Predominance of Dystrophinopathy Genotypes in Mexican Male Patients Presenting as Muscular Dystrophy with A Normal Multiplex Polymerase Chain Reaction DMD Gene Result: A Study Including Targeted Next-Generation Sequencing

 , , , ,
, , , ,
Abstract
1. Introduction
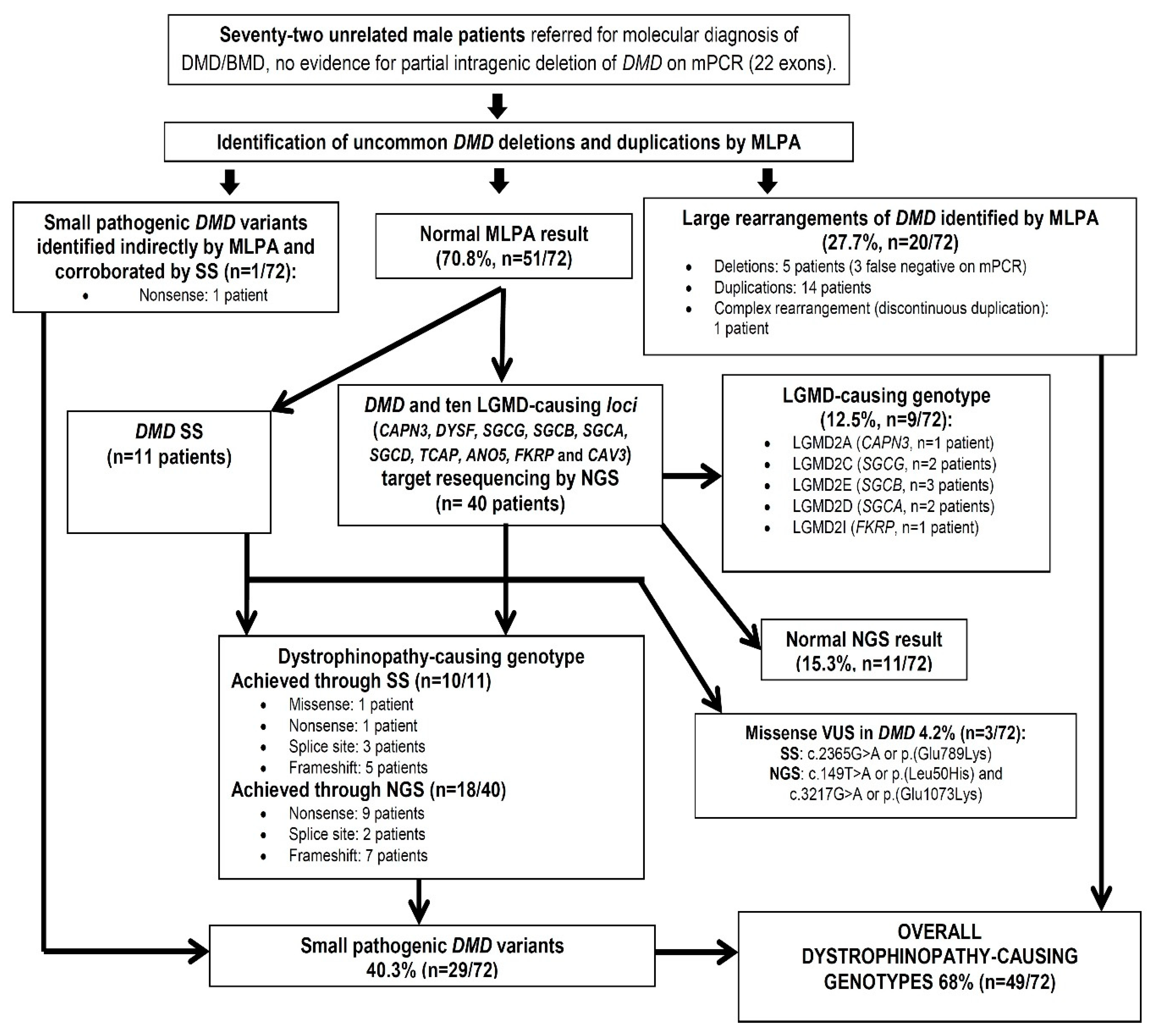
2. Methods
2.1. Patients
2.2. MLPA Analysis
2.3. SS of DMD and Targeted NGS Resequencing of DMD and 10 LGMD-Causing Loci
3. Results and Discussion
3.1. Identification of DMD Genotypes by MLPA
3.1.1. Partial DMD Gene Duplications
3.1.2. Partial DMD Gene Deletions and mPCR False-Negatives
3.1.3. Resulting Reading Frame for DMD Gene Duplications and Deletions
3.1.4. Indirect MLPA Identification of Small DMD Gene Variants
3.2. Small Pathogenic DMD Genotypes Identified through SS and NGS
3.2.1. Small Mutational DMD Spectrum
3.2.2. Missense VUS in the DMD Gene
3.3. Mother Carrier Diagnosis for Overall DMD Pathogenic Genotypes
3.4. LGMD Genotypes Identified through NGS
The Unexpected Absence of Dysferlinopathies
3.5. Comparison of Overall Achieved Muscular Dystrophy Diagnostic Success Rate with other Similar NGS-based Studies
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Angelini, C.; Giaretta, L.; Marozzo, R. An update on diagnostic options and considerations in limb-girdle dystrophies. Expert. Rev. Neurother. 2018, 18, 693–703. [Google Scholar] [CrossRef] [PubMed]
- Iyadurai, S.J.; Kissel, J.T. The Limb-Girdle Muscular Dystrophies and the Dystrophinopathies. Continuum 2016, 22, 1954–1977. [Google Scholar] [CrossRef] [PubMed]
- Aartsma-Rus, A.; Ginjaar, I.B.; Bushby, K. The importance of genetic diagnosis for Duchenne muscular dystrophy. J. Med. Genet. 2016, 53, 145–151. [Google Scholar] [CrossRef]
- Magri, F.; Nigro, V.; Angelini, C.; Mongini, T.; Mora, M.; Moroni, I.; Toscano, A.; D’angelo, M.G.; Tomelleri, G.; Siciliano, G.; et al. The Italian limb girdle muscular dystrophy registry: Relative frequency, clinical features, and differential diagnosis. Muscle Nerv. 2017, 55, 55–68. [Google Scholar] [CrossRef] [PubMed]
- Bengtsson, N.E.; Hall, J.K.; Odom, G.L.; Phelps, M.P.; Andrus, C.R.; Hawkins, R.D.; Hauschka, S.D.; Chamberlain, J.R.; Chamberlain, J.S. Muscle-specific CRISPR/Cas9 dystrophin gene editing ameliorates pathophysiology in a mouse model for Duchenne muscular dystrophy. Nat. Commun. 2017, 8, 14454. [Google Scholar] [CrossRef] [PubMed]
- Nigro, V.; Savarese, M. Next-generation sequencing approaches for the diagnosis of skeletal muscle disorders. Curr. Opin. Neurol. 2016, 29, 621–627. [Google Scholar] [CrossRef]
- Coral-Vazquez, R.; Arenas, D.; Cisneros, B.; Peñaloza, L.; Salamanca, F.; Kofman, S.; Mercado, R.; Montañez, C. Pattern of deletions of the dystrophin gene in Mexican Duchenne/Becker muscular dystrophy patients: The use of new designed primers for the analysis of the major deletion “hot spot” region. Am. J. Med. Genet. 1997, 70, 240–246. [Google Scholar] [CrossRef]
