The BRCA1 c.4096+3A>G Variant Displays Classical Characteristics of Pathogenic BRCA1 Mutations in Hereditary Breast and Ovarian Cancers, But Still Allows Homozygous Viability

{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
3. Results
3.1. Identification of Probands and Estimation of Allelic Frequencies in Cases and Controls
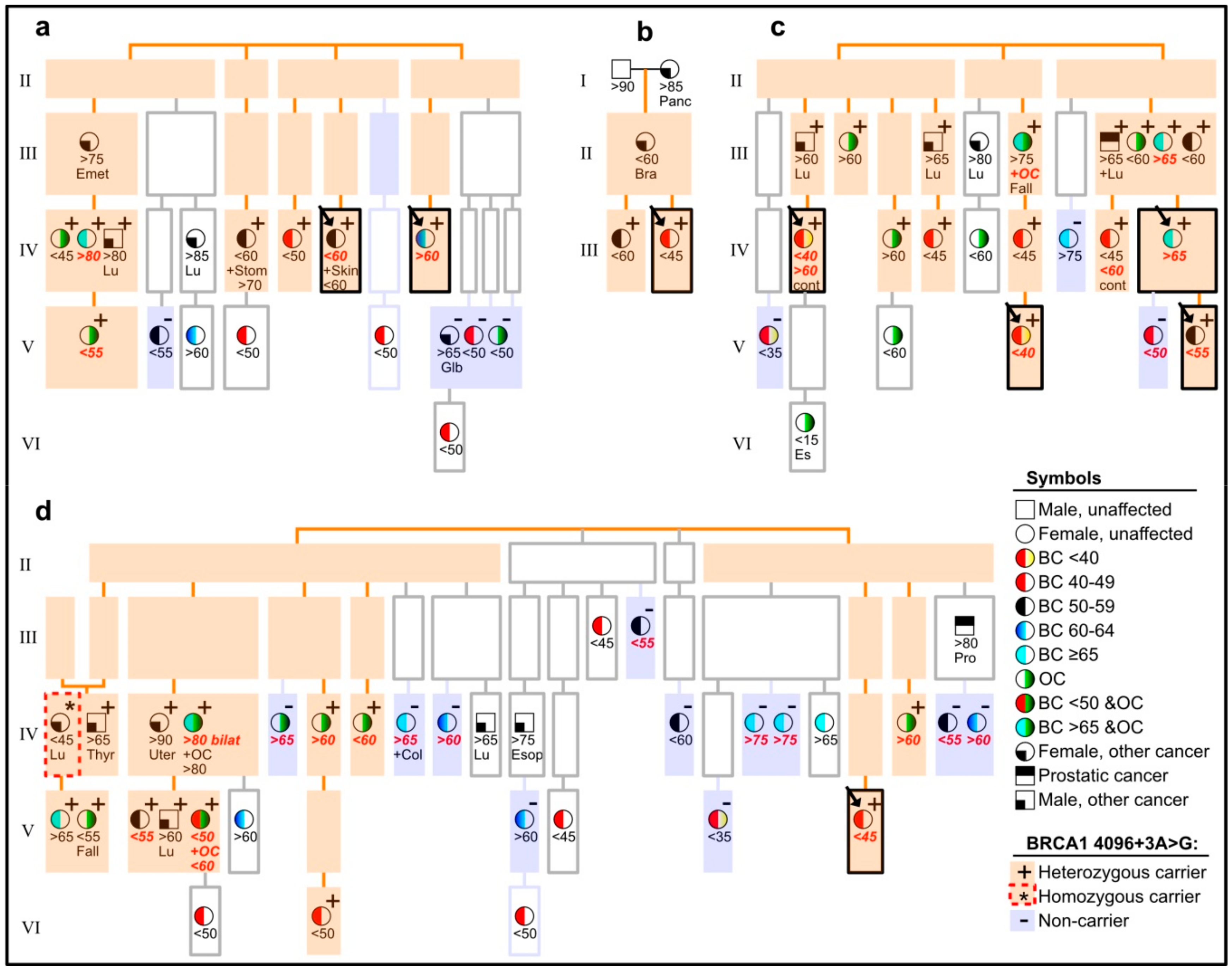
3.2. Pedigrees and Clinical Family History of Probands, and Typing of Affected Relatives
3.3. A Note on Geographical Distribution in Iceland
3.4. Analysis of LOH and Selected Pathological Characteristics
3.5. Instances Where Two Studied Tumors Originated in the Same Person
4. Discussion
5. Conclusions
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Lu, Y.; Ek, W.E.; Whiteman, D.; Vaughan, T.L.; Spurdle, A.B.; Easton, D.F.; Pharoah, P.D.; Thompson, D.J.; Dunning, A.M.; Hayward, N.K.; et al. Most common ‘sporadic’ cancers have a significant germline genetic component. Hum. Mol. Genet. 2014, 23, 6112–6118. [Google Scholar] [CrossRef] [PubMed]
- Garber, J.E.; Offit, K. Hereditary cancer predisposition syndromes. J. Clin. Oncol. 2005, 23, 276–292. [Google Scholar] [CrossRef] [PubMed]
- Miki, Y.; Swensen, J.; Shattuck-Eidens, D.; Futreal, P.A.; Harshman, K.; Tavtigian, S.; Liu, Q.; Cochran, C.; Bennett, L.M.; Ding, W.; et al. A strong candidate for the breast and ovarian cancer susceptibility gene BRCA1. Science 1994, 266, 66–71. [Google Scholar] [CrossRef] [PubMed]
- Wooster, R.; Bignell, G.; Lancaster, J.; Swift, S.; Seal, S.; Mangion, J.; Collins, N.; Gregory, S.; Gumbs, C.; Micklem, G.; et al. Identification of the breast cancer susceptibility gene BRCA2. Nature 1995, 378, 789–792. [Google Scholar] [CrossRef] [PubMed]
- Melchor, L.; Benitez, J. The complex genetic landscape of familial breast cancer. Hum. Genet. 2013, 132, 845–863. [Google Scholar] [CrossRef] [PubMed]
- Kuchenbaecker, K.B.; Hopper, J.L.; Barnes, D.R.; Phillips, K.A.; Mooij, T.M.; Roos-Blom, M.J.; Jervis, S.; van Leeuwen, F.E.; Milne, R.L.; Andrieu, N.; et al. Risks of Breast, Ovarian, and Contralateral Breast Cancer for BRCA1 and BRCA2 Mutation Carriers. JAMA 2017, 317, 2402–2416. [Google Scholar] [CrossRef] [PubMed]
