iTRAQ-Based Protein Profiling Provides Insights into the Mechanism of Light-Induced Anthocyanin Biosynthesis in Chrysanthemum (Chrysanthemum × morifolium)


Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Materials
2.2. Design of Proteomic Libraries
2.3. Protein Sample Extraction and iTRAQ Labeling
2.4. High-Performance Liquid Chromatography (HPLC) Separation
2.5. LC–MS/MS Analysis Based on Q-Exactive
2.6. Data Analysis
3. Results
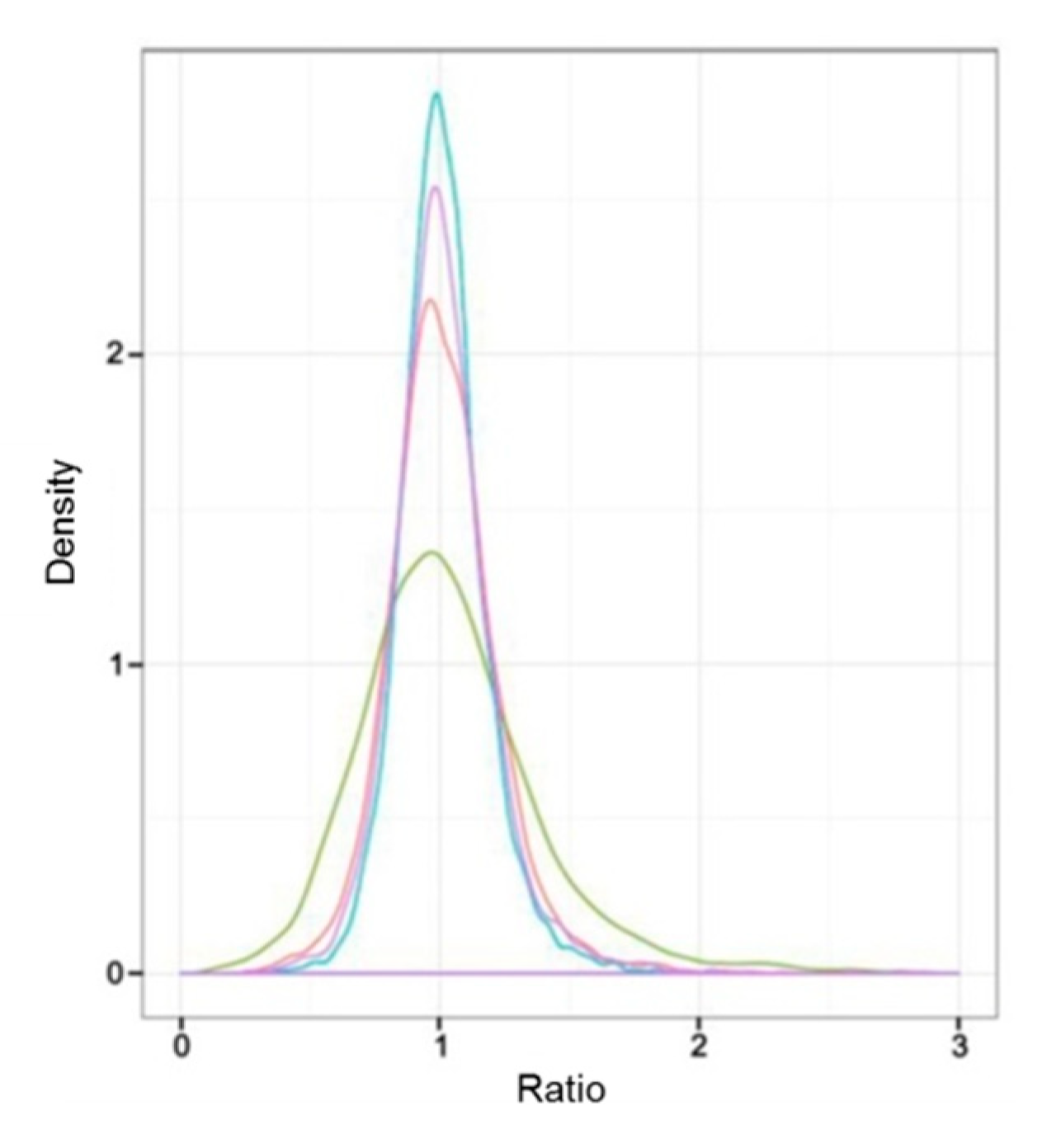
3.1. Overview of Quantitative Proteomics Analysis
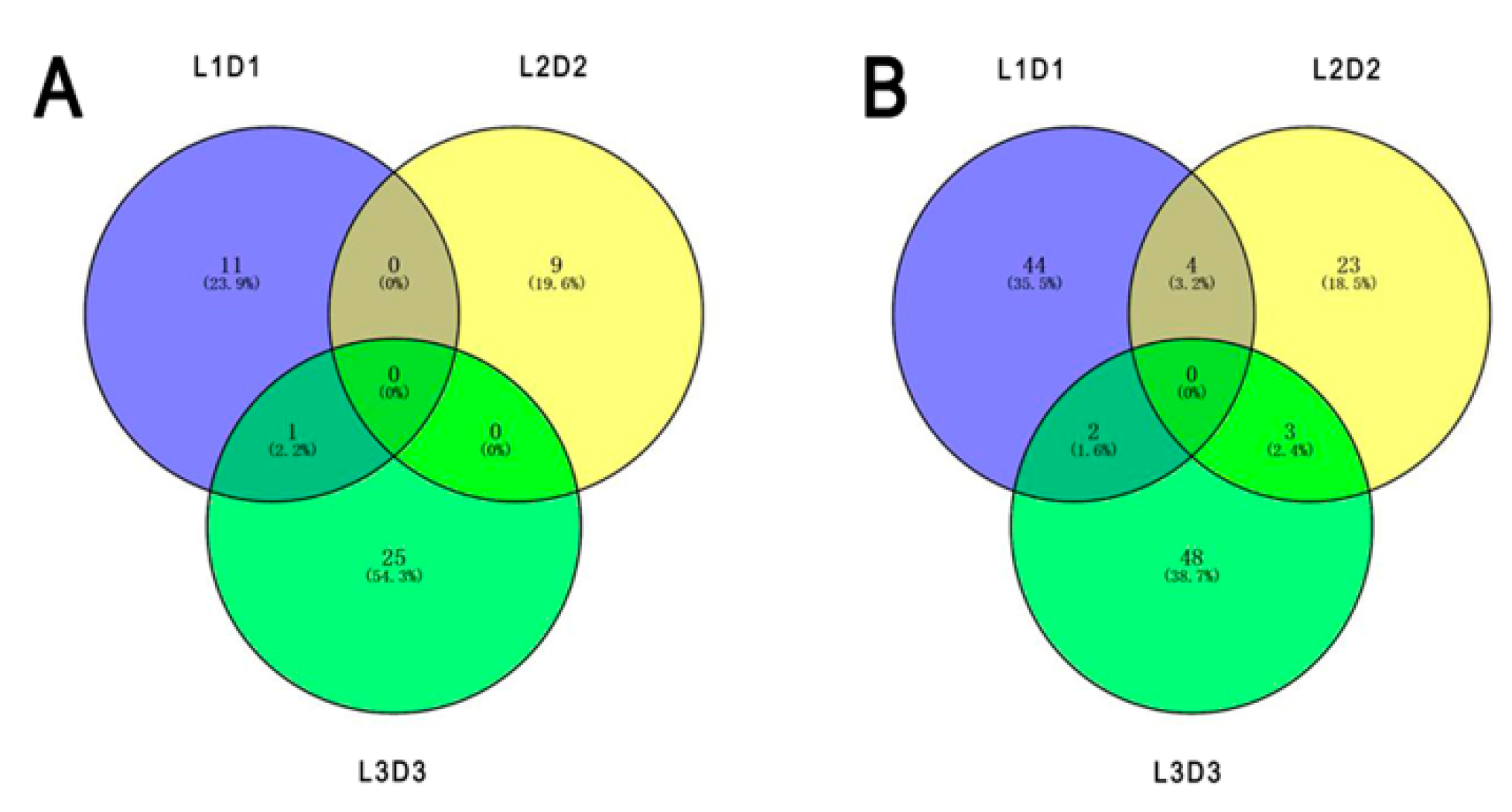
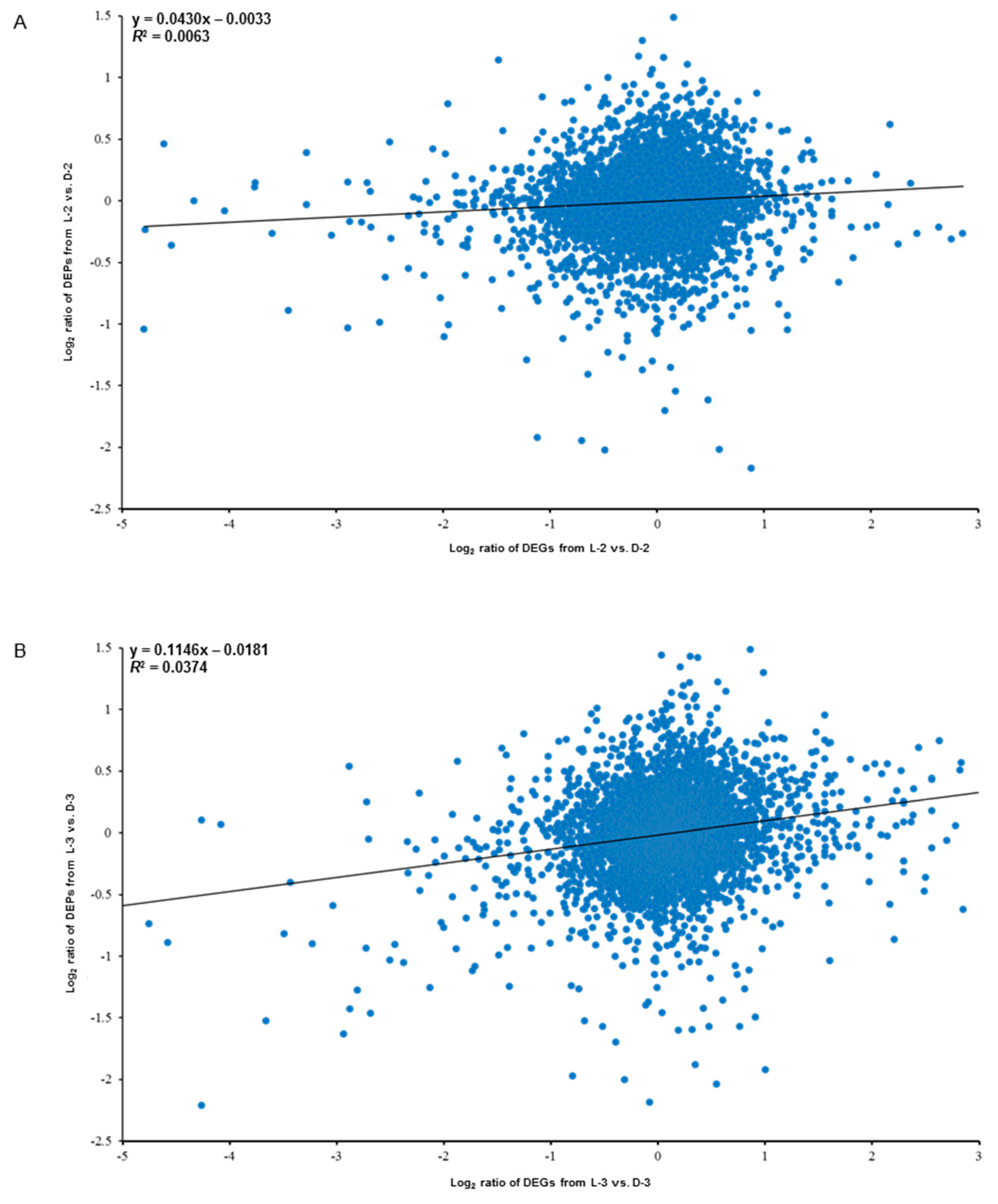
3.2. Correlation Between the Protein Expression and the Capitulum Development under Different Light Treatments
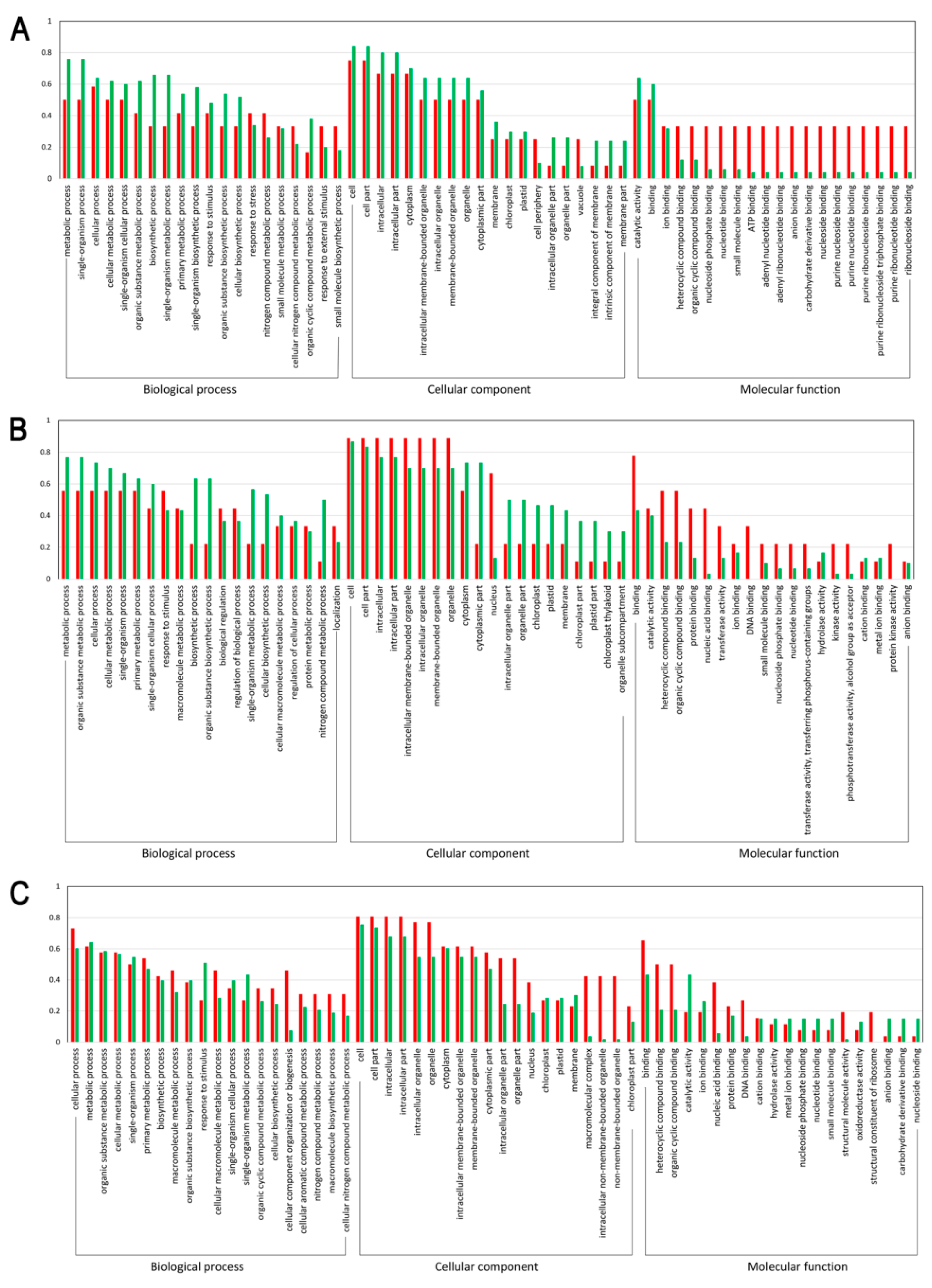
3.3. Functional Enrichment of the DEPs
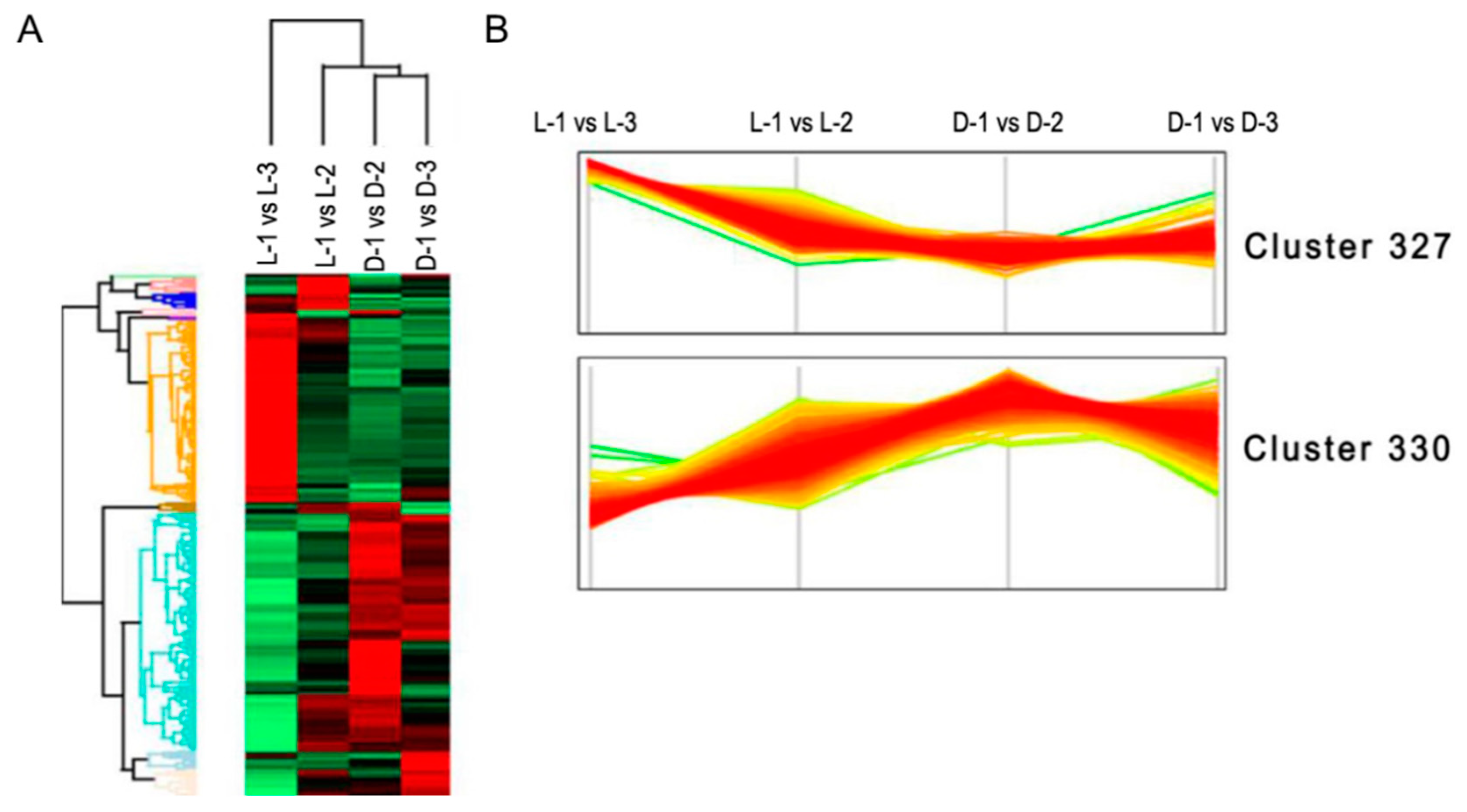
3.4. Clustering and Pathway Enrichment Analyses of the DEPs
3.5. Correlation between the Proteomic and the Transcriptomic Libraries
4. Discussion
4.1. Overview the RNA-Seq and iTRAQ Data
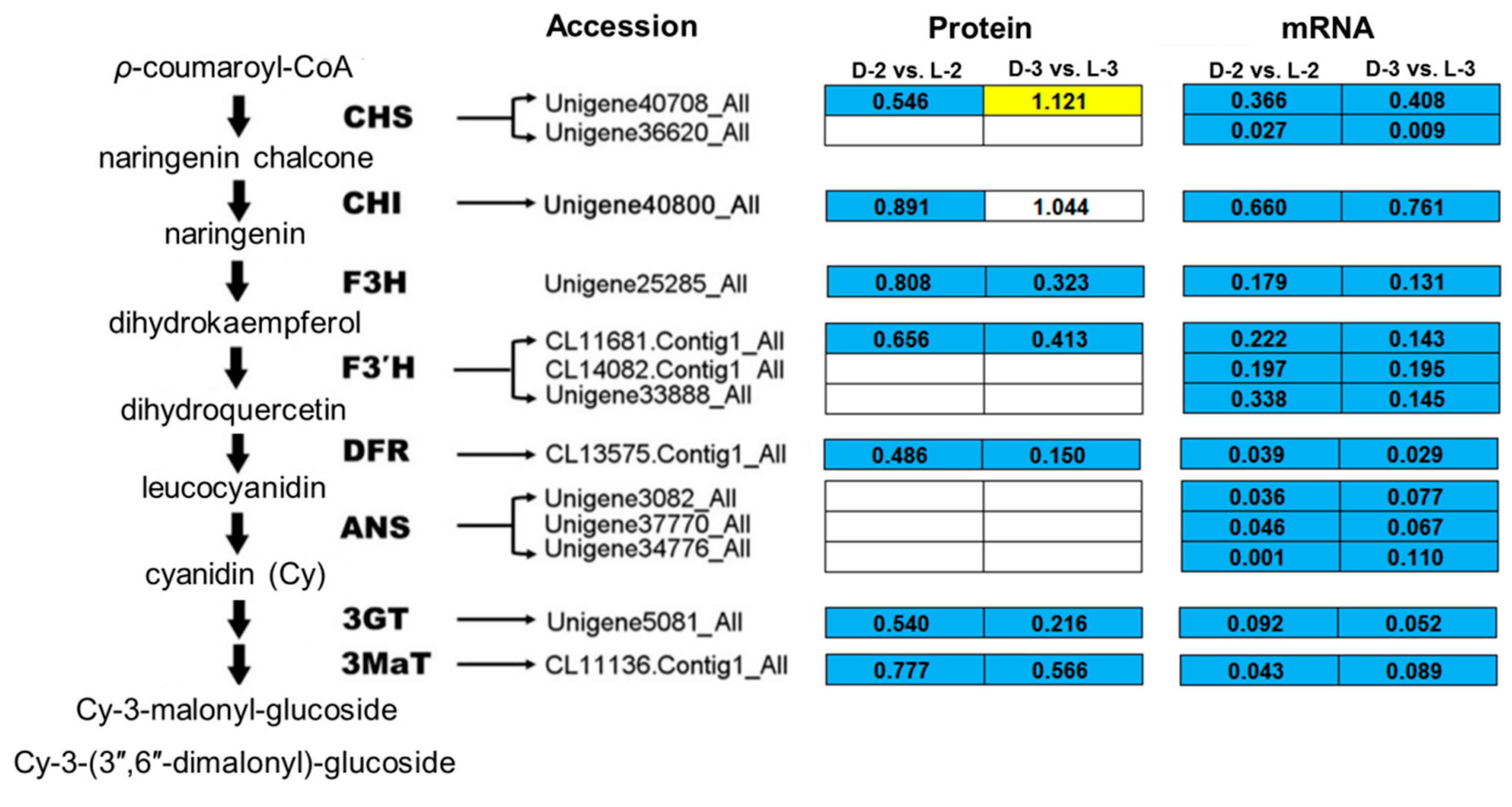
4.2. Proteins Involved in Anthocyanin Biosynthesis
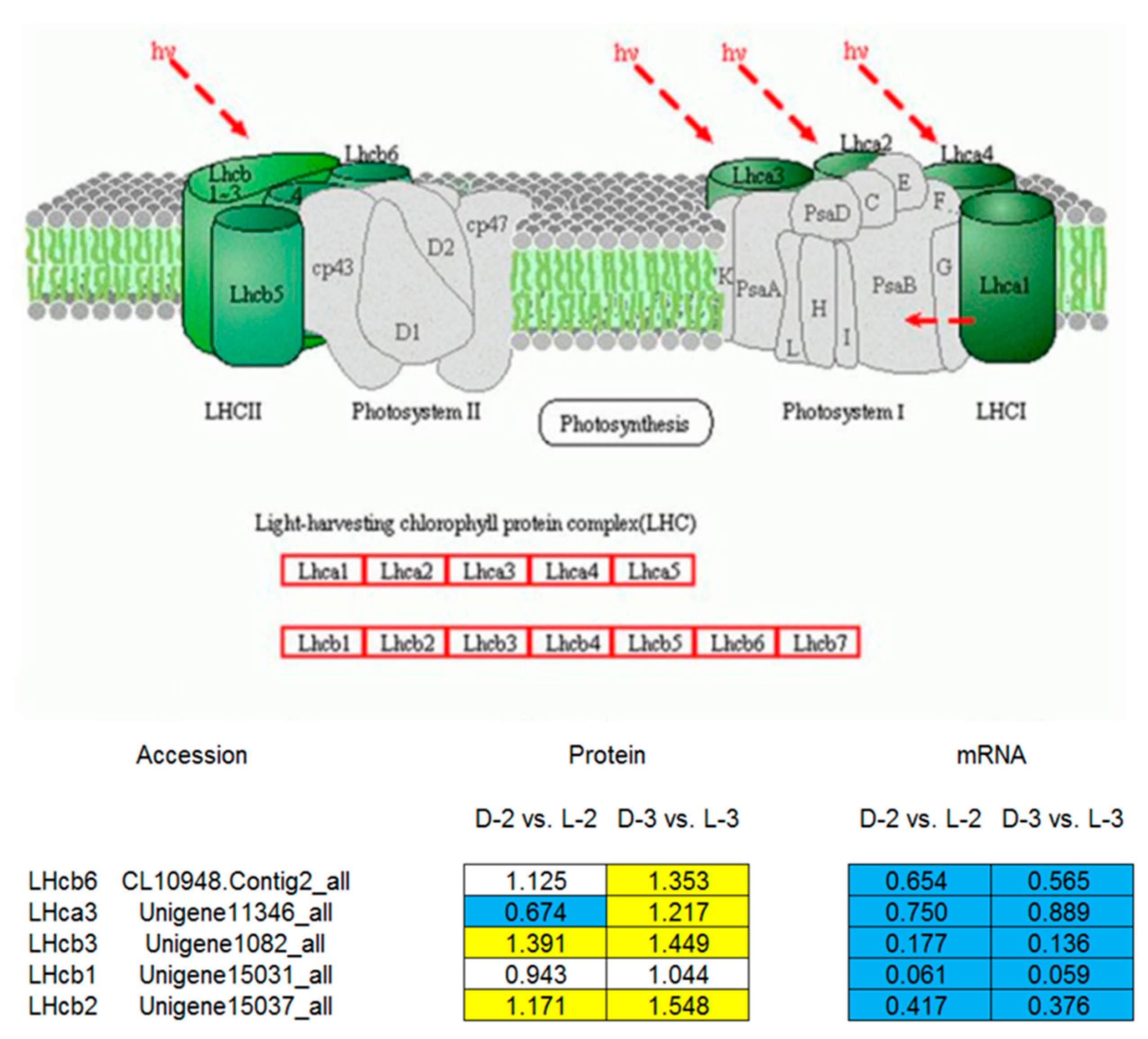
4.3. Proteins Involved in Photosynthesis
4.4. Putative Roles of GDSL Esterase APG
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Silva, V.O.; Freitas, A.A.; Maçanita, A.L.; Quina, F.H. Chemistry and photochemistry of natural plant pigments: The anthocyanins. J. Phys. Org. Chem. 2016, 29, 594–599. [Google Scholar] [CrossRef]
- Somerville, C.; Koornneef, M. A fortunate choice: The history of Arabidopsis as a model plant. Nat. Rev. Genet. 2002, 3, 883. [Google Scholar] [CrossRef] [PubMed]
- Iwashina, T. Contribution to flower colors of flavonoids including anthocyanins: A review. Nat. Prod. Commun. 2015, 10. [Google Scholar] [CrossRef]
- Zhang, H.; Koes, R.; Shang, H.; Fu, Z.; Wang, L.; Dong, X.; Zhang, J.; Passeri, V.; Li, Y.; Jiang, H.; et al. Identification and functional analysis of three new anthocyanin R2R3-MYB genes in Petunia. Plant Direct 2019, 3, e00114. [Google Scholar] [CrossRef]
- Hsu, C.C.; Chen, Y.Y.; Tsai, W.C.; Chen, W.H.; Chen, H.H. Three R2R3-MYB transcription factors regulate distinct floral pigmentation patterning in Phalaenopsis spp. Plant Physiol. 2015, 168, 175–191. [Google Scholar] [CrossRef]
- Li, C.; Qiu, J.; Ding, L.; Huang, M.; Huang, S.; Yang, G.; Yin, J. Anthocyanin biosynthesis regulation of DhMYB2 and DhbHLH1 in Dendrobium hybrids petals. Plant Physiol. Biochem. 2017, 112, 335–345. [Google Scholar] [CrossRef]
- Tanaka, Y.; Ohmiya, A. Seeing is believing: Engineering anthocyanin and carotenoid biosynthetic pathways. Curr. Opin. Biotechnol. 2008, 19, 190–197. [Google Scholar] [CrossRef]
