Developmental and Molecular Changes Underlying the Vernalization-Induced Transition to Flowering in Aquilegia coerulea (James)

 ,
, 
Abstract
1. Introduction
2. Materials and Methods
2.1. Plant Growth and Histology
2.2. Scanning Electron Micrography
2.3. Sample Preparation for RNA-seq
2.4. RNA-seq Library Preparation and Sequencing
2.5. Reference-Based Transcriptome Analysis
2.6. Differentially Expressed Genes and Functional Enrichment Analysis
3. Results and Discussions
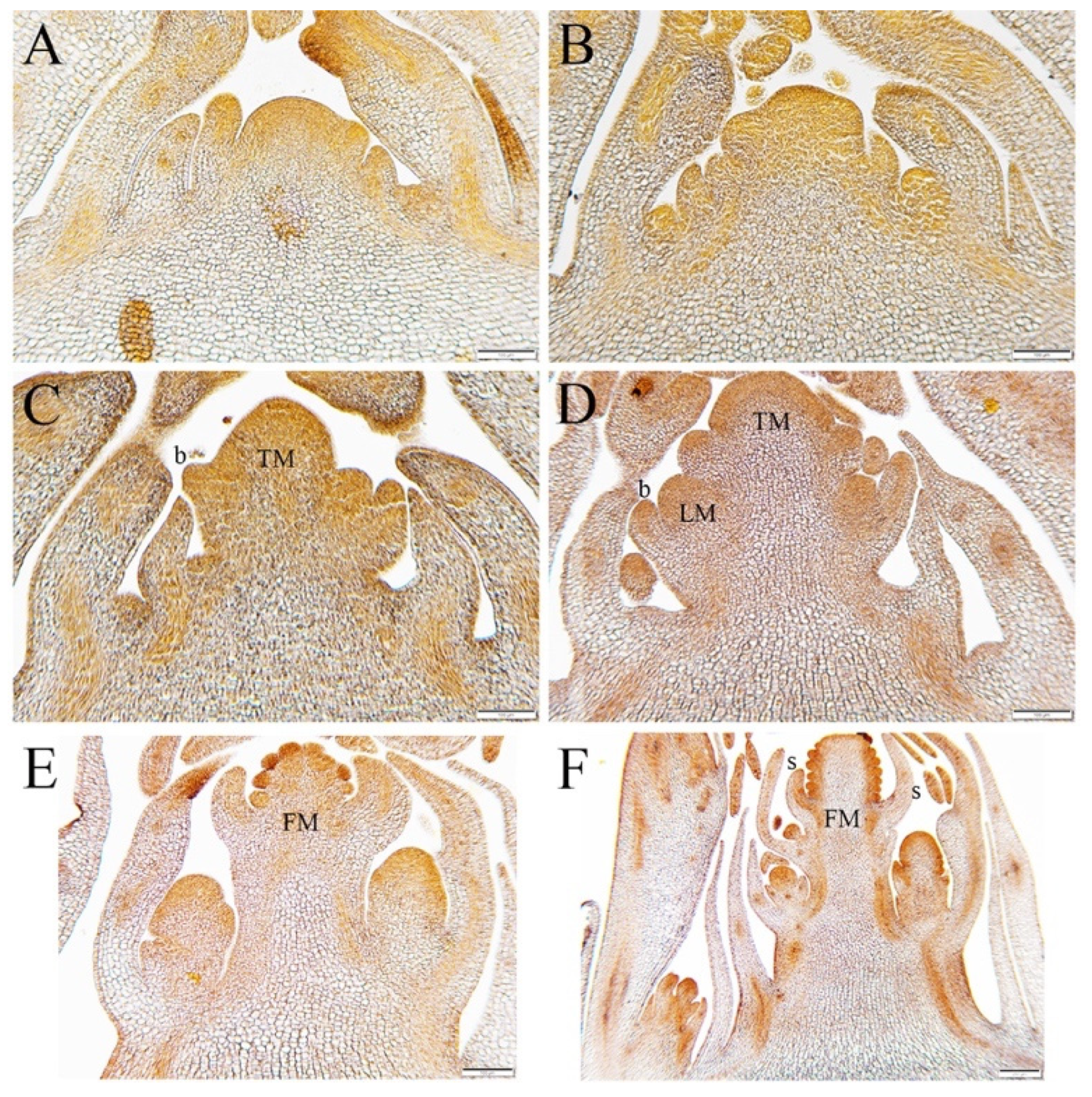
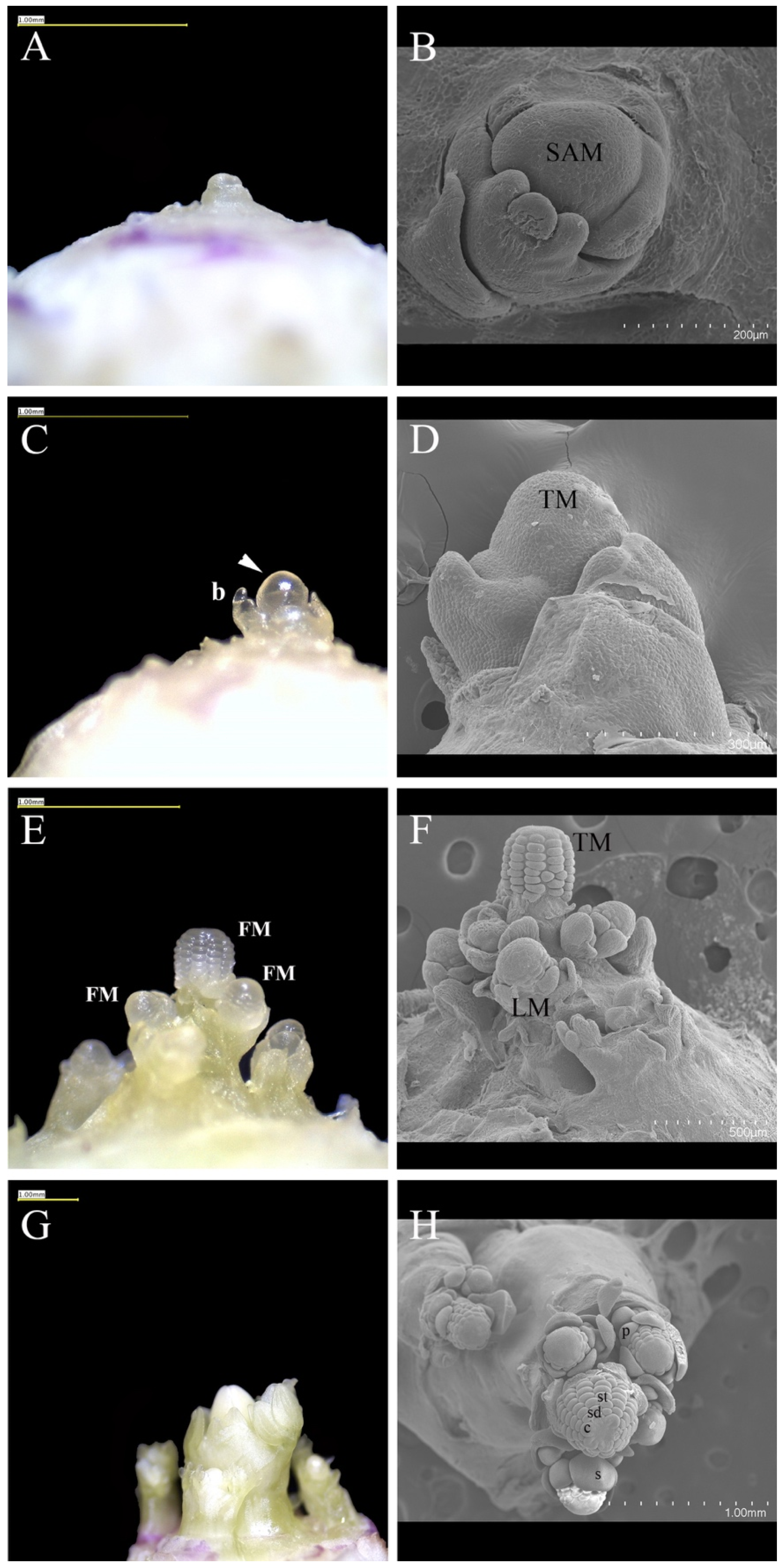
3.1. Vernalization-Induced Developmental Changes in the Aquilegia SAM
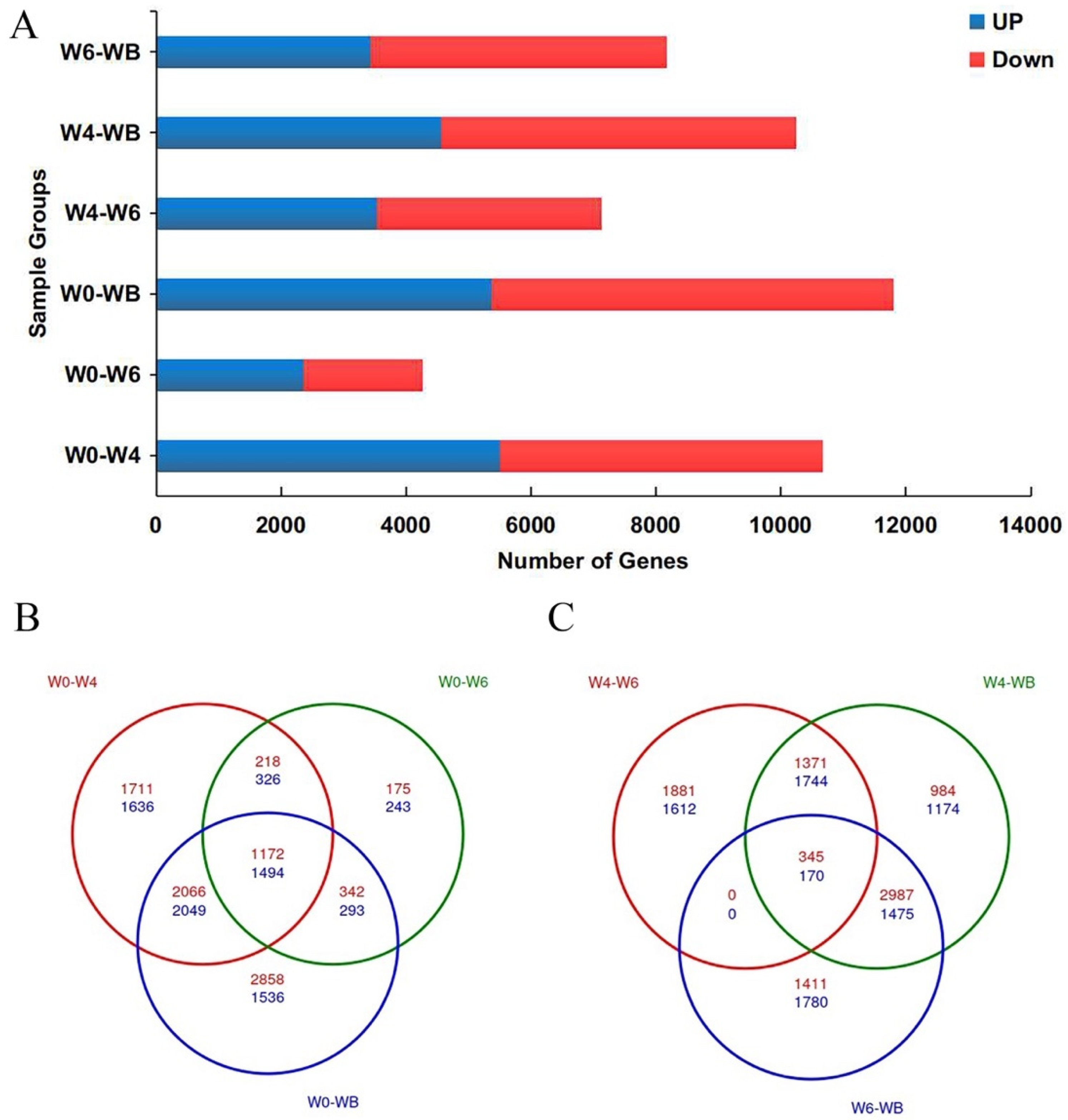
3.2. Transcriptome Sequencing
3.3. GO Enrichment and KEGG Analysis
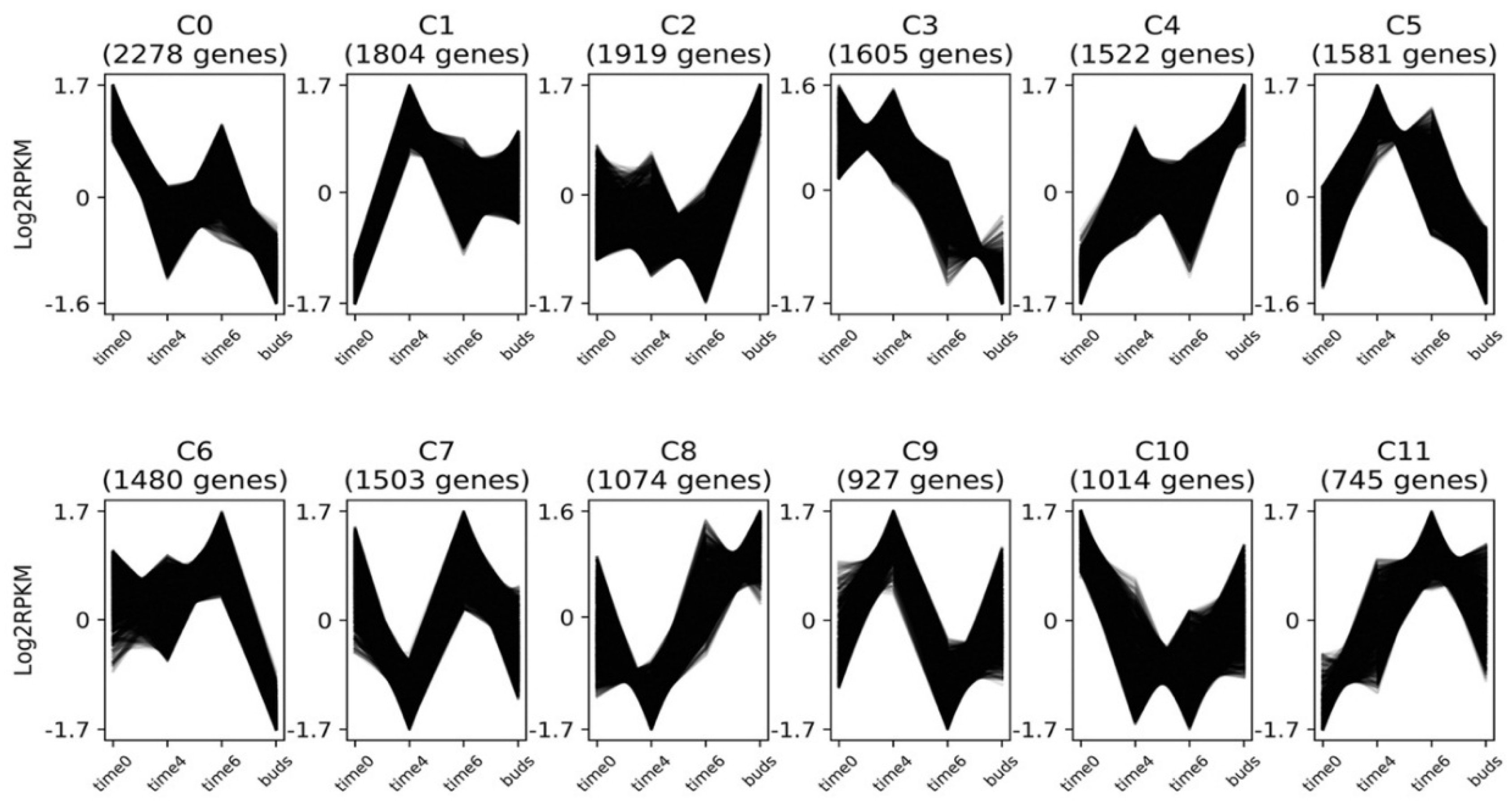
3.4. Gene Expression Clusters
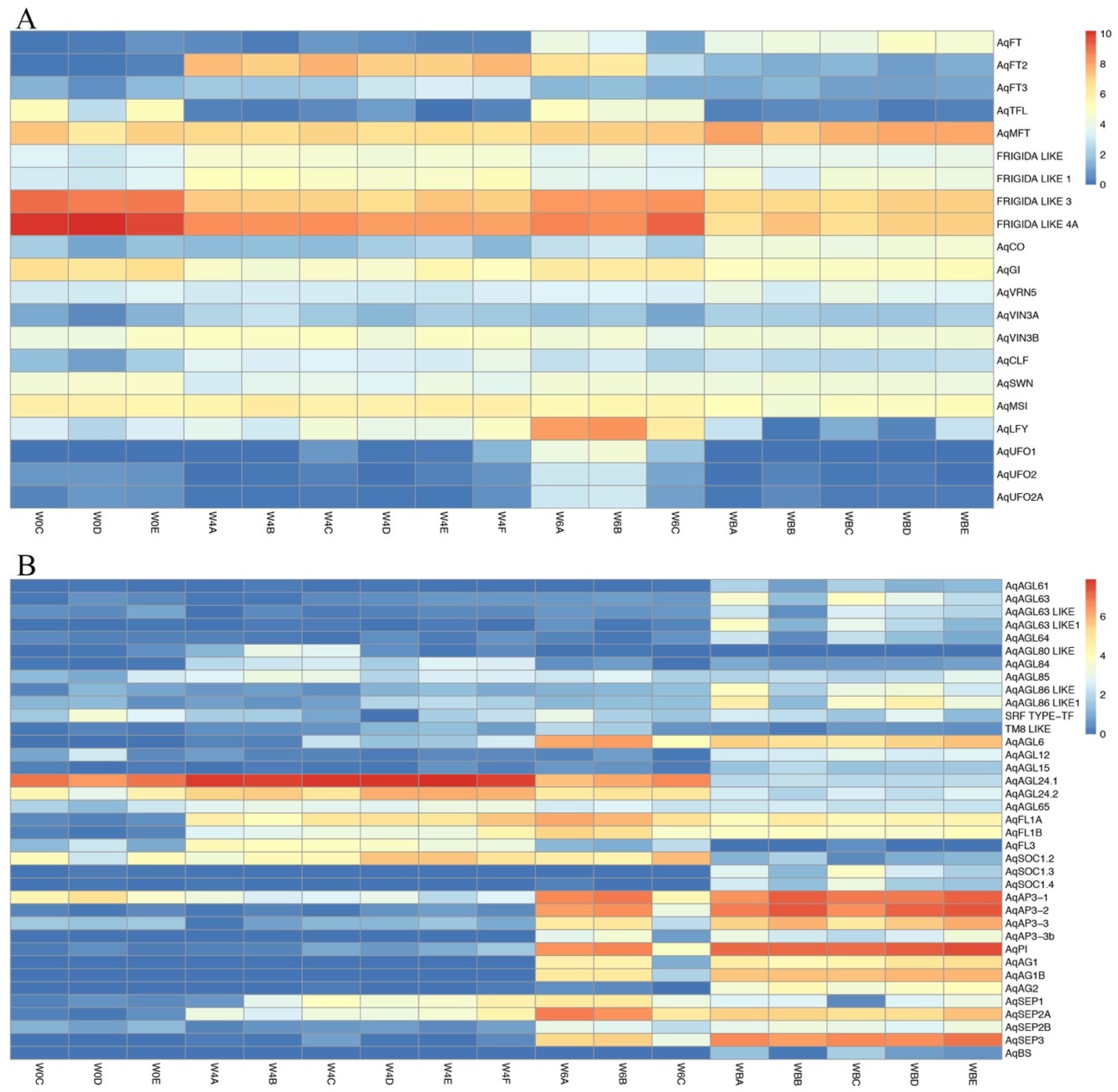
3.4.1. Flowering Time Genes
3.4.2. MADS-Box Genes
4. Conclusions
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Chanderbali, A.S.; Berger, B.A.; Howarth, D.G.; Soltis, D.E.; Soltis, P.S. Evolution of floral diversity: Genomics, genes and gamma. Philos. Trans. R. Soc. B Biol. Sci. 2017, 372, 20150509. [Google Scholar] [CrossRef] [PubMed]
- Kim, D.-H.; Doyle, M.R.; Sung, S.; Amasino, R.M. Vernalization: Winter and the Timing of Flowering in Plants. Annu. Rev. Cell Dev. Biol. 2009, 25, 277–299. [Google Scholar] [CrossRef] [PubMed]
- Simpson, G.G. Arabidopsis, the Rosetta Stone of Flowering Time? Science 2002, 296, 285–289. [Google Scholar] [CrossRef] [PubMed]
- Chouard, P. Vernalization and its Relations to Dormancy. Annu. Rev. Plant Physiol. 1960, 11, 191–238. [Google Scholar] [CrossRef]
- Boss, P.K. Multiple Pathways in the Decision to Flower: Enabling, Promoting, and Resetting. Plant Cell 2004, 16, S18–S31. [Google Scholar] [CrossRef] [PubMed]