- Rebbeck, T.R.; Mitra, N.; Wan, F.; Sinilnikova, O.M.; Healey, S.; McGuffog, L.; Mazoyer, S.; Chenevix-Trench, G.; Easton, D.F.; Antoniou, A.C.; et al. Association of type and location of BRCA1 and BRCA2 mutations with risk of breast and ovarian cancer. JAMA 2015, 313, 1347–1361. [Google Scholar] [CrossRef]
- Milne, R.L.; Antoniou, A.C. Modifiers of breast and ovarian cancer risks for BRCA1 and BRCA2 mutation carriers. Endocr. Relat. Cancer 2016, 23, T69–T84. [Google Scholar] [CrossRef]
- Smith, A.; Moran, A.; Boyd, M.C.; Bulman, M.; Shenton, A.; Smith, L.; Iddenden, R.; Woodward, E.R.; Lalloo, F.; Maher, E.R.; et al. Phenocopies in BRCA1 and BRCA2 families: Evidence for modifier genes and implications for screening. J. Med. Genet. 2007, 44, 10–15. [Google Scholar] [CrossRef]
- Dominguez-Valentin, M.; Evans, D.G.R.; Nakken, S.; Tubeuf, H.; Vodak, D.; Ekstrom, P.O.; Nissen, A.M.; Morak, M.; Holinski-Feder, E.; Martins, A.; et al. Genetic variants of prospectively demonstrated phenocopies in BRCA1/2 kindreds. Hered. Cancer Clin. Pract. 2018, 16, 4. [Google Scholar] [CrossRef]
- Knudson, A.G., Jr. Mutation and cancer: Statistical study of retinoblastoma. Proc. Natl. Acad. Sci. USA 1971, 68, 820–823. [Google Scholar] [CrossRef] [PubMed]
- Smith, S.A.; Easton, D.F.; Evans, D.G.; Ponder, B.A. Allele losses in the region 17q12-21 in familial breast and ovarian cancer involve the wild-type chromosome. Nat. Genet. 1992, 2, 128–131. [Google Scholar] [CrossRef] [PubMed]
- Gudmundsson, J.; Johannesdottir, G.; Bergthorsson, J.T.; Arason, A.; Ingvarsson, S.; Egilsson, V.; Barkardottir, R.B. Different tumor types from BRCA2 carriers show wild-type chromosome deletions on 13q12-q13. Cancer Res. 1995, 55, 4830–4832. [Google Scholar] [PubMed]
- Lakhani, S.R.; Van De Vijver, M.J.; Jacquemier, J.; Anderson, T.J.; Osin, P.P.; McGuffog, L.; Easton, D.F. The pathology of familial breast cancer: Predictive value of immunohistochemical markers estrogen receptor, progesterone receptor, HER-2, and p53 in patients with mutations in BRCA1 and BRCA2. J. Clin. Oncol. 2002, 20, 2310–2318. [Google Scholar] [CrossRef]
- Mavaddat, N.; Barrowdale, D.; Andrulis, I.L.; Domchek, S.M.; Eccles, D.; Nevanlinna, H.; Ramus, S.J.; Spurdle, A.; Robson, M.; Sherman, M.; et al. Pathology of breast and ovarian cancers among BRCA1 and BRCA2 mutation carriers: Results from the Consortium of Investigators of Modifiers of BRCA1/2 (CIMBA). Cancer Epidemiol. Biomark. Prev. 2012, 21, 134–147. [Google Scholar] [CrossRef]
