Intermediate-Salinity Systems at High Altitudes in the Peruvian Andes Unveil a High Diversity and Abundance of Bacteria and Viruses

 ,
,  , ,
, ,
Abstract
1. Introduction
2. Materials and Methods
2.1. Sampling, DNA Extraction, and Sequencing
2.2. Quality Control and Assembly
2.3. Microbial Community Taxonomic Assignments
2.4. Diversity Index
2.5. Genome Reconstruction
2.6. Binning for Putative Genomes
2.7. Functional Analysis and Biogeochemical Cycles
3. Results and Discussion
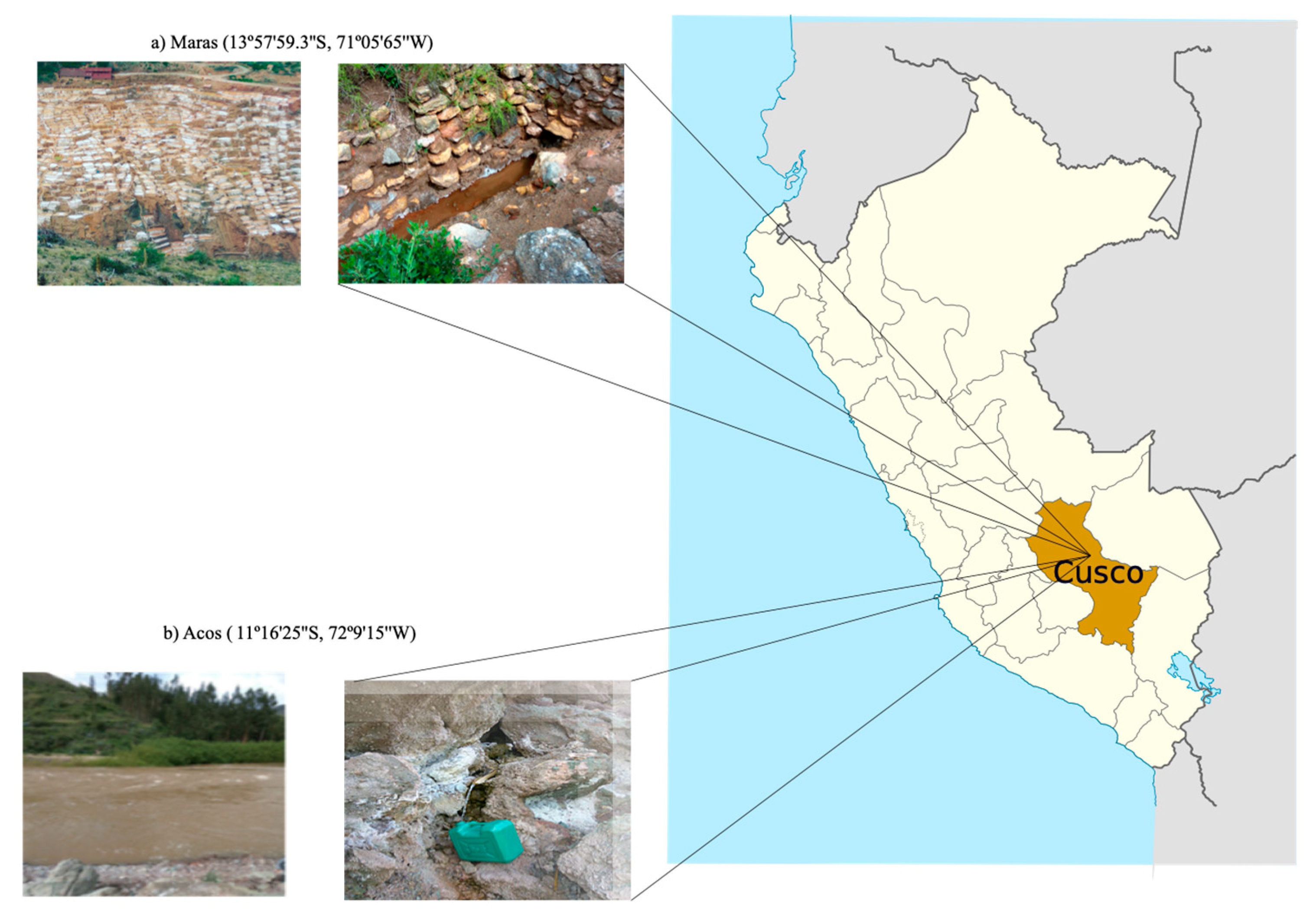
3.1. Site Characterization and Field Sampling
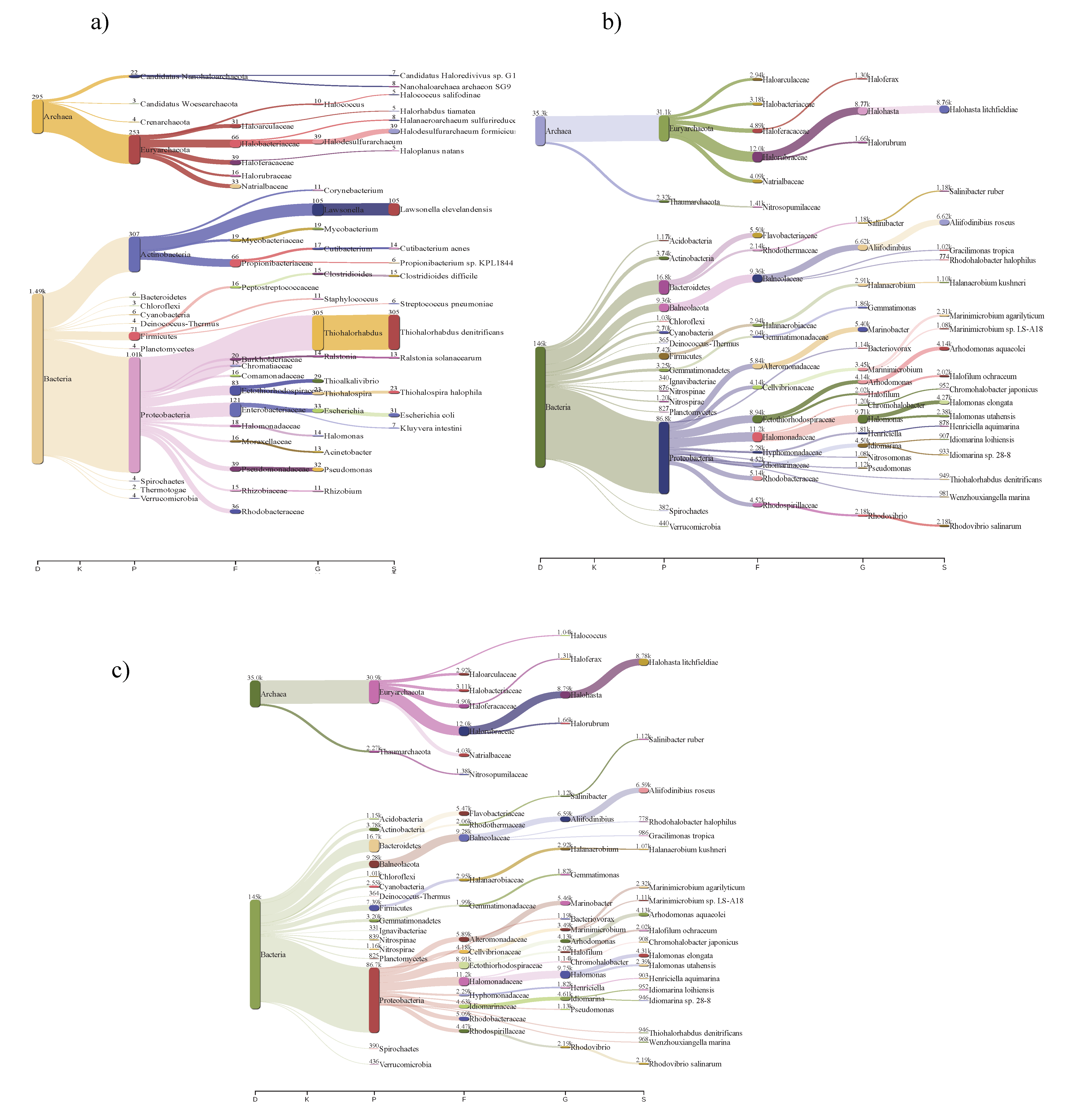
3.2. Community Structures of Intermediate Hypersaline Systems
3.3. Bacterial and Archaeal Community Composition
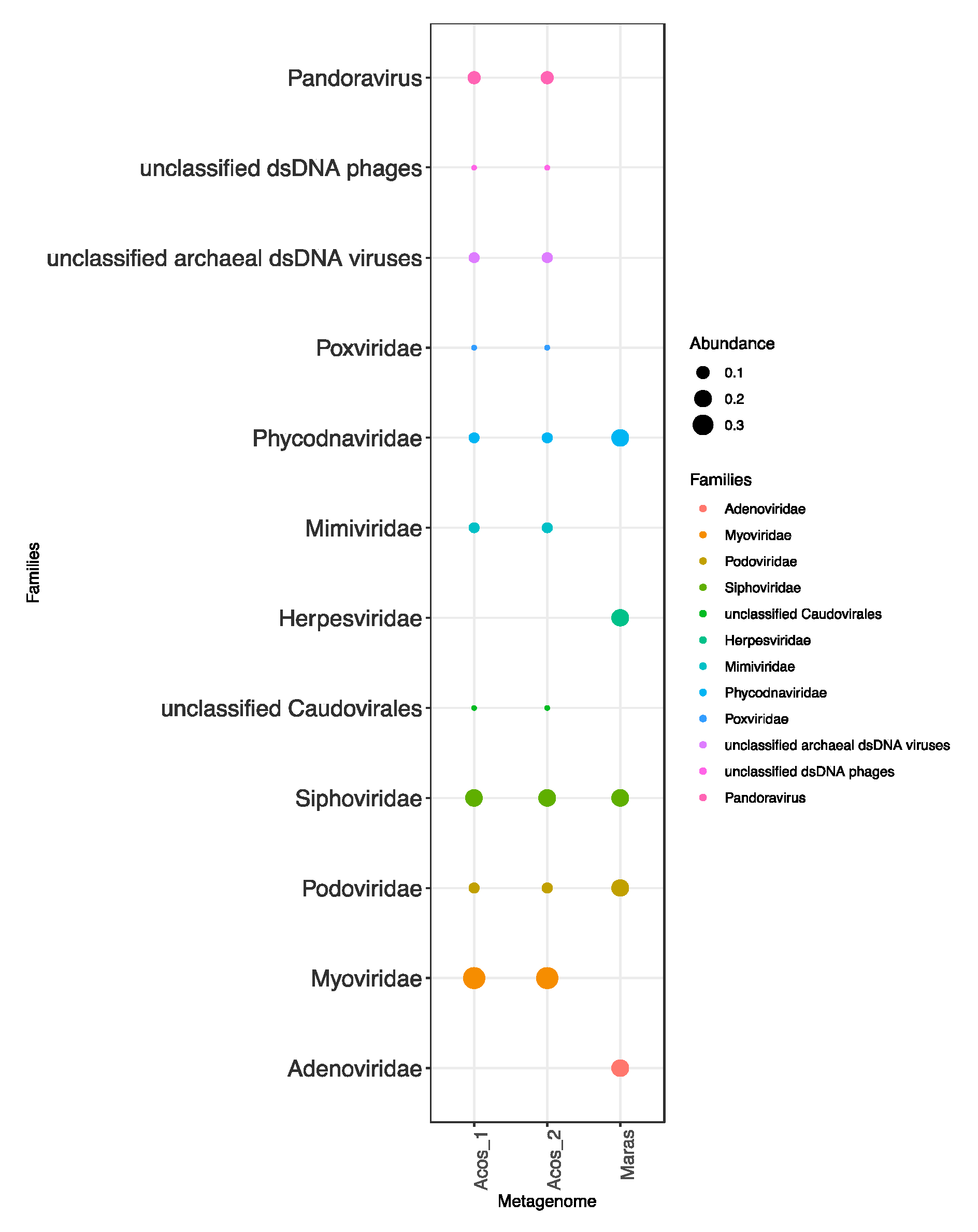
3.4. Composition of the Viral Community
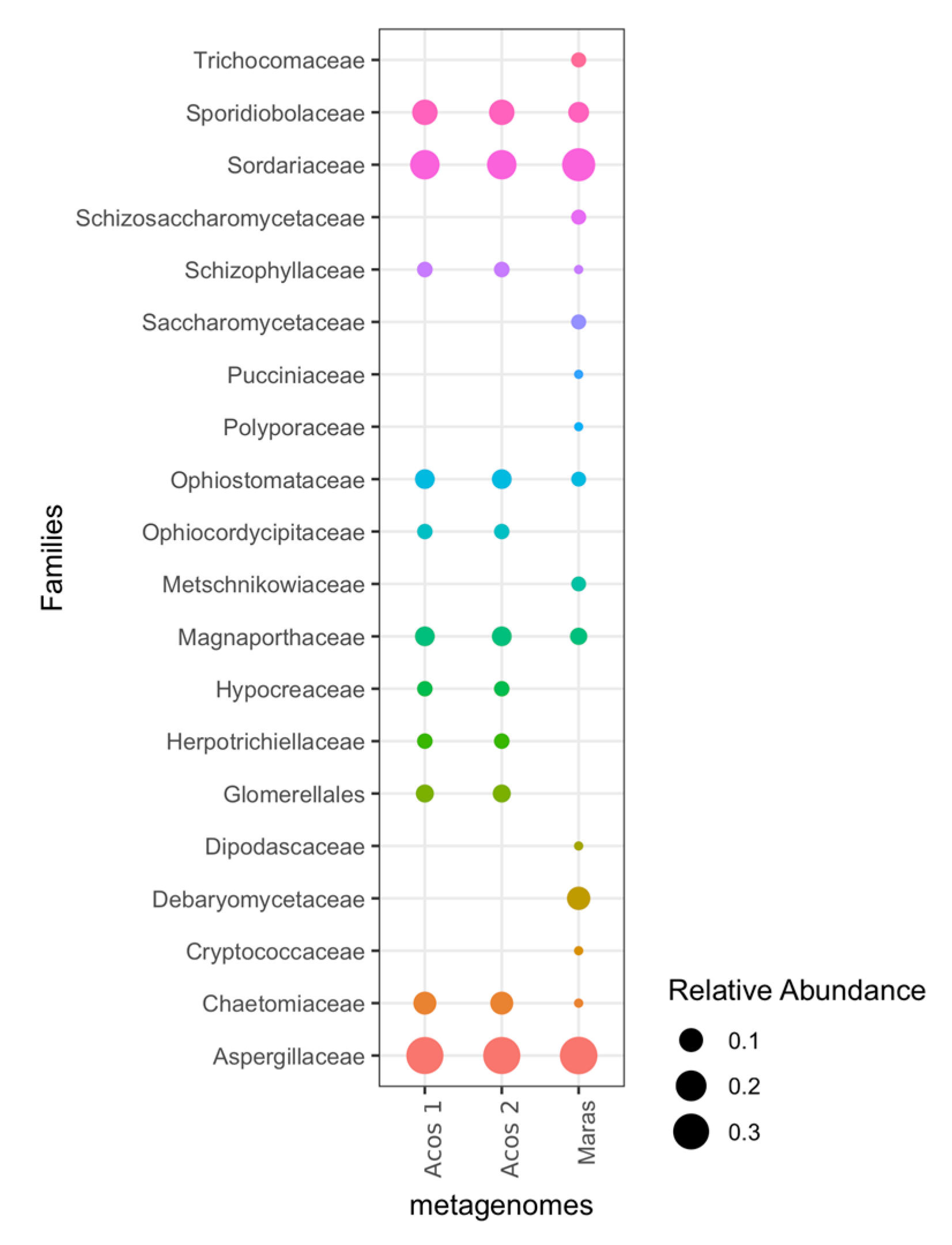
3.5. Composition of Fungal Communities
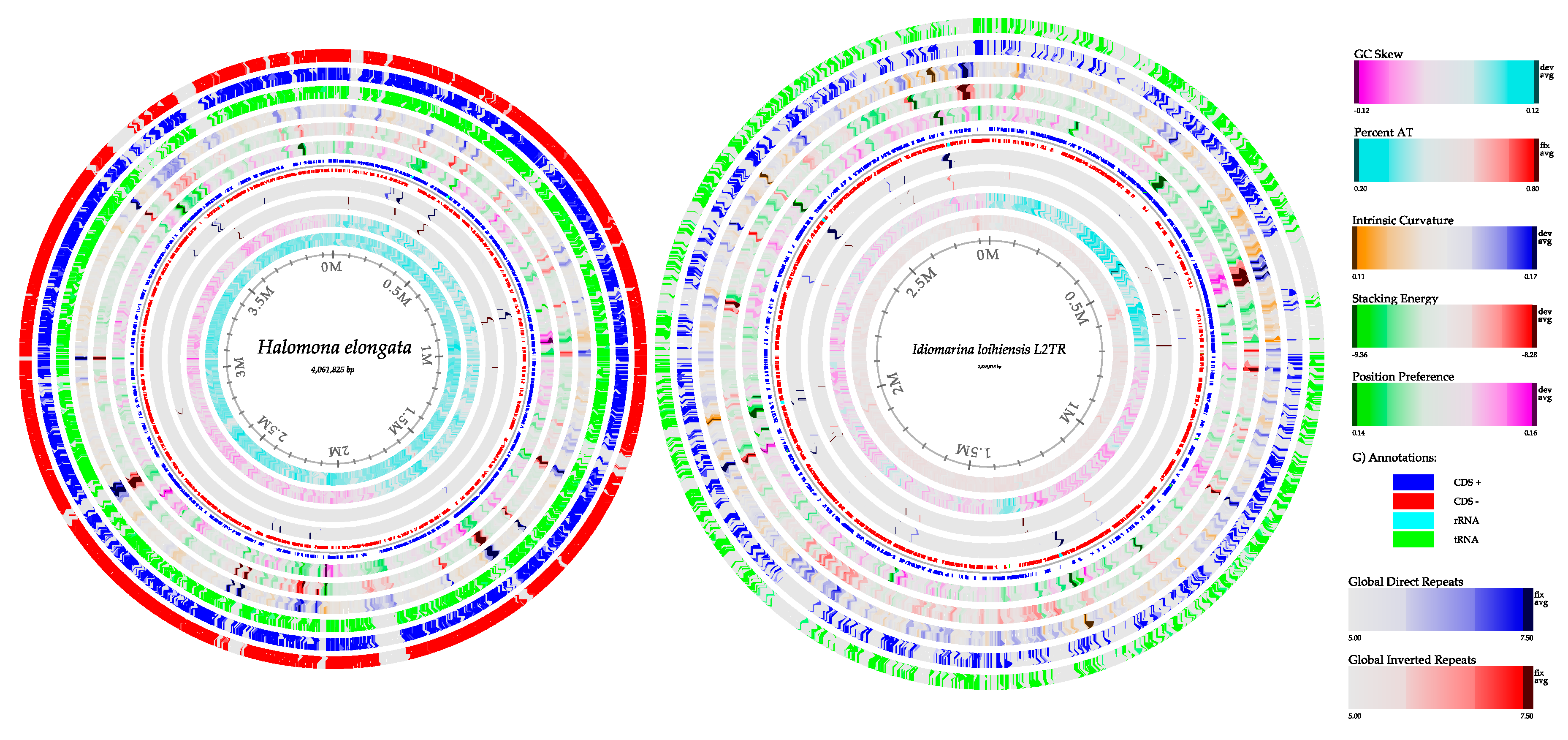
3.6. Genome Reconstruction
3.6.1. Bacterial Genome Reconstruction in A Hypersaline Environment
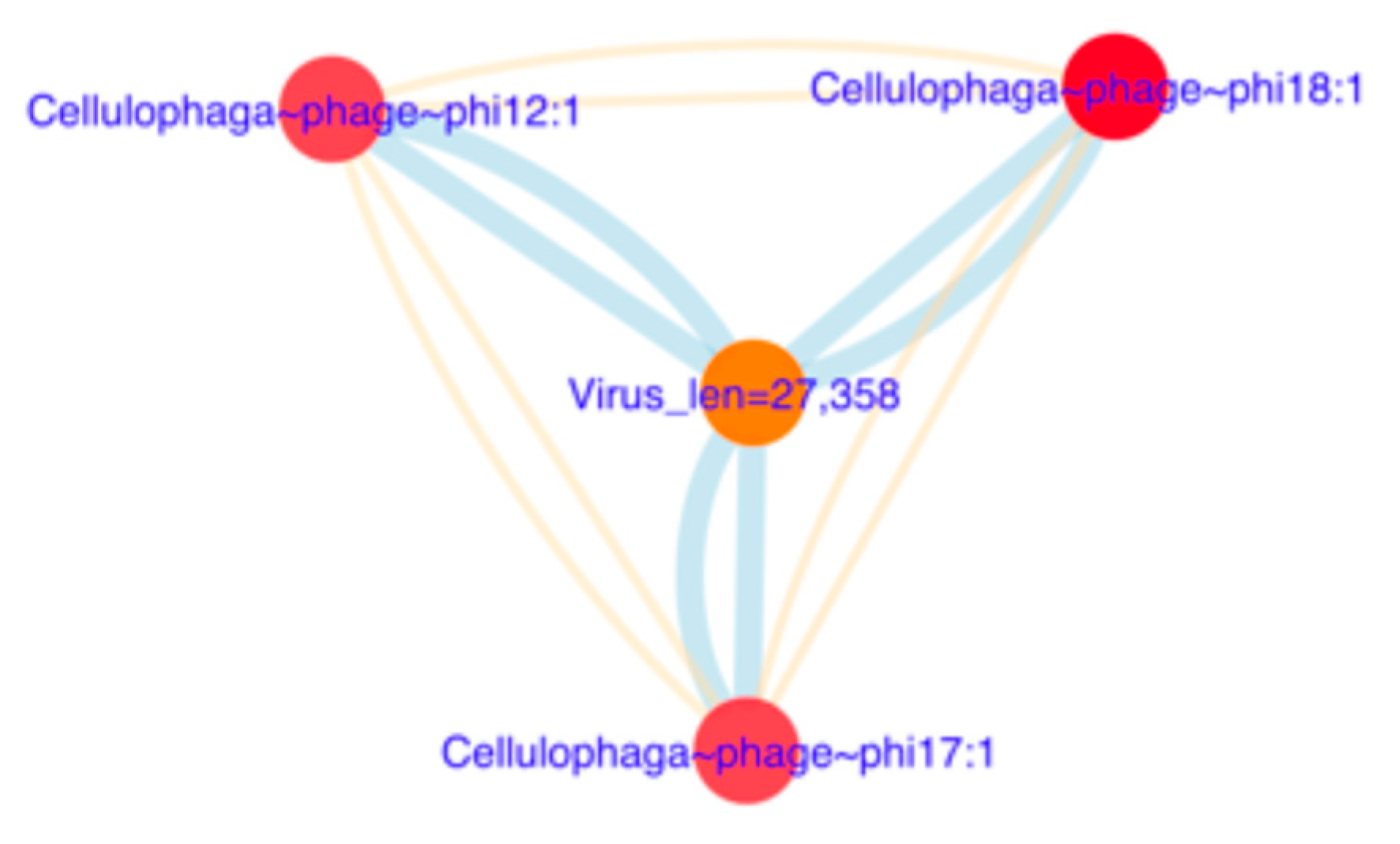
3.6.2. Reconstruction of Viral Genomes
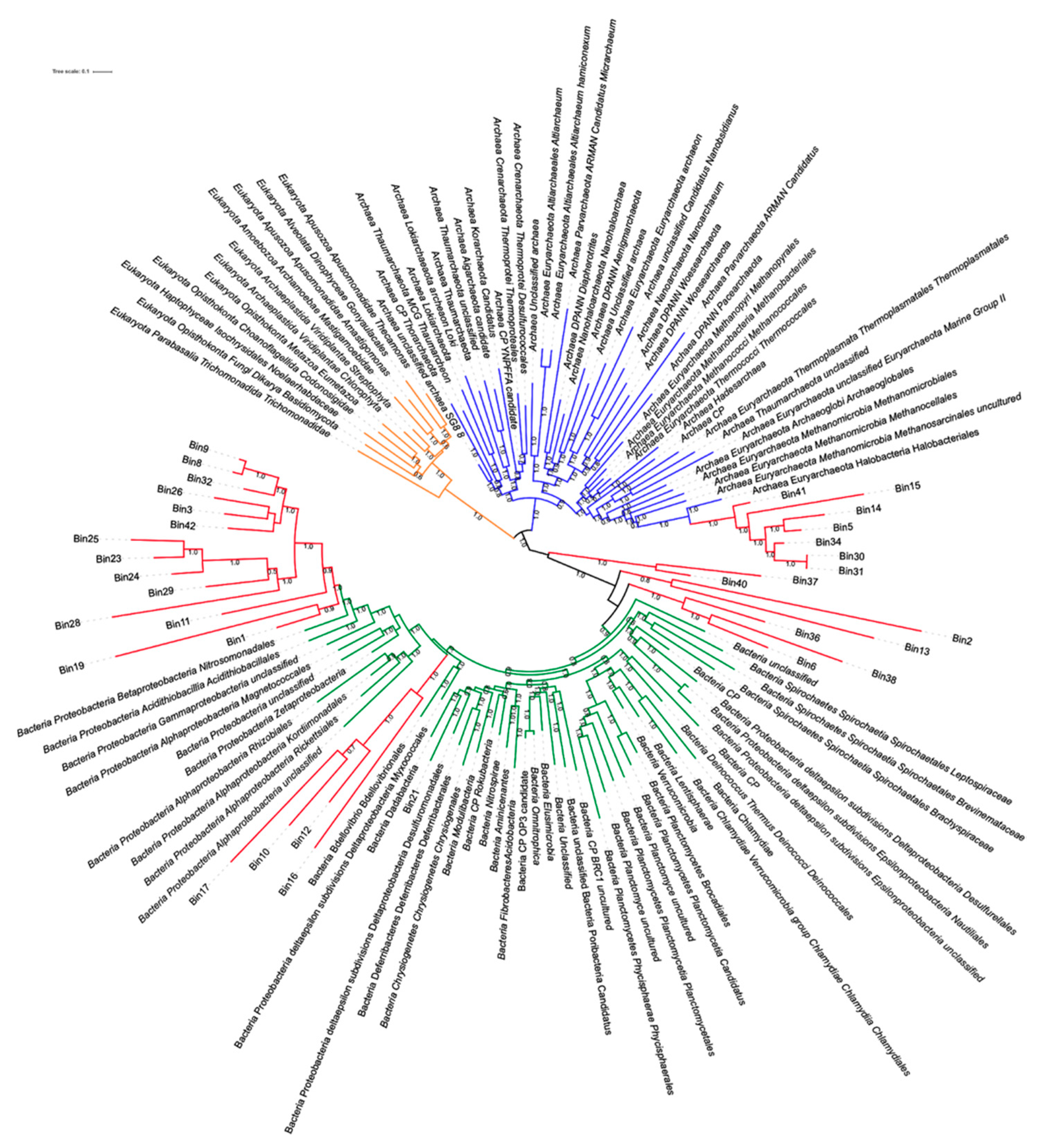
3.6.3. MAGs
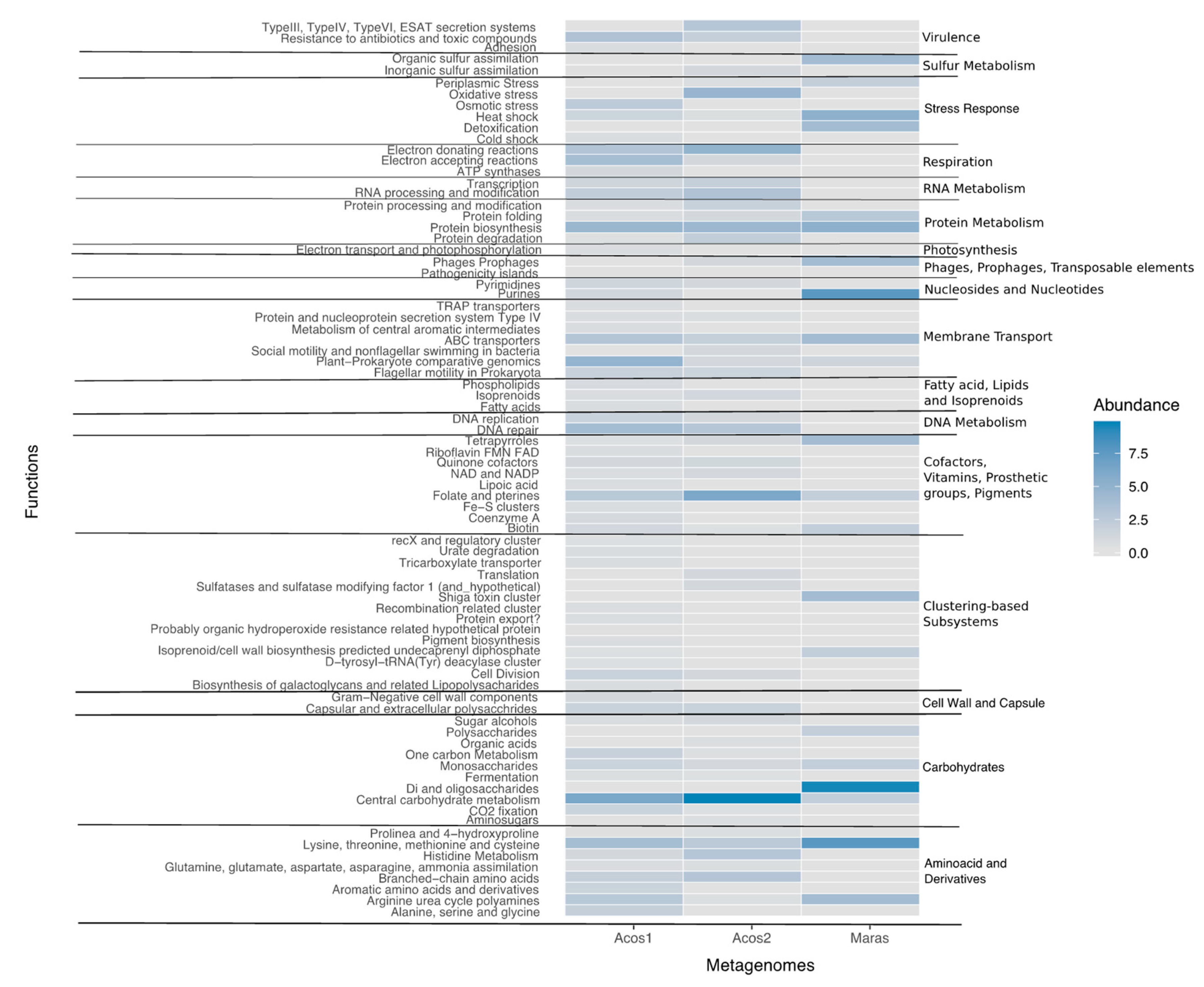
3.7. Functional Community Composition
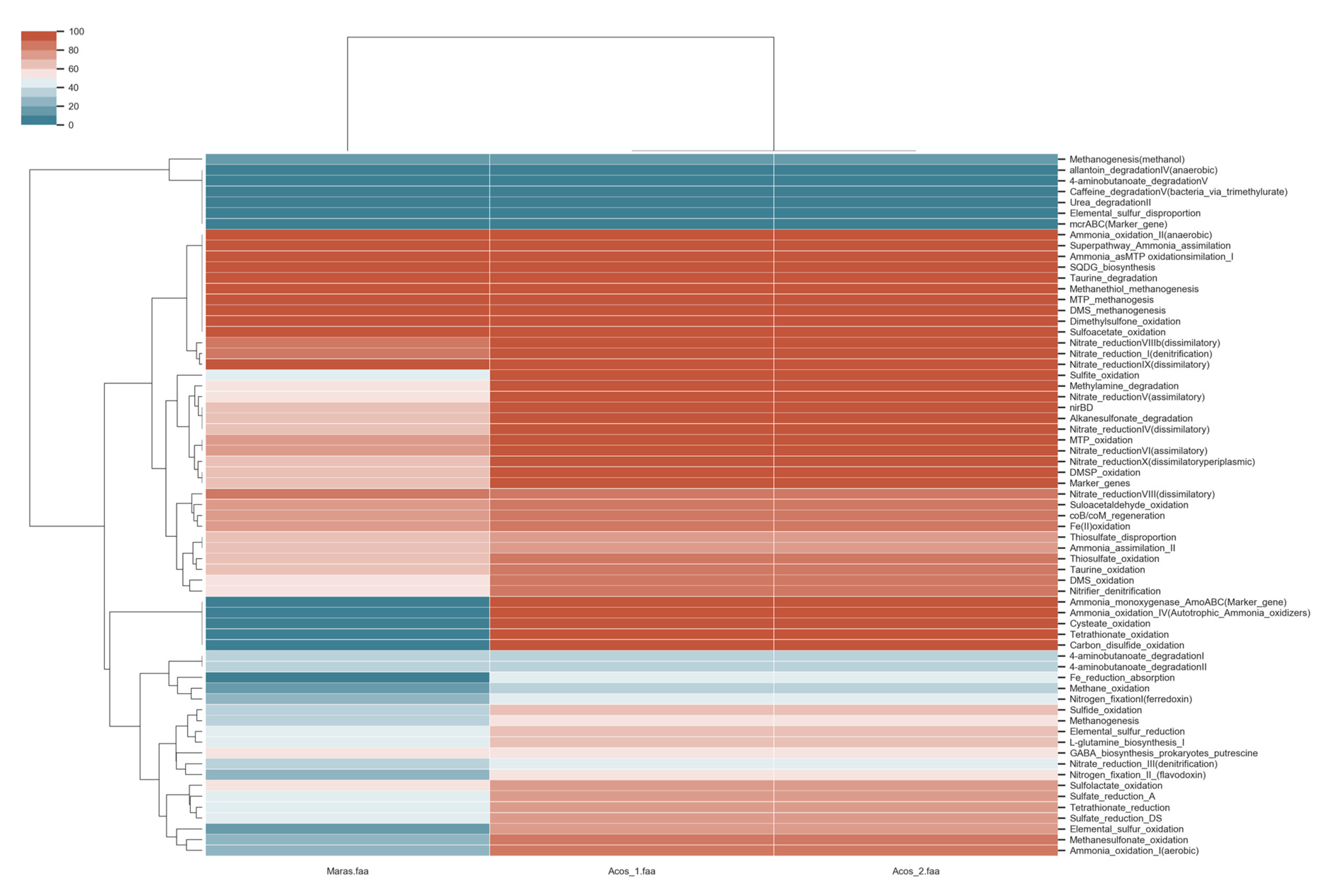
3.8. Metabolic Pathway Involved in Biogeochemical Cycles
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Maturrano, L.; Santos, F.; Rosselló-Mora, R.; Antón, J. Microbial diversity in Maras salterns, a hypersaline environment in the Peruvian Andes. Appl. Environ. Microbiol. 2006, 72, 3887–3895. [Google Scholar] [CrossRef] [PubMed]
- Hernández, L.M. Caracterización de la Microbiota de las Salinas de Maras, un Ambiente Hipersalino de los Andes de Perú. Ph.D. Thesis, Universitat d’Alacant-Universidad de Alicante, Alicante, Spain, 2004. [Google Scholar]
- Grant, W.D. Life at low water activity. Philos. Trans. R. Soc. B Biol. Sci. 2004, 359, 1249–1267. [Google Scholar] [CrossRef] [PubMed]
- Demergasso, C.; Casamayor, E.O.; Chong, G.; Galleguillos, P.; Escudero, L.; Pedrós-Alió, C. Distribution of prokaryotic genetic diversity in athalassohaline lakes of the Atacama Desert, Northern Chile. FEMS Microbiol. Ecol. 2004, 48, 57–69. [Google Scholar] [CrossRef] [PubMed]
- Ventosa, A.; de la Haba, R.R.; Sánchez-Porro, C.; Papke, R.T. Microbial diversity of hypersaline environments: A metagenomic approach. Curr. Opin. Microbiol. 2015, 25, 80–87. [Google Scholar] [CrossRef] [PubMed]
- Naghoni, A.; Emtiazi, G.; Amoozegar, M.A.; Cretoiu, M.S.; Stal, L.J.; Etemadifar, Z.; Shahzadeh Fazeli, S.A.; Bolhuis, H. Microbial diversity in the hypersaline Lake Meyghan, Iran. Sci. Rep. 2017, 7, 11522. [Google Scholar] [CrossRef] [PubMed]
- Fernández, A.B.; Vera-Gargallo, B.; Sánchez-Porro, C.; Ghai, R.; Papke, R.T.; Rodriguez-Valera, F.; Ventosa, A. Comparison of prokaryotic community structure from Mediterranean and Atlantic saltern concentrator ponds by a metagenomic approach. Front. Microbiol. 2014, 5. [Google Scholar] [CrossRef] [PubMed]
- Quillaguamán, J.; Hashim, S.; Bento, F.; Mattiasson, B.; Hatti-Kaul, R. Poly(β-hydroxybutyrate) production by a moderate halophile, Halomonas boliviensis LC1 using starch hydrolysate as substrate. J. Appl. Microbiol. 2005, 99, 151–157. [Google Scholar] [CrossRef]
- Oren, A. Saltern evaporation ponds as model systems for the study of primary production processes under hypersaline conditions. Aquat. Microb. Ecol. 2009, 56, 193–204. [Google Scholar] [CrossRef]
- Ventosa, A.; Fernández, A.B.; León, M.J.; Sánchez-Porro, C.; Rodriguez-Valera, F. The Santa Pola saltern as a model for studying the microbiota of hypersaline environments. Extrem. Life Extreme Cond. 2014, 18, 811–824. [Google Scholar] [CrossRef]
