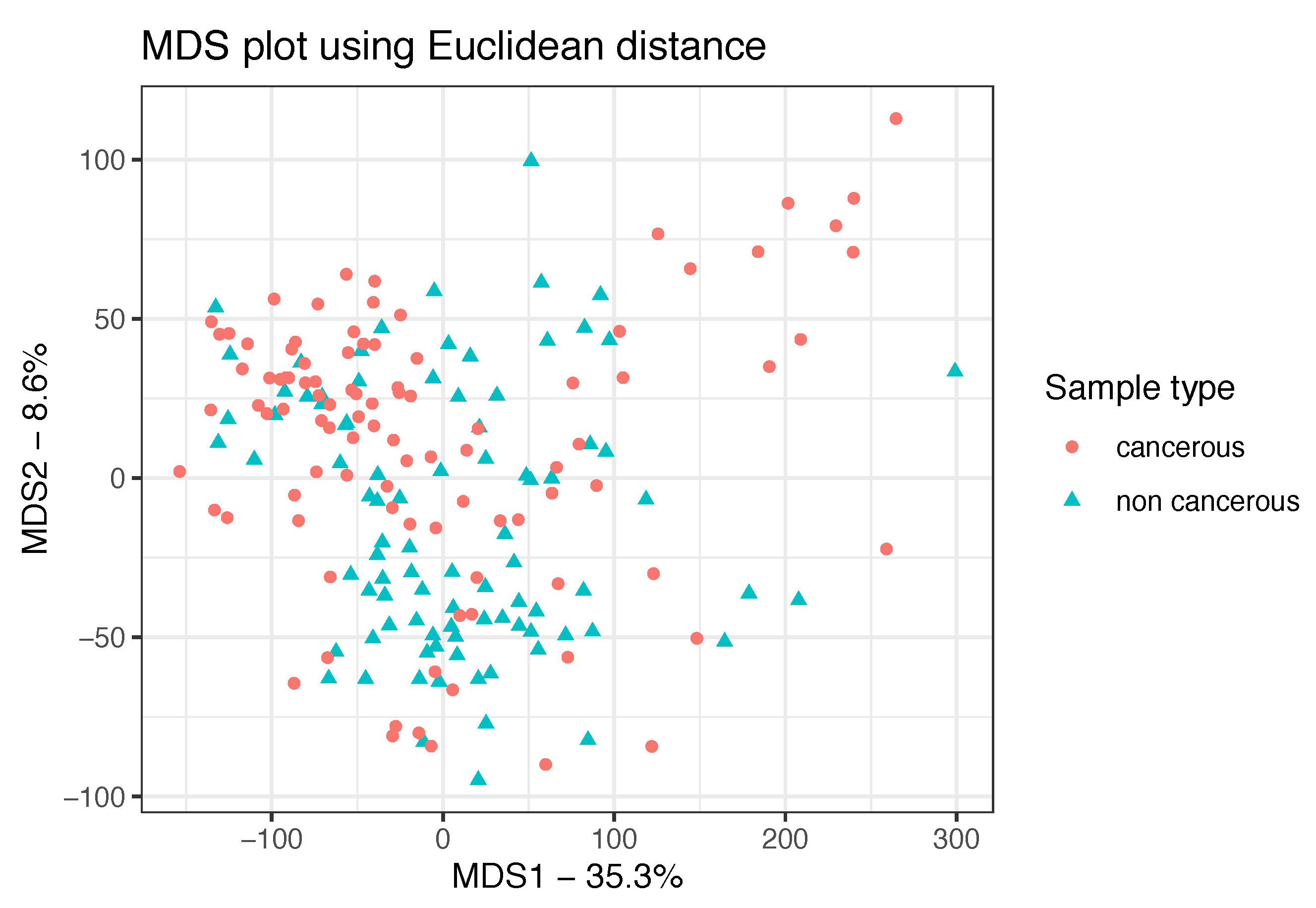
3.1. Data Preprocessing and Exploratory Analyses
3.2. Obtaining Differentially Expressed Genes
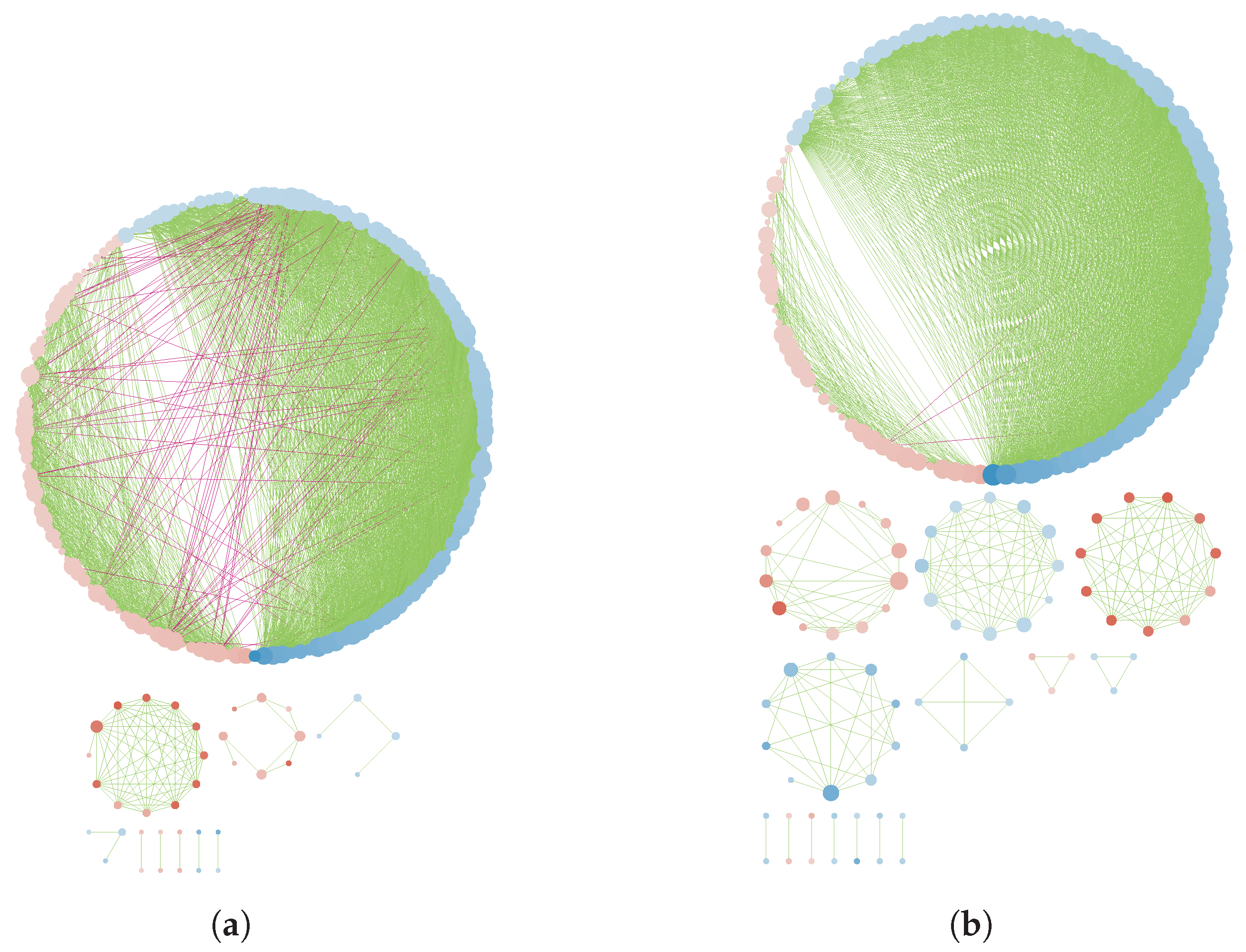
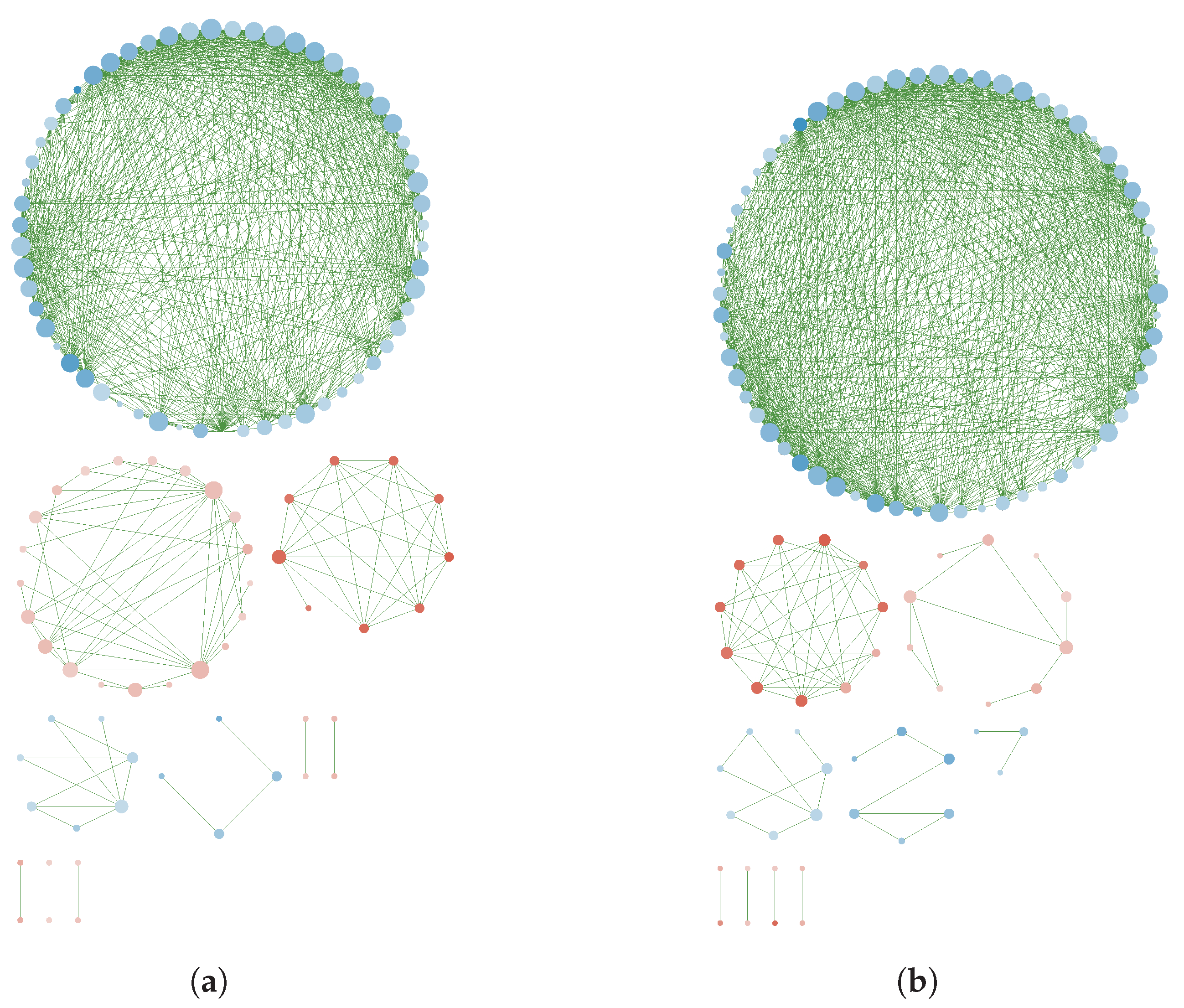
3.3. GN Reconstruction and Topology Analysis
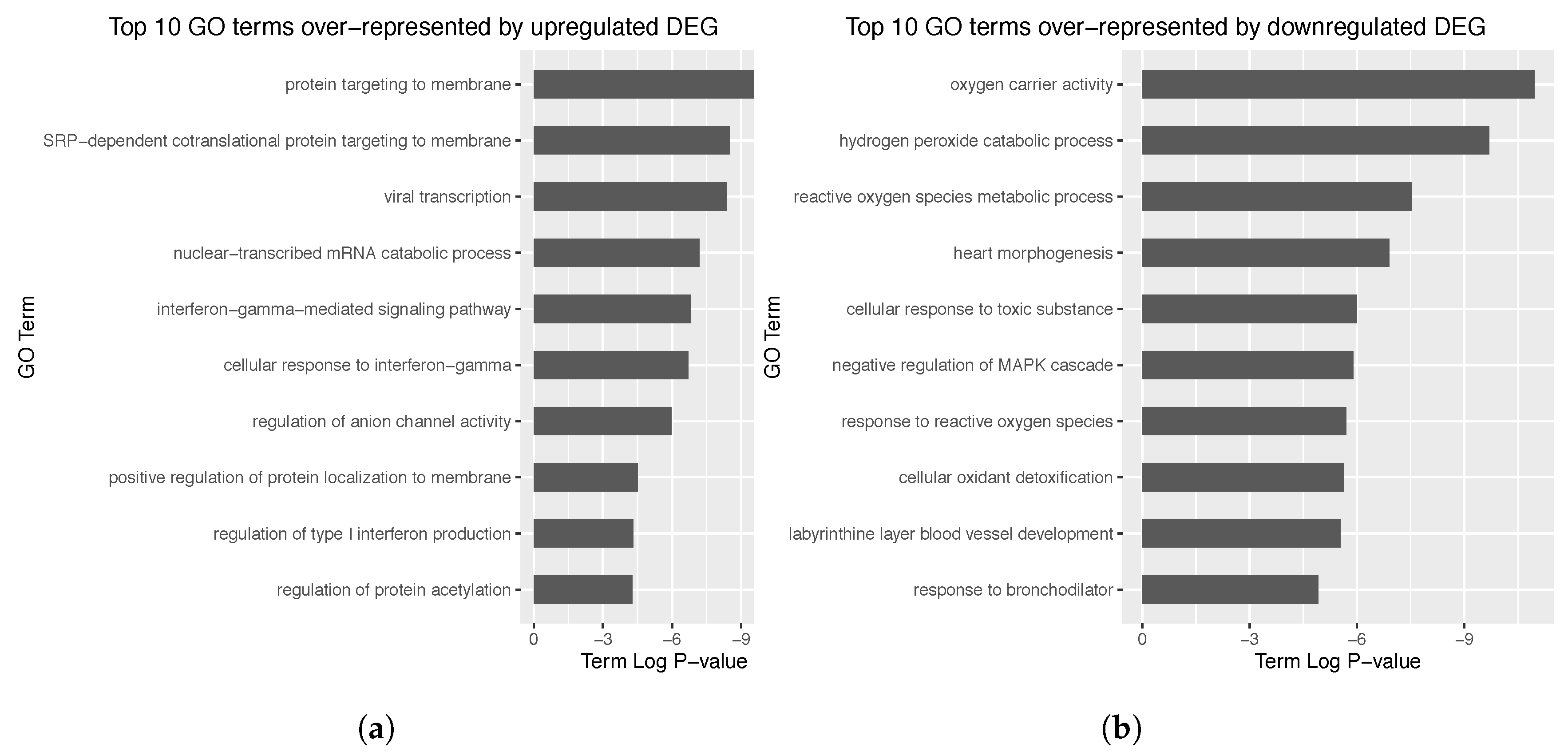
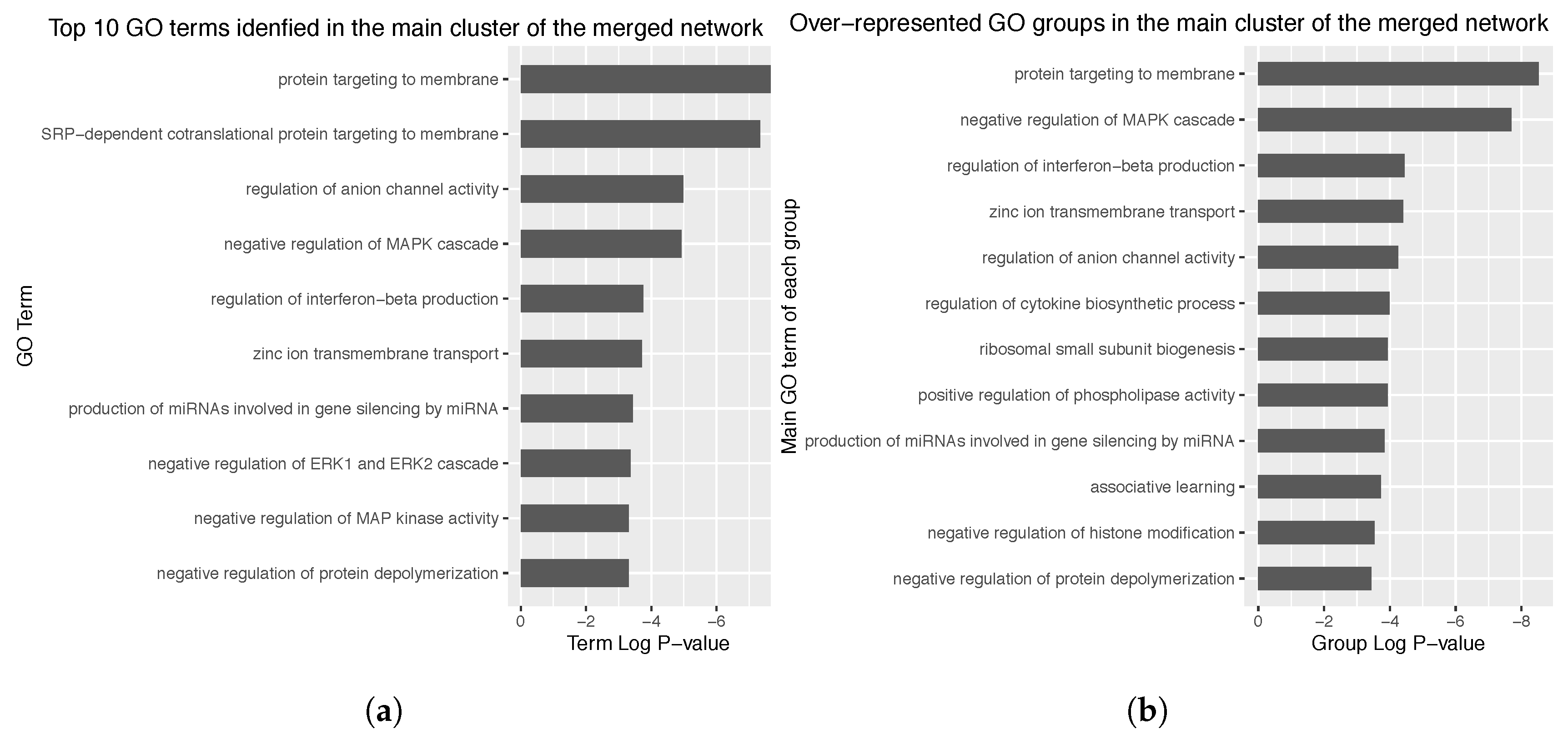
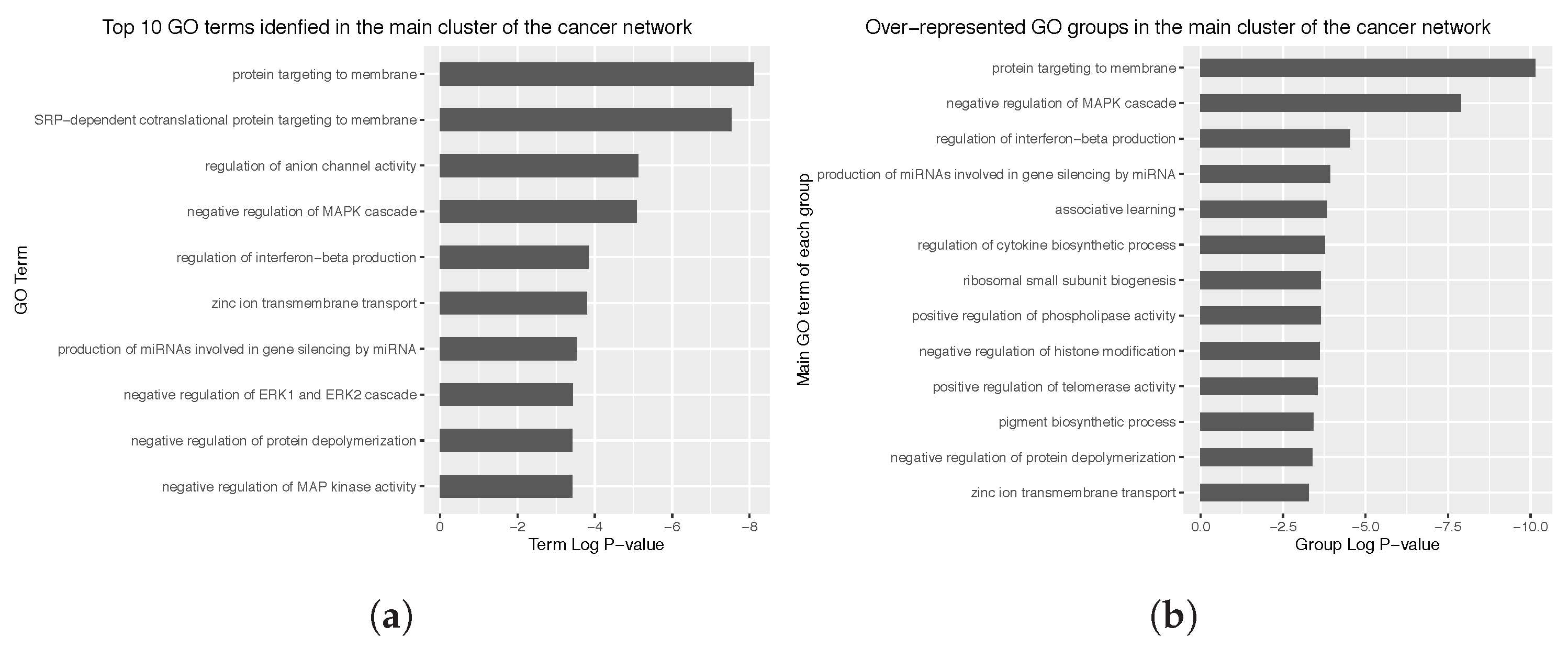
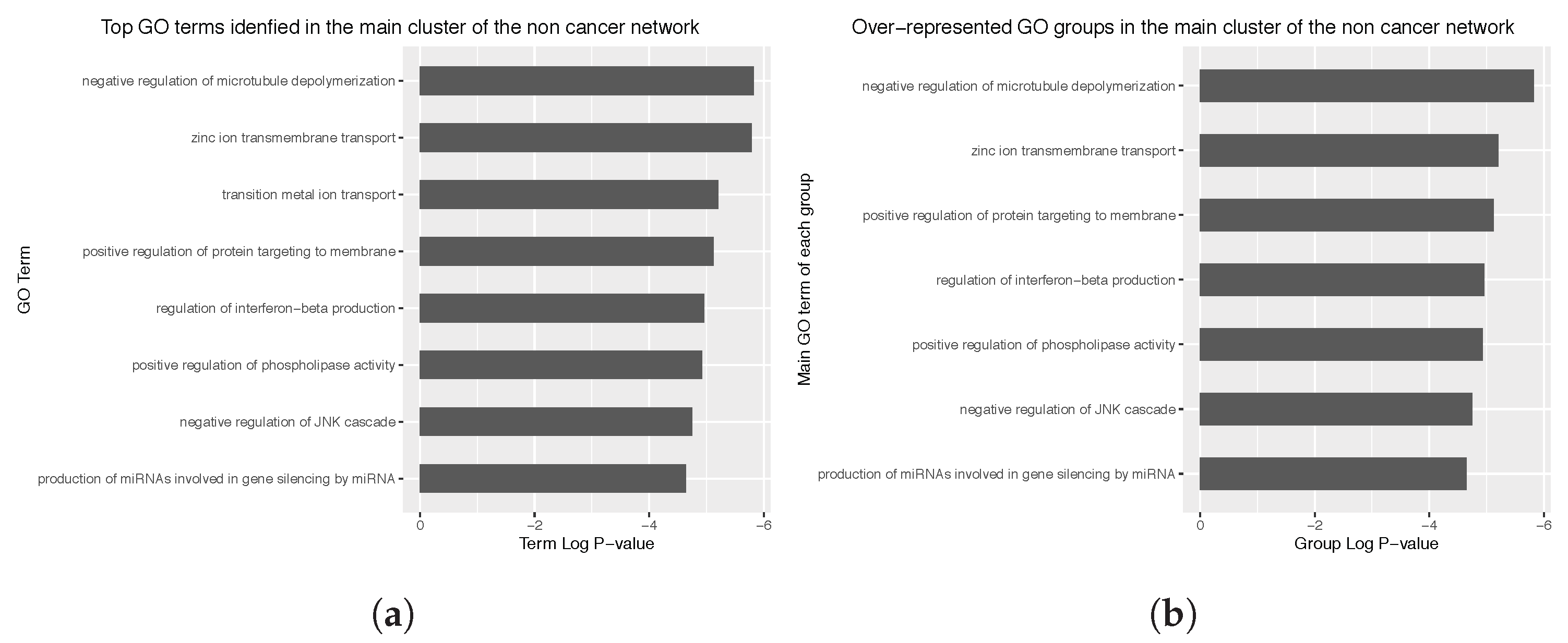
3.4. Enrichment Analysis over the Identified Network Clusters
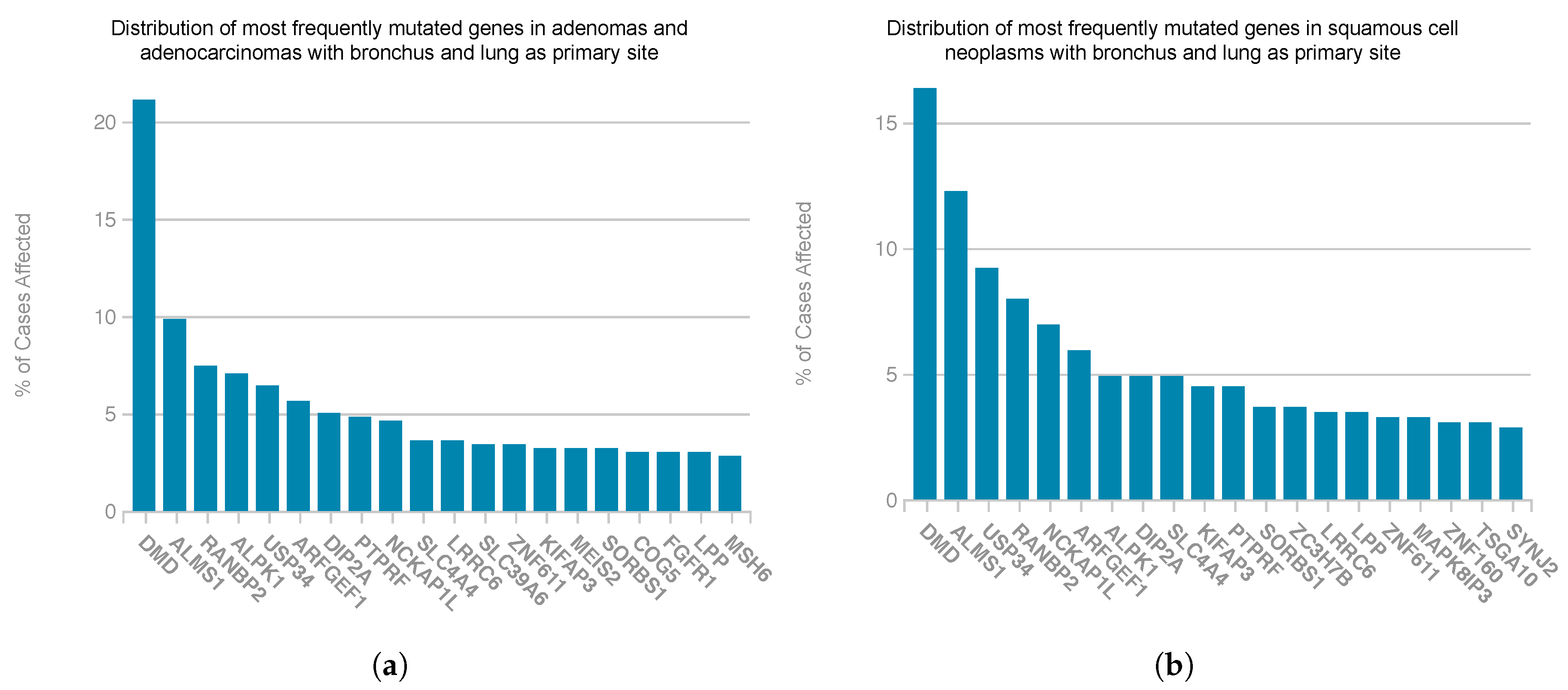
4. Discussion
5. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| DEG | Differentially Expressed Genes |
| FC | Fold Change |
| GO | Gene Ontology |
| GN | Gene Networks |
| PCoA | Principal Coordinates Analysis |
| MDS | Multidimensional scaling |
| T2DM | Type 2 diabetes mellitus |
Appendix A. Definitions of the Used Co-Expression Measures
Appendix A.1. Kendall Co-Expression Measure
Appendix A.2. Spearman Co-Expression Measure
Appendix A.3. Blomqvist Co-Expression Measure
Appendix B. Reconstructed Networks with High Thresholding



Appendix C. Main Over-Represented GO Terms and GO Groups for the Cancer and Non Cancer Networks


Appendix D. Detailed Lists of Sample Type-Exclusive Genes

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Affymetrix ID | Gene Name | Gene Description | Regulation |
|---|---|---|---|
| 202539_s_at | HMGCR | 3-hydroxy-3-methylglutaryl-CoA reductase | Down |
| 211672_s_at | ARPC4-TTLL3 | ARPC4-TTLL3 readthrough | Down |
| 209288_s_at | CDC42EP3 | CDC42 effector protein 3 | Down |
| 213826_s_at | H3F3A | H3 histone family member 3A | Up |
| 220266_s_at | KLF4 | Kruppel like factor 4 | Down |
| 212327_at | LIMCH1 | LIM and calponin homology domains 1 | Down |
| 207480_s_at | MEIS2 | Meis homeobox 2 | Down |
| 217549_at | NCKAP1L | NCK associated protein 1 like | Up |
| 203582_s_at | SPHAR | S-phase response (cyclin related) | Down |
| 216064_s_at | AGA | aspartylglucosaminidase | Down |
| 201942_s_at | CPD | carboxypeptidase D | Down |
| 203492_x_at | CEP57 | centrosomal protein 57 | Down |
| 213753_x_at | EIF5A | eukaryotic translation initiation factor 5A | Up |
| 218343_s_at | GTF3C3 | general transcription factor IIIC subunit 3 | Down |
| 206483_at | LRRC6 | leucine rich repeat containing 6 | Down |
| 218212_s_at | MOCS2 | molybdenum cofactor synthesis 2 | Down |
| 206302_s_at | NUDT4 | nudix hydrolase 4 | Down |
| 208447_s_at | PRPS1 | phosphoribosyl pyrophosphate synthetase 1 | Down |
| 200730_s_at | PTP4A1 | protein tyrosine phosphatase type IVA, member 1 | Down |
| 218276_s_at | SAV1 | salvador family WW domain containing protein 1 | Down |
| 203908_at | SLC4A4 | solute carrier family 4 member 4 | Down |
| 217975_at | TCEAL9 | transcription elongation factor A like 9 | Down |
| 209149_s_at | TM9SF1 | transmembrane 9 superfamily member 1 | Down |
| 204426_at | TMED2 | transmembrane p24 trafficking protein 2 | Down |