- Foulkes, W.D.; Metcalfe, K.; Sun, P.; Hanna, W.M.; Lynch, H.T.; Ghadirian, P.; Tung, N.; Olopade, O.I.; Weber, B.L.; McLennan, J.; et al. Estrogen receptor status in BRCA1- and BRCA2-related breast cancer: The influence of age, grade, and histological type. Clin. Cancer Res. 2004, 10, 2029–2034. [Google Scholar] [CrossRef]
- Tung, N.; Wang, Y.; Collins, L.C.; Kaplan, J.; Li, H.; Gelman, R.; Comander, A.H.; Gallagher, B.; Fetten, K.; Krag, K.; et al. Estrogen receptor positive breast cancers in BRCA1 mutation carriers: Clinical risk factors and pathologic features. Breast Cancer Res. 2010, 12, R12. [Google Scholar] [CrossRef]
- Singer, C.F.; Balmana, J.; Burki, N.; Delaloge, S.; Filieri, M.E.; Gerdes, A.M.; Grindedal, E.M.; Han, S.; Johansson, O.; Kaufman, B.; et al. Genetic counselling and testing of susceptibility genes for therapeutic decision-making in breast cancer—An European consensus statement and expert recommendations. Eur. J. Cancer 2019, 106, 54–60. [Google Scholar] [CrossRef]
- Richards, S.; Aziz, N.; Bale, S.; Bick, D.; Das, S.; Gastier-Foster, J.; Grody, W.W.; Hegde, M.; Lyon, E.; Spector, E.; et al. Standards and guidelines for the interpretation of sequence variants: A joint consensus recommendation of the American College of Medical Genetics and Genomics and the Association for Molecular Pathology. Genet. Med. 2015, 17, 405–424. [Google Scholar] [CrossRef]
- Plon, S.E.; Eccles, D.M.; Easton, D.; Foulkes, W.D.; Genuardi, M.; Greenblatt, M.S.; Hogervorst, F.B.; Hoogerbrugge, N.; Spurdle, A.B.; Tavtigian, S.V.; et al. Sequence variant classification and reporting: Recommendations for improving the interpretation of cancer susceptibility genetic test results. Hum. Mutat. 2008, 29, 1282–1291. [Google Scholar] [CrossRef]
- Spurdle, A.B.; Healey, S.; Devereau, A.; Hogervorst, F.B.; Monteiro, A.N.; Nathanson, K.L.; Radice, P.; Stoppa-Lyonnet, D.; Tavtigian, S.; Wappenschmidt, B.; et al. ENIGMA—Evidence-based network for the interpretation of germline mutant alleles: An international initiative to evaluate risk and clinical significance associated with sequence variation in BRCA1 and BRCA2 genes. Hum. Mutat. 2012, 33, 2–7. [Google Scholar] [CrossRef] [PubMed]
- BRCA Exchange: Facts & Stats. Available online: https://brcaexchange.org/factsheet (accessed on 26 May 2019).