- Haferburg, G.; Gröning, J.A.D.; Schmidt, N.; Kummer, N.-A.; Erquicia, J.C.; Schlömann, M. Microbial diversity of the hypersaline and lithium-rich Salar de Uyuni, Bolivia. Microbiol. Res. 2017, 199, 19–28. [Google Scholar] [CrossRef]
- Emerson, J.B.; Thomas, B.C.; Andrade, K.; Allen, E.E.; Heidelberg, K.B.; Banfield, J.F. Dynamic Viral Populations in Hypersaline Systems as Revealed by Metagenomic Assembly. Appl. Environ. Microbiol. 2012, 78, 6309–6320.11. [Google Scholar] [CrossRef] [PubMed]
- Sarwar, K.; Azam, I.; Tahir, I. Biology and Applications of Halophilic Bacteria and Archea: A Review. Electron. J. Biol. 2015, 11, 98–103. [Google Scholar]
- Bioinformatics, B. FastQC: A Quality Control. Tool for High. Throughput Sequence Data; Babraham Institute: Cambridge, UK, 2011. [Google Scholar]
- Fu, L.; Niu, B.; Zhu, Z.; Wu, S.; Li, W. CD-HIT: Accelerated for clustering the next-generation sequencing data. Bioinformatics 2012, 28, 3150–3152. [Google Scholar] [CrossRef] [PubMed]
- Li, D.; Liu, C.-M.; Luo, R.; Sadakane, K.; Lam, T.-W. MEGAHIT: An ultra-fast single-node solution for large and complex metagenomics assembly via succinct de Bruijn graph. Bioinformatics 2015, 31, 1674–1676. [Google Scholar] [CrossRef] [PubMed]
- Glass, E.M.; Wilkening, J.; Wilke, A.; Antonopoulos, D.; Meyer, F. Using the metagenomics RAST server (MG-RAST) for analyzing shotgun metagenomes. Cold Spring Harb. Protoc. 2010, 2010, Pdb.prot5368. [Google Scholar] [CrossRef]
- Roux, S.; Enault, F.; Hurwitz, B.L.; Sullivan, M.B. VirSorter: Mining viral signal from microbial genomic data. PeerJ 2015, 3, e985. [Google Scholar] [CrossRef]
- Huson, D.H.; Beier, S.; Flade, I.; Górska, A.; El-Hadidi, M.; Mitra, S.; Ruscheweyh, H.-J.; Tappu, R. MEGAN Community Edition-Interactive Exploration and Analysis of Large-Scale Microbiome Sequencing Data. PLoS Comput. Biol. 2016, 12, e1004957. [Google Scholar] [CrossRef] [PubMed]
- McMurdie, P.J.; Holmes, S. phyloseq: An R package for reproducible interactive analysis and graphics of microbiome census data. PLoS ONE 2013, 8, e61217. [Google Scholar] [CrossRef]
- R Development Core Team. R: A Language and Environment for Statistical Computing. R Foundation for Statistical Computing, Vienna. 2011. Available online: http://www.R-project.org (accessed on 30 September 2019).
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef]
- Bushnell, B. BBMap Short-Read Aligner, and Other Bioinformatics Tools; University of California: Berkeley, CA, USA, 2015. [Google Scholar]
- Golosova, O.; Henderson, R.; Vaskin, Y.; Gabrielian, A.; Grekhov, G.; Nagarajan, V.; Oler, A.J.; Quiñones, M.; Hurt, D.; Fursov, M.; et al. Unipro UGENE NGS pipelines and components for variant calling, RNA-seq and ChIP-seq data analyses. PeerJ 2014, 2, e644. [Google Scholar] [CrossRef]
- Minot, S.S.; Krumm, N.; Greenfield, N.B. One Codex: A Sensitive and Accurate Data Platform for Genomic Microbial Identification. Bioinformatics 2015. [Google Scholar] [CrossRef]
- Seemann, T. Prokka: Rapid prokaryotic genome annotation. Bioinformatics 2014, 30, 2068–2069. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Y.; Liang, Y.; Lynch, K.H.; Dennis, J.J.; Wishart, D.S. PHAST: A Fast Phage Search Tool. Nucleic Acids Res. 2011, 39, W347–W352. [Google Scholar] [CrossRef] [PubMed]
- Bolduc, B.; Jang, H.B.; Doulcier, G.; You, Z.-Q.; Roux, S.; Sullivan, M.B. vConTACT: An iVirus tool to classify double-stranded DNA viruses that infect Archaea and Bacteria. PeerJ 2017, 5, e3243. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.-W.; Tang, Y.-H.; Tringe, S.G.; Simmons, B.A.; Singer, S.W. MaxBin: An automated binning method to recover individual genomes from metagenomes using an expectation-maximization algorithm. Microbiome 2014, 2, 26. [Google Scholar] [CrossRef]
