Transcriptome-Based SNP Discovery and Validation in the Hybrid Zone of the Neotropical Annual Fish Genus Austrolebias

 , , ,
, , ,
Abstract
:1. Introduction
2. Materials and Methods
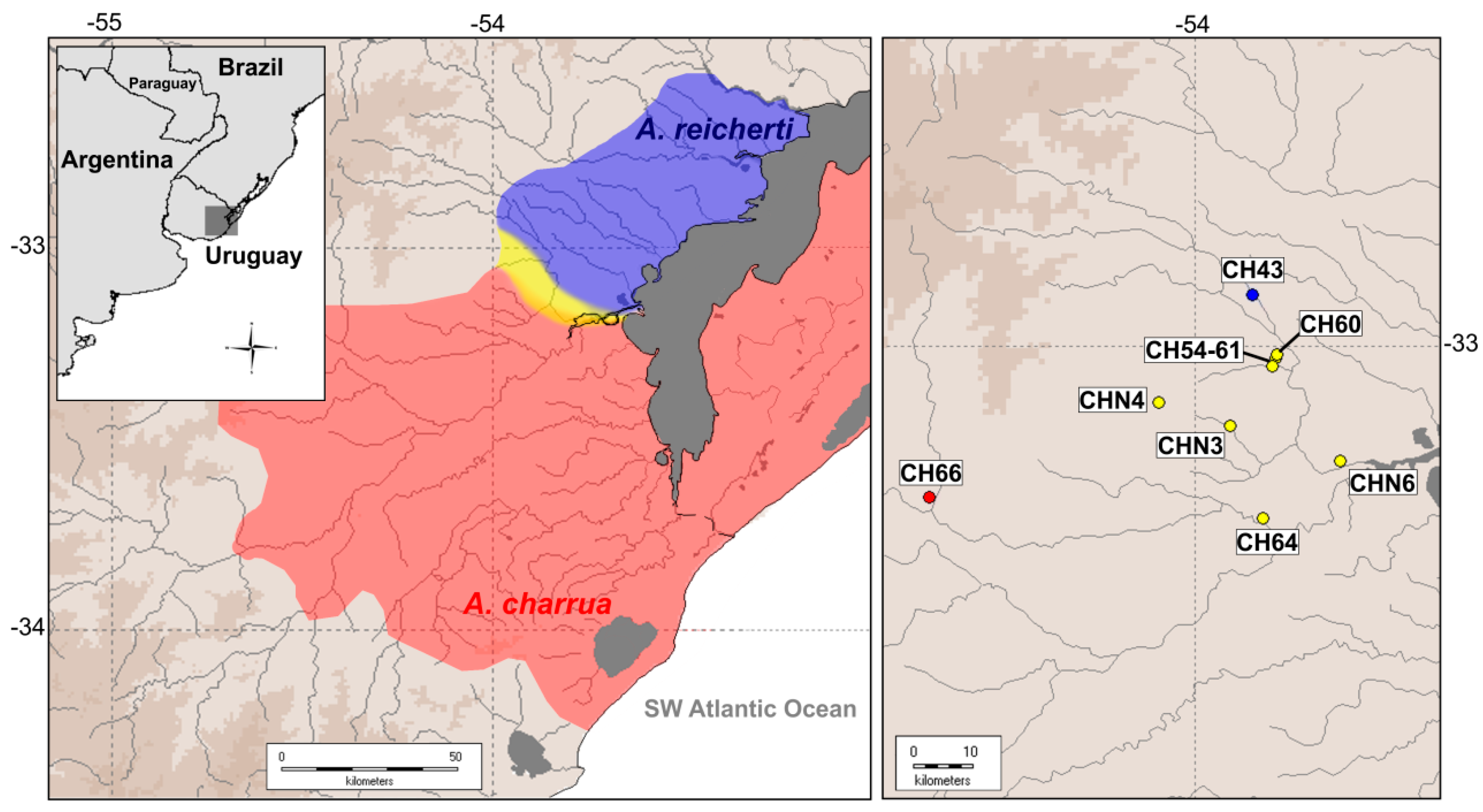
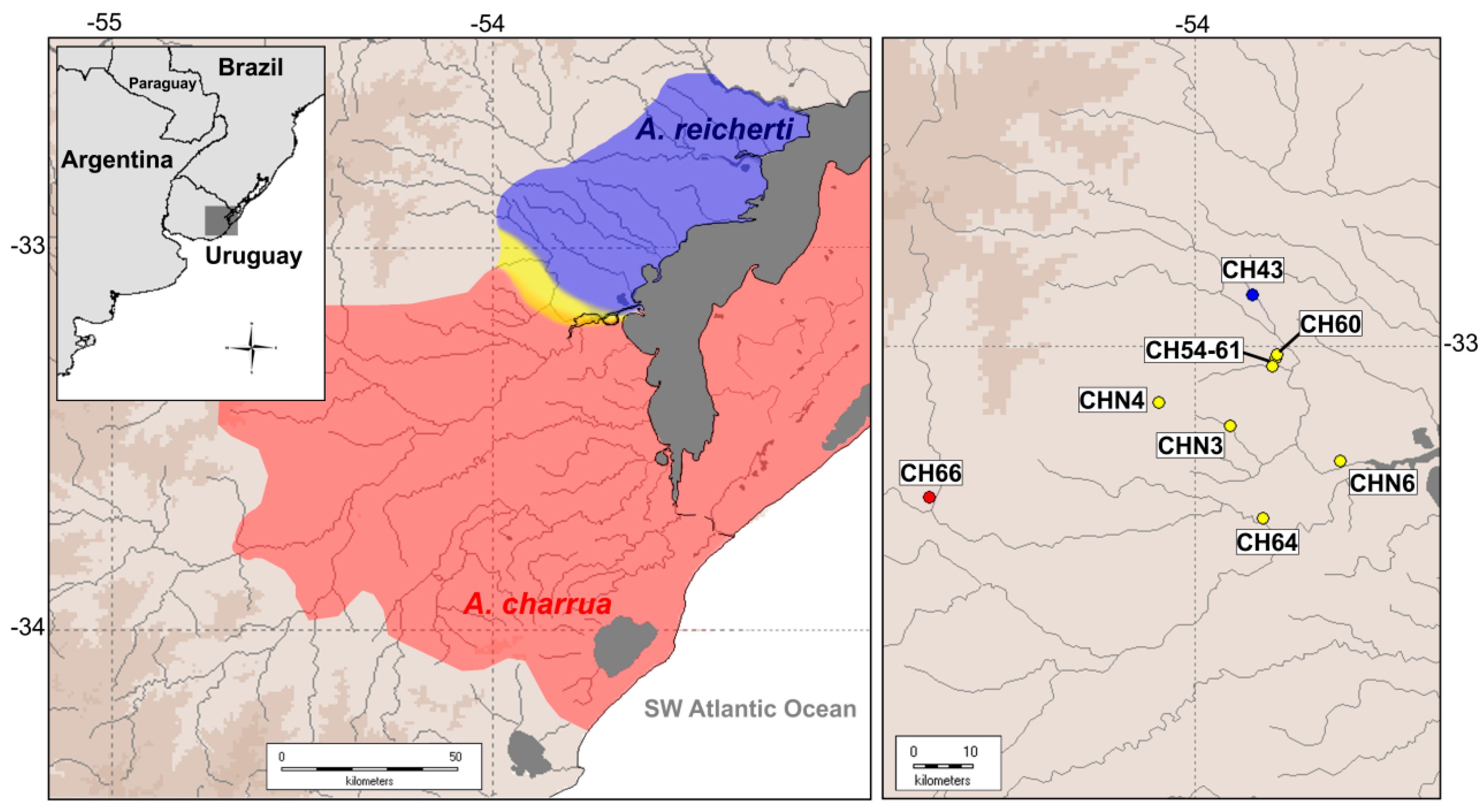
2.1. Re-Mapping of the Hybrid Zone
2.2. Population Genomic Analyses of the Hybrid Zone
2.2.1. Sample Collection for RNA-Seq
2.2.2. Annotation and Mapping of Sequencing Reads
2.2.3. Validation of Fixed-Allele Interspecific Single Nucleotide Polymorphisms (SNPs)
2.2.4. Statistical Analyses Based on SNP Markers
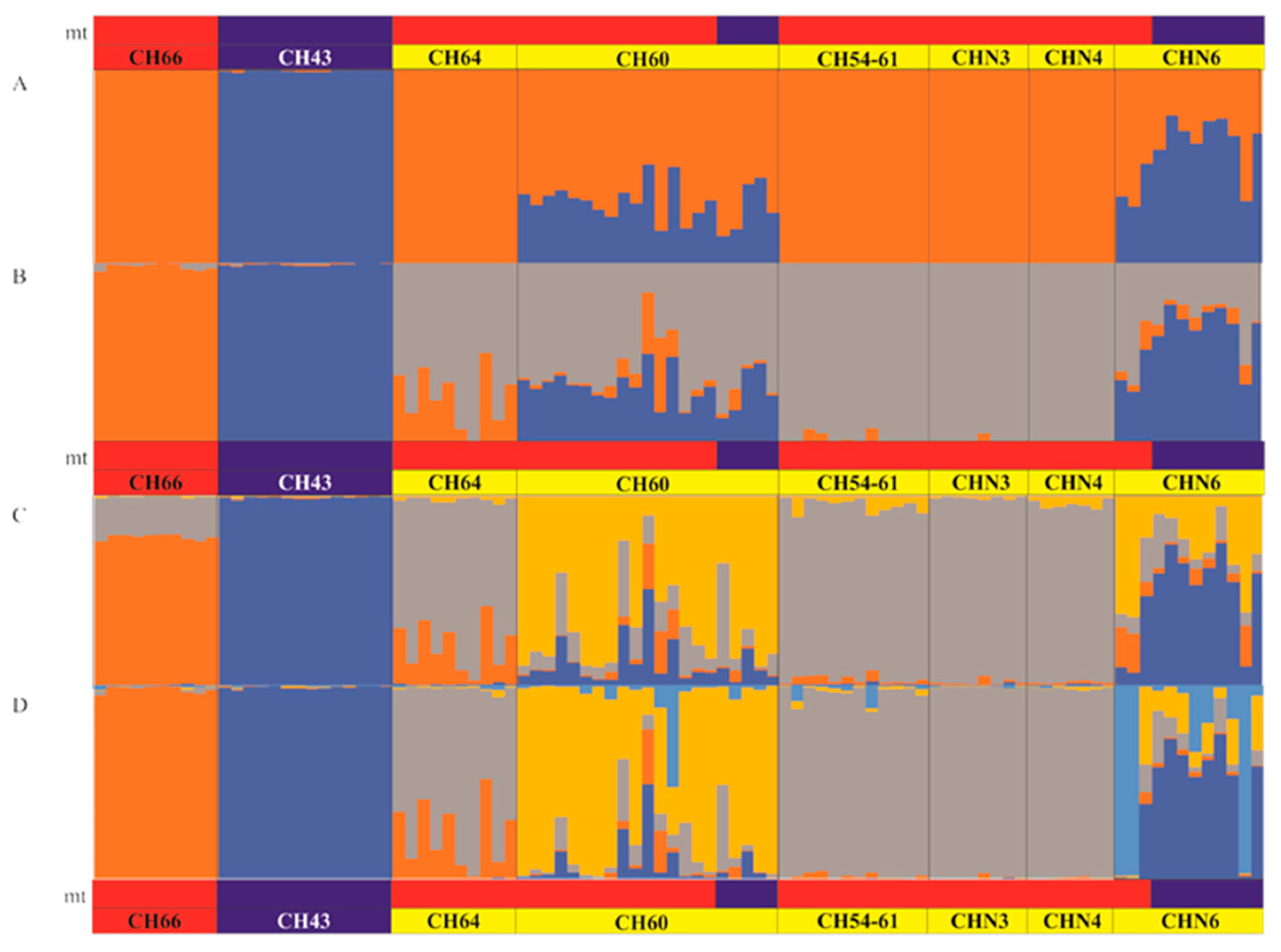
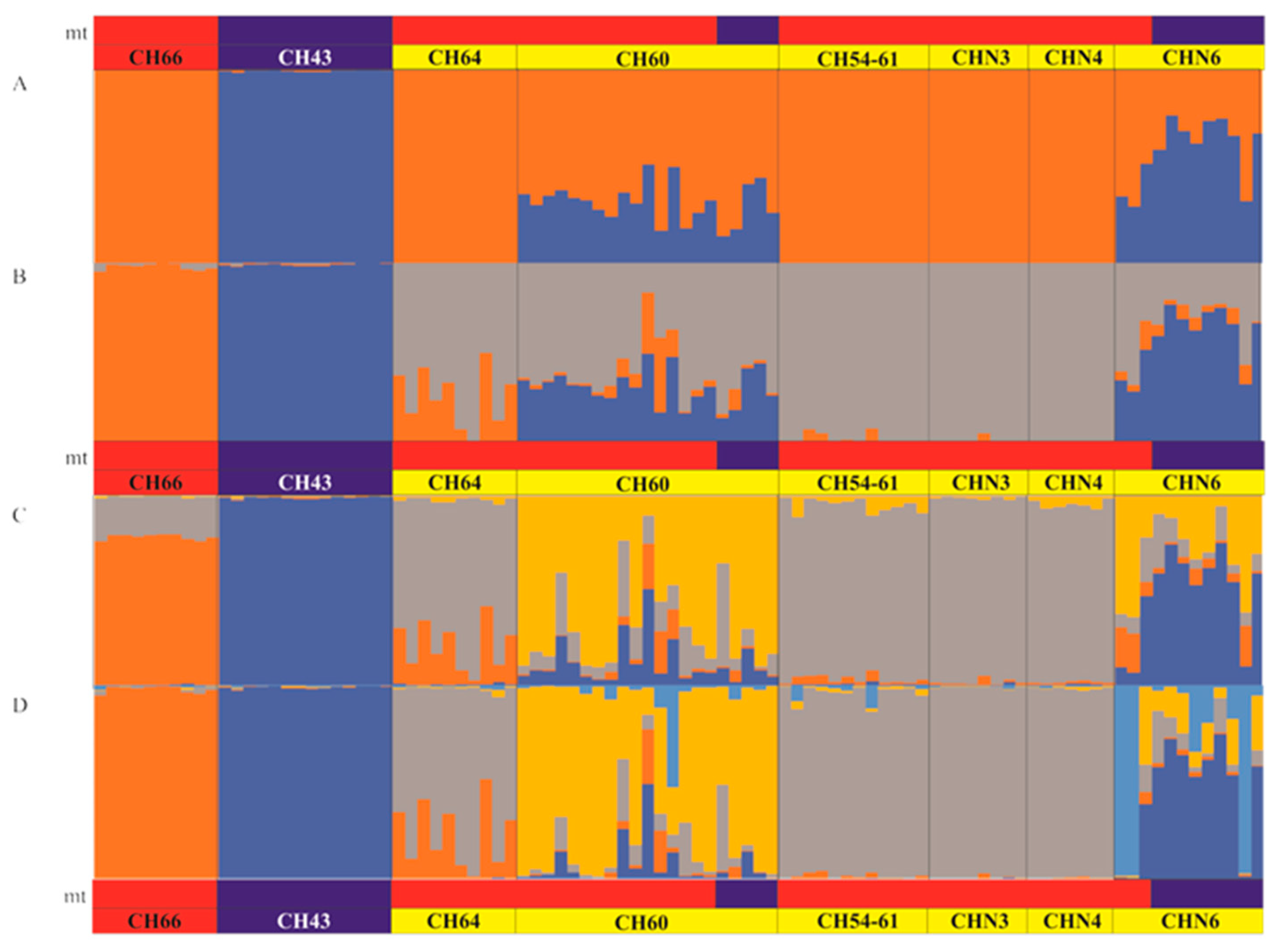
2.2.5. Population Structure and Patterns of Hybridization Based on SNP Markers
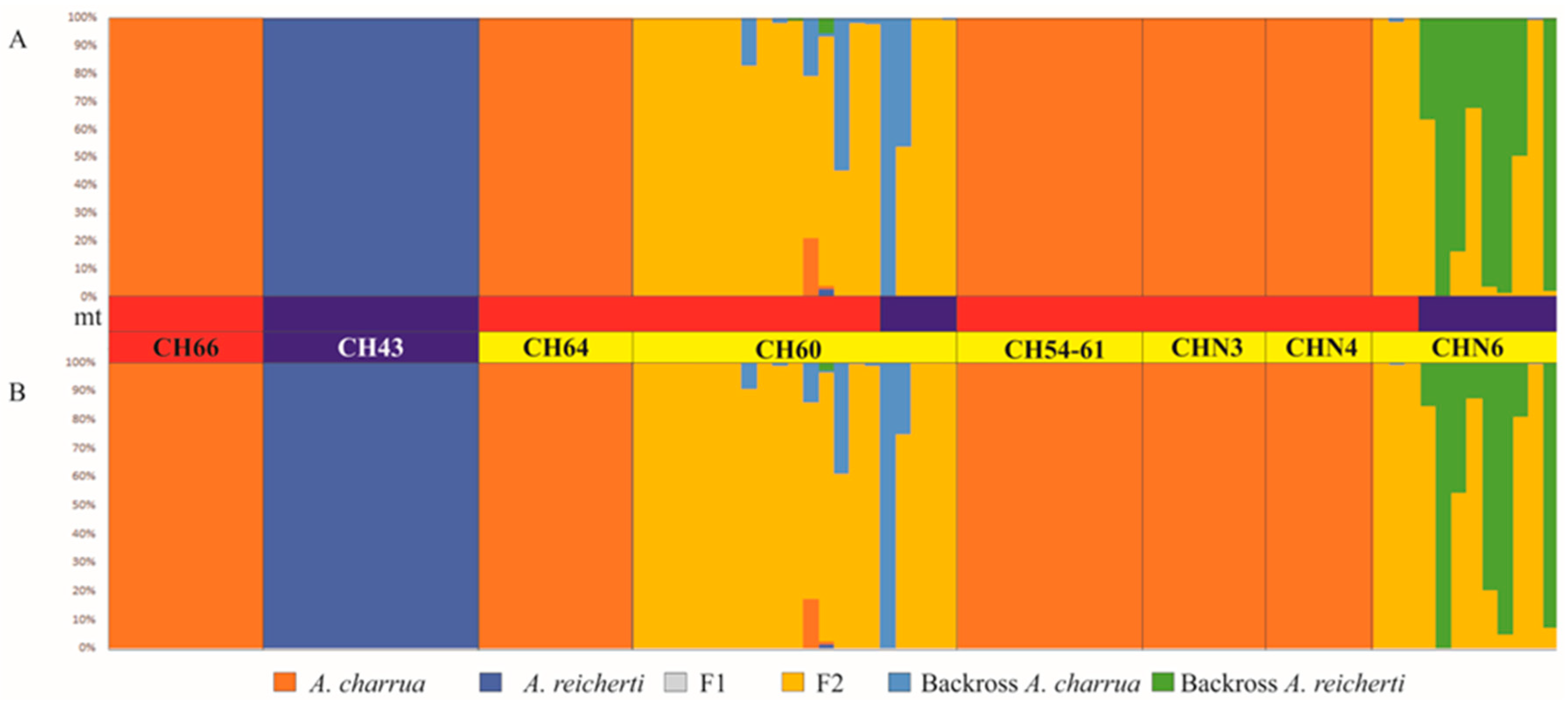
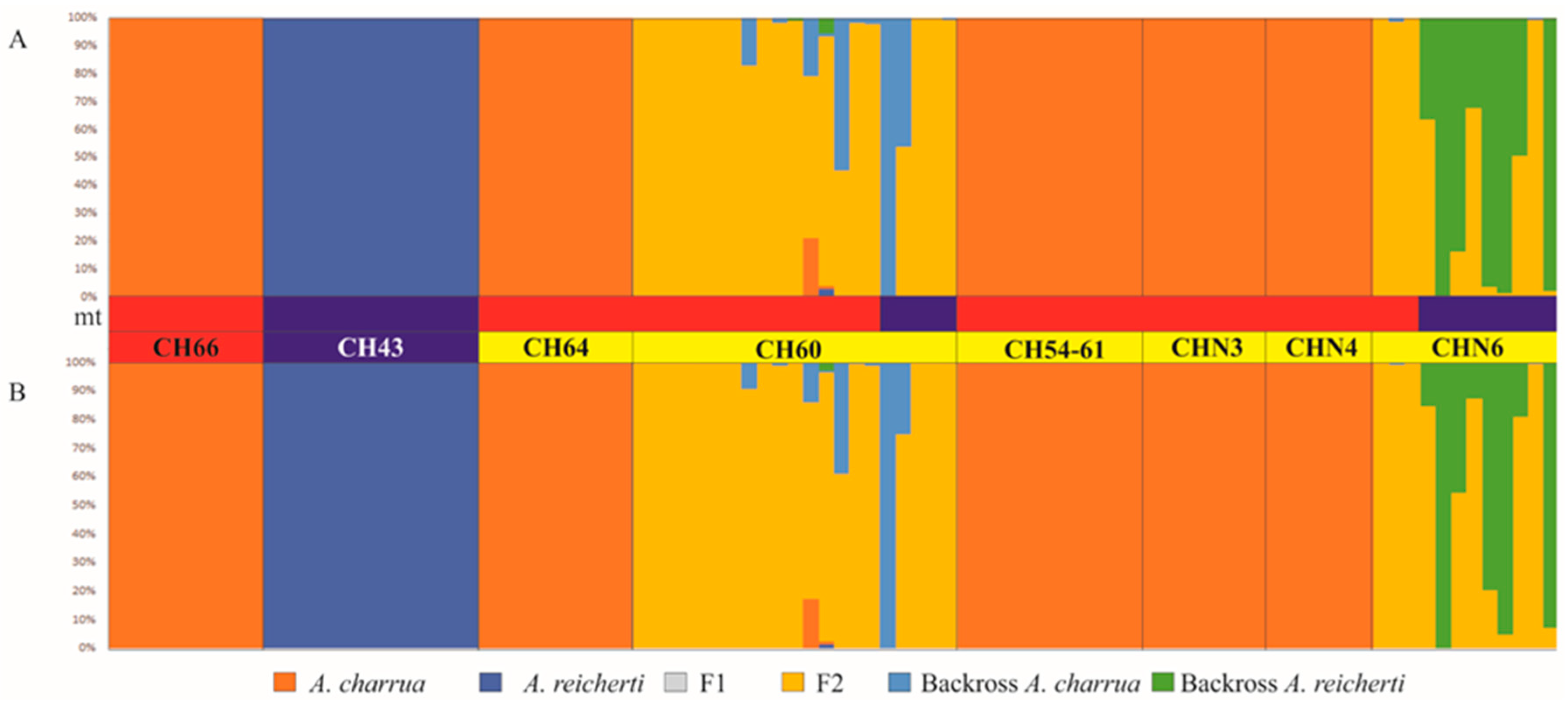
2.2.6. Estimates of Recent Introgression
2.3. Mitochondrial Sequence Analyses
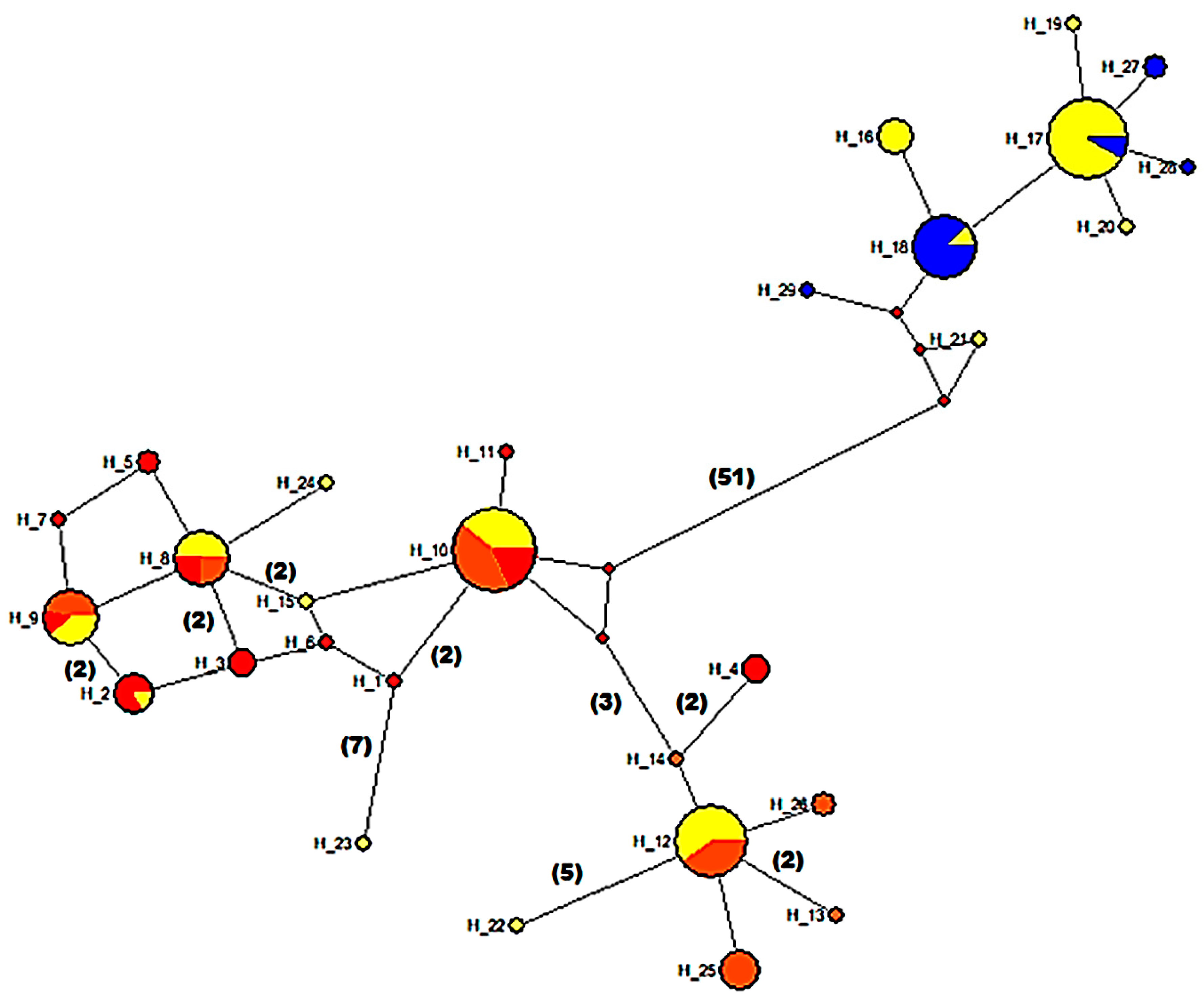
2.3.1. Population Genetic Structure Based on Cytb Gene