| 211689_s_at | TMPRSS2 | transmembrane protease, serine 2 | Down |
| 214007_s_at | TWF1 | twinfilin actin binding protein 1 | Down |
| 211763_s_at | UBE2B | ubiquitin conjugating enzyme E2 B | Down |
| 201337_s_at | VAMP3 | vesicle associated membrane protein 3 | Down |
| Affymetrix ID | Gene Name | Gene Description |
|---|---|---|
| 202539_s_at | HMGCR | 3-hydroxy-3-methylglutaryl-CoA reductase |
| 209288_s_at | CDC42EP3 | CDC42 effector protein 3 |
| 213826_s_at | H3F3A | H3 histone family member 3A |
| 220266_s_at | KLF4 | Kruppel like factor 4 |
| 207480_s_at | MEIS2 | Meis homeobox 2 |
| 217549_at | NCKAP1L | NCK associated protein 1 like |
| 203492_x_at | CEP57 | centrosomal protein 57 |
| 206483_at | LRRC6 | leucine rich repeat containing 6 |
| 208447_s_at | PRPS1 | phosphoribosyl pyrophosphate synthetase 1 |
| 200730_s_at | PTP4A1 | protein tyrosine phosphatase type IVA, member 1 |
| 218276_s_at | SAV1 | salvador family WW domain containing protein 1 |
| 204426_at | TMED2 | transmembrane p24 trafficking protein 2 |
| 211763_s_at | UBE2B | ubiquitin conjugating enzyme E2 B |
| 201337_s_at | VAMP3 | vesicle associated membrane protein 3 |
| Affymetrix ID | Gene Name | Gene Description | Regulation |
|---|---|---|---|
| 212206_s_at | H2AFV | H2A histone family member V | Down |
| 209703_x_at | METTL7A | methyltransferase like 7A | Up |
| 217734_s_at | WDR6 | WD repeat domain 6 | Up |
| 215359_x_at | LOC101060181 | zinc finger protein ZnFP12 | Up |
| 222339_x_at | - | - | Up |
| 220856_x_at | - | - | Up |
| 208082_x_at | - | - | Up |
References
- Delgado, F.M.; Gómez-Vela, F. Computational methods for Gene Regulatory Networks reconstruction and analysis: A review. In Artificial Intelligence in Medicine; Elsevier: Amsterdam, The Netherlands, 2018. [Google Scholar]
- Díaz-Montaña, J.J.; Gómez-Vela, F.; Díaz-Díaz, N. GNC–app: A new Cytoscape app to rate gene networks biological coherence using gene–gene indirect relationships. Biosystems 2018, 166, 61–65. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Vela, F.; Lagares, J.A.; Díaz-Díaz, N. Gene network coherence based on prior knowledge using direct and indirect relationships. Comput. Biol. Chem. 2015, 56, 142–151. [Google Scholar] [CrossRef] [PubMed]
- Darrason, M. Mechanistic and topological explanations in medicine: The case of medical genetics and network medicine. Synthese 2018, 195, 147–173. [Google Scholar] [CrossRef]
- Liang, L.; Gao, L.; Zou, X.P.; Huang, M.L.; Chen, G.; Li, J.J.; Cai, X.Y. Diagnostic significance and potential function of miR-338-5p in hepatocellular carcinoma: A bioinformatics study with microarray and RNA sequencing data. Mol. Med. Rep. 2018, 17, 2297–2312. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Vela, F.; Barranco, C.D.; Díaz-Díaz, N. Incorporating biological knowledge for construction of fuzzy networks of gene associations. Appl. Soft Comput. 2016, 42, 144–155. [Google Scholar] [CrossRef]