- Albert, N.W.; Lewis, D.H.; Zhang, H.; Irving, L.J.; Jameson, P.E.; Davies, K.M. Light-induced vegetative anthocyanin pigmentation in Petunia. J. Exp. Bot. 2009, 60, 2191–2202. [Google Scholar] [CrossRef]
- Jaakola, L.; Hohtola, A. Effect of latitude on flavonoid biosynthesis in plants. Plant Cell Environ. 2010, 33, 1239–1247. [Google Scholar] [CrossRef]
- Jaakola, L. New insights into the regulation of anthocyanin biosynthesis in fruits. Trends Plant Sci. 2013, 18, 477–483. [Google Scholar] [CrossRef]
- Li, Y.Y.; Mao, K.; Zhao, C.; Zhao, X.Y.; Zhang, H.L.; Shu, H.R.; Hao, Y.J. MdCOP1 ubiquitin E3 ligases interact with MdMYB1 to regulate light-induced anthocyanin biosynthesis and red fruit coloration in apple. Plant Physiol. 2012, 160, 1011–1022. [Google Scholar] [CrossRef] [PubMed]
- Viola, I.L.; Camoirano, A.; Gonzalez, D.H. Redox-dependent modulation of anthocyanin biosynthesis by the TCP transcription factor TCP15 during exposure to high light intensity conditions in Arabidopsis. Plant Physiol. 2016, 170, 74–85. [Google Scholar] [CrossRef] [PubMed]
- Lotkowska, M.E.; Tohge, T.; Fernie, A.R.; Xue, G.P.; Balazadeh, S.; Mueller-Roeber, B. The Arabidopsis transcription factor MYB112 promotes anthocyanin formation during salinity and under high light stress. Plant Physiol. 2015, 169, 1862–1880. [Google Scholar] [CrossRef]
- Teixeira da Silva, J.A.; Shinoyama, H.; Aida, R.; Matsushita, Y.; Raj, S.K.; Chen, F. Chrysanthemum biotechnology: Quo vadis? Crit. Rev. Plant Sci. 2013, 32, 21–52. [Google Scholar] [CrossRef]
- Hong, Y.; Bai, X.; Sun, W.; Fu, J. The numerical classification of chrysanthemum flower color phenotype. Acta Hort. Sin. 2012, 39, 1330–1340. [Google Scholar]
- Nakayama, M.; Koshioka, M.; Shibata, M.; Hiradate, S.; Sugie, H.; Yamaguchi, M.A. Identification of cyanidin 3-O-(3″,6″-O-dimalonyl-β-glucopyranoside) as a flower pigment of Chrysanthemum (Dendranthema grandiflorum). Biosci. Biotechnol. Biochem. 1997, 61, 1607–1608. [Google Scholar] [CrossRef]
- Sun, W.; Li, C.H.; Wang, L.S.; Dai, S.L. Analysis of anthocyanins and flavones in different-colored flowers of chrysanthemum. Chin. Bullet. Bot. 2010, 45, 327–336. [Google Scholar]
- Liu, Y.; Chaturvedi, P.; Fu, J.; Cai, Q.; Weckwerth, W.; Yang, P. Induction and quantitative proteomic analysis of cell dedifferentiation during callus formation of lotus (Nelumbo nucifera Gaertn. spp. baijianlian). J. Proteomics 2016, 131, 61–70. [Google Scholar] [CrossRef]
- Wang, J.; Wang, X.R.; Zhou, Q.; Yang, J.M.; Guo, H.X.; Yang, L.J.; Liu, W.Q. iTRAQ protein profile analysis provides integrated insight into mechanisms of tolerance to TMV in tobacco (Nicotiana tabacum). J. Proteomics 2016, 132, 21–30. [Google Scholar] [CrossRef]