- Lopez-Urrutia, E.; Salazar-Rojas, V.; Brito-Elias, L.; Coca-Gonzalez, M.; Silva-Garcia, J.; Sanchez-Marin, D.; Campos-Parra, A.D.; Perez-Plasencia, C. BRCA mutations: Is everything said? Breast Cancer Res. Treat. 2019, 173, 49–54. [Google Scholar] [CrossRef] [PubMed]
- Goldgar, D.E.; Easton, D.F.; Byrnes, G.B.; Spurdle, A.B.; Iversen, E.S.; Greenblatt, M.S.; IARC Unclassified Genetic Variants Working Group. Genetic evidence and integration of various data sources for classifying uncertain variants into a single model. Hum. Mutat. 2008, 29, 1265–1272. [Google Scholar] [CrossRef] [PubMed]
- Osorio, A.; de la Hoya, M.; Rodriguez-Lopez, R.; Martinez-Ramirez, A.; Cazorla, A.; Granizo, J.J.; Esteller, M.; Rivas, C.; Caldes, T.; Benitez, J. Loss of heterozygosity analysis at the BRCA loci in tumor samples from patients with familial breast cancer. Int. J. Cancer 2002, 99, 305–309. [Google Scholar] [CrossRef]
- Chenevix-Trench, G.; Healey, S.; Lakhani, S.; Waring, P.; Cummings, M.; Brinkworth, R.; Deffenbaugh, A.M.; Burbidge, L.A.; Pruss, D.; Judkins, T.; et al. Genetic and histopathologic evaluation of BRCA1 and BRCA2 DNA sequence variants of unknown clinical significance. Cancer Res. 2006, 66, 2019–2027. [Google Scholar] [CrossRef]
- Osorio, A.; Milne, R.L.; Honrado, E.; Barroso, A.; Diez, O.; Salazar, R.; de la Hoya, M.; Vega, A.; Benitez, J. Classification of missense variants of unknown significance in BRCA1 based on clinical and tumor information. Hum. Mutat. 2007, 28, 477–485. [Google Scholar] [CrossRef]
- Hofstra, R.M.; Spurdle, A.B.; Eccles, D.; Foulkes, W.D.; de Wind, N.; Hoogerbrugge, N.; Hogervorst, F.B.; IARC Unclassified Genetic Variants Working Group. Tumor characteristics as an analytic tool for classifying genetic variants of uncertain clinical significance. Hum. Mutat. 2008, 29, 1292–1303. [Google Scholar] [CrossRef]
- Byrjalsen, A.; Steffensen, A.Y.; Hansen, T.V.O.; Wadt, K.; Gerdes, A.M. Classification of the spliceogenic BRCA1 c.4096+3A>G variant as likely benign based on cosegregation data and identification of a healthy homozygous carrier. Clin. Case Rep. 2017, 5, 876–879. [Google Scholar] [CrossRef]
- Gowen, L.C.; Johnson, B.L.; Latour, A.M.; Sulik, K.K.; Koller, B.H. Brca1 deficiency results in early embryonic lethality characterized by neuroepithelial abnormalities. Nat. Genet. 1996, 12, 191–194. [Google Scholar] [CrossRef]
- Hohenstein, P.; Kielman, M.F.; Breukel, C.; Bennett, L.M.; Wiseman, R.; Krimpenfort, P.; Cornelisse, C.; van Ommen, G.J.; Devilee, P.; Fodde, R. A targeted mouse Brca1 mutation removing the last BRCT repeat results in apoptosis and embryonic lethality at the headfold stage. Oncogene 2001, 20, 2544–2550. [Google Scholar] [CrossRef]
- Domchek, S.M.; Tang, J.; Stopfer, J.; Lilli, D.R.; Hamel, N.; Tischkowitz, M.; Monteiro, A.N.; Messick, T.E.; Powers, J.; Yonker, A.; et al. Biallelic deleterious BRCA1 mutations in a woman with early-onset ovarian cancer. Cancer Discov. 2013, 3, 399–405. [Google Scholar] [CrossRef] [PubMed]
- Sawyer, S.L.; Tian, L.; Kahkonen, M.; Schwartzentruber, J.; Kircher, M.; University of Washington Centre for Mendelian Genomics; FORGE Canada Consortium; Majewski, J.; Dyment, D.A.; Innes, A.M.; et al. Biallelic mutations in BRCA1 cause a new Fanconi anemia subtype. Cancer Discov. 2015, 5, 135–142. [Google Scholar] [CrossRef] [PubMed]
- Freire, B.L.; Homma, T.K.; Funari, M.F.A.; Lerario, A.M.; Leal, A.M.; Velloso, E.; Malaquias, A.C.; Jorge, A.A.L. Homozygous loss of function BRCA1 variant causing a Fanconi-anemia-like phenotype, a clinical report and review of previous patients. Eur. J. Med. Genet. 2018, 61, 130–133. [Google Scholar] [CrossRef] [PubMed]
- NCBI ClinVar Database, Entry NM_007294.3(BRCA1):c.4096+3A>G. Available online: https://www.ncbi.nlm.nih.gov/clinvar/variation/37566/ (accessed on 29 May 2019).
