Leishmania Mitochondrial Genomes: Maxicircle Structure and Heterogeneity of Minicircles

 , , ,
, , , 
Abstract
:1. Introduction
2. Materials and Methods
2.1. Parasites, DNA Isolation and Illumina Sequencing
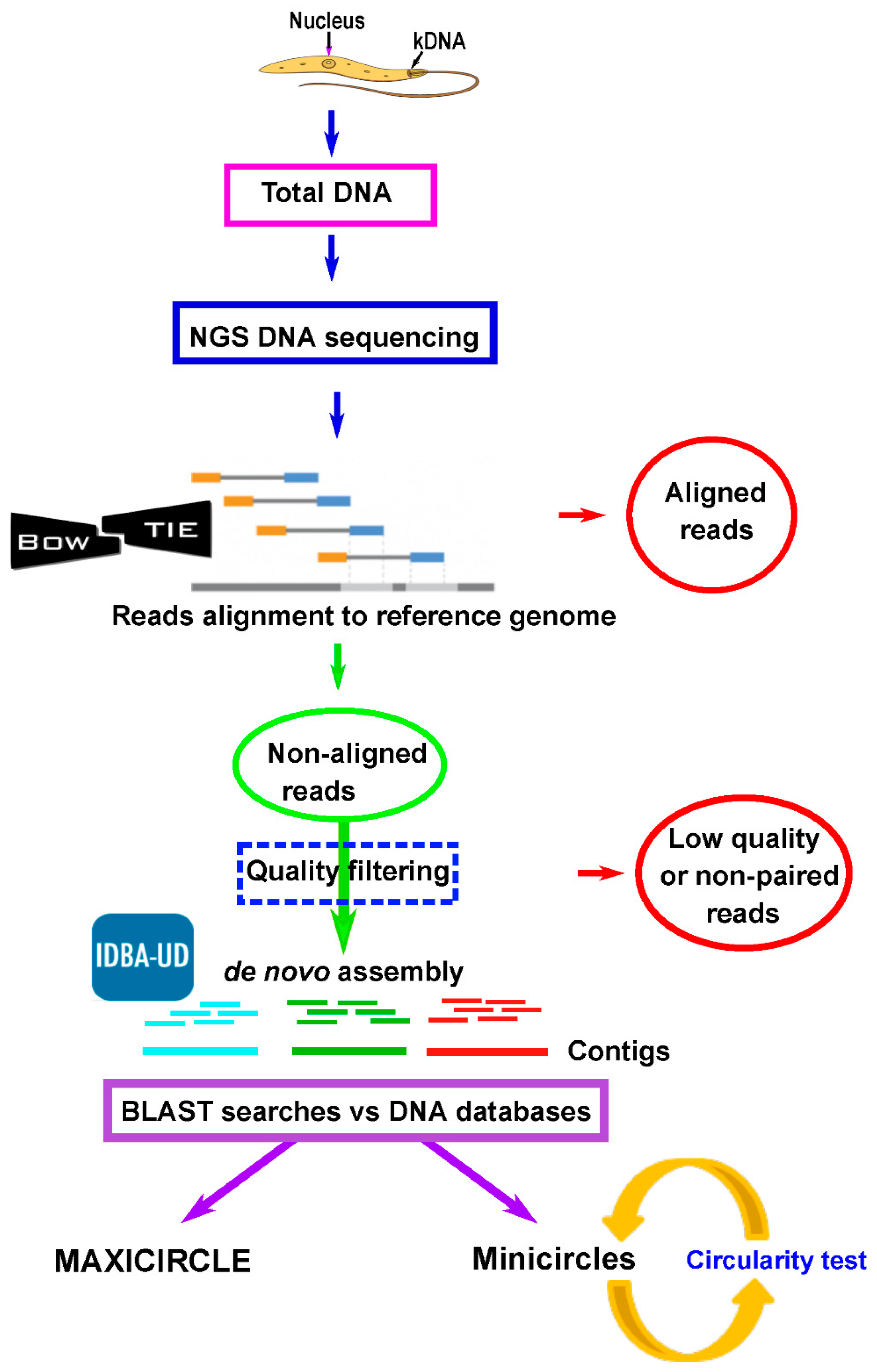
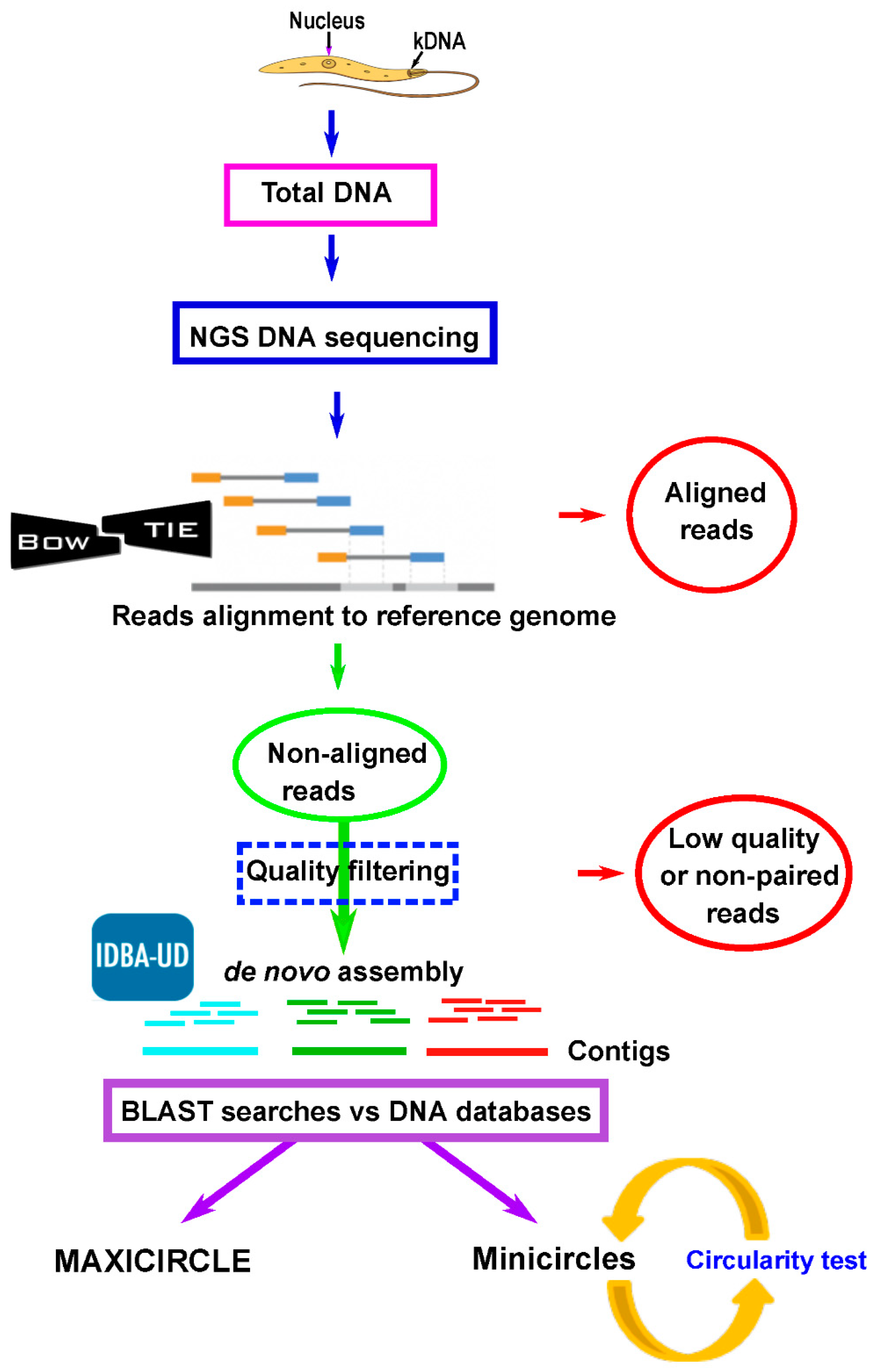
2.2. Assembly of Leishmania Maxicircle and Minicircle Sequences From Total DNA NGS Short Reads
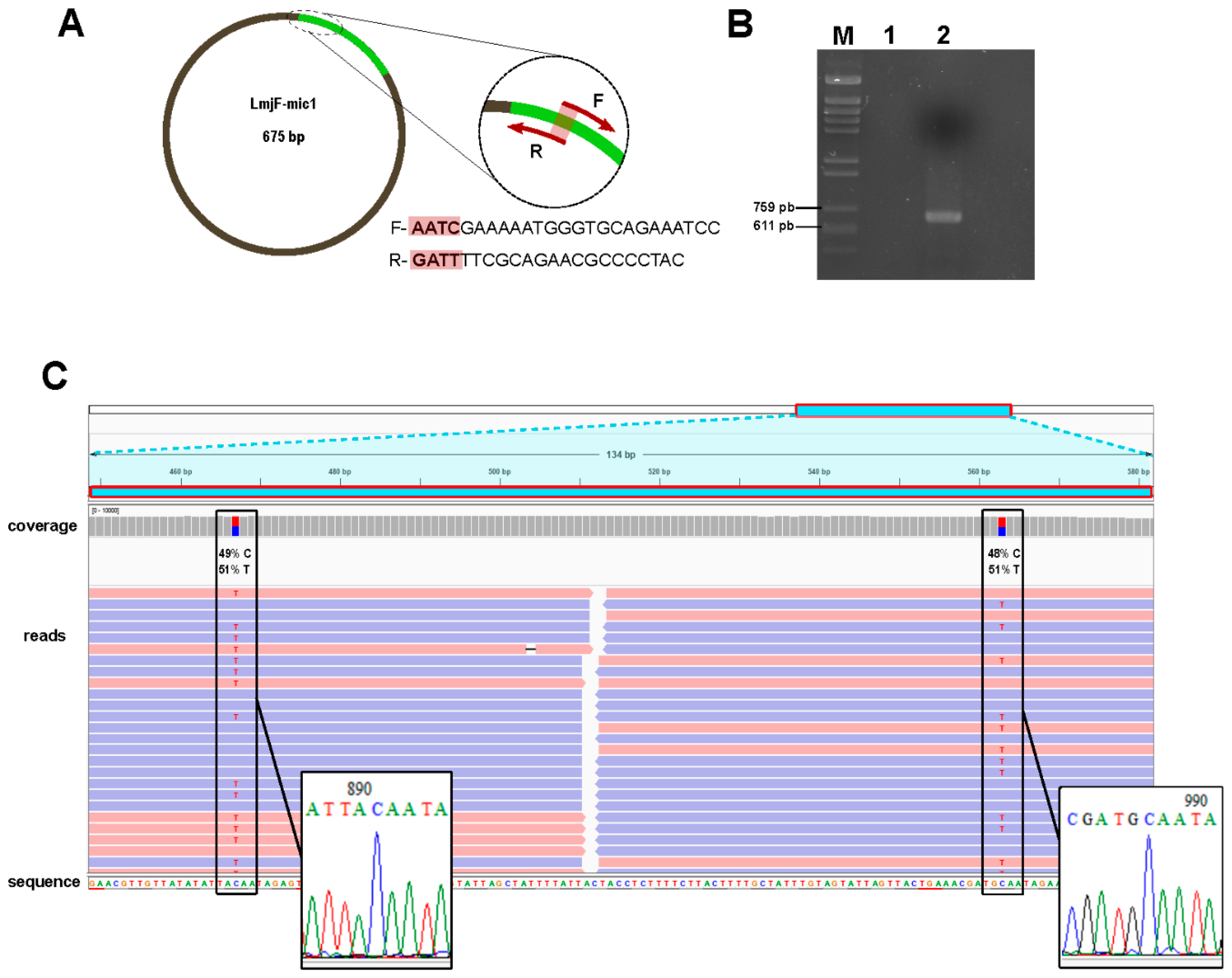
2.3. Validation of Assemblies by PCR Amplification
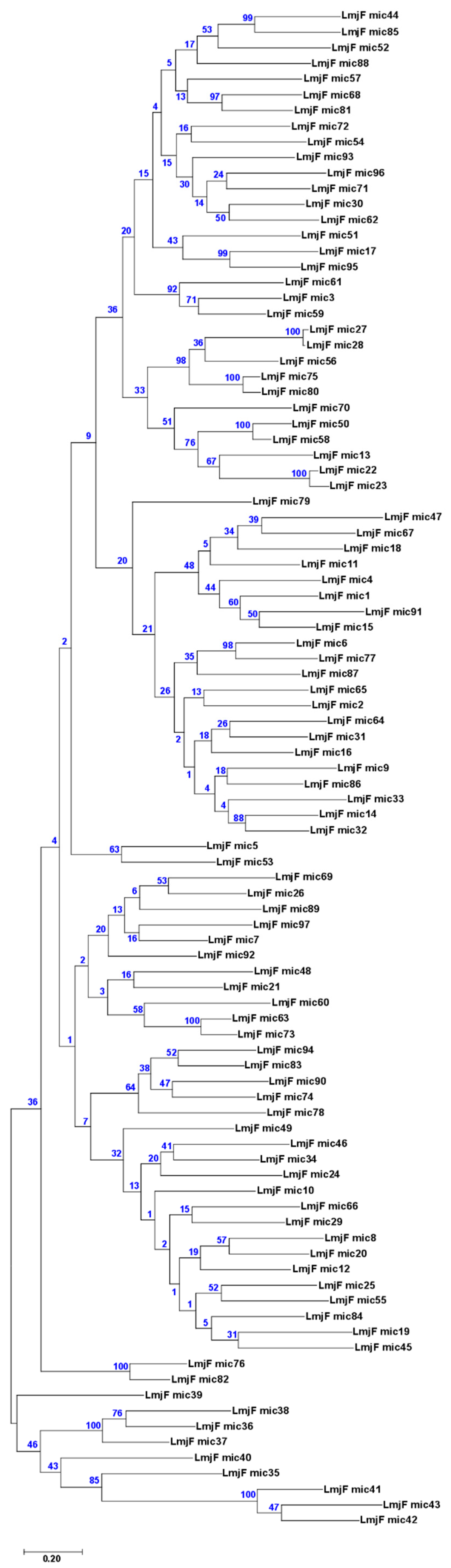
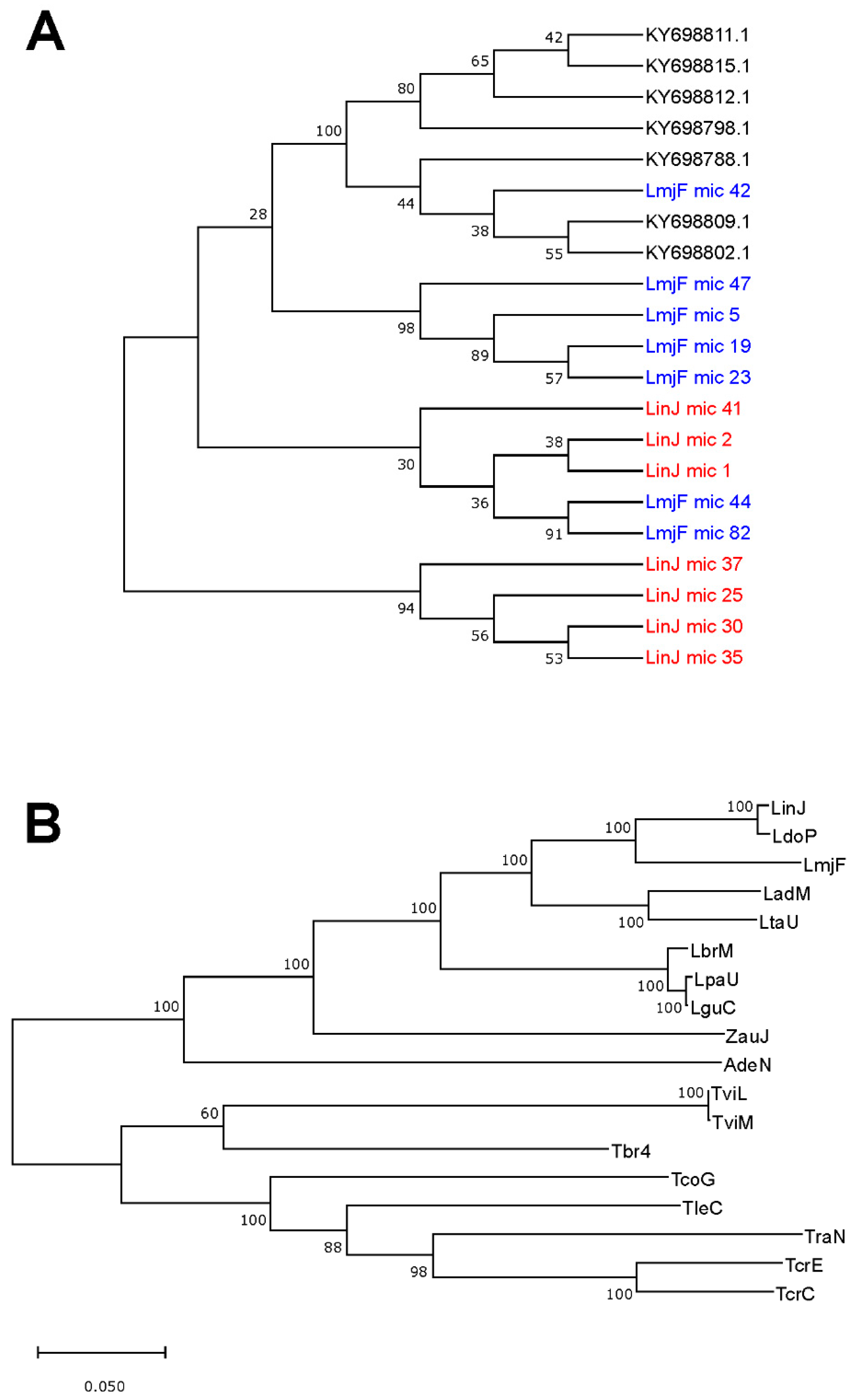
2.4. Phylogenetic Analysis
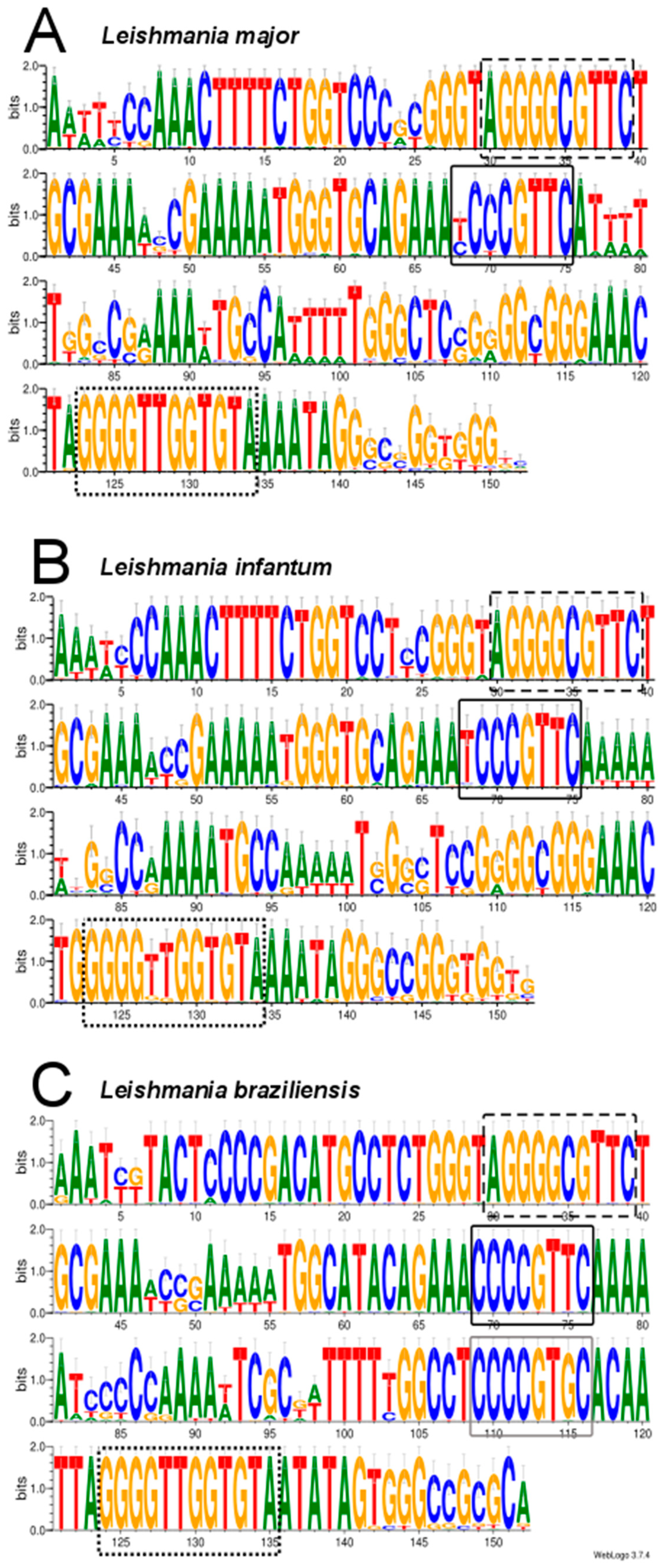
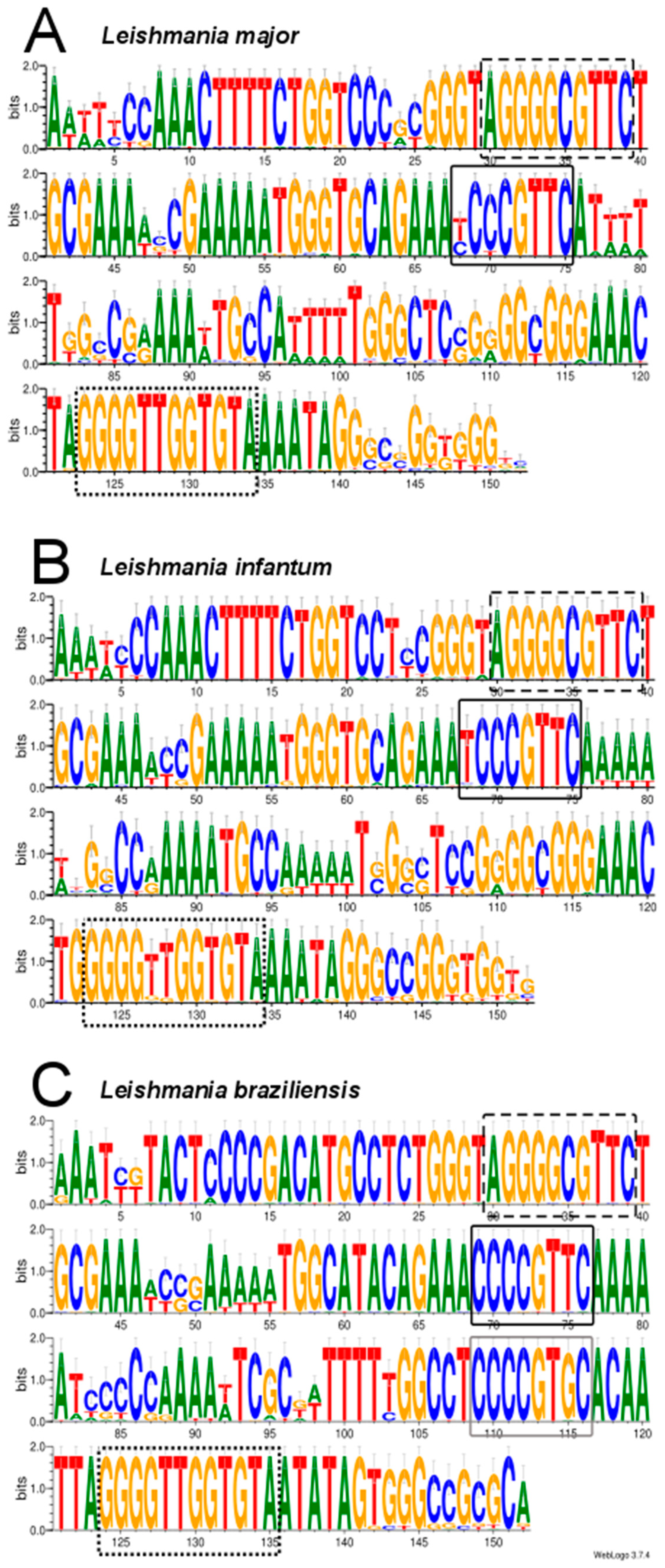
2.5. Analysis of Conserved Motifs in Leishmania Minicircles by LOGOS Software
2.6. Data Availability
3. Results
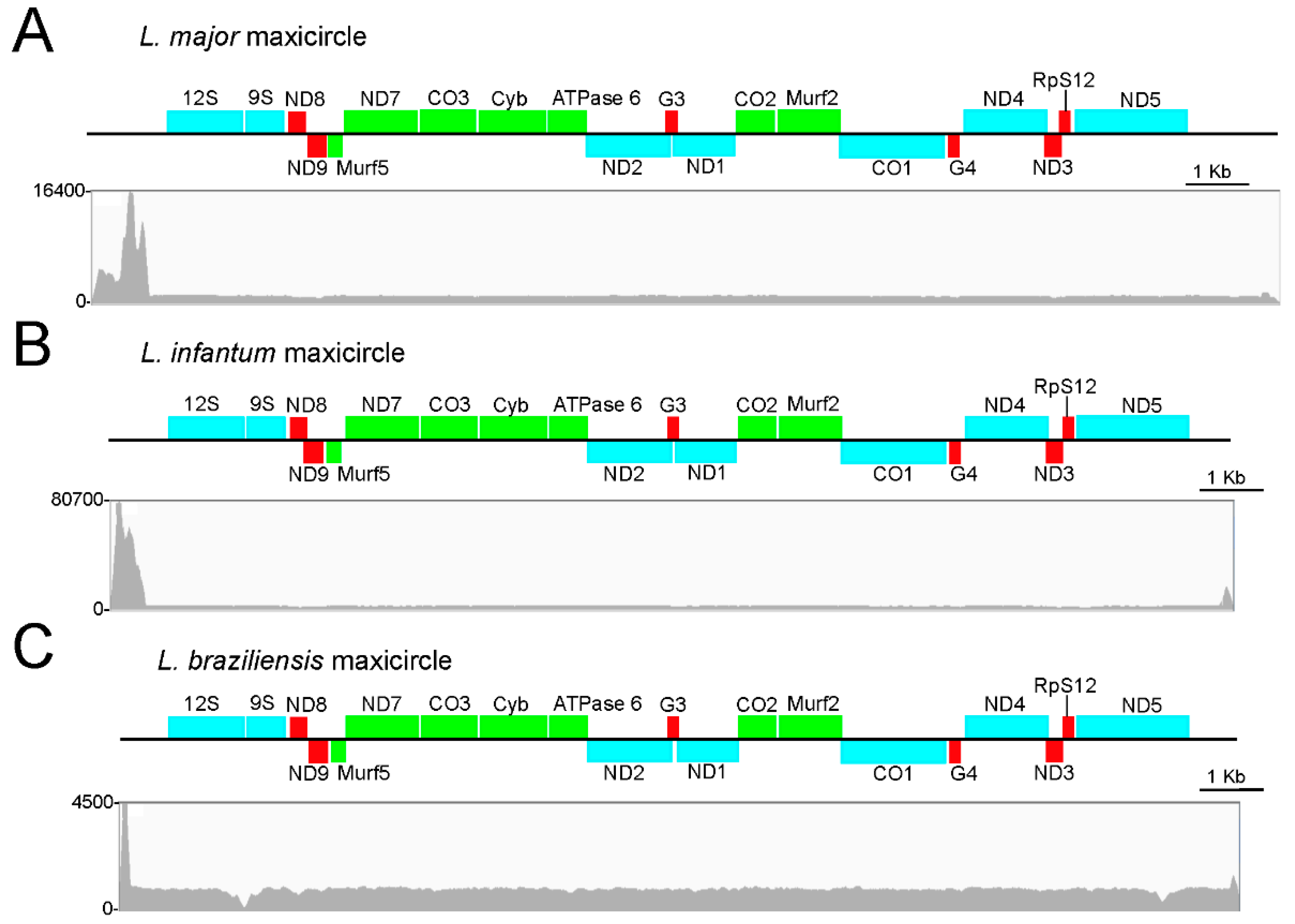
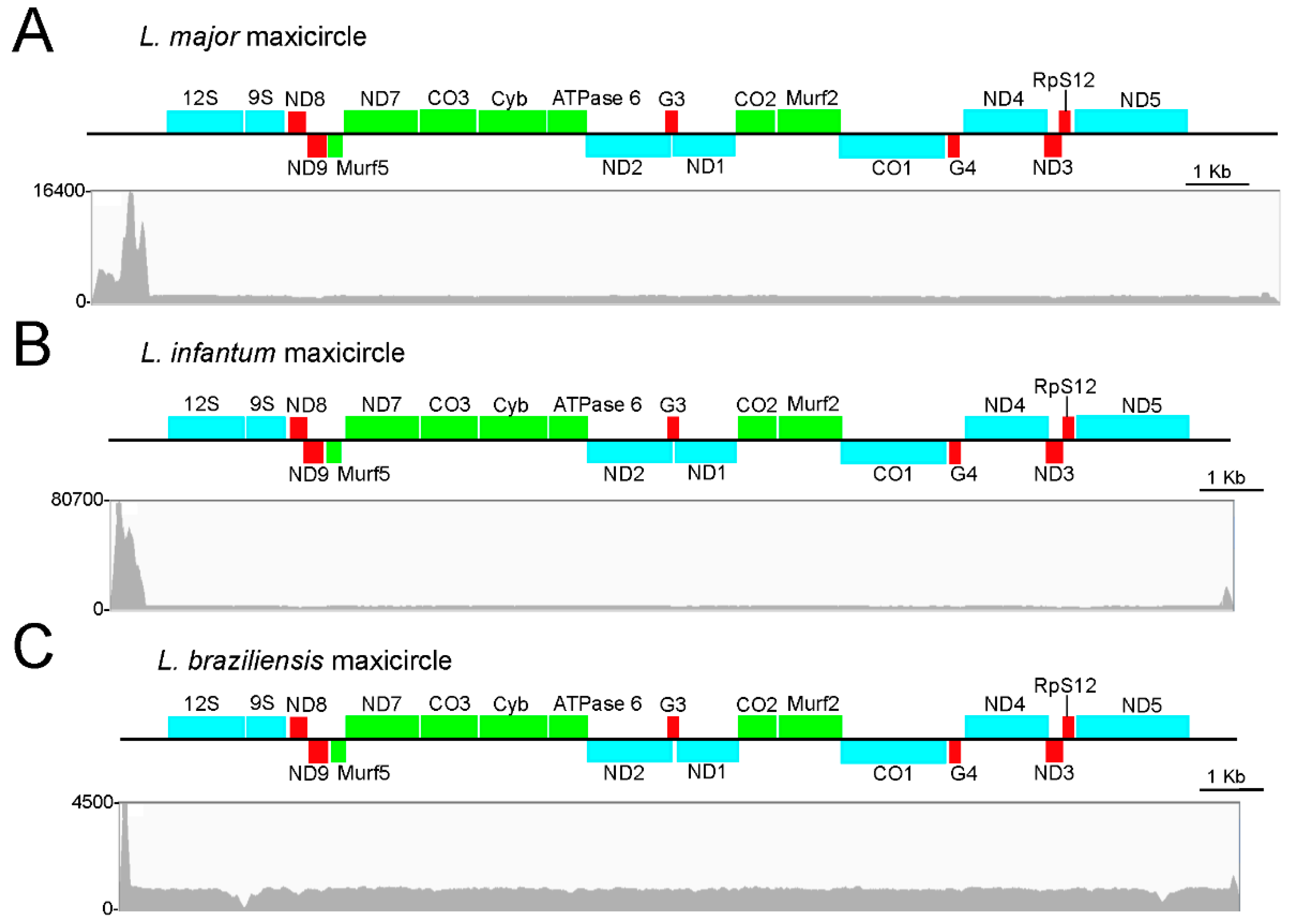
3.1. Assembly and Annotation of the L. major Maxicircle
3.2. Assembly and Annotation of the Maxicircles in L. infantum, L. braziliensis and Other Leishmania Species
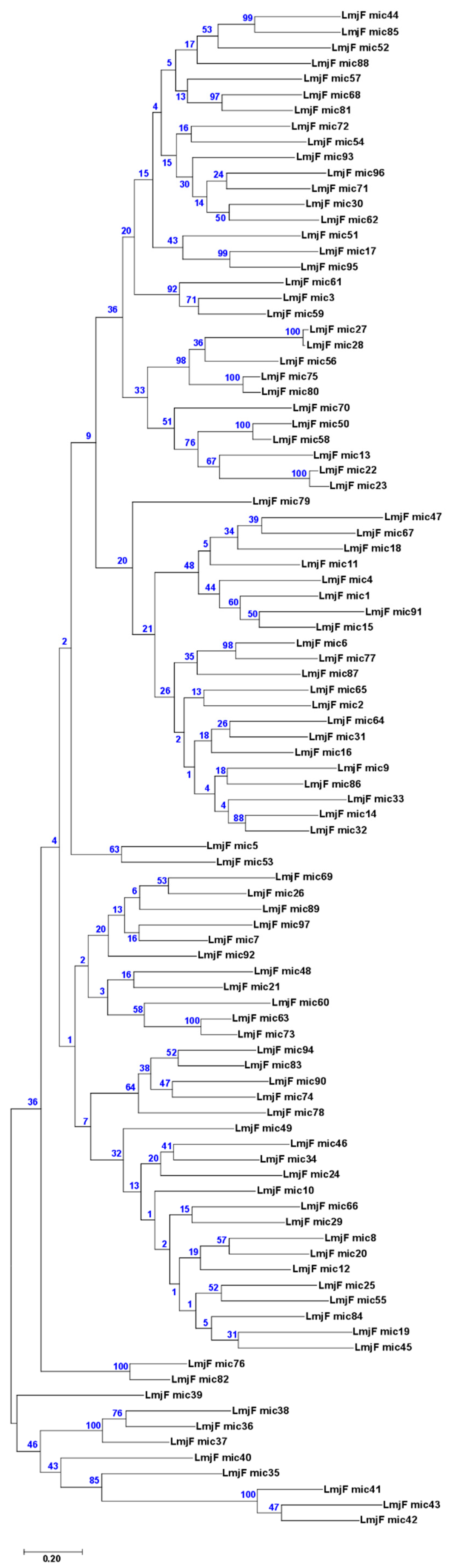
3.3. Assembly of Minicircle Sequences and Analysis of the Minicircle Populations Existing in L. major, L. infantum and L. braziliensis
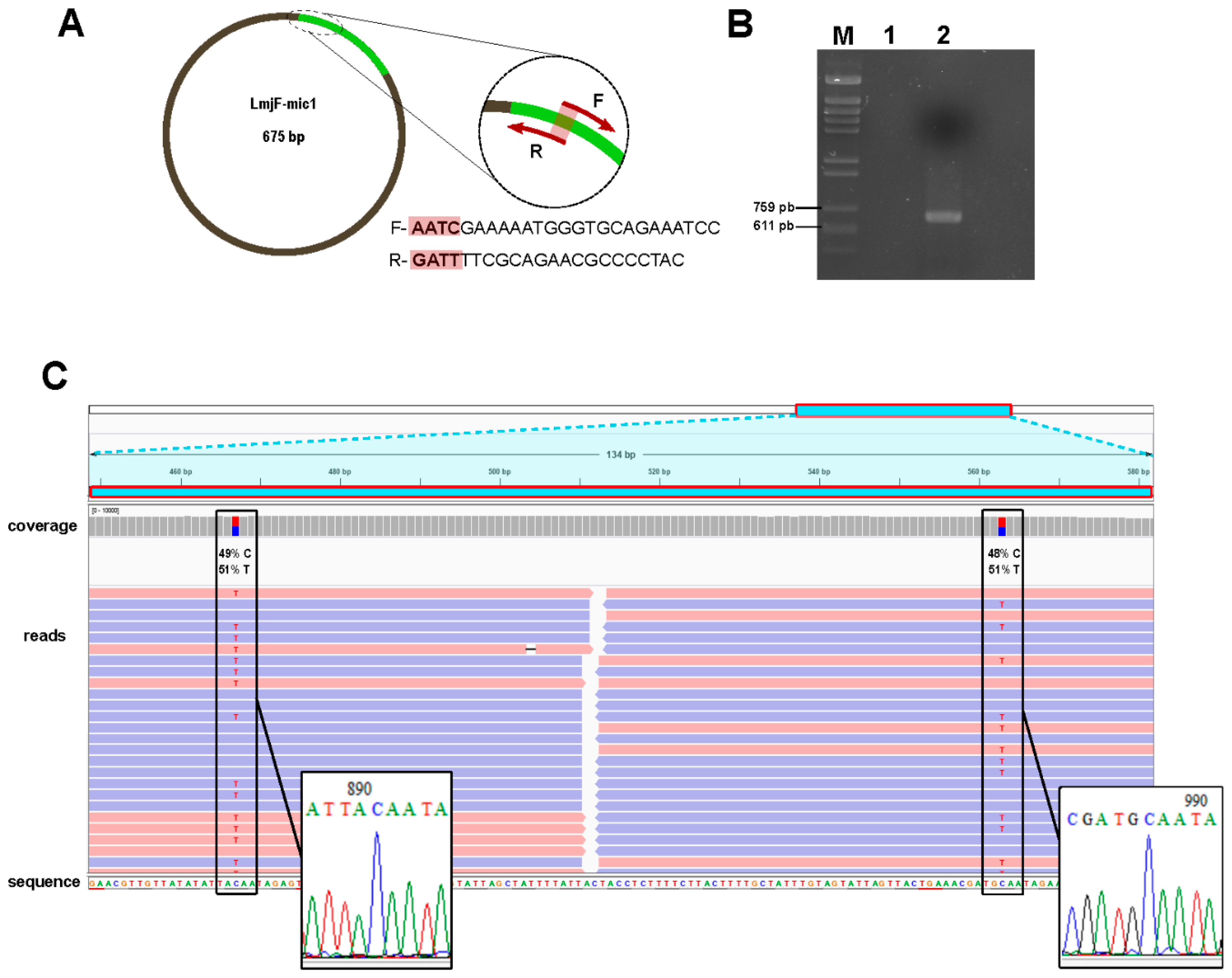
3.4. Experimental Validation of In-Silico Assembled Minicircles
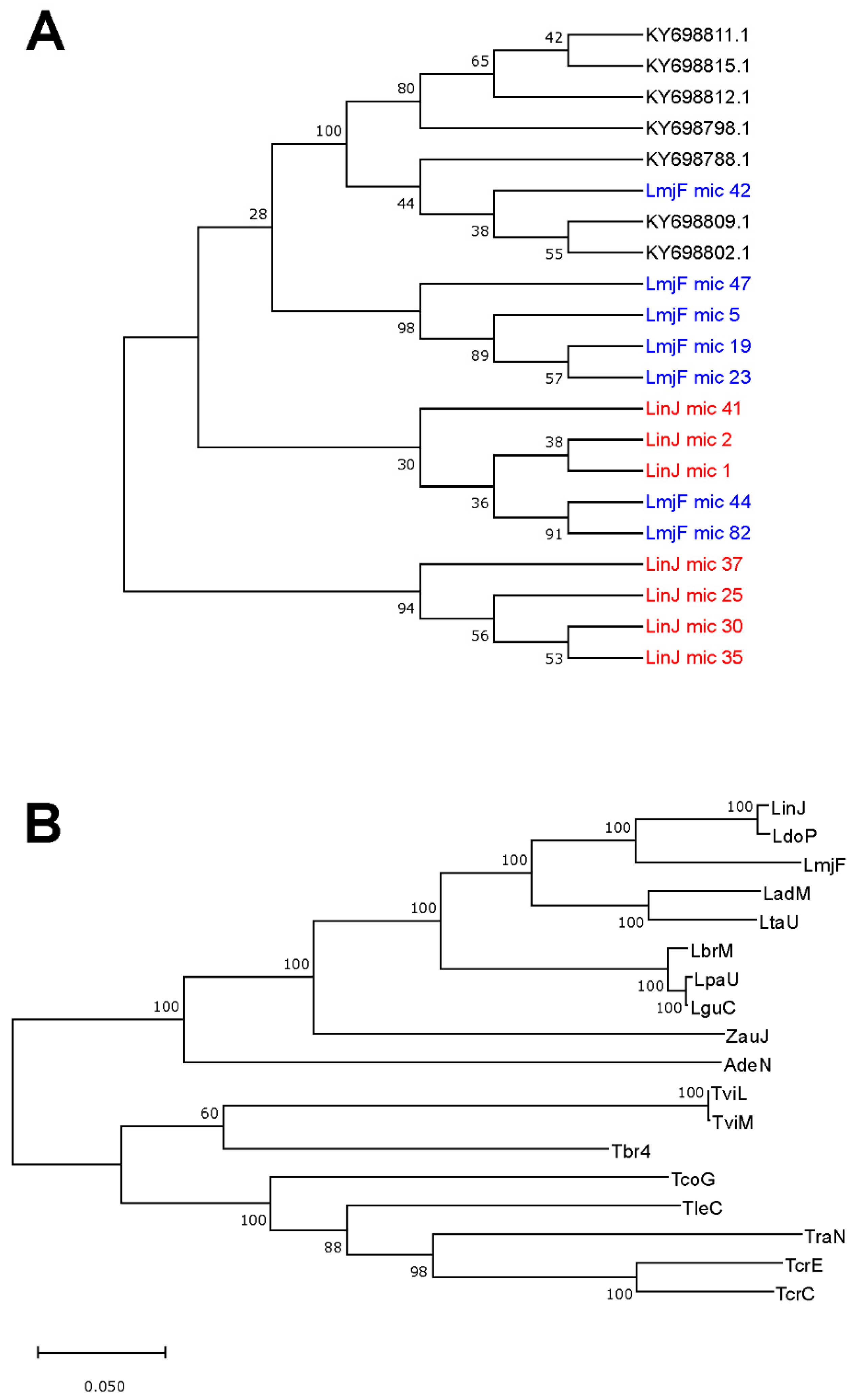
3.5. Usefulness of Minicircle and Maxicircles Sequences for Species Identification and Phylogenetic Analysis
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Moreira, D.; Lopez-Garcia, P.; Vickerman, K. An updated view of kinetoplastid phylogeny using environmental sequences and a closer outgroup: Proposal for a new classification of the class Kinetoplastea. Int. J. Syst. Evol. Microbiol. 2004, 54 Pt 5, 1861–1875. [Google Scholar] [CrossRef]
- Lukes, J.; Butenko, A.; Hashimi, H.; Maslov, D.A.; Votypka, J.; Yurchenko, V. Trypanosomatids Are Much More than Just Trypanosomes: Clues from the Expanded Family Tree. Trends Parasitol. 2018, 34, 466–480. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Stuart, K.; Brun, R.; Croft, S.; Fairlamb, A.; Gurtler, R.E.; McKerrow, J.; Reed, S.; Tarleton, R. Kinetoplastids: Related protozoan pathogens, different diseases. J. Clin. Investig. 2008, 118, 1301–1310. [Google Scholar] [CrossRef] [PubMed]
- Jensen, R.E.; Englund, P.T. Network news: The replication of kinetoplast DNA. Annu. Rev. Microbiol. 2012, 66, 473–491. [Google Scholar] [CrossRef] [PubMed]