- Treumann, A.; Thiede, B. Isobaric protein and peptide quantification: Perspectives and issues. Exp. Rev. Proteomics 2010, 7, 647–653. [Google Scholar] [CrossRef]
- Noirel, J.; Evans, C.; Salim, M.; Mukherjee, J.; Yen Ow, S.; Pandhal, J.; Khoa Pham, T.; Biggs, C.A.; Wright, P.C. Methods in quantitative proteomics: Setting iTRAQ on the right track. Curr. Proteomics 2011, 8, 17–30. [Google Scholar] [CrossRef]
- Evans, C.; Noirel, J.; Ow, S.Y.; Salim, M.; Pereira-Medrano, A.G.; Couto, N.; Pandhal, J.; Smith, D.; Pham, T.K.; Karunakaran, E.; et al. An insight into iTRAQ: Where do we stand now? Anal. Bioanal. Chem. 2012, 404, 1011–1027. [Google Scholar] [CrossRef] [PubMed]
- Chu, P.; Yan, G.X.; Yang, Q.; Zhai, L.N.; Zhang, C.; Zhang, F.Q.; Guan, R.Z. iTRAQ-based quantitative proteomics analysis of Brassica napus leaves reveals pathways associated with chlorophyll deficiency. J. Proteomics 2015, 113, 244–259. [Google Scholar] [CrossRef] [PubMed]
- Xie, H.; Yang, D.H.; Yao, H.; Bai, G.; Zhang, Y.H.; Xiao, B.G. iTRAQ-based quantitative proteomic analysis reveals proteomic changes in leaves of cultivated tobacco (Nicotiana tabacum) in response to drought stress. Biochem. Biophys. Res. Commun. 2016, 469, 768–775. [Google Scholar] [CrossRef] [PubMed]
- Zhou, H.; Yu, Z.; Ye, Z. Key proteins associated to coloured compounds of peach peel using iTRAQ proteomic techniques during development and postharvest. Sci. Hortic. 2018, 239, 123–132. [Google Scholar] [CrossRef]
- Zhou, H.; Yu, Z.; Ye, Z. Effect of bagging duration on peach fruit peel color and key protein changes based on iTRAQ quantitation. Sci. Hortic. 2019, 246, 217–226. [Google Scholar] [CrossRef]
- Hong, Y.; Tang, X.; Huang, H.; Zhang, Y.; Dai, S. Transcriptomic analyses reveal species-specific light-induced anthocyanin biosynthesis in chrysanthemum. BMC Genomics 2015, 16, 202. [Google Scholar] [CrossRef]
- Omar, A.A.; Song, W.Y.; Grosser, J.W. Introduction of Xa21, a Xanthomonas-resistance gene from rice, into ‘Hamlin’ sweet orange [Citrus sinensis (L.) Osbeck] using protoplast-GFP co-transformation or single plasmid transformation. J. Hortic. Sci. Biotechnol. 2007, 82, 914–923. [Google Scholar] [CrossRef]
- Florens, L.; Carozza, M.J.; Swanson, S.K.; Fournier, M.; Coleman, M.K.; Workman, J.L.; Washburn, M.P. Analyzing chromatin remodeling complexes using shotgun proteomics and normalized spectral abundance factors. Methods 2006, 40, 303–311. [Google Scholar] [CrossRef]