- Michaels, S.D.; Amasino, R.M. Memories of winter: Vernalization and the competence to flower. Plant Cell Environ. 2000, 23, 1145–1153. [Google Scholar] [CrossRef]
- Michaels, S.D.; Amasino, R.M. FLOWERING LOCUS C Encodes a Novel MADS Domain Protein That Acts as a Repressor of Flowering. Plant Cell 1999, 11, 949–956. [Google Scholar] [CrossRef]
- Sheldon, C.C.; Rouse, D.T.; Finnegan, E.J.; Peacock, W.J.; Dennis, E.S. The molecular basis of vernalization: The central role of FLOWERING LOCUS C (FLC). Proc. Natl. Acad. Sci. USA 2000, 97, 3753–3758. [Google Scholar] [CrossRef]
- Sheldon, C.C.; Hills, M.J.; Lister, C.; Dean, C.; Dennis, E.S.; Peacock, W.J. Resetting of FLOWERING LOCUS C expression after epigenetic repression by vernalization. Proc. Natl. Acad. Sci. USA 2008, 105, 2214–2219. [Google Scholar] [CrossRef]
- Sheldon, C.C.; Conn, A.B.; Dennis, E.S.; Peacock, W.J. Different Regulatory Regions Are Required for the Vernalization-Induced Repression of FLOWERING LOCUS C and for the Epigenetic Maintenance of Repression. Plant Cell 2002, 14, 2527–2537. [Google Scholar] [CrossRef]
- Yu, H.; Xu, Y.; Tan, E.L.; Kumar, P.P. AGAMOUS-LIKE 24, a dosage-dependent mediator of the flowering signals. Proc. Natl. Acad. Sci. USA 2002, 99, 16336–16341. [Google Scholar] [CrossRef] [PubMed]
- Michaels, S.D.; Ditta, G.; Gustafson-Brown, C.; Pelaz, S.; Yanofsky, M.; Amasino, R.M. AGL24 acts as a promoter of flowering in Arabidopsis and is positively regulated by vernalization. Plant J. Cell Mol. Biol. 2003, 33, 867–874. [Google Scholar] [CrossRef] [PubMed]
- Goslin, K.; Zheng, B.; Serrano-Mislata, A.; Rae, L.; Ryan, P.T.; Kwaśniewska, K.; Thomson, B.; Ó’Maoiléidigh, D.S.; Madueño, F.; Wellmer, F.; et al. Transcription Factor Interplay between LEAFY and APETALA1/CAULIFLOWER during Floral Initiation. Plant Physiol. 2017, 174, 1097–1109. [Google Scholar] [CrossRef] [PubMed]