- Simpson, L.; Douglass, S.M.; Lake, J.A.; Pellegrini, M.; Li, F. Comparison of the Mitochondrial Genomes and Steady State Transcriptomes of Two Strains of the Trypanosomatid Parasite, Leishmania tarentolae. PLoS Negl. Trop. Dis. 2015, 9, e0003841. [Google Scholar] [CrossRef] [PubMed]
- Estevez, A.M.; Simpson, L. Uridine insertion/deletion RNA editing in trypanosome mitochondria—A review. Gene 1999, 240, 247–260. [Google Scholar] [CrossRef]
- Stuart, K.D.; Schnaufer, A.; Ernst, N.L.; Panigrahi, A.K. Complex management: RNA editing in trypanosomes. Trends Biochem. Sci. 2005, 30, 97–105. [Google Scholar] [CrossRef]
- Read, L.K.; Lukes, J.; Hashimi, H. Trypanosome RNA editing: The complexity of getting U in and taking U out. Wiley Interdiscip. Rev. RNA 2016, 7, 33–51. [Google Scholar] [CrossRef]
- Shapiro, T.A.; Englund, P.T. The structure and replication of kinetoplast DNA. Annu. Rev. Microbiol. 1995, 49, 117–143. [Google Scholar] [CrossRef]
- Simpson, L. The mitochondrial genome of kinetoplastid protozoa: Genomic organization, transcription, replication, and evolution. Annu. Rev. Microbiol. 1987, 41, 363–382. [Google Scholar] [CrossRef]
- Simpson, L.; Shaw, J. RNA editing and the mitochondrial cryptogenes of kinetoplastid protozoa. Cell 1989, 57, 355–366. [Google Scholar] [CrossRef]
- Ramrath, D.J.F.; Niemann, M.; Leibundgut, M.; Bieri, P.; Prange, C.; Horn, E.K.; Leitner, A.; Boehringer, D.; Schneider, A.; Ban, N. Evolutionary shift toward protein-based architecture in trypanosomal mitochondrial ribosomes. Science 2018, 362, 7735. [Google Scholar] [CrossRef] [PubMed]
- Duarte, M.; Tomas, A.M. The mitochondrial complex I of trypanosomatids—An overview of current knowledge. J. Bioenerg. Biomembr. 2014, 46, 299–311. [Google Scholar] [CrossRef] [PubMed]
- Thiemann, O.H.; Maslov, D.A.; Simpson, L. Disruption of RNA editing in Leishmania tarentolae by the loss of minicircle-encoded guide RNA genes. EMBO J. 1994, 13, 5689–5700. [Google Scholar] [CrossRef] [PubMed]
- Muhich, M.L.; Neckelmann, N.; Simpson, L. The divergent region of the Leishmania tarentolae kinetoplast maxicircle DNA contains a diverse set of repetitive sequences. Nucleic Acids Res. 1985, 13, 3241–3260. [Google Scholar] [CrossRef] [PubMed]
- Flegontov, P.N.; Guo, Q.; Ren, L.; Strelkova, M.V.; Kolesnikov, A.A. Conserved repeats in the kinetoplast maxicircle divergent region of Leishmania sp. and Leptomonas seymouri. Mol. Genet. Genom. 2006, 276, 322–333. [Google Scholar] [CrossRef] [PubMed]