- González-Herrera, L.; Gamas-Trujillo, P.A.; García-Escalante, M.G.; Castillo-Zapata, I.; Pinto-Escalante, D. Identifying deletions in the dystrophin gene and detecting carriers in families with Duchenne's/Becker's muscular dystrophy. Rev. Neurol. 2009, 48, 66–70. [Google Scholar] [CrossRef]
- López-Hernández, L.B.; Gómez-Díaz, B.; Luna-Angulo, A.B.; Anaya-Segura, M.; Bunyan, D.J.; Zúñiga-Guzman, C.; Escobar-Cedillo, R.E.; Roque-Ramírez, B.; Ruano-Calderón, L.A.; Rangel-Villalobos, H.; et al. Comparison of mutation profiles in the Duchenne muscular dystrophy gene among populations: Implications for potential molecular therapies. Int. J. Mol. Sci. 2015, 16, 5334–5346. [Google Scholar] [CrossRef]
- Rosas-Vargas, H.; Gómez-Díaz, B.; Ruano-Calderón, L.; Fernández-Valverde, F.; Roque-Ramírez, B.; Portillo-Bobadilla, T.; Ordoñez-Razo, R.M.; Minauro-Sanmiguel, F.; Coral-Vázquez, R. Dysferlin homozygous mutation G1418D causes limb-girdle type 2B in a Mexican family. Genet. Test. 2007, 11, 391–396. [Google Scholar] [CrossRef]
- Pantoja-Melendez, C.A.; Miranda-Duarte, A.; Roque-Ramirez, B.; Zenteno, J.C. Epidemiological and Molecular Characterization of a Mexican Population Isolate with High Prevalence of Limb-Girdle Muscular Dystrophy Type 2A Due to a Novel Calpain-3 Mutation. PLoS ONE 2017, 12, e0170280. [Google Scholar] [CrossRef] [PubMed]
- Gómez-Díaz, B.; Rosas-Vargas, H.; Roque-Ramírez, B.; Meza-Espinoza, P.; Ruano-Calderón, L.A.; Fernández-Valverde, F.; Escalante-Bautista, D.; Escobar-Cedillo, R.E.; Sánchez-Chapul, L.; Vargas-Cañas, S.; et al. Immunodetection analysis of muscular dystrophies in Mexico. Muscle Nerv. 2012, 45, 338–345. [Google Scholar] [CrossRef] [PubMed]
- Navarro-Cobos, M.J.; González-Del Angel, A.; Estandia-Ortega, B.; Ruiz-Herrera, A.; Becerra, A.; Vargas-Ramírez, G.; Bermúdez-López, C.; Alcántara-Ortigoza, M.A. Molecular Analysis Confirms that FKRP-Related Disorders Are Underdiagnosed in Mexican Patients with Neuromuscular Diseases. Neuropediatrics 2017, 48, 442–450. [Google Scholar] [CrossRef] [PubMed]
- López-Hernández, L.B.; Gómez-Díaz, B.; Escobar-Cedillo, R.E.; Gama-Moreno, O.; Camacho-Molina, A.; Soto-Valdés, D.M.; Anaya-Segura, M.A.; Luna-Padrón, E.; Zúñiga-Guzmán, C.; Lopez-Hernández, J.A.; et al. Duchenne muscular dystrophy in a developing country: Challenges in management and genetic counseling. Genet. Couns. 2014, 25, 129–141. [Google Scholar]
- Traverso, M.; Malnati, M.; Minetti, C.; Regis, S.; Tedeschi, S.; Pedemonte, M.; Bruno, C.; Biassoni, R.; Zara, F. Multiplex real-time PCR for detection of deletions and duplications in dystrophin gene. Biochem. Biophys. Res. Commun. 2006, 339, 145–150. [Google Scholar] [CrossRef]
- DMD Exonic Deletions/Duplications Reading-Frame Checker Ver. 1.9. Leiden Muscular Dystrophy Pages©. Available online: http://www.dmd.nl/ (accessed on 5 March 2018).