- Gazzani, S.; Gendall, A.R.; Lister, C.; Dean, C. Analysis of the Molecular Basis of Flowering Time Variation in Arabidopsis Accessions. Plant Physiol. 2003, 132, 1107–1114. [Google Scholar] [CrossRef] [PubMed]
- Bouché, F.; Woods, D.P.; Amasino, R.M. Winter Memory throughout the Plant Kingdom: Different Paths to Flowering. Plant Physiol. 2017, 173, 27–35. [Google Scholar] [CrossRef]
- Yan, L. The Wheat VRN2 Gene Is a Flowering Repressor Down-Regulated by Vernalization. Science 2004, 303, 1640–1644. [Google Scholar] [CrossRef]
- Pin, P.A.; Benlloch, R.; Bonnet, D.; Wremerth-Weich, E.; Kraft, T.; Gielen, J.J.L.; Nilsson, O. An Antagonistic Pair of FT Homologs Mediates the Control of Flowering Time in Sugar Beet. Science 2010, 330, 1397–1400. [Google Scholar] [CrossRef]
- Hodges, S.A.; Arnold, M.L. Columbines: A geographically widespread species flock. Proc. Natl. Acad. Sci. USA 1994, 91, 5129–5132. [Google Scholar] [CrossRef]
- Kramer, E.M. Aquilegia: A New Model for Plant Development, Ecology, and Evolution. Annu. Rev. Plant Biol. 2009, 60, 261–277. [Google Scholar] [CrossRef]
- Kramer, E.M.; Hodges, S.A. Aquilegia as a model system for the evolution and ecology of petals. Philos. Trans. R. Soc. B Biol. Sci. 2010, 365, 477–490. [Google Scholar] [CrossRef]
- Filiault, D.L.; Ballerini, E.S.; Mandáková, T.; Aköz, G.; Derieg, N.J.; Schmutz, J.; Jenkins, J.; Grimwood, J.; Shu, S.; Hayes, R.D.; et al. The Aquilegia genome provides insight into adaptive radiation and reveals an extraordinarily polymorphic chromosome with a unique history. eLife 2018, 7, e36426. [Google Scholar] [CrossRef] [PubMed]
- Gould, B.; Kramer, E.M. Virus-induced gene silencing as a tool for functional analyses in the emerging model plant Aquilegia (columbine, Ranunculaceae). Plant Methods 2007, 3, 6. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Guo, C.; Kong, H.; Kramer, E.M. Petal-specific subfunctionalization of an APETALA3 paralog in the Ranunculales and its implications for petal evolution. New Phytol. 2011, 191, 870–883. [Google Scholar] [CrossRef] [PubMed]