- Hug, L.A.; Baker, B.J.; Anantharaman, K.; Brown, C.T.; Probst, A.J.; Castelle, C.J.; Butterfield, C.N.; Hernsdorf, A.W.; Amano, Y.; Ise, K.; et al. A new view of the tree of life. Nat. Microbiol 2016, 1, 16048. [Google Scholar] [CrossRef]
- Katoh, K.; Standley, D.M. MAFFT Multiple Sequence Alignment Software Version 7: Improvements in Performance and Usability. Mol. Biol. Evol. 2013, 30, 772–780. [Google Scholar] [CrossRef]
- Price, M.N.; Dehal, P.S.; Arkin, A.P. FastTree: Computing Large Minimum Evolution Trees with Profiles instead of a Distance Matrix. Mol. Biol. Evol. 2009, 26, 1641–1650. [Google Scholar] [CrossRef]
- Letunic, I.; Bork, P. Interactive tree of life (iTOL) v3: An online tool for the display and annotation of phylogenetic and other trees. Nucleic Acids Res. 2016, 44, W242–W245. [Google Scholar] [CrossRef]
- Hyatt, D.; Chen, G.-L.; LoCascio, P.F.; Land, M.L.; Larimer, F.W.; Hauser, L.J. Prodigal: Prokaryotic gene recognition and translation initiation site identification. BMC Bioinform. 2010, 11, 119. [Google Scholar] [CrossRef]
- Silva, G.G.Z.; Green, K.T.; Dutilh, B.E.; Edwards, R.A. SUPER-FOCUS: A tool for agile functional analysis of shotgun metagenomic data. Bioinformatics 2016, 32, 354–361. [Google Scholar] [CrossRef] [PubMed]
- Wickham, H. ggplot2: Elegant Graphics for Data Analysis; Springer: New York, NY, USA, 2016. [Google Scholar]
- De Anda, V.; Zapata-Peñasco, I.; Poot-Hernandez, A.C.; Eguiarte, L.E.; Contreras-Moreira, B.; Souza, V. MEBS, a software platform to evaluate large (meta)genomic collections according to their metabolic machinery: Unraveling the sulfur cycle. GigaScience 2017, 6. [Google Scholar] [CrossRef] [PubMed]
- Fernandez, A.B.; Ghai, R.; Martin-Cuadrado, A.B.; Sanchez-Porro, C.; Rodriguez-Valera, F.; Ventosa, A. Metagenome Sequencing of Prokaryotic Microbiota from Two Hypersaline Ponds of a Marine Saltern in Santa Pola, Spain. Genome Announc. 2013, 1. [Google Scholar] [CrossRef]
- Fernández, A.B.; Ghai, R.; Martin-Cuadrado, A.-B.; Sánchez-Porro, C.; Rodriguez-Valera, F.; Ventosa, A. Prokaryotic taxonomic and metabolic diversity of an intermediate salinity hypersaline habitat assessed by metagenomics. FEMS Microbiol. Ecol. 2014, 88, 623–635. [Google Scholar] [CrossRef] [PubMed]
- Wang, J.; Yang, D.; Zhang, Y.; Shen, J.; van der Gast, C.; Hahn, M.W.; Wu, Q. Do Patterns of Bacterial Diversity along Salinity Gradients Differ from Those Observed for Macroorganisms? PLoS ONE 2011, 6, e27597. [Google Scholar] [CrossRef] [PubMed]
- Fierer, N.; Jackson, R.B. The diversity and biogeography of soil bacterial communities. Proc. Natl. Acad. Sci. USA 2006, 103, 626–631. [Google Scholar] [CrossRef] [PubMed]
- Pagaling, E.; Wang, H.; Venables, M.; Wallace, A.; Grant, W.D.; Cowan, D.A.; Jones, B.E.; Ma, Y.; Ventosa, A.; Heaphy, S. Microbial Biogeography of Six Salt Lakes in Inner Mongolia, China, and a Salt Lake in Argentina. Appl. Environ. Microbiol. 2009, 75, 5750–5760. [Google Scholar] [CrossRef]
- Mora-Ruiz, M.D.; Cifuentes, A.; Font-Verdera, F.; Pérez-Fernández, C.; Farias, M.E.; González, B.; Orfila, A.; Rosselló-Móra, B. Biogeographical patterns of bacterial and archaeal communities from distant hypersaline environments. Syst. Appl. Microbiol. 2018, 41, 139–150. [Google Scholar] [CrossRef]
- Ghai, R.; Pašić, L.; Fernández, A.B.; Martin-Cuadrado, A.-B.; Mizuno, C.M.; McMahon, K.D.; Papke, R.T.; Stepanauskas, R.; Rodriguez-Brito, B.; Rohwer, F.; et al. New Abundant Microbial Groups in Aquatic Hypersaline Environments. Sci. Rep. 2011, 1, 135. [Google Scholar] [CrossRef]