- Yang, Z.; Li, W.; Su, X.; Ge, P.; Zhou, Y.; Hao, Y.; Shu, H.; Gao, C.; Cheng, S.; Zhu, G.; et al. Early response of radish to heat stress by strand-specific transcriptome and miRNA analysis. Int. J. Mol. Sci. 2019, 20, 3321. [Google Scholar] [CrossRef]
- Takos, A.M.; Jaffé, F.W.; Jacob, S.R.; Bogs, J.; Robinson, S.P.; Walker, A.R. Light-induced expression of a MYB gene regulates anthocyanin biosynthesis in red apples. Plant Physiol. 2006, 142, 1216–1232. [Google Scholar] [CrossRef] [PubMed]
- Ampomah-Dwamena, C.; McGhie, T.; Wibisono, R.; Montefiori, M.; Hellens, R.P.; Allan, A.C. The kiwifruit lycopene beta-cyclase plays a significant role in carotenoid accumulation in fruit. J. Exp. Bot. 2009, 60, 3765–3779. [Google Scholar] [CrossRef] [PubMed]
- Cazzonelli, C.I.; Pogson, B.J. Source to sink: Regulation of carotenoid biosynthesis in plants. Trends Plant Sci. 2010, 15, 266–274. [Google Scholar] [CrossRef] [PubMed]
- Das, P.K.; Geul, B.; Choi, S.B.; Yoo, S.D.; Park, Y.I. Photosynthesis-dependent anthocyanin pigmentation in Arabidopsis. Plant Signal. Behav. 2011, 6, 23–25. [Google Scholar] [CrossRef]
- Hong, Y.; Yang, L.; Li, M.; Dai, S. Comparative analyses of light-induced anthocyanin accumulation and gene expression between the ray florets and leaves in chrysanthemum. Plant Physiol. Biochem. 2016, 103, 120–132. [Google Scholar] [CrossRef]
- Pandey, A.; Mann, M. Proteomics to study genes and genomes. Nature 2000, 405, 837. [Google Scholar] [CrossRef]
- Li, J.M.; Huang, X.S.; Li, L.T.; Zheng, D.M.; Xue, C.; Zhang, S.L.; Wu, J. Proteome analysis of pear reveals key genes associated with fruit development and quality. Planta 2015, 241, 1363–1379. [Google Scholar] [CrossRef]
- Muers, M. Gene expression: Transcriptome to proteome and back to genome. Nat. Rev. Genet. 2011, 12, 518. [Google Scholar] [CrossRef]
- Li, J.; Ren, L.; Gao, Z.; Jiang, M.; Liu, Y.; Zhou, L.; He, Y.; Chen, H. Combined transcriptomic and proteomic analysis constructs a new model for light-induced anthocyanin biosynthesis in eggplant (Solanum melongena L.). Plant Cell Environ. 2017, 40, 3069–3087. [Google Scholar] [CrossRef]
- Luo, X.; Cao, D.; Li, H.; Zhao, D.; Xue, H.; Niu, J.; Chen, L.; Zhang, F.; Cao, S. Complementary iTRAQ-based proteomic and RNA sequencing-based transcriptomic analyses reveal a complex network regulating pomegranate (Punica granatum L.) fruit peel colour. Sci. Rep. 2018, 8, 12362. [Google Scholar] [CrossRef]
- Chen, J.; Liu, S.S.; Kohler, A.; Yan, B.; Luo, H.M.; Chen, X.M.; Guo, S.X. iTRAQ and RNA-Seq analyses provide new insights into regulation mechanism of symbiotic germination of Dendrobium officinale seeds (Orchidaceae). J. Proteome Res. 2017, 16, 2174–2187. [Google Scholar] [CrossRef]