- Sharma, B.; Kramer, E. Sub- and neo-functionalization of APETALA3 paralogs have contributed to the evolution of novel floral organ identity in Aquilegia (columbine, Ranunculaceae). New Phytol. 2013, 197, 949–957. [Google Scholar] [CrossRef] [PubMed]
- Ballerini, E.S.; Kramer, E.M. Environmental and molecular analysis of the floral transition in the lower eudicot Aquilegia formosa. EvoDevo 2011, 2, 4. [Google Scholar] [CrossRef] [PubMed]
- Sharman, B.C. Tannic Acid and Iron Alum with Safranin and Orange G in Studies of the Shoot Apex. Stain Technol. 1943, 18, 105–111. [Google Scholar] [CrossRef]
- Conesa, A.; Madrigal, P.; Tarazona, S.; Gomez-Cabrero, D.; Cervera, A.; McPherson, A.; Szcześniak, M.W.; Gaffney, D.J.; Elo, L.L.; Zhang, X.; et al. A survey of best practices for RNA-seq data analysis. Genome Biol. 2016, 17, 13. [Google Scholar] [CrossRef]
- Kim, D.; Langmead, B.; Salzberg, S.L. HISAT: A fast spliced aligner with low memory requirements. Nat. Methods 2015, 12, 357. [Google Scholar] [CrossRef]
- Lawrence, M.; Huber, W.; Pagès, H.; Aboyoun, P.; Carlson, M.; Gentleman, R.; Morgan, M.T.; Carey, V.J. Software for Computing and Annotating Genomic Ranges. PLoS Comput. Biol. 2013, 9, e1003118. [Google Scholar] [CrossRef]
- Love, M.I.; Huber, W.; Anders, S. Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol. 2014, 15, 550. [Google Scholar] [CrossRef]
- Robinson, M.D.; McCarthy, D.J.; Smyth, G.K. edgeR: A Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics 2010, 26, 139–140. [Google Scholar] [CrossRef]
- Abu-Jamous, B.; Kelly, S. Clust: Automatic extraction of optimal co-expressed gene clusters from gene expression data. Genome Biol. 2018, 19, 172. [Google Scholar] [CrossRef]