- Vera-Gargallo, B.; Ventosa, A. Metagenomic Insights into the Phylogenetic and Metabolic Diversity of the Prokaryotic Community Dwelling in Hypersaline Soils from the Odiel Saltmarshes (SW Spain). Genes 2018, 9, 152. [Google Scholar] [CrossRef]
- Ma, Y.; Galinski, E.A.; Grant, W.D.; Oren, A.; Ventosa, A. Halophiles 2010: Life in Saline Environments. Appl. Environ. Microbiol. 2010, 76, 6971–6981. [Google Scholar] [CrossRef] [PubMed]
- Ventosa, A.; Nieto, J.J.; Oren, A. Biology of Moderately Halophilic Aerobic Bacteria. Microbiol. Mol. Biol. Rev. 1998, 62, 504–544. [Google Scholar] [PubMed]
- Arahal, D.R.; García, M.T.; Vargas, C.; Cánovas, D.; Nieto, J.J.; Ventosa, A. Chromohalobacter salexigens sp. nov., a moderately halophilic species that includes Halomonas elongata DSM 3043 and ATCC 33174. Int. J. Syst. Evol. Microbiol. 2001, 51, 1457–1462. [Google Scholar] [CrossRef] [PubMed]
- Babavalian, H.; Amoozegar, M.A.; Pourbabaee, A.A.; Moghaddam, M.M.; Shakeri, F. Isolation and identification of moderately halophilic bacteria producing hydrolytic enzymes from the largest hypersaline playa in Iran. Microbiology 2013, 82, 466–474. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Tourova, T.P.; Galinski, E.A.; Muyzer, G.; Kuenen, J.G. Thiohalorhabdus denitrificans gen. nov., sp. nov., an extremely halophilic, sulfur-oxidizing, deep-lineage gammaproteobacterium from hypersaline habitats. Int. J. Syst. Evol. Microbiol. 2008, 58, 2890–2897. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Tourova, T.P.; Muyzer, G.; Kuenen, G.J. Thiohalospira halophila gen. nov., sp. nov. and Thiohalospira alkaliphila sp. nov., novel obligately chemolithoautotrophic, halophilic, sulfur-oxidizing gammaproteobacteria from hypersaline habitats. Int. J. Syst. Evol. Microbiol. 2008, 58, 1685–1692. [Google Scholar] [CrossRef]
- Oren, A. Life at high salt concentrations, intracellular KCl concentrations, and acidic proteomes. Front. Microbiol. 2013, 4. [Google Scholar] [CrossRef]
- González-Torres, P.; Gabaldón, T. Genome Variation in the Model Halophilic Bacterium Salinibacter ruber. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef]
- Kennedy, S.P.; Ng, W.V.; Salzberg, S.L.; Hood, L.; DasSarma, S. Understanding the Adaptation of Halobacterium Species NRC-1 to Its Extreme Environment through Computational Analysis of Its Genome Sequence. Genome Res. 2001, 11, 1641–1650. [Google Scholar] [CrossRef]
- Paul, S.; Bag, S.K.; Das, S.; Harvill, E.T.; Dutta, C. Molecular signature of hypersaline adaptation: Insights from genome and proteome composition of halophilic prokaryotes. Genome Biol. 2008, 9, R70. [Google Scholar] [CrossRef]
- Vera Gargallo, B.; Roy Chowdhury, T.; Brown, J.; Fansler, S.J.; Durán Viseras, A.; Sánchez-Porro Álvarez, C.; Bailey, V.L.; Jansson, J.K.; Ventosa, A. Spatial distribution of prokaryotic communities in hypersaline soils. Sci. Rep. 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Mamani, J.I.; Pacheco, K.B.; Elorrieta, P.; Romoacca, P.; Castelan, H.; Davila, S.; Sierra, J.L.; Quispe-Ricalde, M.A. Draft Genome Sequence of Halomonas elongata MH25661 Isolated from a Saline Creek in the Andes of Peru. Microbiol. Resour. Announc. 2019, 8. [Google Scholar] [CrossRef] [PubMed]
- Menes, R.J.; Viera, C.E.; Farías, M.E.; Seufferheld, M.J. Halomonas vilamensis sp. nov., isolated from high-altitude Andean lakes. Int. J. Syst. Evol. Microbiol. 2011, 61, 1211–1217. [Google Scholar] [CrossRef] [PubMed]