- Nallamilli, B.R.; Ankala, A.; Hegde, M. Molecular diagnosis of Duchenne muscular dystrophy. Curr. Protoc. Hum. Genet. 2014, 83, 9–25. [Google Scholar] [CrossRef]
- MacLeod, H.; Pytel, P.; Wollmann, R.; Chelmicka-Schorr, E.; Silver, K.; Anderson, R.B.; Waggoner, D.; McNally, E.M. A novel FKRP mutation in congenital muscular dystrophy disrupts the dystrophin glycoprotein complex. Neuromuscul. Disord. 2007, 17, 285–289. [Google Scholar] [CrossRef]
- Lee, A.J.; Jones, K.A.; Butterfield, R.J.; Cox, M.O.; Konersman, C.G.; Grosmann, C.; Abdenur, J.E.; Boyer, M.; Beson, B.; Wang, C.; et al. Clinical, genetic, and pathologic characterization of FKRP Mexican founder mutation c.1387A>G. Neurol. Genet. 2019, 5, e315. [Google Scholar] [CrossRef]
- Barresi, R. From proteins to genes: Immunoanalysis in the diagnosis of muscular dystrophies. Skelet. Muscle 2011, 1, 24. [Google Scholar] [CrossRef]
- FastQC: A Quality Control Tool for High Throughput Sequence Data. Available online: https://www.bioinformatics.babraham.ac.uk/projects/fastqc/ (accessed on 31 May 2019).
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina sequence data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed]
- FreeBayes: Haplotype-Based Variant Detection from Short-Read Sequencing. Available online: https://arxiv:abs/1207.3907 (accessed on 31 May 2019).
- McKenna, A.; Hanna, M.; Banks, E.; Sivachenko, A.; Cibulskis, K.; Kernytsky, A.; Garimella, K.; Altshuler, D.; Gabriel, S.; Daly, M.; et al. The Genome Analysis Toolkit: A MapReduce framework for analyzing next-generation DNA sequencing data. Genome Res. 2010, 20, 1297–1303. [Google Scholar] [CrossRef] [PubMed]
- Kleinberger, J.; Maloney, K.A.; Pollin, T.I.; Jeng, L.J. An openly available online tool for implementing the ACMG/AMP standards and guidelines for the interpretation of sequence variants. Genet. Med. 2016, 18, 1165. [Google Scholar] [CrossRef] [PubMed]
- Single Nucleotide Polymorphism Database dbSNP. Available online: https://www.ncbi.nlm.nih.gov/projects/SNP/ (accessed on 8 May 2019).
- ClinVar Database. Available online: https://www.ncbi.nlm.nih.gov/clinvar/ (accessed on 8 May 2019).
- Human Gene Mutation Database (HGMD). Available online: http://www.hgmd.cf.ac.uk/ac/all.php (accessed on 8 May 2019).
- The DMD Mutations Database UMD-DMD France. Available online: http://www.umd.be/DMD/W_DMD/index.html (accessed on 8 May 2019).
- Genome Aggregation Database (gnomAD). Available online: https://gnomad.broadinstitute (accessed on 8 May 2019).
- Leiden Open Source Variation Database (LOVD). Available online: http://www.lovd.nl/3.0/home. (accessed on 8 May 2019).