- van Tunen, A.J.; Mur, L.A.; Recourt, K.; Gerats, A.G.; Mol, J.N. Regulation and manipulation of flavonoid gene expression in anthers of petunia: The molecular basis of the Po mutation. Plant Cell 1991, 3, 39–48. [Google Scholar]
- Berenschot, A.S.; Quecini, V. A reverse genetics approach identifies novel mutants in light responses and anthocyanin metabolism in petunia. Physiol. Mol. Biol. Plants 2014, 20, 1–13. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Niu, N.; Cao, Y.; Duan, W.; Wu, B.; Li, S. Proteomic analysis of grape berry skin responding to sunlight exclusion. J. Plant Physiol. 2013, 170, 748–757. [Google Scholar] [CrossRef] [PubMed]
- Maier, A.; Schrader, A.; Kokkelink, L.; Falke, C.; Welter, B.; Iniesto, E.; Rubio, V.; Uhrig, J.F.; Hülskamp, M.; Hoecker, U. Light and the E3 ubiquitin ligase COP1/SPA control the protein stability of the MYB transcription factors PAP 1 and PAP 2 involved in anthocyanin accumulation in Arabidopsis. Plant J. 2013, 74, 638–651. [Google Scholar] [CrossRef] [PubMed]
- Bellafiore, S.; Barneche, F.; Peltier, G.; Rochaix, J.D. State transitions and light adaptation require chloroplast thylakoid protein kinase STN7. Nature 2005, 433, 892. [Google Scholar] [CrossRef]
- Niyogi, K.K.; Li, X.P.; Rosenberg, V.; Jung, H.S. Is PsbS the site of non-photochemical quenching in photosynthesis? J. Exp. Bot. 2004, 56, 375–382. [Google Scholar] [CrossRef]
- Szabó, I.; Bergantino, E.; Giacometti, G.M. Light and oxygenic photosynthesis: Energy dissipation as a protection mechanism against photo-oxidation. EMBO Rep. 2005, 6, 629–634. [Google Scholar] [CrossRef]
- Teramoto, H.; Ono, T.; Minagawa, J. Identification of Lhcb gene family encoding the light-harvesting chlorophyll-a/b proteins of photosystem II in Chlamydomonas reinhardtii. Plant Cell Physiol. 2001, 42, 849–856. [Google Scholar] [CrossRef]
- Jansson, S. A guide to the Lhc genes and their relatives in Arabidopsis. Trends Plant Sci. 1999, 4, 236–240. [Google Scholar] [CrossRef]
- Koziol, A.G.; Borza, T.; Ishida, K.; Keeling, P.; Lee, R.W.; Durnford, D.G. Tracing the evolution of the light-harvesting antennae in chlorophyll a/b-containing organisms. Plant Physiol. 2007, 143, 1802–1816. [Google Scholar] [CrossRef] [PubMed]
- Dittami, S.M.; Michel, G.; Collén, J.; Boyen, C.; Tonon, T. Chlorophyll-binding proteins revisited-a multigenic family of light-harvesting and stress proteins from a brown algal perspective. BMC Evol. Biol. 2010, 10, 365. [Google Scholar] [CrossRef] [PubMed]
- Zhao, H.; Lou, Y.; Sun, H.; Li, L.; Wang, L.; Dong, L.; Gao, Z. Transcriptome and comparative gene expression analysis of Phyllostachys edulis in response to high light. BMC Plant Biol. 2016, 16, 34. [Google Scholar] [CrossRef] [PubMed]