- Xiao-Ran, J.; Jin, Y.; Xiangbin, C.; Guo-Qiang, C. Chapter Eleven—Halomonas and Pathway Engineering for Bioplastics Production. In Methods in Enzymology; Scrutton, N., Ed.; Academic Press: Cambridge, MA, USA, 2018; pp. 309–328. [Google Scholar]
- Adkins, J.P.; Madigan, M.T.; Mandelco, L.; Woese, C.R.; Tanner, R.S. Arhodomonas aquaeolei gen. nov., sp. nov., an aerobic, halophilic bacterium isolated from a subterranean brine. Int. J. Syst. Bacteriol. 1993, 43, 514–520. [Google Scholar] [CrossRef] [PubMed]
- Donachie, S.P.; Hou, S.; Gregory, T.S.; Malahoff, A.; Alam, M. Idiomarina loihiensis sp. nov., a halophilic γ-Proteobacterium from the Lō‘ihi submarine volcano, Hawai’i. Int. J. Syst. Evol. Microbiol. 2003, 53, 1873–1879. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Prado, B.; Del Moral, A.; Quesada, E.; Ríos, R.; Monteoliva-Sanchez, M.; Campos, V.; Ramos-Cormenzana, A. Numerical Taxonomy of Moderately Halophilic Gram-negative Rods Isolated from the Salar de Atacama, Chile. Syst. Appl. Microbiol. 1991, 14, 275–281. [Google Scholar] [CrossRef]
- Minegishi, H.; Echigo, A.; Nagaoka, S.; Kamekura, M.; Usami, R. Halarchaeum acidiphilum gen. nov., sp. nov., a moderately acidophilic haloarchaeon isolated from commercial solar salt. Int. J. Syst. Evol. Microbiol. 2010, 60, 2513–2516. [Google Scholar] [CrossRef]
- Rensing, C. Adaption to life at high salt concentrations in Archaea, Bacteria and Eukarya. Saline Syst. 2005, 1, 6. [Google Scholar] [CrossRef]
- Sorokin, D.Y.; Messina, E.; Smedile, F.; Roman, P.; Damsté, J.S.S.; Ciordia, S.; Mena, M.C.; Ferrer, M.; Golyshin, P.N.; Kublanov, I.V.; et al. Discovery of anaerobic lithoheterotrophic haloarchaea, ubiquitous in hypersaline habitats. ISME J. 2017, 11, 1245–1260. [Google Scholar] [CrossRef]
- Mou, Y.-Z.; Qiu, X.-X.; Zhao, M.-L.; Cui, H.-L.; Oh, D.; Dyall-Smith, M.L. Halohasta litorea gen. nov. sp. nov., and Halohasta litchfieldiae sp. nov., isolated from the Daliang aquaculture farm, China and from Deep Lake, Antarctica, respectively. Extremophiles 2012, 16, 895–901. [Google Scholar] [CrossRef]
- Oren, A. Taxonomy of halophilic Archaea: Current status and future challenges. Extremophiles 2014, 18, 825–834. [Google Scholar] [CrossRef] [PubMed]
- Williams, T.J.; Liao, Y.; Ye, J.; Kuchel, R.P.; Poljak, A.; Raftery, M.J.; Cavicchioli, R. Cold adaptation of the Antarctic haloarchaea Halohasta litchfieldiae and Halorubrum lacusprofundi. Environ. Microbiol. 2017, 19, 2210–2227. [Google Scholar] [CrossRef] [PubMed]
- Fuchsman, C.A.; Collins, R.E.; Rocap, G.; Brazelton, W.J. Effect of the environment on horizontal gene transfer between bacteria and archaea. PeerJ 2017, 5, e3865. [Google Scholar] [CrossRef] [PubMed]
- Pushkarev, A.; Inoue, K.; Larom, S.; Flores-Uribe, J.; Singh, M.; Konno, M.; Tomida, S.; Ito, S.; Nakamura, R.; Tsunoda, S.P.; et al. A distinct abundant group of microbial rhodopsins discovered using functional metagenomics. Nature 2018, 558, 595–599. [Google Scholar] [CrossRef] [PubMed]
- Santos, F.; Yarza, P.; Parro, V.; Meseguer, I.; Rosselló-Móra, R.; Antón, J. Culture-Independent Approaches for Studying Viruses from Hypersaline Environments. Appl. Environ. Microbiol. 2012, 78, 1635–1643. [Google Scholar] [CrossRef]
- Garcia-Heredia, I.; Martin-Cuadrado, A.-B.; Mojica, F.J.M.; Santos, F.; Mira, A.; Antón, J.; Rodriguez-Valera, F. Reconstructing Viral Genomes from the Environment Using Fosmid Clones: The Case of Haloviruses. PLoS ONE 2012, 7, e33802. [Google Scholar] [CrossRef]
- Ramos-Barbero, M.D.; Martínez, J.M.; Almansa, C.; Rodríguez, N.; Villamor, J.; Gomariz, M.; Escudero, C.; dC Rubin, S.; Antón, J.; Martínez-García, M.; et al. Prokaryotic and viral community structure in the singular chaotropic salt lake Salar de Uyuni. Environ. Microbiol. 2019, 21, 2029–2042. [Google Scholar] [CrossRef]