- Yatawara, L.; Le, T.H.; Wickramasinghe, S.; Agatsuma, T. Maxicircle (mitochondrial) genome sequence (partial) of Leishmania major: Gene content, arrangement and composition compared with Leishmania tarentolae. Gene 2008, 424, 80–86. [Google Scholar] [CrossRef] [PubMed]
- Nebohacova, M.; Kim, C.E.; Simpson, L.; Maslov, D.A. RNA editing and mitochondrial activity in promastigotes and amastigotes of Leishmania donovani. Int. J. Parasitol. 2009, 39, 635–644. [Google Scholar] [CrossRef] [Green Version]
- Maslov, D.A. Complete set of mitochondrial pan-edited mRNAs in Leishmania mexicana amazonensis LV78. Mol. Biochem. Parasitol. 2010, 173, 107–114. [Google Scholar] [CrossRef] [Green Version]
- Nocua, P.; Ramirez, C.; Requena, J.M.; Puerta, C.J. Secuencia parcial del genoma del maxicírculo de Leishmania braziliensis, comparación con otros tripanosomátidos. Univ. Sci. 2011, 16, 29–50. [Google Scholar] [CrossRef]
- Akhoundi, M.; Downing, T.; Votypka, J.; Kuhls, K.; Lukes, J.; Cannet, A.; Ravel, C.; Marty, P.; Delaunay, P.; Kasbari, M.; et al. Leishmania infections: Molecular targets and diagnosis. Mol. Asp. Med. 2017, 57, 1–29. [Google Scholar] [CrossRef] [PubMed]
- Rogers, M.B.; Hilley, J.D.; Dickens, N.J.; Wilkes, J.; Bates, P.A.; Depledge, D.P.; Harris, D.; Her, Y.; Herzyk, P.; Imamura, H.; et al. Chromosome and gene copy number variation allow major structural change between species and strains of Leishmania. Genome Res. 2011, 21, 2129–2142. [Google Scholar] [CrossRef] [PubMed]
- Singh, N.; Chikara, S.; Sundar, S. SOLiD sequencing of genomes of clinical isolates of Leishmania donovani from India confirm leptomonas co-infection and raise some key questions. PLoS ONE 2013, 8, e55738. [Google Scholar] [CrossRef] [PubMed]
- Real, F.; Vidal, R.O.; Carazzolle, M.F.; Mondego, J.M.; Costa, G.G.; Herai, R.H.; Wurtele, M.; de Carvalho, L.M.; Carmona e Ferreira, R.; Mortara, R.A.; et al. The genome sequence of Leishmania (Leishmania) amazonensis: Functional annotation and extended analysis of gene models. DNA Res. 2013, 20, 567–581. [Google Scholar] [CrossRef] [PubMed]
- Llanes, A.; Restrepo, C.M.; Del Vecchio, G.; Anguizola, F.J.; Lleonart, R. The genome of Leishmania panamensis: Insights into genomics of the L. (Viannia) subgenus. Sci. Rep. 2015, 5, 8550. [Google Scholar] [CrossRef] [PubMed]
- Imamura, H.; Downing, T.; Van den Broeck, F.; Sanders, M.J.; Rijal, S.; Sundar, S.; Mannaert, A.; Vanaerschot, M.; Berg, M.; De Muylder, G.; et al. Evolutionary genomics of epidemic visceral leishmaniasis in the Indian subcontinent. eLife 2016, 5, e12613. [Google Scholar] [CrossRef] [PubMed]