- Gómez-Vela, F.; Rodriguez-Baena, D.S.; Vázquez-Noguera, J.L. Structure Optimization for Large Gene Networks Based on Greedy Strategy. Comput. Math. Methods Med. 2018, 2018. [Google Scholar] [CrossRef]
- Zhao, W.; Langfelder, P.; Fuller, T.; Dong, J.; Li, A.; Hovarth, S. Weighted gene coexpression network analysis: State of the art. J. Biopharm. Stat. 2010, 20, 281–300. [Google Scholar] [CrossRef]
- Kumari, S.; Nie, J.; Chen, H.S.; Ma, H.; Stewart, R.; Li, X.; Lu, M.Z.; Taylor, W.M.; Wei, H. Evaluation of gene association methods for coexpression network construction and biological knowledge discovery. PLoS ONE 2012, 7, e50411. [Google Scholar] [CrossRef]
- De Siqueira Santos, S.; Takahashi, D.Y.; Nakata, A.; Fujita, A. A comparative study of statistical methods used to identify dependencies between gene expression signals. Briefings Bioinform. 2013, 15, 906–918. [Google Scholar] [CrossRef]
- D’haeseleer, P.; Liang, S.; Somogyi, R. Genetic network inference: From co-expression clustering to reverse engineering. Bioinformatics 2000, 16, 707–726. [Google Scholar] [CrossRef]
- Emamjomeh, A.; Robat, E.S.; Zahiri, J.; Solouki, M.; Khosravi, P. Gene co-expression network reconstruction: A review on computational methods for inferring functional information from plant-based expression data. Plant Biotechnol. Rep. 2017, 11, 71–86. [Google Scholar] [CrossRef]
- Liu, Z.P. Quantifying gene regulatory relationships with association measures: A comparative study. Front. Genet. 2017, 8, 96. [Google Scholar] [CrossRef] [PubMed]
- Zhong, R.; Allen, J.D.; Xiao, G.; Xie, Y. Ensemble-based network aggregation improves the accuracy of gene network reconstruction. PLoS ONE 2014, 9, e106319. [Google Scholar] [CrossRef] [PubMed]
- Hecker, M.; Lambeck, S.; Toepfer, S.; Van Someren, E.; Guthke, R. Gene regulatory network inference: Data integration in dynamic models—A review. Biosystems 2009, 96, 86–103. [Google Scholar] [CrossRef]
- Wang, Z.; Xu, P.; Chen, B.; Zhang, Z.; Zhang, C.; Zhan, Q.; Huang, S.; Xia, Z.; Peng, W. Identifying circRNA-associated-ceRNA networks in the hippocampus of Aβ1-42-induced Alzheimer’s disease-like rats using microarray analysis. Aging (Albany NY) 2018, 10, 775. [Google Scholar] [CrossRef]
- Jemal, A.; Bray, F.; Center, M.M.; Ferlay, J.; Ward, E.; Forman, D. Global cancer statistics. CA Cancer J. Clin. 2011, 61, 69–90. [Google Scholar] [CrossRef]
- WHO. International Programme on Chemical Safety: Biomarkers in Risk Assessment: Validity and Validation, 2001; WHO: Genewa, Switzerland, 2015. [Google Scholar]
- Murray, J.F.; Nadel, J.A. Murray & Nadel’s Textbook of Respiratory Medicine; Elsevier Saunders: Amsterdam, The Netherlands, 2016. [Google Scholar]
- Stewart, B.; Wild, C.P. World Cancer Report 2014; WHO: Genewa, Switzerland, 2014. [Google Scholar]
- Peng, G.; Tisch, U.; Adams, O.; Hakim, M.; Shehada, N.; Broza, Y.Y.; Billan, S.; Abdah-Bortnyak, R.; Kuten, A.; Haick, H. Diagnosing lung cancer in exhaled breath using gold nanoparticles. Nat. Nanotechnol. 2009, 4, 669. [Google Scholar] [CrossRef]