- Weitz, J.S.; Wilhelm, S.W. Ocean viruses and their effects on microbial communities and biogeochemical cycles. F1000 Biol. Rep. 2012, 4. [Google Scholar] [CrossRef]
- Roux, S.; Adriaenssens, E.M.; Dutilh, B.E.; Koonin, E.V.; Kropinski, A.M.; Krupovic, M.; Kuhn, J.H.; Lavigne, R.; Brister,, J.R.; Varsani, A.; et al. Minimum Information about an Uncultivated Virus Genome (MIUViG). Nat. Biotechnol. 2019, 37, 29–37. [Google Scholar] [CrossRef]
- Castelán-Sánchez, H.G.; Lopéz-Rosas, I.; García-Suastegui, W.A.; Peralta, R.; Dobson, A.D.W.; Batista-García, R.A.; Dávila-Ramos, S. Extremophile deep-sea viral communities from hydrothermal vents: Structural and functional analysis. Mar. Genom. 2019, 46, 16–28. [Google Scholar] [CrossRef]
- Antunes, A.; Ngugi, D.K.; Stingl, U. Microbiology of the Red Sea (and other) deep-sea anoxic brine lakes. Environ. Microbiol. Rep. 2011, 3, 416–433. [Google Scholar] [CrossRef] [PubMed]
- Claverie, J.-M.; Abergel, C. Mimiviridae: An Expanding Family of Highly Diverse Large dsDNA Viruses Infecting a Wide Phylogenetic Range of Aquatic Eukaryotes. Viruses 2018, 10, 506. [Google Scholar] [CrossRef] [PubMed]
- Yau, S.; Lauro, F.M.; DeMaere, M.Z.; Brown, M.V.; Thomas, T.; Raftery, M.J.; Andrews-Pfannkoch, C.; Lewis, M.; Hoffman, J.M.; Gibson, J.A.; et al. Virophage control of antarctic algal host-virus dynamics. Proc. Natl. Acad. Sci. USA 2011, 108, 6163–6168. [Google Scholar] [CrossRef] [PubMed]
- Le Romancer, M.; Gaillard, M.; Geslin, C.; Prieur, D. Viruses in extreme environments. In Life in Extreme Environments; Amils, R., Ellis-Evans, C., Hinghofer-Szalkay, H., Eds.; Springer Netherlands: Dordrecht, The Netherlands, 2007; pp. 99–113. [Google Scholar]
- Buchalo, A.S.; Nevo, E.; Wasser, S.P.; Oren, A.; Molitoris, H.P. Fungal life in the extremely hypersaline water of the Dead Sea: ®first records. Proc. R. Soc. Lond. Ser. B Biol. Sci. 1998, 265, 1461–1465. [Google Scholar] [CrossRef]
- Gunde-Cimerman, N.; Zalar, P. Extremely Halotolerant and Halophilic Fungi Inhabit Brine in Solar Salterns around the Globe. Food Technol. Biotechnol. 2014, 52, 170–179. [Google Scholar]
- Butinar, L.; Sonjak, S.; Zalar, P.; Plemenitaš, A.; Gunde-Cimerman, N. Melanized halophilic fungi are eukaryotic members of microbial communities in hypersaline waters of solar salterns. Bot. Mar. 2005, 48. [Google Scholar] [CrossRef]
- Lahav, R.; Fareleira, P.; Nejidat, A.; Abeliovich, A. The Identification and Characterization of Osmotolerant Yeast Isolates from Chemical Wastewater Evaporation Ponds. Microb. Ecol. 2002, 43, 388–396. [Google Scholar] [CrossRef]
- Asem, A.; Eimanifar, A.; Wink, M. Update of “Biodiversity of the Hypersaline Urmia Lake National Park (NW Iran)”. Diversity 2016, 8, 6. [Google Scholar] [CrossRef]
- Tkavc, R.; Matrosova, V.Y.; Grichenko, O.E.; Gostinčar, C.; Volpe, R.P.; Klimenkova, P.; Gaidamakova, E.K.; Zhou, C.E.; Stewart, B.J.; Lyman, M.G.; et al. Prospects for Fungal Bioremediation of Acidic Radioactive Waste Sites: Characterization and Genome Sequence of Rhodotorula taiwanensis MD1149. Front. Microbiol. 2018, 8. [Google Scholar] [CrossRef]
- Jünemann, S.; Kleinbölting, N.; Jaenicke, S.; Henke, C.; Hassa, J.; Nelkner, J.; Stolze, Y.; Albaum, S.P.; Schlüter, A.; Goesmann, A.; et al. Bioinformatics for NGS-based metagenomics and the application to biogas research. J. Biotechnol. 2017, 261, 10–23. [Google Scholar] [CrossRef]
- McNair, K.; Edwards, R.A. Genome Peek—An online tool for prokaryotic genome and metagenome analysis. PeerJ 2015, 3, e1025. [Google Scholar] [CrossRef] [PubMed]