- Ghouila, A.; Guerfali, F.Z.; Atri, C.; Bali, A.; Attia, H.; Sghaier, R.M.; Mkannez, G.; Dickens, N.J.; Laouini, D. Comparative genomics of Tunisian Leishmania major isolates causing human cutaneous leishmaniasis with contrasting clinical severity. Infect. Genet. Evol. 2017, 50, 110–120. [Google Scholar] [CrossRef]
- Teixeira, D.G.; Monteiro, G.R.G.; Martins, D.R.A.; Fernandes, M.Z.; Macedo-Silva, V.; Ansaldi, M.; Nascimento, P.R.P.; Kurtz, M.A.; Streit, J.A.; Ximenes, M.; et al. Comparative analyses of whole genome sequences of Leishmania infantum isolates from humans and dogs in northeastern Brazil. Int. J. Parasitol. 2017, 47, 655–665. [Google Scholar] [CrossRef] [Green Version]
- Valdivia, H.O.; Almeida, L.V.; Roatt, B.M.; Reis-Cunha, J.L.; Pereira, A.A.S.; Gontijo, C.; Fujiwara, R.T.; Reis, A.B.; Sanders, M.J.; Cotton, J.A.; et al. Comparative genomics of canine-isolated Leishmania (Leishmania) amazonensis from an endemic focus of visceral leishmaniasis in Governador Valadares, southeastern Brazil. Sci. Rep. 2017, 7, 40804. [Google Scholar] [CrossRef]
- Gonzalez-de la Fuente, S.; Peiro-Pastor, R.; Rastrojo, A.; Moreno, J.; Carrasco-Ramiro, F.; Requena, J.M.; Aguado, B. Resequencing of the Leishmania infantum (strain JPCM5) genome and de novo assembly into 36 contigs. Sci. Rep. 2017, 7, 18050. [Google Scholar] [CrossRef]
- Zackay, A.; Cotton, J.A.; Sanders, M.; Hailu, A.; Nasereddin, A.; Warburg, A.; Jaffe, C.L. Genome wide comparison of Ethiopian Leishmania donovani strains reveals differences potentially related to parasite survival. PLoS Genet. 2018, 14, e1007133. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez-de la Fuente, S.; Camacho, E.; Peiro-Pastor, R.; Rastrojo, A.; Carrasco-Ramiro, F.; Aguado, B.; Requena, J.M. Complete and de novo assembly of the Leishmania braziliensis (M2904) genome. Memorias do Instituto Oswaldo Cruz 2018, 114, e180438. [Google Scholar] [CrossRef] [PubMed]
- Burza, S.; Croft, S.L.; Boelaert, M. Leishmaniasis. Lancet 2018, 392, 951–970. [Google Scholar] [CrossRef]
- Requena, J.M.; Lopez, M.C.; Jimenez-Ruiz, A.; de la Torre, J.C.; Alonso, C. A head-to-tail tandem organization of hsp70 genes in Trypanosoma cruzi. Nucleic Acids Res. 1988, 16, 1393–1406. [Google Scholar] [CrossRef] [PubMed]