- Lalic, T.; Vossen, R.H.; Coffa, J.; Schouten, J.P.; Guc-Scekic, M.; Radivojevic, D.; Djurisic, M.; Breuning, M.H.; White, S.J.; den Dunnen, J.T. Deletion and duplication screening in the DMD gene using MLPA. Eur. J. Hum. Genet. 2005, 13, 1231–1234. [Google Scholar] [CrossRef] [PubMed]
- Kohli, S.; Saxena, R.; Thomas, E.; Singh, J.; Verma, I.C. Gene changes in Duchenne muscular dystrophy: Comparison of multiplex PCR and multiplex ligation-dependent probe amplification techniques. Neurol. India 2010, 58, 852–856. [Google Scholar] [CrossRef]
- Janssen, B.; Hartmann, C.; Scholz, V.; Jauch, A.; Zschocke, J. MLPA analysis for the detection of deletions, duplications and complex rearrangements in the dystrophin gene: Potential and pitfalls. Neurogenetics 2005, 6, 29–35. [Google Scholar] [CrossRef]
- Dastur, R.S.; Kachwala, M.Y.; Khadilkar, S.V.; Hegde, M.R.; Gaitonde, P.S. Identification of deletions and duplications in the Duchenne muscular dystrophy gene and female carrier status in western India using combined methods of multiplex polymerase chain reaction and multiplex ligation-dependent probe amplification. Neurol. India 2011, 59, 803–809. [Google Scholar] [CrossRef]
- Nouri, N.; Fazel-Najafabadi, E.; Salehi, M.; Hosseinzadeh, M.; Behnam, M.; Ghazavi, M.R.; Sedghi, M. Evaluation of multiplex ligation-dependent probe amplification analysis versus multiplex polymerase chain reaction assays in the detection of dystrophin gene rearrangements in an Iranian population subset. Adv. Biomed. Res. 2014, 3, 72. [Google Scholar] [CrossRef]
- Mohammed, F.; Elshafey, A.; Al-Balool, H.; Alaboud, H.; Ali, M.A.B.; Baqer, A.; Bastaki, L. Mutation spectrum analysis of Duchenne/Becker muscular dystrophy in 68 families in Kuwait: The era of personalized medicine. PLoS ONE 2018, 13, e0197205. [Google Scholar] [CrossRef]
- Bladen, C.L.; Salgado, D.; Monges, S.; Foncuberta, M.E.; Kekou, K.; Kosma, K.; Dawkins, H.; Lamont, L.; Roy, A.J.; Chamova, T.; et al. The TREAT-NMD DMD Global Database: Analysis of more than 7000 Duchenne muscular dystrophy mutations. Hum. Mutat. 2015, 36, 395–402. [Google Scholar] [CrossRef]
- de Almeida, P.A.D.; Machado-Costa, M.C.; Manzoli, G.N.; Ferreira, L.S.; Rodrigues, M.C.S.; Bueno, L.S.M.; Saute, J.A.M.; Pinto Vairo, F.; Matte, U.S.; Siebert, M.; et al. Genetic profile of Brazilian patients with dystrophinopathies. Clin. Genet. 2017, 92, 199–203. [Google Scholar] [CrossRef] [PubMed]
- Luce, L.N.; Carcione, M.; Mazzanti, C.; Ferrer, M.; Szijan, I.; Giliberto, F. Small mutation screening in the DMD gene by whole exome sequencing of an argentine Duchenne/Becker muscular dystrophies cohort. Neuromuscul. Disord. 2018, 28, 986–995. [Google Scholar] [CrossRef] [PubMed]
- Singh, B.; Mandal, K.; Lallar, M.; Narayanan, D.L.; Mishra, S.; Gambhir, P.S.; Phadke, S.R. Next Generation Sequencing in Diagnosis of MLPA Negative Cases Presenting as Duchenne/Becker Muscular Dystrophies. Indian J. Pediatr. 2018, 85, 309–310. [Google Scholar] [CrossRef] [PubMed]
- Juan-Mateu, J.; Gonzalez-Quereda, L.; Rodriguez, M.J.; Baena, M.; Verdura, E.; Nascimento, A.; Ortez, C.; Baiget, M.; Gallano, P. DMD Mutations in 576 Dystrophinopathy Families: A Step Forward in Genotype-Phenotype Correlations. PLoS ONE 2015, 10, e0135189. [Google Scholar] [CrossRef] [PubMed]
- Flanigan, K.M.; von Niederhausern, A.; Dunn, D.M.; Alder, J.; Mendell, J.R.; Weiss, R.B. Rapid direct sequence analysis of the dystrophin gene. Am. J. Hum. Genet. 2003, 72, 931–939. [Google Scholar] [CrossRef]
- MutationTaster Program. Available online: http://www.mutationtaster (accessed on 1 January 2019).
- PolyPhen Program. Available online: http://genetics.bwh.harvard.edu/pph2/ (accessed on 1 January 2019).
- Pmut Program. Available online: http://mmb.pcb.ub.es/PMut/ (accessed on 1 January 2019).
- MutPred2 Program. Available online: http://mutpred.mutdb (accessed on 1 January 2019).
- PROVEAN Program. Available online: http://provean.jcvi:index.php (accessed on 1 January 2019).
- SIFT Program. Available online: http://sift.bii.a-star.edu.sg/ (accessed on 1 January 2019).