- Kindzierski, V.; Raschke, S.; Knabe, N.; Siedler, F.; Scheffer, B.; Pflüger-Grau, K.; Pfeiffer, F.; Oesterhelt, D.; Marin-Sanguino, A.; Kunte, H.J. Osmoregulation in the Halophilic Bacterium Halomonas elongata: A Case Study for Integrative Systems Biology. PLoS ONE 2017, 12, e0168818. [Google Scholar] [CrossRef] [PubMed]
- Ren, M.; Zhang, Z.; Wang, X.; Zhou, Z.; Chen, D.; Zeng, H.; Zhao, S.; Chen, L.; Hu, Y.; Zhang, C.; et al. Diversity and Contributions to Nitrogen Cycling and Carbon Fixation of Soil Salinity Shaped Microbial Communities in Tarim Basin. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Mobberley, J.M.; Authement, R.N.; Segall, A.M.; Paul, J.H. The Temperate Marine Phage ΦHAP-1 of Halomonas aquamarina Possesses a Linear Plasmid-Like Prophage Genome. J. Virol. 2008, 82, 6618–6630. [Google Scholar] [CrossRef] [PubMed]
- Oren, A. Microbial life at high salt concentrations: Phylogenetic and metabolic diversity. Saline Syst. 2008, 4, 2. [Google Scholar] [CrossRef]
- Rhodes, M.E.; Spear, J.R.; Oren, A.; House, C.H. Differences in lateral gene transfer in hypersaline versus thermal environments. BMC Evol. Biol. 2011, 11, 199. [Google Scholar] [CrossRef]
- Reed, C.J.; Lewis, H.; Trejo, E.; Winston, V.; Evilia, C. Protein Adaptations in Archaeal Extremophiles. Archaea 2013, 2013. [Google Scholar] [CrossRef]
- Becker, E.A.; Seitzer, P.M.; Tritt, A.; Larsen, D.; Krusor, M.; Yao, A.I.; Wu, D.; Madern, D.; Eisen, J.A.; Darling, A.E.; et al. Phylogenetically Driven Sequencing of Extremely Halophilic Archaea Reveals Strategies for Static and Dynamic Osmo-response. PLOS Genet. 2014, 10, e1004784. [Google Scholar] [CrossRef]
- Srivastava, P.; Kowshik, M. Mechanisms of Metal Resistance and Homeostasis in Haloarchaea. Archaea 2013, 2013, 732864. [Google Scholar] [CrossRef]
- Spooner, R.; Yilmaz, Ö. The Role of Reactive-Oxygen-Species in Microbial Persistence and Inflammation. Int. J. Mol. Sci. 2011, 12, 334–352. [Google Scholar] [CrossRef]
- Lipson, D.A.; Haggerty, J.M.; Srinivas, A.; Raab, T.K.; Sathe, S.; Dinsdale, E.A. Metagenomic Insights into Anaerobic Metabolism along an Arctic Peat Soil Profile. PLoS ONE 2013, 8, e64659. [Google Scholar] [CrossRef] [PubMed]
- Jones, D.L.; Baxter, B.K. DNA Repair and Photoprotection: Mechanisms of Overcoming Environmental Ultraviolet Radiation Exposure in Halophilic Archaea. Front. Microbiol. 2017, 8. [Google Scholar] [CrossRef] [PubMed]
- Plominsky, A.M.; Henríquez-Castillo, C.; Delherbe, N.; Podell, S.; Ramirez-Flandes, S.; Ugalde, J.A.; Santibañez, J.F.; van den Engh, G.; Hanselmann, K.; Ulloa, O.; et al. Distinctive Archaeal Composition of an Artisanal Crystallizer Pond and Functional Insights Into Salt-Saturated Hypersaline Environment Adaptation. Front. Microbiol. 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Kinoshita, E.; van der Linden, E.; Sanchez, H.; Wyman, C. RAD50, an SMC family member with multiple roles in DNA break repair: How does ATP affect function? Chromosome Res. Int J. Mol. Supramol. Evol. Asp. Chromosome Biol. 2009, 17, 277–288. [Google Scholar] [CrossRef]
- de Souza, R.F.; Iyer, L.M.; Aravind, L. Diversity and evolution of chromatin proteins encoded by DNA viruses. Biochim. Biophys. Acta 2010, 1799, 302–318. [Google Scholar] [CrossRef]
- Ramírez-Orozco, M.; Serrano-Pinto, V.; Ochoa-Álvarez, N.A.; Makarov, R.Y.; Martínez-Díaz, S.F. Genome sequence analysis of the Vibrio parahaemolyticus lytic bacteriophage VPMS1. Arch. Virol. 2013, 158, 2409–2413. [Google Scholar] [CrossRef]
- Gao, E.-B.; Huang, Y.; Ning, D. Metabolic Genes within Cyanophage Genomes: Implications for Diversity and Evolution. Genes 2016, 7, 80. [Google Scholar] [CrossRef]