- Conesa, A.; Götz, S. Blast2GO: A Comprehensive Suite for Functional Analysis in Plant Genomics. Int. J. Plant Genomics 2008, 2008, 1–12. [Google Scholar] [CrossRef]
- Yu, G.; Wang, L.-G.; Han, Y.; He, Q.-Y. clusterProfiler: An R Package for Comparing Biological Themes Among Gene Clusters. OMICS J. Integr. Biol. 2012, 16, 284–287. [Google Scholar] [CrossRef]
- Benjamini, Y.; Hochberg, Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. J. R. Stat. Soc. Ser. B Methodol. 1995, 57, 289–300. [Google Scholar] [CrossRef]
- Pabón-Mora, N.; Sharma, B.; Holappa, L.D.; Kramer, E.M.; Litt, A. The Aquilegia FRUITFULL-like genes play key roles in leaf morphogenesis and inflorescence development. Plant J. 2013, 74, 197–212. [Google Scholar] [CrossRef]
- Karlgren, A.; Gyllenstrand, N.; Källman, T.; Sundström, J.F.; Moore, D.; Lascoux, M.; Lagercrantz, U. Evolution of the PEBP Gene Family in Plants: Functional Diversification in Seed Plant Evolution. Plant Physiol. 2011, 156, 1967–1977. [Google Scholar] [CrossRef]
- Harig, L.; Beinecke, F.A.; Oltmanns, J.; Muth, J.; Müller, O.; Rüping, B.; Twyman, R.M.; Fischer, R.; Prüfer, D.; Noll, G.A. Proteins from the FLOWERING LOCUS T-like subclade of the PEBP family act antagonistically to regulate floral initiation in tobacco. Plant J. 2012, 72, 908–921. [Google Scholar] [CrossRef]
- Alvarez, J.; Guli, C.L.; Yu, X.-H.; Smyth, D.R. terminal flower: A gene affecting inflorescence development in Arabidopsis thaliana. Plant J. 1992, 2, 103–116. [Google Scholar] [CrossRef]
- Johanson, U. Molecular Analysis of FRIGIDA, a Major Determinant of Natural Variation in Arabidopsis Flowering Time. Science 2000, 290, 344–347. [Google Scholar] [CrossRef]
- Michaels, S.D.; Bezerra, I.C.; Amasino, R.M. FRIGIDA-related genes are required for the winter-annual habit in Arabidopsis. Proc. Natl. Acad. Sci. USA 2004, 101, 3281–3285. [Google Scholar] [CrossRef]