- Aggarwal, A.; Guo, D.L.; Hoshida, Y.; Yuen, S.T.; Chu, K.M.; So, S.; Boussioutas, A.; Chen, X.; Bowtell, D.; Aburatani, H.; et al. Topological and Functional Discovery in a Gene Coexpression Meta-Network of Gastric Cancer. Cell Tumor Stem Cell Biol. 2006, 66, 232–241. [Google Scholar] [CrossRef]
- Ma, S.; Shi, M.; Li, Y.; Yi, D.; Shia, B.C. Incorporating gene co-expression network in identification of cancer prognosis markers. BMC Bioinform. 2010, 11, 271. [Google Scholar] [CrossRef]
- Clarke, C.; Madden, S.F.; Doolan, P.; Aherne, S.T.; Joyce, H.; O’Driscoll, L.; Gallagher, W.M.; Hennessy, B.T.; Moriarty, M.; Crown, J.; et al. Correlating transcriptional networks to breast cancer survival: A large-scale coexpression analysis. Carcinogenesis 2013, 34, 2300–2308. [Google Scholar] [CrossRef]
- Chang, X.; Shi, L.; Gao, F.; Russin, J.; Zeng, L.; He, S.; Chen, T.C.; Giannotta, S.L.; Weisenberger, D.J.; Zada, G.; et al. Genomic and transcriptome analysis revealing an oncogenic functional module in meningiomas. Neurosurg. Focus 2013, 35, E3. [Google Scholar] [CrossRef] [PubMed]
- Yang, Y.; Han, L.; Yuan, Y.; Li, J.; Hei, N.; Liang, H. Gene co-expression network analysis reveals common system-level properties of prognostic genes across cancer types. Nat. Commun. 2014, 5, 3231. [Google Scholar] [CrossRef] [PubMed]
- Liu, R.; Cheng, Y.; Yu, J.; Lv, Q.L.; Zhou, H.H. Identification and validation of gene module associated with lung cancer through coexpression network analysis. Gene 2015, 563, 56–62. [Google Scholar] [CrossRef] [PubMed]
- Yang, L.; Li, Y.; Wei, Z.; Chang, X. Coexpression network analysis identifies transcriptional modules associated with genomic alterations in neuroblastoma. Biochim. Biophys. Acta (BBA) Mol. Basis Dis. 2018, 1864, 2341–2348. [Google Scholar] [CrossRef] [PubMed]
- Xu, B.; Lv, W.; Li, X.; Zhang, L.; Lin, J. Prognostic genes of hepatocellular carcinoma based on gene coexpression network analysis. J. Cell. Biochem. 2019, 120, 11616–11623. [Google Scholar] [CrossRef] [PubMed]
- Spira, A.; Beane, J.E.; Shah, V.; Steiling, K.; Liu, G.; Schembri, F.; Gilman, S.; Dumas, Y.M.; Calner, P.; Sebastiani, P.; et al. Airway epithelial gene expression in the diagnostic evaluation of smokers with suspect lung cancer. Nat. Med. 2007, 13, 361. [Google Scholar] [CrossRef]
- Gustafson, A.M.; Soldi, R.; Anderlind, C.; Scholand, M.B.; Qian, J.; Zhang, X.; Cooper, K.; Walker, D.; McWilliams, A.; Liu, G.; et al. Airway PI3K pathway activation is an early and reversible event in lung cancer development. Sci. Transl. Med. 2010, 2, 26ra25–26ra25. [Google Scholar] [CrossRef]
- Edgar, R.; Domrachev, M.; Lash, A.E. Gene Expression Omnibus: NCBI gene expression and hybridization array data repository. Nucleic Acids Res. 2002, 30, 207–210. [Google Scholar] [CrossRef]