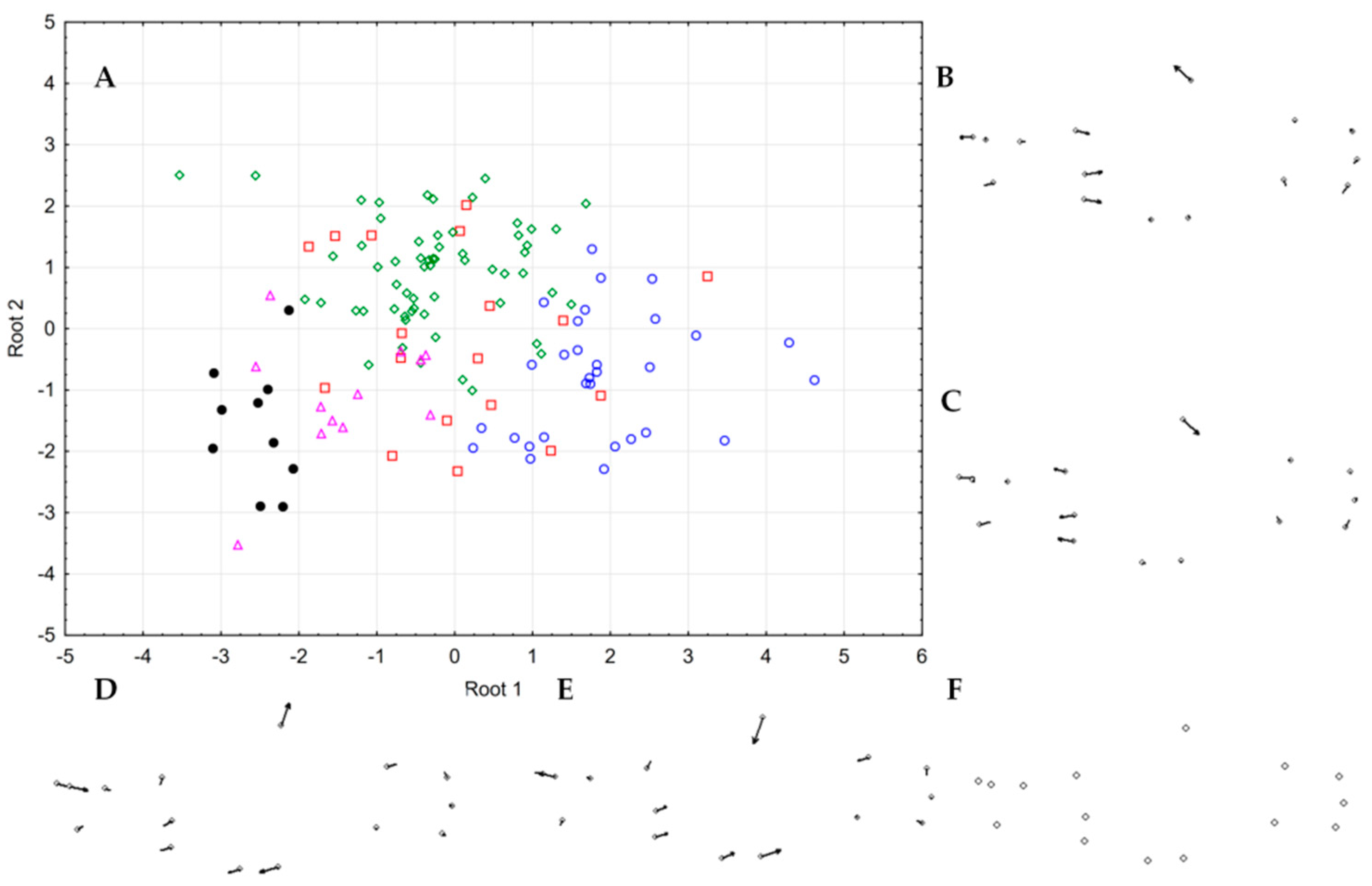
2.4. Morpho-Meristic Analyses
3. Results
3.1. Transcriptome Sequencing, SNP Detection and Annotation
3.2. SNP Validation by the iPLEX Assay Design
3.3. Genetic Diversity Parameters Based on 106 SNPs
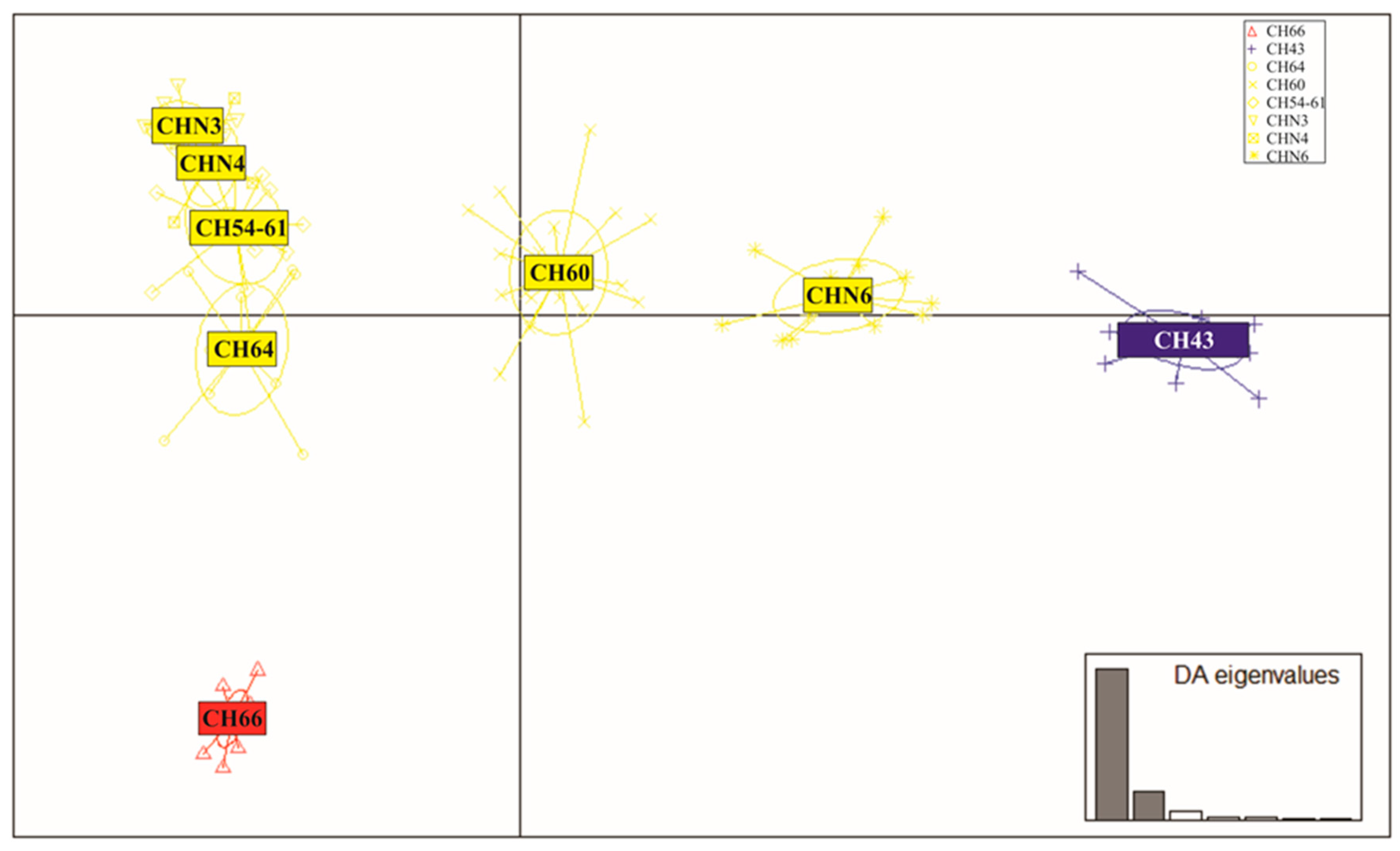
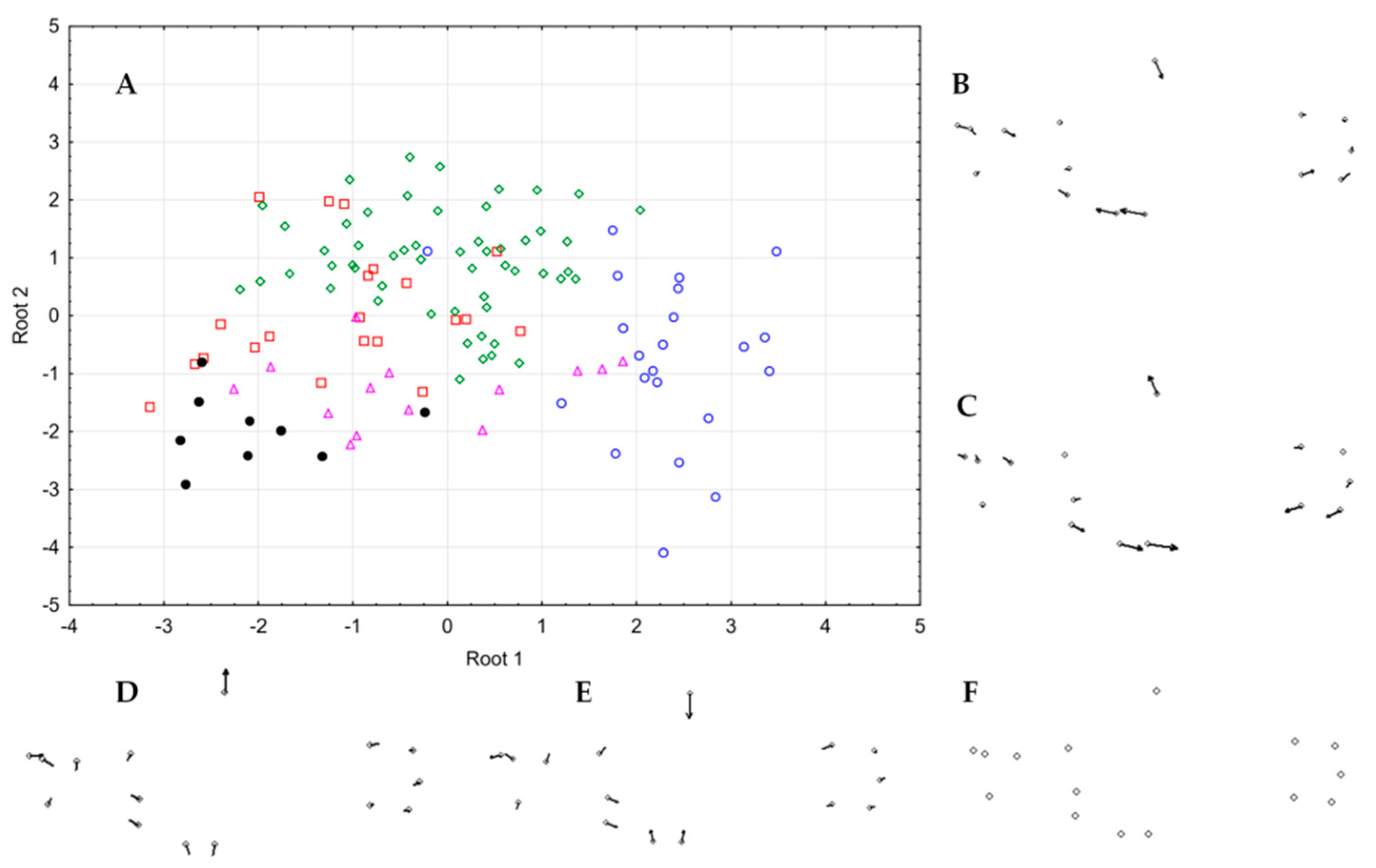
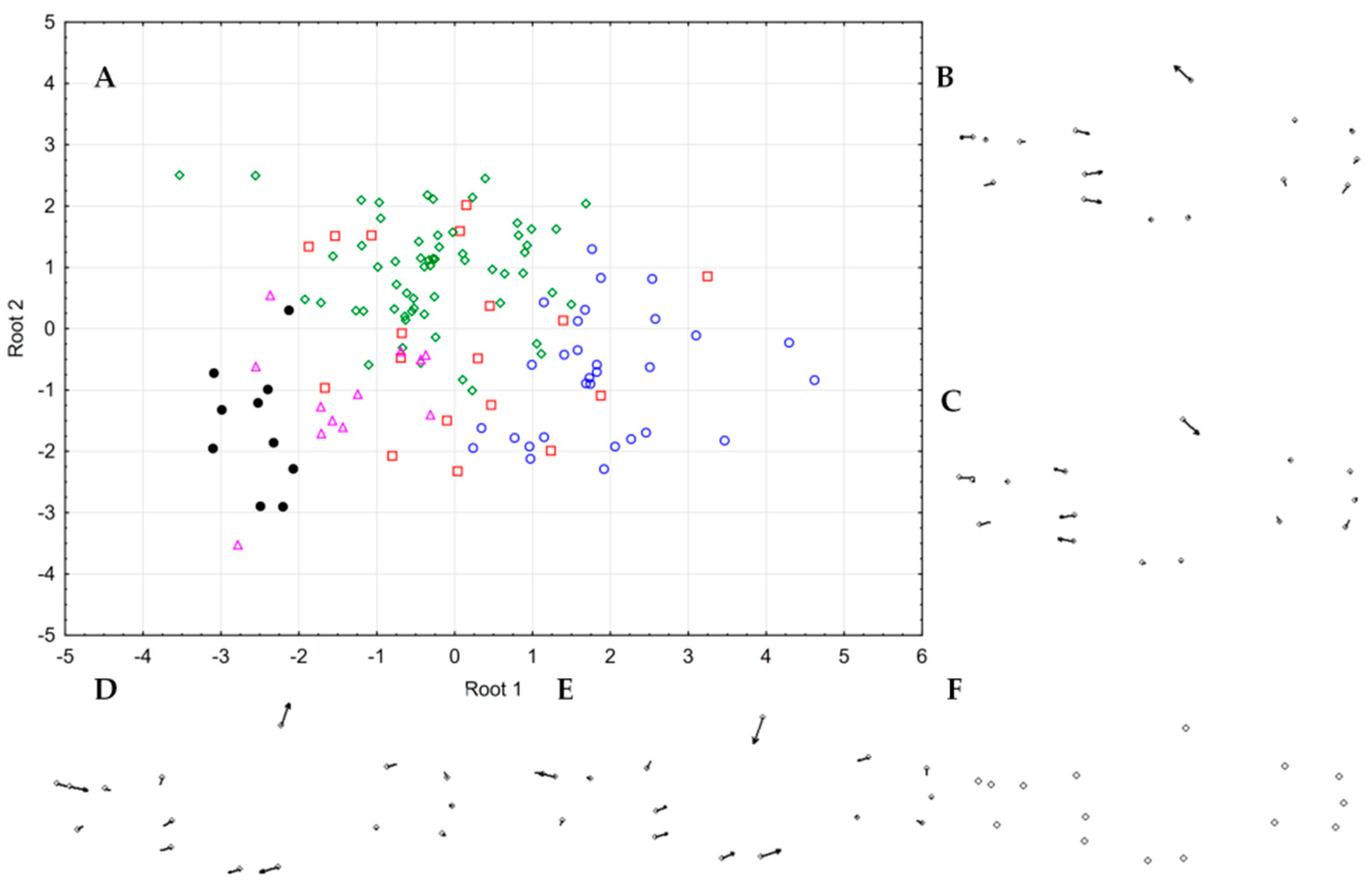
3.4. Patterns of Hybridization and Population Genetics Structure Based on 103 SNPs
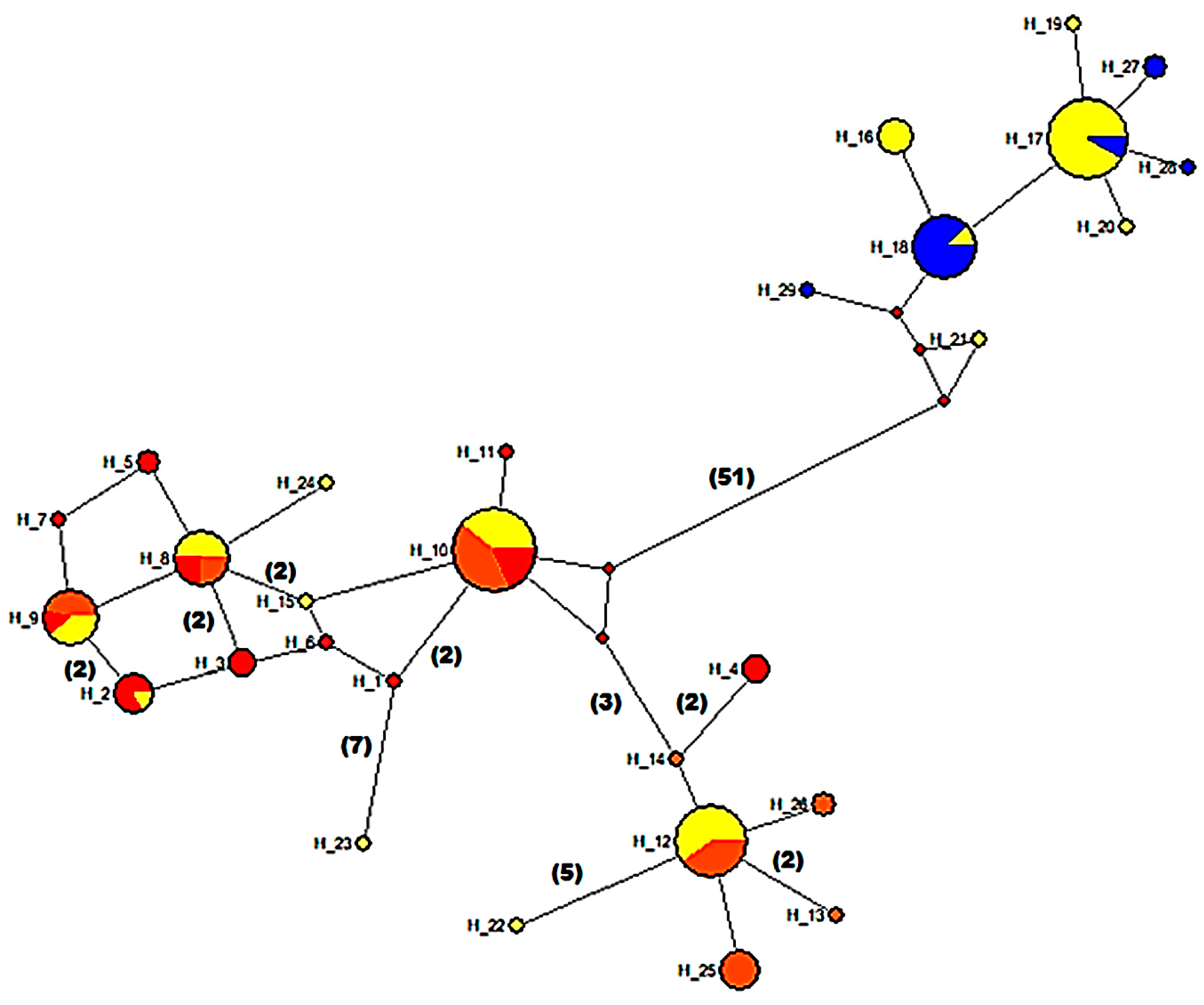
3.5. Mitochondrial Sequence Variation and Population Analyses
3.6. Morphology
4. Discussion
4.1. Geographic Delimitation of the Present Hybrid Zone
4.2. Structure of the Hybrid Zone
4.3. Dynamics Underlying Patterns and Processes in the Hybrid Zone
4.4. Further Prospects and Conservation Remarks
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Pante, E.; Rohfritsch, A.; Becquet, V.; Belkhir, K.; Bierne, N.; García, P. SNP Detection from De Novo Transcriptome Sequencing in the Bivalve Macoma balthica: Marker Development for Evolutionary Studies. PLoS ONE 2013, 8, c15–c19. [Google Scholar] [CrossRef]