- Choi, K.; Kim, J.; Hwang, H.-J.; Kim, S.; Park, C.; Kim, S.Y.; Lee, I. The FRIGIDA Complex Activates. Transcription of FLC, a Strong Flowering Repressor in Arabidopsis, by Recruiting Chromatin Modification Factors. Plant Cell 2011, 23, 289–303. [Google Scholar] [CrossRef]
- Sawa, M.; Kay, S.A. GIGANTEA directly activates Flowering Locus T in Arabidopsis thaliana. Proc. Natl. Acad. Sci. USA 2011, 108, 11698–11703. [Google Scholar] [CrossRef]
- Suárez-López, P.; Wheatley, K.; Robson, F.; Onouchi, H.; Valverde, F.; Coupland, G. CONSTANS mediates between the circadian clock and the control of flowering in Arabidopsis. Nature 2001, 410, 1116–1120. [Google Scholar] [CrossRef]
- Yanovsky, M.J.; Kay, S.A. Molecular basis of seasonal time measurement in Arabidopsis. Nature 2002, 419, 308–312. [Google Scholar] [CrossRef]
- Mizoguchi, T.; Wright, L.; Fujiwara, S.; Cremer, F.; Lee, K.; Onouchi, H.; Mouradov, A.; Fowler, S.; Kamada, H.; Putterill, J.; et al. Distinct Roles of GIGANTEA in Promoting Flowering and Regulating Circadian Rhythms in Arabidopsis. Plant Cell 2005, 17, 2255–2270. [Google Scholar] [CrossRef]
- Hennig, L.; Derkacheva, M. Diversity of Polycomb group complexes in plants: Same rules, different players? Trends Genet. 2009, 25, 414–423. [Google Scholar] [CrossRef]
- Köhler, C.; Hennig, L. Regulation of cell identity by plant Polycomb and trithorax group proteins. Curr. Opin. Genet. Dev. 2010, 20, 541–547. [Google Scholar] [CrossRef]
- Gleason, E.J.; Kramer, E.M. Conserved roles for Polycomb Repressive Complex 2 in the regulation of lateral organ development in Aquilegia × coerulea ‘Origami’. BMC Plant Biol. 2013, 13, 185. [Google Scholar] [CrossRef]
- Gleason, E.J.; Kramer, E.M. Characterization of Aquilegia Polycomb Repressive Complex 2 homologs reveals absence of imprinting. Gene 2012, 507, 54–60. [Google Scholar] [CrossRef]