- Henderson, D.M.; Lee, A.; Ervasti, J.M. Disease-causing missense mutations in actin binding domain 1 of dystrophin induce thermodynamic instability and protein aggregation. Proc. Natl. Acad. Sci. USA 2010, 107, 9632–9637. [Google Scholar] [CrossRef]
- Koczok, K.; Merő, G.; Szabó, G.P.; Madar, L.; Gombos, É.; Ajzner, É.; Mótyán, J.A.; Hortobágyi, T.; Balogh, I. A novel point mutation affecting Asn76 of dystrophin protein leads to dystrophinopathy. Neuromuscul. Disord. 2018, 28, 129–136. [Google Scholar] [CrossRef]
- Grimm, T.; Kress, W.; Meng, G.; Müller, C.R. Risk assessment and genetic counseling in families with Duchenne muscular dystrophy. Acta Myol. 2012, 31, 179–183. [Google Scholar]
- Giugliano, T.; Fanin, M.; Savarese, M.; Piluso, G.; Angelini, C.; Nigro, V. Identification of an intragenic deletion in the SGCB gene through a re-evaluation of negative next generation sequencing results. Neuromuscul. Disord. 2016, 26, 367–369. [Google Scholar] [CrossRef]
- Vainzof, M.; Anderson, L.V.; McNally, E.M.; Davis, D.B.; Faulkner, G.; Valle, G.; Moreira, E.S.; Pavanello, R.C.; Passos-Bueno, M.R.; Zatz, M. Dysferlin protein analysis in limb-girdle muscular dystrophies. J. Mol. Neurosci. 2001, 17, 71–80. [Google Scholar] [CrossRef]
- Fanin, M.; Angelini, C. Progress and challenges in diagnosis of dysferlinopathy. Muscle Nerv. 2016, 54, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Kitamura, Y.; Kondo, E.; Urano, M.; Aoki, R.; Saito, K. Target resequencing of neuromuscular disease-related genes using next-generation sequencing for patients with undiagnosed early-onset neuromuscular disorders. J. Hum. Genet. 2016, 61, 931–942. [Google Scholar] [CrossRef] [PubMed]
- Inashkina, I.; Jankevics, E.; Stavusis, J.; Vasiljeva, I.; Viksne, K.; Micule, I.; Strautmanis, J.; Naudina, M.S.; Cimbalistiene, L.; Kucinskas, V.; et al. Robust genotyping tool for autosomal recessive type of limb-girdle muscular dystrophies. BMC Musculoskelet. Disord. 2016, 17, 200. [Google Scholar] [CrossRef] [PubMed]
- Kress, W.; Rost, S.; Kolokotronis, K.; Meng, G.; Pluta, N.; Müller-Reible, C. The Genetic Approach: Next-Generation Sequencing-Based Diagnosis of Congenital and Infantile Myopathies/Muscle Dystrophies. Neuropediatrics 2017, 48, 242–246. [Google Scholar] [CrossRef] [PubMed]
- Monies, D.; Alhindi, H.N.; Almuhaizea, M.A.; Abouelhoda, M.; Alazami, A.M.; Goljan, E.; Alyounes, B.; Jaroudi, D.; AlIssa, A.; Alabdulrahman, K.; et al. A first-line diagnostic assay for limb-girdle muscular dystrophy and other myopathies. Hum. Genomics 2016, 10, 32. [Google Scholar] [CrossRef] [PubMed]


{kind=link}
| Genotype | Patient ID | Available Relevant Clinical Data | NMD Familial History | Relatives’ Genotype Status | |
|---|---|---|---|---|---|
| Hemizygous DMD gene deletions identified by MLPA A | |||||
| 1 | c.531-?_960 + ?del (DELETION OF EXON 7 TO 9: out-of-frame) | DMD-386 | NA. False negative on Multiplex PCR assay (DMD exon 8 deletion unnoticed). | ABSENT | Non-carrier mother |
| 2 | c.2169-?_2292 + ?del (DELETION OF EXON 18: out-of-frame) | DMD-1834 | DMD phenotype (still ambulant at 9 yr), HyperCKemia, MP-EMG. | PRESENT | Carrier mother, two normal homozygous sisters. |
| 3 | c.2804-? 4071 + ?del (DELETION OF EXON 22 TO 29: out-of-frame) | DMD-1302 | NA | PRESENT | Carrier mother, two healthy hemizygous brothers. |
| 4 | c.4234-?_6290 + ?del (DELETION OF EXON 31 TO 43: out-of-frame) | DMD-1355 | NA. False negative on Multiplex PCR assay (DMD exon 43 deletion unnoticed). | ABSENT | Non-carrier mother |
| 5 | c.6439-?_6614 + ?del (DELETION OF EXON 45: out-of-frame) | DMD-128 | NA. False negative on Multiplex PCR assay (DMD exon 45 deletion unnoticed). | ABSENT | NA |
| Hemizygous DMD gene duplications identified by MLPA A | |||||
| 1 | c.32-?_93 + ?dup (DUPLICATION OF EXON 2: out-of-frame) | DMD-943 | HyperCKemia, dystrophic changes in muscle biopsy. | ABSENT | Non-carrier mother |
| 2 | c.32-?_93 + ?dup (DUPLICATION OF EXON 2: out-of-frame) | DMD-1432 | NA | ABSENT | Non-carrier mother |
| 3 | c.94-?_357 + ?dup (DUPLICATION OF EXON 3 TO 5: in-frame) | DMD-752 | BMD phenotype, hyperCKemia. | ABSENT | Non-carrier mother |