- Laroche, J.; Mortain-Bertrand, A.; Falkowski, P.G. Light intensity-induced changes in cab mRNA and light harvesting complex II apoprotein levels in the unicellular chlorophyte Dunaliella tertiolecta. Plant Physiol. 1991, 97, 147–153. [Google Scholar] [CrossRef]
- Tanaka, R.; Koshino, Y.; Sawa, S.; Ishiguro, S.; Okada, K.; Tanaka, A. Overexpression of chlorophyllide a oxygenase (CAO) enlarges the antenna size of photosystem II in Arabidopsis thaliana. Plant J. 2001, 26, 365–373. [Google Scholar] [CrossRef]
- Masuda, T.; Tanaka, A.; Melis, A. Chlorophyll antenna size adjustments by irradiance in Dunaliella salina involve coordinate regulation of chlorophyll a oxygenase (CAO) and Lhcb gene expression. Plant Mol. Biol. 2003, 51, 757–771. [Google Scholar] [CrossRef]
- Kunugi, M.; Satoh, S.; Ihara, K.; Shibata, K.; Yamagishi, Y.; Kogame, K.; Obokata, J.; Takabayashi, A.; Tanaka, A. Evolution of green plants accompanied changes in light-harvesting systems. Plant Cell Physiol. 2016, 57, 1231–1243. [Google Scholar] [CrossRef]
- Yamamoto, Y.Y.; Nakamura, M.; Kondo, Y.; Tsuji, H.; Obokata, J. Early light-response of psaD, psaE and psaH gene families of photosystem I in Nicotiana sylvestris: PSI-D has an isoform of very quick response. Plant Cell Physiol. 1995, 36, 727–732. [Google Scholar]
- Kley, J.; Schmidt, B.; Boyanov, B.; Stolt-Bergner, P.C.; Kirk, R.; Ehrmann, M.; Knopf, R.R.; Naveh, L.; Adam, Z.; Clausen, T. Structural adaptation of the plant protease Deg1 to repair photosystem II during light exposure. Nat. Struct. Mol. Biol. 2011, 18, 728. [Google Scholar] [CrossRef]
- Roose, J.L.; Frankel, L.K.; Mummadisetti, M.P.; Bricker, T.M. The extrinsic proteins of photosystem II: Update. Planta 2016, 243, 889–908. [Google Scholar] [CrossRef]
- Akoh, C.C.; Lee, G.C.; Liaw, Y.C.; Huang, T.H.; Shaw, J.F. GDSL family of serine esterases/lipases. Prog. Lipid Res. 2004, 43, 534–552. [Google Scholar] [CrossRef] [PubMed]
- Ling, H. Sequence analysis of GDSL lipase gene family in Arabidopsis thaliana. Pak. J. Biol. Sci. 2008, 11, 763. [Google Scholar] [CrossRef] [PubMed]
- Chepyshko, H.; Lai, C.P.; Huang, L.M.; Liu, J.H.; Shaw, J.F. Multifunctionality and diversity of GDSL esterase/lipase gene family in rice (Oryza sativa L. japonica) genome: New insights from bioinformatics analysis. BMC Genomics 2012, 13, 309. [Google Scholar]
- Mayfield, J.A.; Fiebig, A.; Johnstone, S.E.; Preuss, D. Gene families from the Arabidopsis thaliana pollen coat proteome. Science 2001, 292, 2482–2485. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, K.; Shimada, T.; Kondo, M.; Tamai, A.; Mori, M.; Nishimura, M.; Hara-Nishimura, I. Ectopic expression of an esterase, which is a candidate for the unidentified plant cutinase, causes cuticular defects in Arabidopsis thaliana. Plant Cell Physiol. 2009, 51, 123–131. [Google Scholar] [CrossRef]