- Borg, A.; Haile, R.W.; Malone, K.E.; Capanu, M.; Diep, A.; Torngren, T.; Teraoka, S.; Begg, C.B.; Thomas, D.C.; Concannon, P.; et al. Characterization of BRCA1 and BRCA2 deleterious mutations and variants of unknown clinical significance in unilateral and bilateral breast cancer: The WECARE study. Hum. Mutat. 2010, 31, E1200–E1240. [Google Scholar] [CrossRef] [PubMed]
- Beissel, J.M.; Kendrick, M.L.; Podratz, K.C.; Bakkum-Gamez, J.N. Pancreaticoduodenectomy in optimal primary cytoreduction of epithelial ovarian cancer: A case report and review of the literature. Gynecol. Oncol. Rep. 2014, 10, 25–27. [Google Scholar] [CrossRef] [PubMed]
- Song, H.; Cicek, M.S.; Dicks, E.; Harrington, P.; Ramus, S.J.; Cunningham, J.M.; Fridley, B.L.; Tyrer, J.P.; Alsop, J.; Jimenez-Linan, M.; et al. The contribution of deleterious germline mutations in BRCA1, BRCA2 and the mismatch repair genes to ovarian cancer in the population. Hum. Mol. Genet. 2014, 23, 4703–4709. [Google Scholar] [CrossRef] [PubMed]
- Wappenschmidt, B.; Becker, A.A.; Hauke, J.; Weber, U.; Engert, S.; Kohler, J.; Kast, K.; Arnold, N.; Rhiem, K.; Hahnen, E.; et al. Analysis of 30 putative BRCA1 splicing mutations in hereditary breast and ovarian cancer families identifies exonic splice site mutations that escape in silico prediction. PLoS ONE 2012, 7, e50800. [Google Scholar] [CrossRef]
- The Exome Aggregation Consortium (ExAC), searching for rs80358015 and inspecting its hg19 chromosomal location 17-41243449. Available online: http://exac.broadinstitute.org/region/17-41243429-41243469 (accessed on 30 May 2019).
- Genome Aggregation Database (gnomAD), searching for rs80358015 and inspecting its hg19 chromosomal location 17-41243449. Available online: https://gnomad.broadinstitute.org/region/17-41243429-41243469 (accessed on 30 May 2019).
- Exome Variant Server, NHLBI GO Exome Sequencing Project (ESP), Seattle, WA. Available online: http://evs.gs.washington.edu/EVS/ (accessed on 30 May 2019).
- Auer, P.L.; Reiner, A.P.; Wang, G.; Kang, H.M.; Abecasis, G.R.; Altshuler, D.; Bamshad, M.J.; Nickerson, D.A.; Tracy, R.P.; Rich, S.S.; et al. Guidelines for Large-Scale Sequence-Based Complex Trait Association Studies: Lessons Learned from the NHLBI Exome Sequencing Project. Am. J. Hum. Genet. 2016, 99, 791–801. [Google Scholar] [CrossRef]
- ElShamy, W.M.; Livingston, D.M. Identification of BRCA1-IRIS, a BRCA1 locus product. Nat. Cell Biol. 2004, 6, 954–967. [Google Scholar] [CrossRef]
- Colombo, M.; Blok, M.J.; Whiley, P.; Santamarina, M.; Gutierrez-Enriquez, S.; Romero, A.; Garre, P.; Becker, A.; Smith, L.D.; De Vecchi, G.; et al. Comprehensive annotation of splice junctions supports pervasive alternative splicing at the BRCA1 locus: A report from the ENIGMA consortium. Hum. Mol. Genet. 2014, 23, 3666–3680. [Google Scholar] [CrossRef]
- Wang, Y.; Bernhardy, A.J.; Cruz, C.; Krais, J.J.; Nacson, J.; Nicolas, E.; Peri, S.; van der Gulden, H.; van der Heijden, I.; O’Brien, S.W.; et al. The BRCA1-Delta11q Alternative Splice Isoform Bypasses Germline Mutations and Promotes Therapeutic Resistance to PARP Inhibition and Cisplatin. Cancer Res. 2016, 76, 2778–2790. [Google Scholar] [CrossRef] [PubMed]