| 4 | c.94-?_530 + ?dup (DUPLICATION OF EXON 3 TO 6: out-of-frame) | DMD-899 | NA | PRESENT | Carrier mother |
| 5 | c.94-?_960 + ?dup (DUPLICATION OF EXON 3 TO 9: in-frame) | DMD-640 | BMD phenotype, hyperCKemia, MP-EMG, dystrophic changes in muscle biopsy. | ABSENT | NA |
| 6 | c.1993-?_2803 + ?dup (DUPLICATION OF EXON 17 TO 21: out-of-frame) | DMD-425 | NA | ABSENT | Carrier mother |
| 7 | c.4072-?_6290 + ?dup (DUPLICATION OF EXON 30 TO 43: out-of-frame) | DMD-1561 | DMD phenotype, hyperCKemia, MP-EMG, dystrophic changes and abnormal immunoanalysis pattern of dystrophin in muscle biopsy. | ABSENT | Non-carrier mother |
| 8 | c.4675-?_6290 + ?dup (DUPLICATION OF EXON 34 TO 43: out-of-frame) | DMD-1430 | NA | PRESENT | Carrier mother |
| 9 | c.6291-?_8217 + ?dup (DUPLICATION OF EXON 44 TO 55: out-of-frame) | DMD-907 | NA | PRESENT | Carrier mother and non-carrier sister. |
| 10 | c.6615-?_7200 + ?dup (DUPLICATION OF EXON 46 TO 49: out-of-frame) | DMD-1749 | DMD phenotype, hyperCKemia, MP-EMG, dystrophic changes in muscle biopsy. | PRESENT | Carrier mother and two carrier sisters. |
| 11 | c.7543-?_7660 + ?dup (DUPLICATION OF EXON 52: out-of-frame) | DMD-1191 | Still ambulant at 14 yr without corticosteroid therapy, hyperCKemia, MP-EMG, abnormal immunoanalysis pattern of dystrophin in muscle biopsy. | ABSENT | Non-carrier mother |
| 12 | c.7543-?_7660 + ?dup (DUPLICATION OF EXON 52: out-of-frame) | DMD-1585 | DMD phenotype, hyperCKemia, MP-EMG, dystrophic changes in muscle biopsy. | ABSENT | NA |
| 13 | c.7543-?_7660 + ?dup (DUPLICATION OF EXON 52: out-of-frame) | DMD-1751 | Still ambulant at 17 yr, hyperCKemia, MP-EMG. Deflazacort therapy initiated at 16 yr. | ABSENT | Non-carrier mother |
| 14 | c.8938-?_9807 + ?dup (DUPLICATION OF EXON 60 TO 67: in-frame) | DMD-1460 | DMD phenotype | PRESENT | Carrier mother |
| Hemizygous DMD gene complex rearrangements identified by MLPAA | |||||
| 1 | c.[5587-?_7309 + ?dup;9225-?_* 2691 + ?dup] (DISCONTINUOUS DUPLICATION OF EXONS 45 TO 50 AND 63 TO 79: Unpredictable frame rule effect) | DMD-1872 | Probable BMD phenotype (still ambulant at 12 yr), hyperCKemia, MP-EMG. No corticosteroid therapy. | ABSENT | NA |
| Hemizygous DMD genotypes for small variants identified indirectly by MLPA and Sanger sequencing B | |||||
| 1 | c.2707G > T or p.(Gly903 *). LOVD DB-ID: DMD_001252. | DMD-1803 | Still ambulant at 8 yr, hyperCKemia. Apparent DMD exon 21 deletion in MLPA assay, which was discarded and correctly annotated by Sanger sequencing. | PRESENT | Carrier mother |
| Hemizygous DMD genotypes for small variants identified by Sanger sequencing B | |||||
| Missense | |||||
| 1 | c.494A > T or p.(Asp165Val). LOVD DB-ID: DMD_000547 | DMD-1852 | Still ambulant at 9 yr with deflazacort therapy, hyperCKemia and dystrophin absence by immunoanalysis. | ABSENT | Non-carrier mother |
| Non-Sense | |||||
| 2 | c.4758G > A or p.(Trp1586*). ClinVar: RCV000711459.1 | DMD-1187 | DMD phenotype, hyperCKemia, dystrophic changes in muscle biopsy with dystrophin absence by immunoanalysis. | ABSENT | Carrier mother and normal homozygous sister. |
| Splicing | |||||
| 3 | c.2292 + 2T > G. ClinVar: RCV000585714.1 | DMD-1372 | DMD phenotype, hyperCKemia, MP-EMG. | PRESENT | Carrier mother and affected hemizygous brother; normal homozygous sister. |
| 4 | c.2622 + 1G > A. dbSNP: rs398123901 | DMD-1890 | Still ambulant at 8 yr, hyperCKemia, dystrophic changes in muscle biopsy with dystrophin absence by immunoanalysis. | PRESENT | Carrier mother and affected hemizygous brother. |
| 5 | c.7661-1G > A. LOVD DB-ID: DMD_000263 | DMD-1793 | DMD phenotype, hyperCKemia, dystrophic changes in muscle biopsy with “abnormal” dystrophin pattern by immunoanalysis. | ABSENT | NA |
| Frameshift | |||||
| 6 | c.294del or p.(Asp98Glufs * 3) (novel) | DMD-1801 | Still ambulant at 9 yr, hyperCKemia, MP-EMG. | ABSENT | NA |
| 7 | c.2281_2285del or p.(Glu761Serfs * 10). dbSNP: rs398123881 | DMD-1789 | DMD phenotype, hyperCKemia, dystrophic changes in muscle biopsy with dystrophin absence by immunoanalysis. | ABSENT | Non-carrier mother |