- Langmead, B.; Salzberg, S.L. Fast gapped-read alignment with Bowtie 2. Nat. Methods 2012, 9, 357–359. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Quinlan, A.R.; Hall, I.M. BEDTools: A flexible suite of utilities for comparing genomic features. Bioinformatics 2010, 26, 841–842. [Google Scholar] [CrossRef]
- Peng, Y.; Leung, H.C.; Yiu, S.M.; Chin, F.Y. IDBA-UD: A de novo assembler for single-cell and metagenomic sequencing data with highly uneven depth. Bioinformatics 2012, 28, 1420–1428. [Google Scholar] [CrossRef]
- Treangen, T.J.; Sommer, D.D.; Angly, F.E.; Koren, S.; Pop, M. Next generation sequence assembly with AMOS. Curr. Protoc. Bioinform. 2011, 33, 11.8.1–11.8.18. [Google Scholar] [CrossRef]
- Tamura, K.; Nei, M. Estimation of the number of nucleotide substitutions in the control region of mitochondrial DNA in humans and chimpanzees. Mol. Biol. Evol. 1993, 10, 512–526. [Google Scholar]
- Kumar, S.; Stecher, G.; Li, M.; Knyaz, C.; Tamura, K. MEGA X: Molecular Evolutionary Genetics Analysis across Computing Platforms. Mol. Biol. Evol. 2018, 35, 1547–1549. [Google Scholar] [CrossRef]
- Crooks, G.E.; Hon, G.; Chandonia, J.M.; Brenner, S.E. WebLogo: A sequence logo generator. Genome Res. 2004, 14, 1188–1190. [Google Scholar] [CrossRef]
- Alonso, G.; Rastrojo, A.; Lopez-Perez, S.; Requena, J.M.; Aguado, B. Resequencing and assembly of seven complex loci to improve the Leishmania major (Friedlin strain) reference genome. Parasites Vectors 2016, 9, 74. [Google Scholar] [CrossRef]
- Flegontov, P.N.; Strelkova, M.V.; Kolesnikov, A.A. The Leishmania major maxicircle divergent region is variable in different isolates and cell types. Mol. Biochem. Parasitol. 2006, 146, 173–179. [Google Scholar] [CrossRef] [PubMed]
- Lin, R.H.; Lai, D.H.; Zheng, L.L.; Wu, J.; Lukes, J.; Hide, G.; Lun, Z.R. Analysis of the mitochondrial maxicircle of Trypanosoma lewisi, a neglected human pathogen. Parasites Vectors 2015, 8, 665. [Google Scholar] [CrossRef] [PubMed]
- Sloof, P.; de Haan, A.; Eier, W.; van Iersel, M.; Boel, E.; van Steeg, H.; Benne, R. The nucleotide sequence of the variable region in Trypanosoma brucei completes the sequence analysis of the maxicircle component of mitochondrial kinetoplast DNA. Mol. Biochem. Parasitol. 1992, 56, 289–299. [Google Scholar] [CrossRef]
- Botero, A.; Kapeller, I.; Cooper, C.; Clode, P.L.; Shlomai, J.; Thompson, R.C.A. The kinetoplast DNA of the Australian trypanosome, Trypanosoma copemani, shares features with Trypanosoma cruzi and Trypanosoma lewisi. Int. J. Parasitol. 2018, 48, 691–700. [Google Scholar] [CrossRef]