- Ihaka, R.; Gentleman, R. R: A language for data analysis and graphics. J. Comput. Graph. Stat. 1996, 5, 299–314. [Google Scholar]
- Gruvberger, S.; Ringnér, M.; Chen, Y.; Panavally, S.; Saal, L.H.; Borg, Å.; Fernö, M.; Peterson, C.; Meltzer, P.S. Estrogen receptor status in breast cancer is associated with remarkably distinct gene expression patterns. Cancer Res. 2001, 61, 5979–5984. [Google Scholar]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: Berlin, Germany, 2016. [Google Scholar]
- Smyth, G.K. Limma: Linear models for microarray data. In Bioinformatics and Computational Biology Solutions Using R and Bioconductor; Springer: Berlin, Germany, 2005; pp. 397–420. [Google Scholar]
- Diboun, I.; Wernisch, L.; Orengo, C.A.; Koltzenburg, M. Microarray analysis after RNA amplification can detect pronounced differences in gene expression using limma. BMC Genom. 2006, 7, 252. [Google Scholar] [CrossRef] [PubMed]
- Ben-Dov, I.Z.; Whalen, V.M.; Goilav, B.; Max, K.E.; Tuschl, T. Cell and microvesicle urine microRNA deep sequencing profiles from healthy individuals: Observations with potential impact on biomarker studies. PLoS ONE 2016, 11, e0147249. [Google Scholar] [CrossRef] [PubMed]
- Thissen, D.; Steinberg, L.; Kuang, D. Quick and easy implementation of the Benjamini-Hochberg procedure for controlling the false positive rate in multiple comparisons. J. Educ. Behav. Stat. 2002, 27, 77–83. [Google Scholar] [CrossRef]
- Marbach, D.; Mattiussi, C.; Floreano, D. Combining multiple results of a reverse engineering algorithm: Application to the DREAM five gene network challenge. Ann. N. Y. Acad. Sci. 2009, 1158, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Zainal, A.; Maarof, M.A.; Shamsuddin, S.M. Ensemble classifiers for network intrusion detection system. J. Inf. Assur. Secur. 2009, 4, 217–225. [Google Scholar]
- Altay, G.; Emmert-Streib, F. Revealing differences in gene network inference algorithms on the network level by ensemble methods. Bioinformatics 2010, 26, 1738–1744. [Google Scholar] [CrossRef] [PubMed]
- Martin, A.J.; Dominguez, C.; Contreras-Riquelme, S.; Holmes, D.S.; Perez-Acle, T. Graphlet Based Metrics for the comparison of gene regulatory networks. PLoS ONE 2016, 11, e0163497. [Google Scholar] [CrossRef]
- Mukaka, M.M. A guide to appropriate use of correlation coefficient in medical research. Malawi Med. J. 2012, 24, 69–71. [Google Scholar]
- Cooke, D.; Clarke, G. A Basic Course in Statistics; Arnold: London, UK, 1989. [Google Scholar]
- Kendall, M.G. Rank Correlation Methods; American Psychological Association: Washington, DC, USA, 1948. [Google Scholar]
- Mindrila, D.; Balentyne, P. Scatterplots and correlation. Retrieved From. 2017. Available online: https://www.westga.edu/academics/research/vrc/assets/docs/scatterplots_and_correlation_notes.pdf (accessed on 15 October 2019).
- Csardi, G.; Nepusz, T. The igraph software package for complex network research. InterJ. Complex Syst. 2006, 1695, 1–9. [Google Scholar]