| 8 | c.6128_6131del or p.(Asp2043Valfs * 29). dbSNP: rs863225006 | DMD-1837 | Still ambulant at 11 yr, hyperCKemia. | PRESENT | Carrier mother |
| 9 | c.6446dup or p.(Asp2150Glyfs * 73) (novel) | DMD-1847 | Still ambulant at 9 yr, 9 mo; hyperCKemia, dystrophic changes in muscle biopsy with dystrophin absence by immunoanalysis. | ABSENT | Carrier mother |
| 10 | c.9204_9207del or p.(Asn3068Lysfs * 20). dbSNP: rs863225015 | DMD-1777 | DMD phenotype, hyperCKemia, MP-EMG. | ABSENT | Carrier mother |
| Variants of unknown significance | |||||
| 11 | c.2365G > A or p.(Glu789Lys). dbSNP: rs763844939 | DMD-1313 | BMD phenotype (died age 34 yr, unknown cause) | ABSENT | Heterozygous mother. Two normal homozygous sisters. |
| Hemizygous DMD genotypes for small variants identified by NGS B | |||||
| Non-sense | |||||
| 1 | c.583C > T or p.(Arg195 *). dbSNP: rs398123999 | DMD-1395 | HyperCKemia. | ABSENT | Non-carrier mother |
| 2 | c.2704C > T or p.(Gln902 *). LOVD DB-ID: DMD_003328 | DMD-941 | NA | ABSENT | Non-carrier mother |
| 3 | c.2926G > T or p.(Glu976 *) (novel) | DMD-627 | NA | PRESENT | Carrier status confirmed in mother, sister and niece. Hemizygous affected nephew. |
| 4 | c.3268C > T or p.(Gln1090 *). ClinVar: RCV000630553.1 | DMD-1665 | DMD phenotype, hyperCKemia, MP-EMG, dystrophic changes in muscle biopsy with dystrophin absence by immunoanalysis. | ABSENT | Carrier mother |
| 5 | c.3274A > T or p.(Arg1092 *). LOVD DB-ID: DMD_000335 | DMD-1042 | NA | PRESENT | Carrier mother and affected hemizygous brother. |
| 6 | c.4757G > A or p.(Trp1586 *). LOVD DB-ID: DMD_000571 | DMD-1061 | HyperCKemia, dystrophic changes in muscle biopsy. | ABSENT | Non-carrier mother |
| 7 | c.5140G > T or p.(Glu1714 *). dbSNP: rs886042747 | DMD-1565 | HyperCkemia, dystrophic changes in muscle biopsy with dystrophin absence by immunoanalysis. | ABSENT | Carrier mother |
| 8 | c.8744G > A or p.(Trp2915 *). HGMD: CM066784 | DMD-983 | NA | PRESENT | Carrier mother and affected hemizygous brother. |
| 9 | c.10171C > T or p.(Arg3391 *). dbSNP: rs398123832 | DMD-1884 | DMD phenotype. | ABSENT | NA |
| Splicing | |||||
| 10 | c.2168 + 1G > T LOVD DB-ID: DMD_046199 | DMD-465 | HyperCKemia. | ABSENT | Carrier mother |
| 11 | c.8937 + 2T > C. LOVD DB-ID: DMD_002149 | DMD-757 | Died age 33 yr (unknown cause) | PRESENT | Carrier mother and maternal aunt. |
| Frameshift | |||||
| 12 | c.1374dup or p.(Glu459Argfs * 4). LOVD DB-ID: DMD_000109 | DMD-495 | NA | PRESENT | Carrier mother |
| 13 | c.2054dup or p.(Thr686Asnfs * 34) (novel) | DMD-1800 | Still ambulant at 8 yr, hyperCKemia, MP-EMG. | ABSENT | NA |
| 14 | c.2125dup or p.(Gln709Profs * 11) (novel) | DMD-491 | NA | ABSENT | Non-carrier mother |
| 15 | c.4856_4857del or p.(Lys1619Argfs * 3). HGMD: CD084923 | DMD-749 | HyperCKemia. | ABSENT | Non-carrier mother |
| 16 | c.5864_5886delinsTGAGAGCAAG or p.(Arg1955Leufs * 24) (novel) C | DMD-1579 | DMD phenotype, died age 14 yr, hyperCKemia, MP-EMG, dystrophic changes in muscle biopsy with dystrophin absence by immunoanalysis. | PRESENT | Carrier status confirmed in mother and half-sister. Normal homozygous maternal aunt. |
| 17 | c.8374_8375del or p.(Lys2792Valfs * 5). dbSNP: rs398124070 | DMD-1531 | HyperCKemia. | PRESENT | Carrier mother |
| 18 | c.10453del or p.(Leu3485 *). dbSNP: rs886043375 | DMD-1451 | DMD phenotype, hyperCKemia. | PRESENT | Carrier status confirmed in mother and two sisters. |
| Variants of unknown significance | |||||
| 19 | c.149T > A o p.(Leu50His). ClinVar: RCV000630527.2 | DMD-1918 | Proximal muscle weakness, still ambulant at 3 yr, hyperCKemia, MP-EMG. | ABSENT | Heterozygous mother |
| 20 | c.3217G > A or p.(Glu1073Lys). dbSNP: rs398123931 | DMD-1236 | DMD phenotype, hyperCKemia, MP-EMG, dystrophic changes in muscle biopsy with “abnormal” dystrophin pattern by immunoanalysis. | ABSENT | Heterozygous mother and sister. |
| LGMD (type and phenotype MIM number) genotypes identified by NGSB | |||||
| 1 | CAPN3 (LGMD2A or R1, MIM#253600): Homozygous NM_000070.2:c.2290del or p.(Asp764Thrfs * 12). dbSNP: rs886044527 | DMD-945 | “BMD phenotype”. | ABSENT | Carrier mother and sister. Father NA. |