- Goulet, B.E.; Roda, F.; Hopkins, R. Hybridization in plants: Old ideas, new techniques. Plant Physiol. 2017, 173, 65–78. [Google Scholar] [CrossRef] [PubMed]
- Barton, N.; Hewitt, G. Analysis of hybrid zones. Annu. Rev. Ecol. Syst. 1985, 16, 113–148. [Google Scholar] [CrossRef]
- Harrison, R.G. Hybrid zones: Windows on the evolutionary process. Oxford Surv. Evol. Biol. 1990, 7, 69–128. [Google Scholar]
- Jiggins, C.D.; Mallet, J. Bimodal hybrid zones and speciation. Trends Ecol. Evol. 2000, 15, 250–255. [Google Scholar] [CrossRef]
- Harrison, H.B.; Berumen, M.L.; Saenz-Agudelo, P.; Salas, E.; Williamson, D.H.; Jones, G.P. Widespread hybridization and bidirectional introgression in sympatric species of coral reef fish. Mol. Ecol. 2017, 26, 5692–5704. [Google Scholar] [CrossRef]
- Albert, J.; Reis, R. (Eds.) Historical Biogeography of Neotropical Freshwater Fishes; University of California Press: Berkeley, CA, USA, 2011. [Google Scholar]
- Culumber, Z.C.; Fisher, H.S.; Tobler, M.; Mateos, M.; Barber, P.H.; Sorenson, M.D.; Rosenthal, G.G. Replicated hybrid zones of Xiphophorus swordtails along an elevational gradient. Mol. Ecol. 2011, 2, 342–356. [Google Scholar] [CrossRef] [PubMed]
- García, G.; Gutiérrez, V.; Ríos, N.; Papa, N.; Serra, S.; Loureiro, M. Characterization of a hybrid zone between two annual killifish genus Austrolebias from the Biosphere Reserve and Ramsar Sites in South America. Hydrobiologia, MS under review.
- Costa, W.J.E.M. Trophic radiation in the South American annual killifish genus Austrolebias (Cyprinodontiformes: Rivulidae). Ichthyol. Explor. Freshw. 2009, 20, 179–191. [Google Scholar]
- Costa, W.J.E.M.; Amorim, P.F.; Mattos, J.L.O. Molecular phylogeny and timing of diversification in South American Cynolebiini seasonal killifishes. Mol. Phylogenet. Evol. 2017, 116, 61–68. [Google Scholar] [CrossRef]