- Kolaczyk, E.D.; Csárdi, G. Statistical Analysis of Network Data with R; Springer: Berlin, Germany, 2014; Volume 65. [Google Scholar]
- Contreras-Lopez, O.; Moyano, T.C.; Soto, D.C.; Gutiérrez, R.A. Step-by-step construction of gene co-expression networks from high-throughput arabidopsis RNA sequencing data. In Root Development; Springer: Berlin, Germany, 2018; pp. 275–301. [Google Scholar]
- Godsil, C.; Royle, G.F. Algebraic Graph Theory; Springer Science & Business Media: Berlin, Germany, 2013; Volume 207. [Google Scholar]
- Parikshak, N.N.; Gandal, M.J.; Geschwind, D.H. Systems biology and gene networks in neurodevelopmental and neurodegenerative disorders. Nat. Rev. Genet. 2015, 16, 441. [Google Scholar] [CrossRef] [PubMed]
- Rizzolo, K.; Kumar, A.; Kakihara, Y.; Phanse, S.; Minic, Z.; Snider, J.; Stagljar, I.; Zilles, S.; Babu, M.; Houry, W.A. Systems analysis of the genetic interaction network of yeast molecular chaperones. Mol. Omics 2018, 14, 82–94. [Google Scholar] [CrossRef] [PubMed]
- Shannon, P.; Markiel, A.; Ozier, O.; Baliga, N.S.; Wang, J.T.; Ramage, D.; Amin, N.; Schwikowski, B.; Ideker, T. Cytoscape: A software environment for integrated models of biomolecular interaction networks. Genome Res. 2003, 13, 2498–2504. [Google Scholar] [CrossRef] [PubMed]
- Nepomuceno-Chamorro, I.A.; Aguilar-Ruiz, J.S.; Riquelme, J.C. Inferring gene regression networks with model trees. BMC Bioinform. 2010, 11, 517. [Google Scholar] [CrossRef] [PubMed]
- Morris, J.H.; Apeltsin, L.; Newman, A.M.; Baumbach, J.; Wittkop, T.; Su, G.; Bader, G.D.; Ferrin, T.E. clusterMaker: A multi-algorithm clustering plugin for Cytoscape. BMC Bioinform. 2011, 12, 436. [Google Scholar] [CrossRef] [PubMed]
- Su, G.; Kuchinsky, A.; Morris, J.H.; States, D.J.; Meng, F. GLay: Community structure analysis of biological networks. Bioinformatics 2010, 26, 3135–3137. [Google Scholar] [CrossRef] [PubMed]
- Milenković, T.; Pržulj, N. Uncovering biological network function via graphlet degree signatures. Cancer Inform. 2008, 6, S680. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Bioinformatics enrichment tools: Paths toward the comprehensive functional analysis of large gene lists. Nucleic Acids Res. 2008, 37, 1–13. [Google Scholar] [CrossRef]
- Bindea, G.; Mlecnik, B.; Hackl, H.; Charoentong, P.; Tosolini, M.; Kirilovsky, A.; Fridman, W.H.; Pagès, F.; Trajanoski, Z.; Galon, J. ClueGO: A Cytoscape plug-in to decipher functionally grouped gene ontology and pathway annotation networks. Bioinformatics 2009, 25, 1091–1093. [Google Scholar] [CrossRef]
- Bindea, G.; Galon, J.; Mlecnik, B. CluePedia Cytoscape plugin: Pathway insights using integrated experimental and in silico data. Bioinformatics 2013, 29, 661–663. [Google Scholar] [CrossRef]
- Huang, D.W.; Sherman, B.T.; Lempicki, R.A. Systematic and integrative analysis of large gene lists using DAVID bioinformatics resources. Nat. Protoc. 2009, 4, 44. [Google Scholar] [CrossRef] [PubMed]