| 2 | FKRP (LGMD2I or R9, MIM#607155): Compound Heterozygous NM_001039885.2:c.[826C > A];[1387A > G] or p.[Leu276Ile];[Asn463Asp]. dbSNP: rs28937900 and rs121908110, respectively. | DMD-786 | “BMD phenotype”, hyperCKemia, MP-EMG and died at age 15 yr due to dilated cardiomyopathy confirmed by post-mortem study. | PRESENT (suggestive for autosomal recessive muscular dystrophy trait) | Carrier mother p.[Asn463Asp]; [=]; father NA. One compound heterozygous sister also deceased at 30 yr due to dilated cardiomyopathy. |
| 3 | SGCA (LGMD2D or R3, MIM#608099): Homozygous NM_000023.2: c.229C >T or p.(Arg77Cys). dbSNP: rs28933693 | DMD-1421 | “BMD phenotype”, hyperCKemia, MP-EMG, dystrophic changes in muscle biopsy with “abnormal” dystrophin pattern by immunoanalysis. | ABSENT | Obligate carrier status confirmed in both parents (first-grade cousins) and one sister. Two normal homozygous siblings. |
| 4 | SGCA (LGMD2D or R3, MIM#608099): Homozygous NM_000023.2: c.696del or p.(Tyr233Thrfs * 15) (novel) | DMD-423 | NA | ABSENT | NA |
| 5 | SGCB (LGMD2E or R4, MIM#604286): Homozygous NM_000232.4:c.323T > G or p.(Leu108Arg). dbSNP: rs104893870 | DMD-1825 | Still ambulant at 11 yr, hyperCKemia, dystrophic changes in muscle biopsy. | PRESENT | Carrier mother and one affected homozygous brother. Non-consanguineous parents, but they came from an inbreeding community (~700 inhabitants, Ejutla, Oaxaca, Mexico). |
| 6 | SGCB (LGMD2E or R4, MIM#604286): Homozygous NM_000232.4: c.499G > A or p.(Gly167Ser). dbSNP: rs779516489 | DMD-621 | HyperCKemia. | PRESENT | Carrier mother; father NA. Homozygous affected brother. |
| 7 | SGCB (LGMD2E or R4, MIM#604286): Homozygous NG_008891.1(NM_000232.4): c.622-2A > G. dbSNP: rs780596734 | DMD-954 | HyperCKemia, dystrophic changes in muscle biopsy. | ABSENT | Carrier mother; father NA. |
| 8 | SGCG (LGMD2C or R5, MIM#253700): Homozygous NG_008759.1(NM_000231.2): c.241_297 + 1169del (partial deletion of exon and intron 3, novel)D | DMD-1762 | “DMD phenotype”, hyperCKemia, MP-EMG. Bilateral retinoblastoma. | ABSENT | NA. Family history of hereditary retinoblastoma. |
| 9 | SGCG (LGMD2C or R5, MIM#253700): Homozygous NM_000231.2: c.752del or p.(Thr251Serfs * 29). dbSNP: rs886042749 | DMD-820 | HyperCKemia. | PRESENT | Carrier mother; father NA. |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Alcántara-Ortigoza, M.A.; Reyna-Fabián, M.E.; González-del Angel, A.; Estandia-Ortega, B.; Bermúdez-López, C.; Cruz-Miranda, G.M.; Ruíz-García, M. Predominance of Dystrophinopathy Genotypes in Mexican Male Patients Presenting as Muscular Dystrophy with A Normal Multiplex Polymerase Chain Reaction DMD Gene Result: A Study Including Targeted Next-Generation Sequencing. Genes 2019, 10, 856. https://doi.org/10.3390/genes10110856
Alcántara-Ortigoza MA, Reyna-Fabián ME, González-del Angel A, Estandia-Ortega B, Bermúdez-López C, Cruz-Miranda GM, Ruíz-García M. Predominance of Dystrophinopathy Genotypes in Mexican Male Patients Presenting as Muscular Dystrophy with A Normal Multiplex Polymerase Chain Reaction DMD Gene Result: A Study Including Targeted Next-Generation Sequencing. Genes. 2019; 10(11):856. https://doi.org/10.3390/genes10110856
Chicago/Turabian StyleAlcántara-Ortigoza, Miguel Angel, Miriam Erandi Reyna-Fabián, Ariadna González-del Angel, Bernardette Estandia-Ortega, Cesárea Bermúdez-López, Gabriela Marisol Cruz-Miranda, and Matilde Ruíz-García. 2019. "Predominance of Dystrophinopathy Genotypes in Mexican Male Patients Presenting as Muscular Dystrophy with A Normal Multiplex Polymerase Chain Reaction DMD Gene Result: A Study Including Targeted Next-Generation Sequencing" Genes 10, no. 11: 856. https://doi.org/10.3390/genes10110856
APA StyleAlcántara-Ortigoza, M. A., Reyna-Fabián, M. E., González-del Angel, A., Estandia-Ortega, B., Bermúdez-López, C., Cruz-Miranda, G. M., & Ruíz-García, M. (2019). Predominance of Dystrophinopathy Genotypes in Mexican Male Patients Presenting as Muscular Dystrophy with A Normal Multiplex Polymerase Chain Reaction DMD Gene Result: A Study Including Targeted Next-Generation Sequencing. Genes, 10(11), 856. https://doi.org/10.3390/genes10110856