- Goodrich, J.; Puangsomlee, P.; Martin, M.; Long, D.; Meyerowitz, E.M.; Coupland, G. A Polycomb-group gene regulates homeotic gene expression in Arabidopsis. Nature 1997, 386, 44–51. [Google Scholar] [CrossRef]
- Jiang, D.; Wang, Y.; Wang, Y.; He, Y. Repression of FLOWERING LOCUS C and FLOWERING LOCUS T by the Arabidopsis Polycomb Repressive Complex 2 Components. PLoS ONE 2008, 3, e3404. [Google Scholar] [CrossRef]
- Lopez-Vernaza, M.; Yang, S.; Müller, R.; Thorpe, F.; de Leau, E.; Goodrich, J. Antagonistic Roles of SEPALLATA3, FT and FLC Genes as Targets of the Polycomb Group Gene CURLY LEAF. PLoS ONE 2012, 7, e30715. [Google Scholar] [CrossRef]
- Levin, J.Z.; Meyerowitz, E.M. UFO: An Arabidopsis gene involved in both floral meristem and floral organ development. Plant Cell 1995, 7, 529–548. [Google Scholar]
- Gramzow, L.; Theissen, G. A hitchhiker’s guide to the MADS world of plants. Genome Biol. 2010, 11, 214. [Google Scholar] [CrossRef]
- Sharma, B.; Kramer, E.M. The MADS-Box Gene Family of the Basal Eudicot and Hybrid Aquilegia coerulea ‘Origami’ (Ranunculaceae). Ann. Mo. Bot. Gard. 2014, 99, 313–322. [Google Scholar] [CrossRef][Green Version]
- Shore, P.; Sharrocks, A.D. The MADS-Box Family of Transcription Factors. Eur. J. Biochem. 1995, 229, 1–13. [Google Scholar] [CrossRef]
- Purugganan, M.D.; Rounsley, S.D.; Schmidt, R.J.; Yanofsky, M.F. Molecular evolution of flower development: Diversification of the plant MADS-box regulatory gene family. Genetics 1995, 140, 345–356. [Google Scholar]
- Kramer, E.M.; Holappa, L.; Gould, B.; Jaramillo, M.A.; Setnikov, D.; Santiago, P.M. Elaboration of B Gene Function to Include the Identity of Novel Floral Organs in the Lower Eudicot Aquilegia. Plant Cell 2007, 19, 750–766. [Google Scholar] [CrossRef]
- Sharma, B.; Kramer, E.M. Aquilegia B gene homologs promote petaloidy of the sepals and maintenance of the C domain boundary. EvoDevo 2017, 8, 22. [Google Scholar] [CrossRef]