- Jiao, X.; Sherman, B.T.; Huang, D.W.; Stephens, R.; Baseler, M.W.; Lane, H.C.; Lempicki, R.A. DAVID-WS: A stateful web service to facilitate gene/protein list analysis. Bioinformatics 2012, 28, 1805–1806. [Google Scholar] [CrossRef]
- Grossman, R.L.; Heath, A.P.; Ferretti, V.; Varmus, H.E.; Lowy, D.R.; Kibbe, W.A.; Staudt, L.M. Toward a shared vision for cancer genomic data. New Engl. J. Med. 2016, 375, 1109–1112. [Google Scholar] [CrossRef] [PubMed]
- Weinstein, J.N.; Collisson, E.A.; Mills, G.B.; Shaw, K.R.M.; Ozenberger, B.A.; Ellrott, K.; Shmulevich, I.; Sander, C.; Stuart, J.M.; Network, C.G.A.R.; et al. The cancer genome atlas pan-cancer analysis project. Nat. Genet. 2013, 45, 1113. [Google Scholar] [CrossRef] [PubMed]
- Apiwattanakul, M.; Milone, M.; Pittock, S.J.; Kryzer, T.J.; Fryer, J.P.; O’toole, O.; Mckeon, A.; Lennon, V.A. Signal recognition particle immunoglobulin g detected incidentally associates with autoimmune myopathy. Muscle Nerve 2016, 53, 925–932. [Google Scholar] [CrossRef]
- Ren, Y.G.; Wagner, K.W.; Knee, D.A.; Aza-Blanc, P.; Nasoff, M.; Deveraux, Q.L. Differential regulation of the TRAIL death receptors DR4 and DR5 by the signal recognition particle. Mol. Biol. Cell 2004, 15, 5064–5074. [Google Scholar] [CrossRef]
- Zhong, B.; Bian, L.; Wang, G.; Zhou, Y.; Chen, Y.; Peng, F. Identification of key genes involved in HER2-positive breast cancer. Eur. Rev. Med. Pharmacol. Sci. 2016, 20, 664–672. [Google Scholar]
- Fahrmann, J.F.; Grapov, D.; Wanichthanarak, K.; DeFelice, B.C.; Salemi, M.R.; Rom, W.N.; Gandara, D.R.; Phinney, B.S.; Fiehn, O.; Pass, H.; et al. Integrated metabolomics and proteomics highlight altered nicotinamide and polyamine pathways in lung adenocarcinoma. Carcinogenesis 2017, 38, 271–280. [Google Scholar] [CrossRef]
- Currie, C.J.; Poole, C.D.; Jenkins-Jones, S.; Gale, E.A.; Johnson, J.A.; Morgan, C.L. Mortality after incident cancer in people with and without type 2 diabetes: Impact of metformin on survival. Diabetes Care 2012, 35, 299–304. [Google Scholar] [CrossRef]
- Currie, C.; Poole, C.; Gale, E. The influence of glucose-lowering therapies on cancer risk in type 2 diabetes. Diabetologia 2009, 52, 1766–1777. [Google Scholar] [CrossRef]
- Evans, J.M.; Donnelly, L.A.; Emslie-Smith, A.M.; Alessi, D.R.; Morris, A.D. Metformin and reduced risk of cancer in diabetic patients. Bmj 2005, 330, 1304–1305. [Google Scholar] [CrossRef] [PubMed]
- Hsu, P.P.; Sabatini, D.M. Cancer cell metabolism: Warburg and beyond. Cell 2008, 134, 703–707. [Google Scholar] [CrossRef] [PubMed]
- Hamanaka, R.B.; Chandel, N.S. Targeting glucose metabolism for cancer therapy. J. Exp. Med. 2012, 209, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Hirsch, H.A.; Iliopoulos, D.; Tsichlis, P.N.; Struhl, K. Metformin selectively targets cancer stem cells, and acts together with chemotherapy to block tumor growth and prolong remission. Cancer Res. 2009, 69, 7507–7511. [Google Scholar] [CrossRef]