- Chock, K.L.; Allison, J.M.; Shimizu, Y.; ElShamy, W.M. BRCA1-IRIS overexpression promotes cisplatin resistance in ovarian cancer cells. Cancer Res. 2010, 70, 8782–8791. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Shimizu, Y.; Luk, H.; Horio, D.; Miron, P.; Griswold, M.; Iglehart, D.; Hernandez, B.; Killeen, J.; ElShamy, W.M. BRCA1-IRIS overexpression promotes formation of aggressive breast cancers. PLoS ONE 2012, 7, e34102. [Google Scholar] [CrossRef] [PubMed]
- Blanchard, Z.; Paul, B.T.; Craft, B.; ElShamy, W.M. BRCA1-IRIS inactivation overcomes paclitaxel resistance in triple negative breast cancers. Breast Cancer Res. 2015, 17, 5. [Google Scholar] [CrossRef] [PubMed]
- Paul, B.T.; Blanchard, Z.; Ridgway, M.; ElShamy, W.M. BRCA1-IRIS inactivation sensitizes ovarian tumors to cisplatin. Oncogene 2015, 34, 3036–3052. [Google Scholar] [CrossRef] [PubMed]
- ElShamy, W.M. The role of BRCA1-IRIS in ovarian cancer formation, drug resistance and progression. Oncoscience 2016, 3, 145–146. [Google Scholar] [CrossRef] [PubMed]
- Tammaro, C.; Raponi, M.; Wilson, D.I.; Baralle, D. BRCA1 exon 11 alternative splicing, multiple functions and the association with cancer. Biochem. Soc. Trans. 2012, 40, 768–772. [Google Scholar] [CrossRef]
- Li, D.; Harlan-Williams, L.M.; Kumaraswamy, E.; Jensen, R.A. BRCA1-No Matter How You Splice It. Cancer Res. 2019, 79, 2091–2098. [Google Scholar] [CrossRef]
- Seo, A.; Steinberg-Shemer, O.; Unal, S.; Casadei, S.; Walsh, T.; Gumruk, F.; Shalev, S.; Shimamura, A.; Akarsu, N.A.; Tamary, H.; et al. Mechanism for survival of homozygous nonsense mutations in the tumor suppressor gene BRCA1. Proc. Natl. Acad. Sci. USA 2018, 115, 5241–5246. [Google Scholar] [CrossRef]
- Pfeffer, C.M.; Ho, B.N.; Singh, A.T.K. The Evolution, Functions and Applications of the Breast Cancer Genes BRCA1 and BRCA2. Cancer Genomics Proteomics 2017, 14, 293–298. [Google Scholar] [CrossRef][Green Version]
- Takaoka, M.; Miki, Y. BRCA1 gene: Function and deficiency. Int. J. Clin. Oncol. 2018, 23, 36–44. [Google Scholar] [CrossRef] [PubMed]
- Yi, Y.W.; Kang, H.J.; Bae, I. BRCA1 and Oxidative Stress. Cancers 2014, 6, 771–795. [Google Scholar] [CrossRef] [PubMed]
- Coene, E.D.; Hollinshead, M.S.; Waeytens, A.A.; Schelfhout, V.R.; Eechaute, W.P.; Shaw, M.K.; Van Oostveldt, P.M.; Vaux, D.J. Phosphorylated BRCA1 is predominantly located in the nucleus and mitochondria. Mol. Biol. Cell 2005, 16, 997–1010. [Google Scholar] [CrossRef] [PubMed]
- Paull, T.T.; Cortez, D.; Bowers, B.; Elledge, S.J.; Gellert, M. Direct DNA binding by Brca1. Proc. Natl. Acad. Sci. USA 2001, 98, 6086–6091. [Google Scholar] [CrossRef]
- Naseem, R.; Webb, M. Analysis of the DNA binding activity of BRCA1 and its modulation by the tumour suppressor p53. PLoS ONE 2008, 3, e2336. [Google Scholar] [CrossRef]
- Masuda, T.; Xu, X.; Dimitriadis, E.K.; Lahusen, T.; Deng, C.X. “DNA Binding Region” of BRCA1 Affects Genetic Stability through modulating the Intra-S-Phase Checkpoint. Int. J. Biol. Sci. 2016, 12, 133–143. [Google Scholar] [CrossRef]
- Yamane, K.; Tsuruo, T. Conserved BRCT regions of TopBP1 and of the tumor suppressor BRCA1 bind strand breaks and termini of DNA. Oncogene 1999, 18, 5194–5203. [Google Scholar] [CrossRef]