- Loureiro, M.; de Sá, R.O. Osteological analysis of the killifish genus Cynolebias (Cyprinodontiformes: Rivulidae). J. Morphol 1998, 238, 245–262. [Google Scholar] [CrossRef]
- García, G. Multiple simultaneous speciation in killifishes of the Cynolebias adloffi species complex (Cyprinodontiformes, Rivulidae) from phylogeography and chromosome data. J. Zool. Syst. Evol. Res. 2006, 44, 75–87. [Google Scholar] [CrossRef]
- García, G.; Gutiérrez, V.; Ríos, N.; Turner, B.; Santiñaque, F.; López-Carro, B.; Folle, G. Burst speciation processes and genomic expansion in the neotropical annual killifish genus Austrolebias (Cyprinodontiformes, Rivulidae). Genetica 2014, 142, 87–98. [Google Scholar] [CrossRef] [PubMed]
- García, G. Chromosome Evolution, Divergent Meiotic Pathways, and Speciation in the Family Rivulidae. In Annual fishes: Life History Strategy, Diversity, and Evolution; Berois, N., García, G., de Sá, R.O., Eds.; CRC Press: Boca Raton, FL, USA, 2016; Volume 16, pp. 281–293. [Google Scholar]
- García, G.; Gutiérrez, V.; Ríos, N.; de Sá, R.O. Comparative Phylogeographic Patterns in Austrolebias from Different South American Basins. In Annual Fishes: Life History Strategy, Diversity, and Evolution; Berois, N., García, G., de Sá, R.O., Eds.; CRC Press: Boca Raton, FL, USA, 2016; Volume 15, pp. 253–273. [Google Scholar]
- García, G.; Ríos, N.; Gutiérrez, V. Next-generation sequencing detects repetitive elements expansion in giant genomes of annual killifish genus Austrolebias (Cyprinodontiformes, Rivulidae). Genetica 2015, 143, 353–360. [Google Scholar] [CrossRef] [PubMed]
- Valdivieso, C.; Pereiro, L.; Di Genova, A.; Gajardo, F.; García, G.; Arezo, M.J.; Papa, N.; Gutiérrez, V.; Berois, N.; Nardocci, G.; et al. The genome sequence of the annual killifish Austrolebias charrua reveals mechanisms of genomic expansion via retrolelements. In Abstract Book IX Latin American Society For Developmental Biology, In Proceedings of the International Meeting of the LASDB, Medellín, Colombia, 9–13 October 2017; p. 192.