- Chen, C.C.; Fu, S.F.; Lee, Y.I.; Lin, C.Y.; Lin, W.C.; Huang, H.J. Transcriptome analysis of age-related gain of callus-forming capacity in Arabidopsis hypocotyls. Plant Cell Physiol. 2012, 53, 1457–1469. [Google Scholar] [CrossRef]
- Park, J.J.; Jin, P.; Yoon, J.; Yang, J.I.; Jeong, H.J.; Ranathunge, K.; Schreiber, L.; Franke, R.; Lee, I.J.; An, G. Mutation in Wilted Dwarf and Lethal 1 (WDL1) causes abnormal cuticle formation and rapid water loss in rice. Plant Mol. Biol. 2010, 74, 91–103. [Google Scholar] [CrossRef]
- Riemann, M.; Gutjahr, C.; Korte, A.; Riemann, M.; Danger, B.; Muramatsu, T.; Bayer, U.; Waller, F.; Furuya, M.; Nick, P. GER1, a GDSL motif-encoding gene from rice is a novel early light-and jasmonate-induced gene. Plant Biol. 2007, 9, 32–40. [Google Scholar] [CrossRef]
- Takano, M.; Inagaki, N.; Xie, X.; Yuzurihara, N.; Hihara, F.; Ishizuka, T.; Yano, M.; Nishimura, M.; Miyao, A.; Hirochika, H.; et al. Distinct and cooperative functions of phytochromes A, B, and C in the control of deetiolation and flowering in rice. Plant Cell 2005, 17, 3311–3325. [Google Scholar] [CrossRef]
- Hong, Y.; Li, M.; Dai, S. Ectopic expression of multiple chrysanthemum (Chrysanthemum × morifolium) R2R3-MYB transcription factor genes regulates anthocyanin accumulation in tobacco. Genes 2019, 10, 777. [Google Scholar] [CrossRef]
- Yang, Y.; Yang, X.; Jang, Z.; Chen, Z.; Ruo, X.; Jin, W.; Wu, Y.; Shi, X.; Xu, M. UV RESISTANCE LOCUS 8 from Chrysanthemum morifolium Ramat (CmUVR8) plays important roles in UV-B signal transduction and UV-B-induced accumulation of flavonoids. Front. Plant Sci. 2018, 9, 955. [Google Scholar] [CrossRef] [PubMed]









{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| No. | Expressive Pattern of Proteins | Sample Intersection | Encoding Unigenes | Species | Functional Annotation in NCBI-nr | Ko ID |
|---|---|---|---|---|---|---|
| 1 | Upregulated | L-1, D-1, L-3, D-3 | Unigene28439_All_1 | Acanthopanax senticosus | SSR sequence | – |
| 2 | Downregulated | L-1, D-1, L-2, D-2 | CL9080.Contig2_All | Glycine max | Protease Do-like 8 (PS II) | K01362 |
| 3 | Unigene6234_All | Asteraceae spp. | psaA (PS I) | K02689 | ||
| 4 | Unigene7244_All | Vitis vinifera | Auxin-induced protein 5NG4 | – | ||
| 5 | Unigene29413_All | Nicotiana tabacum | psaH (PS II) | K02695 | ||
| 6 | L-2, D-2, L-3, D-3 | CL972.Contig2_All | Taraxacum mongolicum | TO41-1rc mRNA | – | |
| 7 | CL13575.Contig1_All | Chrysanthemum × morifolium | DFR | K13082 | ||