- Ouchi, T. BRCA1 phosphorylation: Biological consequences. Cancer Biol. Ther. 2006, 5, 470–475. [Google Scholar] [CrossRef]
- Christou, C.M.; Kyriacou, K. BRCA1 and Its Network of Interacting Partners. Biology 2013, 2, 40–63. [Google Scholar] [CrossRef]
- Luo, A.; Zhang, K.; Zhao, Y.; Zhu, Z.; Fu, L.; Dong, J.T. ZNF121 interacts with ZBRK1 and BRCA1 to regulate their target genes in mammary epithelial cells. FEBS Open Bio 2018, 8, 1943–1952. [Google Scholar] [CrossRef]
- Hu, Y.F.; Miyake, T.; Ye, Q.; Li, R. Characterization of a novel trans-activation domain of BRCA1 that functions in concert with the BRCA1 C-terminal (BRCT) domain. J. Biol. Chem. 2000, 275, 40910–40915. [Google Scholar] [CrossRef] [PubMed]
- Daza-Martin, M.; Starowicz, K.; Jamshad, M.; Tye, S.; Ronson, G.E.; MacKay, H.L.; Chauhan, A.S.; Walker, A.K.; Stone, H.R.; Beesley, J.F.J.; et al. Isomerization of BRCA1–BARD1 promotes replication fork protection. Nature 2019, 571, 521–527. [Google Scholar] [CrossRef] [PubMed]
- Bochar, D.A.; Wang, L.; Beniya, H.; Kinev, A.; Xue, Y.; Lane, W.S.; Wang, W.; Kashanchi, F.; Shiekhattar, R. BRCA1 is associated with a human SWI/SNF-related complex: Linking chromatin remodeling to breast cancer. Cell 2000, 102, 257–265. [Google Scholar] [CrossRef]
- Harte, M.T.; O’Brien, G.J.; Ryan, N.M.; Gorski, J.J.; Savage, K.I.; Crawford, N.T.; Mullan, P.B.; Harkin, D.P. BRD7, a subunit of SWI/SNF complexes, binds directly to BRCA1 and regulates BRCA1-dependent transcription. Cancer Res. 2010, 70, 2538–2547. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Kim, Y.S.; Yang, X.P.; Li, L.P.; Liao, G.; Xia, F.; Jetten, A.M. The ubiquitin-interacting motif containing protein RAP80 interacts with BRCA1 and functions in DNA damage repair response. Cancer Res. 2007, 67, 6647–6656. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Watkins, T.; Reddy, A.; Reddy, E.S.; Rao, V.N. A novel mechanism whereby BRCA1/1a/1b fine tunes the dynamic complex interplay between SUMO-dependent/independent activities of Ubc9 on E2-induced ERalpha activation/repression and degradation in breast cancer cells. Int. J. Oncol. 2009, 34, 939–949. [Google Scholar] [CrossRef]
- Fabbro, M.; Rodriguez, J.A.; Baer, R.; Henderson, B.R. BARD1 induces BRCA1 intranuclear foci formation by increasing RING-dependent BRCA1 nuclear import and inhibiting BRCA1 nuclear export. J. Biol. Chem. 2002, 277, 21315–21324. [Google Scholar] [CrossRef]
- Qin, Y.; Xu, J.; Aysola, K.; Begum, N.; Reddy, V.; Chai, Y.; Grizzle, W.E.; Partridge, E.E.; Reddy, E.S.; Rao, V.N. Ubc9 mediates nuclear localization and growth suppression of BRCA1 and BRCA1a proteins. J. Cell. Physiol. 2011, 226, 3355–3367. [Google Scholar] [CrossRef]
- Arason, A.; Gunnarsson, H.; Johannesdottir, G.; Jonasson, K.; Bendahl, P.O.; Gillanders, E.M.; Agnarsson, B.A.; Jonsson, G.; Pylkas, K.; Mustonen, A.; et al. Genome-wide search for breast cancer linkage in large Icelandic non-BRCA1/2 families. Breast Cancer Res. 2010, 12, R50. [Google Scholar] [CrossRef]