- Montaña, J.R.; Bossi, J. Geomorfología de los humedales de la cuenca de la Laguna Merín en el departamento de Rocha. UDELAR. Revista Facultad de Agronomía. Montevideo Uruguay 1995, 2, 1–32. [Google Scholar]
- Sprechman, P. The paleoecology and paleogeography and stratigraphy of the Uruguayan coastal area during the neogene and quaternary. Zitteliana 1978, 4, 3–72. [Google Scholar]
- García, G.; Loureiro, M.; Berois, N.; Arezo, M.J.; Casanova, G.; Clivio, G.; Olivera, A. Pattern of differentiation in the annual killifish genus Austrolebias (Cyprinodontiformes; Rivulidae) from a biosphere reserve site in South America: A multidisciplinary approach. Biol. J. Linn. Soc. 2009, 98, 620–635. [Google Scholar] [CrossRef]
- Price, T.D.; Bouvier, M.M. The evolution of F1 postzygotic incompatibilities in birds. Evol. Int. J. Org. Evol. 2002, 56, 2083–2089. [Google Scholar] [CrossRef]
- Seehausen, O. Hybridization and adaptive radiation. Trend. Ecol. Evol. 2004, 19, 198–406. [Google Scholar] [CrossRef]
- Gourbière, S.; Mallet, J. Are species real? The shape of the species boundary with exponential failure, reinforcement, and the “missing snowball”. Evolution 2010, 64, 1–24. [Google Scholar] [CrossRef]
- Vähä, J.P.; Primmer, C.R. Efficiency of model-based Bayesian methods for detecting hybrid individuals under different hybridization scenarios and with different numbers of loci. Mol. Ecol. 2006, 15, 63–72. [Google Scholar] [CrossRef] [PubMed]
- Larson, E.L.; White, T.A.; Ross, C.L.; Harrison, R.G. Gene flow and the maintenance of species boundaries. Mol. Ecol. 2014, 23, 1668–1678. [Google Scholar] [CrossRef] [PubMed]
- De Witt, P.; Pespeni, M.H.; Palumbi, S.R. SNP genotyping and population genomics from. expressed sequences–current advances and future possibilities. Mol. Ecol. 2015, 24, 2310–2323. [Google Scholar] [CrossRef] [PubMed]
- Li, C.; Gowan, S.; Anil, A.; Beck, B.H.; Thondga, W.; Kucuktas, H.; Kaltenboeck, L.; Peatman, E. Discovery and validation of gene-linked diagnostic SNP markers for assessing hybridization between Largemouth. bass (Micropterus salmoides) and Florida bass (M. floridanus). Mol. Ecol. Resour. 2014. [Google Scholar] [CrossRef] [PubMed]
- Bolger, A.M.; Lohse, M.; Usadel, B. Trimmomatic: A flexible trimmer for Illumina Sequence Data. Bioinformatics 2014, 30, 2114–2120. [Google Scholar] [CrossRef] [PubMed]
- Gajardo, F.; Di Genova, A.; Valdivieso, C.; Pereiro, L.; Arezo, M.J.; Nardocci, G.; Rojas, N.; Gutiérrez, V.; Cumplido, N.; Marina, R.; et al. Accelerated genome expansion in an annual South American killifish. Manuscript in preparation.
- Langmead, B.; Salzberg, S. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, H.; Handsaker, B.; Wysoker, A.; Fennell, T.; Ruan, J.; Homer, N.; Marth, G.; Abecasis, G.; Durbin, R. 1000 Genome Project Data Processing Subgroup. The Sequence Alignment/Map format and SAMtools. Bioinformatics 2009, 25, 2078–2079. [Google Scholar] [CrossRef]
- Medrano, J.F.; Aasen, E.; Sharrow, L. DNA extraction from nucleated red blood cells. BioTechniques 1990, 8, 43. [Google Scholar]
- Raymond, M.; Rousset, F. Genepop (Version-1.2) population-genetics software for exact tests and ecumenicism. J. Hered. 1995, 86, 248–249. [Google Scholar] [CrossRef]
- Excoffier, L.; Lischer, H.E.L. ARLEQUIN suite ver. 3.5: A new series of programs to perform population genetics analyses under Linux and windows. Mol. Ecol. Res. 2010, 10, 564–567. [Google Scholar] [CrossRef]
- Goudet, J. FSTAT, a Program to Estimate and Test Gene Diversities and Fixation Indices (Version 2.9. 3). 2001. Available online: unil.ch/izea/softwares/Fstat.html (accessed on 13 September 2019).
- Weir, B.S.; Cockerham, C.C. Estimating F-statistics for the analysis of population structure. Evolution 1984, 38, 1358–1370. [Google Scholar] [PubMed]
- Guo SThompson, E. Performing the exact test for Hardy Weinberg Proportion for multiple alleles. Biometrics 1992, 48, 361–372. [Google Scholar]
- Bonferroni, C. Teoria statistica delle classi e calcolo delle probabilitá. Pubbl. Del R Ist. Super. Di Sci. Econ. E Commer. Di Firenze 1936, 8, 3–62. [Google Scholar]
- Jombart, T. Adegenet: A R package for the multivariate analysis of genetic markers. Bioinformatics 2008, 24, 1403–1405. [Google Scholar] [CrossRef]
- Pritchard, J.K.; Stephens, M.; Donnelly, P. Inference of population structure using multilocus genotype data. Genetics 2000, 155, 945–959. [Google Scholar]
- Kopelman, N.M.; Mayzel, J.; Jakobsson, M.; Rosenberg, N.A. C LUMPAK: A program for identifying clustering modes and packaging population structure inferences across K. Mol. Ecol. Res. 2015, 15, 1179–1191. [Google Scholar] [CrossRef]
- Evanno, G.; Regnaut, S.; Goudet, J. Detecting the number of clusters of individuals using the software STRUCTURE: A simulation study. Mol. Ecol. 2005, 14, 2611–2620. [Google Scholar] [CrossRef]
- Earl, D.A.; von Holdt, B.M. Structure Harvester: A website and program for visualizing STRUCTURE output and implementing the Evanno method. Conserv. Genet. Resour. 2012, 4, 359–361. [Google Scholar] [CrossRef]
- Anderson, E.C.; Thompson, E.A. A model-based method for identifying species hybrids using multilocus genetic data. Genetics 2002, 160, 1217–1229. [Google Scholar]
- Palumbi, S.; Martin, A.; Romano, S.; McMillan, W.O.; Stice, L.; Grabowski, G. The Simple Fool’s Guide to PCR; Department of Zoology and Kewalo Marine Laboratory, Univ. Hawaii: Honolulu, HI, USA, 1991. [Google Scholar]
- Kumar, K.; Stecher, G.; Tamura, K. MEGA7: Molecular Evolutionary Genetics Analysis Version 7.0 for Bigger Datasets. Mol. Biol. Evol. 2016, 33, 1870–1874. [Google Scholar] [CrossRef] [Green Version]
- Librado, P.; Rozas, J. DnaSP v5: A software for comprehensive analysis of DNA polymorphism data. Bioinformatics 2009, 25, 1451–1452. [Google Scholar] [CrossRef] [PubMed]
- Kimura, M. A simple method for estimating evolutionary rate of base substitutions through comparative studies of nucleotide sequences. J. Mol. Evol. 1980, 16, 111–120. [Google Scholar] [CrossRef] [PubMed]
- Nei, M. Molecular Evolutionary Genetics; Columbia University Press: New York, NY, USA, 1987; p. 179. [Google Scholar]
- Tajima, F. Statistical method for testing the neutral mutation hypothesis by DNA polymorphism. Genetics 1989, 123, 585–595. [Google Scholar] [PubMed]
- Bandelt, H.; Forster, P.; Rohl, A. Median-joining networks for inferring intraspecific phylogenies. Mol. Biol. Evol. 1999, 16, 37–48. [Google Scholar] [CrossRef] [PubMed]
- Excoffier, L.; Smouse, P.E.; Quattro, J.M. Analysis of molecular variance inferred from metric distances among DNA haplotypes: Application to human mitochondrial DNA restriction data. Genetics 1992, 131, 479–491. [Google Scholar] [PubMed]
- Bookstein, F.L. Morphometric Tools for Landmark Data: Geometry and Biology; Cambridge University Press: New York, NY, USA, 1991. [Google Scholar]
- D’Anatro, A.; Loureiro, M. Geographic variation in the annual killifish Austrolebias luteoflammulatus Vaz Ferreira, Sierra & Scaglia (Cyprinodontiformes, Rivulidae). J. Fish Biol. 2005, 67, 849–865. [Google Scholar]
- Rohlf, F.J. Morphometrics at SUNY Stony Brook. 2003. Available online: http://life.bio.sunysb.edu/morph (accessed on 13 September 2019).