- Lake, J.A.; de la Cruz, V.F.; Ferreira, P.C.G.; Morel, C.; Simpson, L. Evolution of parasitism: Kinetoplastid protozoan history reconstructed from mitochondrial rRNA gene sequences. Proc. Natl. Acad. Sci. USA 1988, 85, 4779–4783. [Google Scholar] [CrossRef]
- Coughlan, S.; Mulhair, P.; Sanders, M.; Schonian, G.; Cotton, J.A.; Downing, T. The genome of Leishmania adleri from a mammalian host highlights chromosome fission in Sauroleishmania. Sci. Rep. 2017, 7, 43747. [Google Scholar] [CrossRef]
- Coughlan, S.; Taylor, A.S.; Feane, E.; Sanders, M.; Schonian, G.; Cotton, J.A.; Downing, T. Leishmania naiffi and Leishmania guyanensis reference genomes highlight genome structure and gene evolution in the Viannia subgenus. R. Soc. Open Sci. 2018, 5, 172212. [Google Scholar] [CrossRef] [Green Version]
- Ray, D.S. Conserved sequence blocks in kinetoplast minicircles from diverse species of trypanosomes. Mol. Cell. Biol. 1989, 9, 1365–1367. [Google Scholar] [CrossRef]
- Milman, N.; Motyka, S.A.; Englund, P.T.; Robinson, D.; Shlomai, J. Mitochondrial origin-binding protein UMSBP mediates DNA replication and segregation in trypanosomes. Proc. Natl. Acad. Sci. USA 2007, 104, 19250–19255. [Google Scholar] [CrossRef] [Green Version]
- Kocher, A.; Valiere, S.; Banuls, A.L.; Murienne, J. High-throughput sequencing of kDNA amplicons for the analysis of Leishmania minicircles and identification of Neotropical species. Parasitology 2018, 145, 585–594. [Google Scholar] [CrossRef]
- Domingo, E.; Perales, C. Quasispecies and virus. Eur. Biophys. J. 2018, 47, 443–457. [Google Scholar] [CrossRef]
- Kaufer, A.; Barratt, J.; Stark, D.; Ellis, J. The complete coding region of the maxicircle as a superior phylogenetic marker for exploring evolutionary relationships between members of the Leishmaniinae. Infect. Genet. Evol. 2019, 70, 90–100. [Google Scholar] [CrossRef]
- Barratt, J.; Kaufer, A.; Peters, B.; Craig, D.; Lawrence, A.; Roberts, T.; Lee, R.; McAuliffe, G.; Stark, D.; Ellis, J. Isolation of Novel Trypanosomatid, Zelonia australiensis sp. nov. (Kinetoplastida: Trypanosomatidae) Provides Support for a Gondwanan Origin of Dixenous Parasitism in the Leishmaniinae. PLoS Negl. Trop. Dis. 2017, 11, e0005215. [Google Scholar] [CrossRef]
- Huang, T. Next generation sequencing to characterize mitochondrial genomic DNA heteroplasmy. Curr. Protoc. Hum. Genet. 2011, 71, 19.8.1–19.8.12. [Google Scholar]