- Cabello, P. Nitrate reduction and the nitrogen cycle in archaea. Microbiology 2004, 150, 3527–3546. [Google Scholar] [CrossRef]
- DasSarma, S.; DasSarma, P. Halophiles. eLS 2017. [Google Scholar] [CrossRef]
- He, T.; Li, H.; Zhang, X. Deep-Sea Hydrothermal Vent Viruses Compensate for Microbial Metabolism in Virus-Host Interactions. MBio 2017, 8. [Google Scholar] [CrossRef]
- Andrei, A.-Ş.; Banciu, H.L.; Oren, A. Living with salt: Metabolic and phylogenetic diversity of archaea inhabiting saline ecosystems. FEMS Microbiol. Lett. 2012, 330, 1–9. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Beer, L.L.; Whitman, W.B. Sulfur metabolism in archaea reveals novel processes. Environ. Microbiol. 2012, 14, 2632–2644. [Google Scholar] [CrossRef] [PubMed]
- Yau, S.; Lauro, F.M.; Williams, T.J.; DeMaere, M.Z.; Brown, M.V.; Rich, J.; Gibson, J.A.; Cavicchioli, R. Metagenomic insights into strategies of carbon conservation and unusual sulfur biogeochemistry in a hypersaline Antarctic lake. ISME J. 2013, 7, 1944–1961. [Google Scholar] [CrossRef] [PubMed]












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Data Set | Salinity | pH | Number of Paired-End Reads | Number of Contigs Assembled | Sequences | Taxonomical Classification | ||||
|---|---|---|---|---|---|---|---|---|---|---|
| Classified | Unclassified | Bacteria | Archaea | Eukarya | Viruses | |||||
| Acos 1 | 19% | 7.9 | 63,387,998 | 257,314 | 71% | 29% | 57% | 14% | 2% | 0.2% |
| Acos 2 | 19% | 7.9 | 79,304,621 | 256,430 | 71% | 29% | 57% | 16% | 2% | 0.2% |
| Maras | 23% | 7.0 | 56,086,809 | 2650 | 70% | 30% | 56% | 11% | 1% | 1.32% |
| Features | Halomonas elongata (Acos 1) | Halomonas elongata (Acos 2) | Idiomarina loihiensis (Acos 1) | Idiomarina loihiensis (Acos 2) |
|---|---|---|---|---|
| Length size (bp) | 3,768,127 | 3,763,770 | 2,111,175 | 2,227,077 |
| % GC | 64 | 64 | 47.2 | 47.3 |
| CDS | 4564 | 4678 | 4060 | 3871 |
| tRNAs | 65 | 69 | 55 | 56 |
| tmRNA | 1 | 1 | 1 | 1 |
| rRNAs | 12 | 12 | 12 | 13 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Castelán-Sánchez, H.G.; Elorrieta, P.; Romoacca, P.; Liñan-Torres, A.; Sierra, J.L.; Vera, I.; Batista-García, R.A.; Tenorio-Salgado, S.; Lizama-Uc, G.; Pérez-Rueda, E.; et al. Intermediate-Salinity Systems at High Altitudes in the Peruvian Andes Unveil a High Diversity and Abundance of Bacteria and Viruses. Genes 2019, 10, 891. https://doi.org/10.3390/genes10110891
Castelán-Sánchez HG, Elorrieta P, Romoacca P, Liñan-Torres A, Sierra JL, Vera I, Batista-García RA, Tenorio-Salgado S, Lizama-Uc G, Pérez-Rueda E, et al. Intermediate-Salinity Systems at High Altitudes in the Peruvian Andes Unveil a High Diversity and Abundance of Bacteria and Viruses. Genes. 2019; 10(11):891. https://doi.org/10.3390/genes10110891
Chicago/Turabian StyleCastelán-Sánchez, Hugo Gildardo, Paola Elorrieta, Pedro Romoacca, Arturo Liñan-Torres, José Luis Sierra, Ingrid Vera, Ramón Alberto Batista-García, Silvia Tenorio-Salgado, Gabriel Lizama-Uc, Ernesto Pérez-Rueda, and et al. 2019. "Intermediate-Salinity Systems at High Altitudes in the Peruvian Andes Unveil a High Diversity and Abundance of Bacteria and Viruses" Genes 10, no. 11: 891. https://doi.org/10.3390/genes10110891
APA StyleCastelán-Sánchez, H. G., Elorrieta, P., Romoacca, P., Liñan-Torres, A., Sierra, J. L., Vera, I., Batista-García, R. A., Tenorio-Salgado, S., Lizama-Uc, G., Pérez-Rueda, E., Quispe-Ricalde, M. A., & Dávila-Ramos, S. (2019). Intermediate-Salinity Systems at High Altitudes in the Peruvian Andes Unveil a High Diversity and Abundance of Bacteria and Viruses. Genes, 10(11), 891. https://doi.org/10.3390/genes10110891