- Harris, T.B.; Launer, L.J.; Eiriksdottir, G.; Kjartansson, O.; Jonsson, P.V.; Sigurdsson, G.; Thorgeirsson, G.; Aspelund, T.; Garcia, M.E.; Cotch, M.F.; et al. Age, Gene/Environment Susceptibility-Reykjavik Study: Multidisciplinary applied phenomics. Am. J. Epidemiol. 2007, 165, 1076–1087. [Google Scholar] [CrossRef]
- Sigurdardottir, L.G.; Jonasson, J.G.; Stefansdottir, S.; Jonsdottir, A.; Olafsdottir, G.H.; Olafsdottir, E.J.; Tryggvadottir, L. Data quality at the Icelandic Cancer Registry: Comparability, validity, timeliness and completeness. Acta Oncol. 2012, 51, 880–889. [Google Scholar] [CrossRef] [PubMed]
- Pukkala, E.; Engholm, G.; Hojsgaard Schmidt, L.K.; Storm, H.; Khan, S.; Lambe, M.; Pettersson, D.; Olafsdottir, E.; Tryggvadottir, L.; Hakanen, T.; et al. Nordic Cancer Registries—An overview of their procedures and data comparability. Acta Oncol. 2018, 57, 440–455. [Google Scholar] [CrossRef] [PubMed]
- University of California Santa Cruz (UCSC) Genome Browser Gateway, Marker D17S855. Available online: http://genome.ucsc.edu/cgi-bin/hgc?hgsid=731572807_GFHLiW2UabhS2HW338rF9jqD4AiK&c=chr17&l=41104743&r=41304894&o=41204743&t=41204894&g=stsMap&i=D17S855 (accessed on 24 June 2019).
- University of California Santa Cruz (UCSC) Genome Browser Gateway, Marker D17S579. Available online: http://genome.ucsc.edu/cgi-bin/hgc?hgsid=731572807_GFHLiW2UabhS2HW338rF9jqD4AiK&c=chr17&l=42706811&r=42907263&o=42806811&t=42807263&g=stsMap&i=D17S579 (accessed on 24 June 2019).
- Anderson, L.A.; Friedman, L.; Osborne-Lawrence, S.; Lynch, E.; Weissenbach, J.; Bowcock, A.; King, M.C. High-density genetic map of the BRCA1 region of chromosome 17q12-q21. Genomics 1993, 17, 618–623. [Google Scholar] [CrossRef] [PubMed]




© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Arason, A.; Agnarsson, B.A.; Johannesdottir, G.; Johannsson, O.T.; Hilmarsdottir, B.; Reynisdottir, I.; Barkardottir, R.B. The BRCA1 c.4096+3A>G Variant Displays Classical Characteristics of Pathogenic BRCA1 Mutations in Hereditary Breast and Ovarian Cancers, But Still Allows Homozygous Viability. Genes 2019, 10, 882. https://doi.org/10.3390/genes10110882
Arason A, Agnarsson BA, Johannesdottir G, Johannsson OT, Hilmarsdottir B, Reynisdottir I, Barkardottir RB. The BRCA1 c.4096+3A>G Variant Displays Classical Characteristics of Pathogenic BRCA1 Mutations in Hereditary Breast and Ovarian Cancers, But Still Allows Homozygous Viability. Genes. 2019; 10(11):882. https://doi.org/10.3390/genes10110882
Chicago/Turabian StyleArason, Adalgeir, Bjarni A Agnarsson, Gudrun Johannesdottir, Oskar Th Johannsson, Bylgja Hilmarsdottir, Inga Reynisdottir, and Rosa B Barkardottir. 2019. "The BRCA1 c.4096+3A>G Variant Displays Classical Characteristics of Pathogenic BRCA1 Mutations in Hereditary Breast and Ovarian Cancers, But Still Allows Homozygous Viability" Genes 10, no. 11: 882. https://doi.org/10.3390/genes10110882
APA StyleArason, A., Agnarsson, B. A., Johannesdottir, G., Johannsson, O. T., Hilmarsdottir, B., Reynisdottir, I., & Barkardottir, R. B. (2019). The BRCA1 c.4096+3A>G Variant Displays Classical Characteristics of Pathogenic BRCA1 Mutations in Hereditary Breast and Ovarian Cancers, But Still Allows Homozygous Viability. Genes, 10(11), 882. https://doi.org/10.3390/genes10110882