- Heller, G.; Babinsky, V.N.; Ziegler, B.; Weinzierl, M.; Noll, C.; Altenberger, C.; Müllauer, L.; Dekan, G.; Grin, Y.; Lang, G.; et al. Genome-wide CpG island methylation analyses in non-small cell lung cancer patients. Carcinogenesis 2012, 34, 513–521. [Google Scholar] [CrossRef]
- Teng, Y.; Qin, H.; Bahassan, A.; Bendzunas, N.G.; Kennedy, E.J.; Cowell, J.K. The WASF3–NCKAP1–CYFIP1 complex is essential for breast cancer metastasis. Cancer Res. 2016, 76, 5133–5142. [Google Scholar] [CrossRef]
- Xiao, C.Z.; Wei, W.; Guo, Z.X.; Zhang, M.Y.; Zhang, Y.F.; Wang, J.H.; Shi, M.; Wang, H.Y.; Guo, R.P. MicroRNA-34c-3p promotes cell proliferation and invasion in hepatocellular carcinoma by regulation of NCKAP1 expression. J. Cancer Res. Clin. Oncol. 2017, 143, 263–273. [Google Scholar] [CrossRef]
- Luce, L.N.; Abbate, M.; Cotignola, J.; Giliberto, F. Non-myogenic tumors display altered expression of dystrophin (DMD) and a high frequency of genetic alterations. Oncotarget 2017, 8, 145. [Google Scholar] [CrossRef]
- Duggan, D.J.; Bittner, M.; Chen, Y.; Meltzer, P.; Trent, J.M. Expression profiling using cDNA microarrays. Nat. Genet. 1999, 21, 10. [Google Scholar] [CrossRef]
- Ben-Dor, A.; Shamir, R.; Yakhini, Z. Clustering gene expression patterns. J. Comput. Biol. 1999, 6, 281–297. [Google Scholar] [CrossRef]
- Majd, H.A.; Talebi, A.; Gilany, K.; Khayyer, N. Two-Way Gene Interaction From Microarray Data Based on Correlation Methods. Iran. Red Crescent Med. J. 2016, 18. [Google Scholar]
- Blomqvist, N. On a measure of dependence between two random variables. Ann. Math. Stat. 1950, 21, 593–600. [Google Scholar] [CrossRef]





© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Delgado-Chaves, F.M.; Gómez-Vela, F.; García-Torres, M.; Divina, F.; Vázquez Noguera, J.L. Computational Inference of Gene Co-Expression Networks for the identification of Lung Carcinoma Biomarkers: An Ensemble Approach. Genes 2019, 10, 962. https://doi.org/10.3390/genes10120962
Delgado-Chaves FM, Gómez-Vela F, García-Torres M, Divina F, Vázquez Noguera JL. Computational Inference of Gene Co-Expression Networks for the identification of Lung Carcinoma Biomarkers: An Ensemble Approach. Genes. 2019; 10(12):962. https://doi.org/10.3390/genes10120962
Chicago/Turabian StyleDelgado-Chaves, Fernando M., Francisco Gómez-Vela, Miguel García-Torres, Federico Divina, and José Luis Vázquez Noguera. 2019. "Computational Inference of Gene Co-Expression Networks for the identification of Lung Carcinoma Biomarkers: An Ensemble Approach" Genes 10, no. 12: 962. https://doi.org/10.3390/genes10120962
APA StyleDelgado-Chaves, F. M., Gómez-Vela, F., García-Torres, M., Divina, F., & Vázquez Noguera, J. L. (2019). Computational Inference of Gene Co-Expression Networks for the identification of Lung Carcinoma Biomarkers: An Ensemble Approach. Genes, 10(12), 962. https://doi.org/10.3390/genes10120962