- Moreira, D.A.; Furtado, C.; Parente, T.E. The use of transcriptomic next-generation sequencing data to assemble mitochondrial genomes of Ancistrus spp. (Loricariidae). Gene 2015, 573, 171–175. [Google Scholar] [CrossRef]
- Wang, T.; Zhang, S.; Pei, T.; Yu, Z.; Liu, J. Tick mitochondrial genomes: Structural characteristics and phylogenetic implications. Parasites Vectors 2019, 12, 451. [Google Scholar] [CrossRef]
- Urrea, D.A.; Triana-Chavez, O.; Alzate, J.F. Mitochondrial genomics of human pathogenic parasite Leishmania (Viannia) panamensis. PeerJ 2019, 7, e7235. [Google Scholar] [CrossRef]
- Degrave, W.; Fernandes, O.; Thiemann, O.; Wincker, P.; Britto, C.; Cardoso, A.; Pereira, J.B.; Bozza, M.; Lopes, U.; Morel, C. Detection of Trypanosoma cruzi and Leishmania using the polymerase chain reaction. Memórias do Instituto Oswaldo Cruz 1994, 89, 367–368. [Google Scholar] [CrossRef] [Green Version]
- Jara, M.; Adaui, V.; Valencia, B.M.; Martinez, D.; Alba, M.; Castrillon, C.; Cruz, M.; Cruz, I.; Van der Auwera, G.; Llanos-Cuentas, A.; et al. Real-time PCR assay for detection and quantification of Leishmania (Viannia) organisms in skin and mucosal lesions: Exploratory study of parasite load and clinical parameters. J. Clin. Microbiol. 2013, 51, 1826–1833. [Google Scholar] [CrossRef]
- Jaber, H.T.; Hailu, A.; Pratlong, F.; Lami, P.; Bastien, P.; Jaffe, C.L. Analysis of genetic polymorphisms and tropism in East African Leishmania donovani by Amplified Fragment Length Polymorphism and kDNA minicircle sequencing. Infect. Genet. Evol. 2018, 65, 80–90. [Google Scholar] [CrossRef]
- Foulet, F.; Botterel, F.; Buffet, P.; Morizot, G.; Rivollet, D.; Deniau, M.; Pratlong, F.; Costa, J.M.; Bretagne, S. Detection and identification of Leishmania species from clinical specimens by using a real-time PCR assay and sequencing of the cytochrome B gene. J. Clin. Microbiol. 2007, 45, 2110–2115. [Google Scholar] [CrossRef]
- Asato, Y.; Oshiro, M.; Myint, C.K.; Yamamoto, Y.; Kato, H.; Marco, J.D.; Mimori, T.; Gomez, E.A.L.; Hashiguchi, Y.; Uezato, H. Phylogenic analysis of the genus Leishmania by cytochrome b gene sequencing. Exp. Parasitol. 2009, 121, 352–361. [Google Scholar] [CrossRef]
- Kuhls, K.; Mauricio, I. Phylogenetic Studies. Methods Mol. Biol. 2019, 1971, 9–68. [Google Scholar]

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Coordinates (strand: Plus (+) or minus (−)) | |||
|---|---|---|---|
| Gene a | L. major | L. infantum | L. braziliensis |
| 12S rRNA | 1302-2462 (+) | 993-2156 (+) | 815-1974 (+) |
| 9S rRNA | 2498-3107 (+) | 2181-2790 (+) | 2005-2615 (+) |