- Twyford, A.D.; Ennos, R.A. Next-generation hybridization and introgression. Heredity 2012, 108, 179–189. [Google Scholar] [CrossRef] [PubMed]
- Brown, W.L.; Wilson, E.O. Character displacement. Syst. Zool. 1956, 5, 49–64. [Google Scholar] [CrossRef]
- Pfennig, K.S.; Pfennig, D.W. Character displacement: Ecological and reproductive responses to a common evolutionary problem. Quart. Rev. Biol. 2009, 84, 253–276. [Google Scholar] [CrossRef]
- Sobel, J.M.; Chen, G.F.; Watt, L.R.; Schemske, D.W. The biology of speciation. Evolution 2010, 64, 295–315. [Google Scholar] [CrossRef]
- Loureiro, M.; García, G. Austrolebias reicherti Loureiro and García, a valid species of annual fish (Cyprinodontiformes: Rivulidae) from Uruguay. Zootaxa 2008, 1940, 1–15. [Google Scholar] [CrossRef]
- Passos, C.; Tassino, B.; Rosenthal, G.G.; Reichard, M. Reproductive Behavior and Sexual Selection in Annual Fishes. In Annual fishes: Life history Strategy, Diversity, and Evolution; Berois, N., García, G., de Sá, R.O., Eds.; CRC Press: Boca Raton, FL, USA, 2016; Volume 12, pp. 207–229. [Google Scholar]
- Abbott, R.J. Plant speciation across environmental gradients and the occurrence and nature of hybrid zones. J. Syst. Evol. 2017, 55, 238–258. [Google Scholar] [CrossRef]
- Bracco, R.; Inda, H.; del Puerto, L.; Capdepont, I.; Panario, D.; García-Rodríguez, F. A reply to Relative sea level during the Holocene in Uruguay. Palaeogeogr. Palaeoclimatol. Palaeoecol. 2014, 401, 166–170. [Google Scholar] [CrossRef]
- Kagawa, K.; Takimoto, G. Hybridization can promote adaptive radiation by means of transgressive segregation. Ecol. Lett. 2017, 21, 264–274. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hamilton, J.A.; Miller, J.M. Adaptive introgression as a resource for management and genetic conservation in a changing climate; 2015 Society for Conservation Biology. Conserv. Biol. 2016, 30, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Becker, M.; Gruenheit, N.; Steel, M.; Voelckel, C.; Deusch, O.; Heenan, P.; McLenachan, P.A.; Kardailsky, O.; Leigh, J.W.; Lockhard, P. Hybridization may facilitate in situ survival of endemic species through periods of climate change. Nat. Clim. Chang. 2013, 3, 1039–1043. [Google Scholar] [CrossRef]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Population | N | He (s.d) | p | FIS (s.d) |
|---|---|---|---|---|
| CH64 | 10 | 0.373 (0.130) | 0.753 | −0.023 (0.651) |
| CH66 | 10 | 0.280 (0.186) | 0.237 | −0.338 (0.996) |
| CH43 | 14 | 0.243 (0.141) | 0.001 | 0.141 (0.073) |
| CH60 | 21 | 0.331 (0.159) | 0.000 | −0.100 (0.944) |
| CH54-61 | 12 | 0.340 (0.161) | 0.460 | 0.019 (0.456) |
| CHN3 | 8 | 0.386 (0.146) | 0.751 | −0.029 (0.621) |
| CHN4 | 7 | 0.349 (0.153) | 0.981 | −0.019 (0.617) |
| CHN6 | 12 | 0.303 (0.143) | 0.007 | −0.033 (0.671) |
| Hypothesis | Source of Variation | df | Sum of Squares | Variance Components | Percentage of Variation | Φ Statistics |
|---|---|---|---|---|---|---|
| a | Among groups | 3 | 217.221 | 1.41953 Va | 26.25 | ΦCT = −0.263 (0.001) |
| Among population within groups | 4 | 39.714 | 0.29998 Vb | 5.55 | ΦSC = 0.075 (0.000) | |
| Among individuals within populations | 86 | 304.043 | −0.15252 Vc | −2.82 | ΦIS = −0.041 (0.885) | |
| Within populations | 94 | 361.000 | 3.84043 Vd | 71.02 | ΦIT = 0.290 (0.000) | |
| b | Among groups | 2 | 1423.097 | 15.54290 Va | 65.39 | ΦCT = 0.653 (0.010) |
| Among population within groups | 5 | 144.291 | 1.24626 Vb | 5.24 | ΦSC = 0.151 (0.000) | |
| Within populations | 147 | 1026.096 | 6.98024 Vc | 29.37 | ΦST = 0.706 (0.000) |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 | CH66 | 0.00000 | |||||||
| 2 | CH43 | 0.639 | 0.00000 | ||||||
| 3 | CH54-61 | 0.240 | 0.515 | 0.00000 | |||||
| 4 | CHN4 | 0.385 | 0.605 | 0.009 | 0.00000 | ||||
| 5 | CHN3 | 0.401 | 0.613 | 0.068 | 0.100 | 0.00000 | |||
| 6 | CH64 | 0.149 | 0.524 | 0.040 | 0.145 | 0.161 | 0.00000 | ||
| 7 | CH60 | 0.310 | 0.255 | 0.127 | 0.173 | 0.241 | 0.169 | 0.00000 | |
| 8 | CHN6 | 0.357 | 0.152 | 0.207 | 0.267 | 0.304 | 0.225 | 0.016 | 0.00000 |
| Population | N | HpN | h (s.d) | π (s.d) | K2P | D |
|---|---|---|---|---|---|---|
| CH64 | 11 | 4 | 0.745 | 0.002 | 0.002 | 0.628 |
| (0.098) | (0.001) | (0.001) | p > 0.10 | |||
| CH66 | 16 | 7 | 0.858 | 0.006 (0.001) | 0.007 | 1.024 |
| (0.057) | (0.002) | (NS) | ||||
| CH43 | 20 | 6 | 0.579 | 0.001 | 0.001 | −1.332 |
| (0.015) | (0.000) | (0.001) | p > 0.10 | |||
| CH60 | 35 | 8 | 0.800 | 0.004 | 0.028 | 1.164 |
| (0.001) | (0.000) | (0.004) | (NS) | |||
| CH54-61 | 21 | 8 | 0.865 | 0.005 | 0.005 | 0.357 |
| (0.002) | (0.000) | (0.001) | p > 0.10 | |||
| CHN3 | 12 | 6 | 0.873 | 0.003 | 0.002 | 1.803 |
| (0.071) | (0.000) | (0.001) | (NS) | |||
| CHN4 | 7 | 4 | 0.810 | 0,005 | 0.006 | 0.885 |
| (0.130) | (0.001) | (0.002) | p > 0.10 | |||
| CHN6 | 37 | 13 | 0.701 | 0.032 | 0.038 | 0.759 |
| (0.005) | (0.006) | (0.005) | p > 0.10 |
| 1 | 2 | 3 | 4 | 5 | 6 | 7 | 8 | ||
|---|---|---|---|---|---|---|---|---|---|
| 1 | CH66 | 0.00000 | |||||||
| 2 | CH43 | 0.96138 | 0.00000 | ||||||
| 3 | CHN6 | 0.62306 | 0.17907 | 0.00000 | |||||
| 4 | CH54-61 | 0.61678 | 0.96075 | 0.62750 | 0.00000 | ||||
| 5 | CH60 | 0.29516 | 0.73899 | 0.41500 | 0.11801 | 0.00000 | |||
| 6 | CH64 | 0.61543 | 0.98576 | 0.59722 | 0.40308 | 0.08874 | 0.00000 | ||
| 7 | CHN3 | 0.60930 | 0.98489 | 0.59850 | 0.42342 | 0.09494 | -0.06163 | 0.00000 | |
| 8 | CHN4 | 0.48559 | 0.96341 | 0.55235 | 0.03462 | 0.05959 | 0.42159 | 0.41856 | 0.00000 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
García, G.; Ríos, N.; Gutiérrez, V.; Serra, S.; Loureiro, M. Transcriptome-Based SNP Discovery and Validation in the Hybrid Zone of the Neotropical Annual Fish Genus Austrolebias. Genes 2019, 10, 789. https://doi.org/10.3390/genes10100789
García G, Ríos N, Gutiérrez V, Serra S, Loureiro M. Transcriptome-Based SNP Discovery and Validation in the Hybrid Zone of the Neotropical Annual Fish Genus Austrolebias. Genes. 2019; 10(10):789. https://doi.org/10.3390/genes10100789
Chicago/Turabian StyleGarcía, Graciela, Néstor Ríos, Verónica Gutiérrez, Sebastián Serra, and Marcelo Loureiro. 2019. "Transcriptome-Based SNP Discovery and Validation in the Hybrid Zone of the Neotropical Annual Fish Genus Austrolebias" Genes 10, no. 10: 789. https://doi.org/10.3390/genes10100789