| 8 | CL2096.Contig1_All | N. tabacum | Non-specific lipid-transfer protein | – | ||
| 9 | L-1, D-1, L-3, D-3 | Unigene36275_All | V. vinifera | ABC transporter A family member 3 | K05643 | |
| 10 | Unigene21072_All | Solanum lycopersicum | d-galactonate dehydratase | K01684 |
| No. | Dispersion | Accession | Species | Functional Annotation in NCBI-nr |
|---|---|---|---|---|
| 1 | Log2 ratio of DEP < −1 and Log2 ratio of DEG < −2 | CL13575.Contig1_All | Chrysanthemum × morifolium | DFR |
| 2 | CL11681.Contig1_All | C. × morifolium | F3’H | |
| 3 | Unigene23847_All | C. × morifolium | F5H | |
| 4 | Unigene5081_All | Lobelia erinus | 3GT | |
| 5 | Unigene25285_All | Nicotiana tabacum | F3H | |
| 6 | CL4884.Contig1_All | Ricinus communis | APG | |
| 7 | Log2 ratio of DEP > 0.5 and Log2 ratio of DEG > 2 | Unigene20184_All | Vitis vinifera | GA3OX4 |
| 8 | CL6029.Contig1_All | Actinidia deliciosa | Polygalacturonase inhibitor | |
| 9 | Unigene37908_All | Elaeis guineensis | Auxin-repressed protein | |
| 10 | Log2 ratio of DEP < −1.5 and −0.5 < Log2 ratio of DEG < 0.5 | CL11963.Contig1_All | Helianthus annuus | 17.6 kDa HSP |
| 11 | Unigene37058_All | H. annuus | Nonspecific lipid-transfer protein | |
| 12 | CL4402.Contig1_All | Lactuca sativa | HSP90 | |
| 13 | CL4402.Contig3_All | Solanum lycopersicum | HSP83 | |
| 14 | Unigene10675_All | V. vinifera | 17.4 kDa HSP | |
| 15 | CL1186.Contig4_All | Ageratina adenophora | 17.7 kDa HSP | |
| 16 | Unigene28001_All | V. vinifera | HSP83 | |
| 17 | Unigene40423_All | R. communis | Putative HSP |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hong, Y.; Li, M.; Dai, S. iTRAQ-Based Protein Profiling Provides Insights into the Mechanism of Light-Induced Anthocyanin Biosynthesis in Chrysanthemum (Chrysanthemum × morifolium). Genes 2019, 10, 1024. https://doi.org/10.3390/genes10121024
Hong Y, Li M, Dai S. iTRAQ-Based Protein Profiling Provides Insights into the Mechanism of Light-Induced Anthocyanin Biosynthesis in Chrysanthemum (Chrysanthemum × morifolium). Genes. 2019; 10(12):1024. https://doi.org/10.3390/genes10121024
Chicago/Turabian StyleHong, Yan, Mengling Li, and Silan Dai. 2019. "iTRAQ-Based Protein Profiling Provides Insights into the Mechanism of Light-Induced Anthocyanin Biosynthesis in Chrysanthemum (Chrysanthemum × morifolium)" Genes 10, no. 12: 1024. https://doi.org/10.3390/genes10121024
APA StyleHong, Y., Li, M., & Dai, S. (2019). iTRAQ-Based Protein Profiling Provides Insights into the Mechanism of Light-Induced Anthocyanin Biosynthesis in Chrysanthemum (Chrysanthemum × morifolium). Genes, 10(12), 1024. https://doi.org/10.3390/genes10121024