| ND8 | 3200-3450 (+) | 2885-3198 (+) | 2688-2878 (+) |
| ND9 | 3511-3802 (−) | 3185-3524 (−) | 3086-3313 (−) |
| MURF5 | 3777-4082 (−) | 3496-3792 (−) | 3333-3632 (−) |
| ND7 | 4095-5271 (+) | 3806-4979 (+) | 3650-4802 (+) |
| CO3 | 5275-6155 (+) | 4999-5879 (+) | 4884-5719 (+) |
| CYb | 6166-7270 (+) | 5889-6998 (+) | 5743-6830 (+) |
| ATPase 6 | 7336-7934 (+) | 7061-7660 (+) | 6878-7467 (+) |
| ND2 | 7939-9272 (−) | 7665-8996 (−) | 7482-8807 (−) |
| G3 | 9232-9397 (+) | 8958-9120 (+) | 8775-8941 (+) |
| ND1 | 9361-10302 (−) | 9085-10026 (−) | 8930-9855 (−) |
| CO2 | 10311-10939 (+) | 10029-10657 (+) | 9862-10490 (+) |
| MURF2 | 10931-12009 (+) | 10649-11728 (+) | 10482-11573 (+) |
| CO1 | 12000-13649 (−) | 11719-13368 (−) | 11564-13196 (−) |
| G4 | 13698-13861 (−) | 13372-13493 (−) | 13292-13459 (−) |
| ND4 | 13987-15299 (+) | 13705-15017 (+) | 13512-14823 (+) |
| G5 (ND3) | 15282-15524 (−) | 15000-15218 (−) | 14806-15004 (−) |
| RPS12 | 15474-15670 (+) | 15258-15425 (+) | 15036-15191 (+) |
| ND5 | 15751-17522 (+) | 15493-17265 (+) | 15253-17024 (+) |
| Minicircle Type | Circular | Linear/Partial | Uncertain | Total | ||
|---|---|---|---|---|---|---|
| Species | Number | Size Range | Mean Size | |||
| L. major (Friedlin) | 97 | 660–876 | 691 | 7 | - | 104 |
| L. infantum (JPCM5) | 49 | 775–832 | 797 | 28 | 15 | 92 |
| L. braziliensis (M2904) | 3 | 741–749 | 744 | 3 | 22 | 28 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Camacho, E.; Rastrojo, A.; Sanchiz, Á.; González-de la Fuente, S.; Aguado, B.; Requena, J.M. Leishmania Mitochondrial Genomes: Maxicircle Structure and Heterogeneity of Minicircles. Genes 2019, 10, 758. https://doi.org/10.3390/genes10100758
Camacho E, Rastrojo A, Sanchiz Á, González-de la Fuente S, Aguado B, Requena JM. Leishmania Mitochondrial Genomes: Maxicircle Structure and Heterogeneity of Minicircles. Genes. 2019; 10(10):758. https://doi.org/10.3390/genes10100758
Chicago/Turabian StyleCamacho, Esther, Alberto Rastrojo, África Sanchiz, Sandra González-de la Fuente, Begoña Aguado, and Jose M. Requena. 2019. "Leishmania Mitochondrial Genomes: Maxicircle Structure and Heterogeneity of Minicircles" Genes 10, no. 10: 758. https://doi.org/10.3390/genes10100758
APA StyleCamacho, E., Rastrojo, A., Sanchiz, Á., González-de la Fuente, S., Aguado, B., & Requena, J. M. (2019). Leishmania Mitochondrial Genomes: Maxicircle Structure and Heterogeneity of Minicircles. Genes, 10(10), 758. https://doi.org/10.3390/genes10100758