- Zahn, L.M.; Kong, H.; Leebens-Mack, J.H.; Kim, S.; Soltis, P.S.; Landherr, L.L.; Soltis, D.E.; dePamphilis, C.W.; Ma, H. The Evolution of the SEPALLATA Subfamily of MADS-Box Genes: A Preangiosperm Origin with Multiple Duplications Throughout Angiosperm History. Genetics 2005, 169, 2209–2223. [Google Scholar] [CrossRef]
- Rijpkema, A.S.; Zethof, J.; Gerats, T.; Vandenbussche, M. The petunia AGL6 gene has a SEPALLATA-like function in floral patterning. Plant J. 2009, 60, 1–9. [Google Scholar] [CrossRef]
- Wang, P.; Liao, H.; Zhang, W.; Yu, X.; Zhang, R.; Shan, H.; Duan, X.; Yao, X.; Kong, H. Flexibility in the structure of spiral flowers and its underlying mechanisms. Nat. Plants 2016, 2, 15188. [Google Scholar] [CrossRef]
- Bemer, M.; Wolters-Arts, M.; Grossniklaus, U.; Angenent, G.C. The MADS Domain Protein DIANA Acts Together with AGAMOUS-LIKE80 to Specify the Central Cell in Arabidopsis Ovules. Plant Cell 2008, 20, 2088–2101. [Google Scholar] [CrossRef]
- Steffen, J.G.; Kang, I.-H.; Portereiko, M.F.; Lloyd, A.; Drews, G.N. AGL61 Interacts with AGL80 and Is Required for Central Cell Development in Arabidopsis. Plant Physiol. 2008, 148, 259–268. [Google Scholar] [CrossRef]







{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Pairwise Comparisons | GO Category | GO Term | Description |
|---|---|---|---|
| W0-W4 up | BP | GO:0070647 | Protein modification by small protein conjugation or removal |
| W0-W6 up | BP | GO:0010033 | Response to organic substance |
| W0-WB up | BP | GO:0070647 | Protein modification by small protein conjugation or removal |
| W4-W6 up | BP | GO:0055086 | Nucleobase-containing small molecule metabolic process |
| W4-WB up | BP | GO:0034660 | ncRNA metabolic process |
| W6-WB up | BP | GO:0048856 | Anatomical structure development |
| W0-W4 down | BP | GO:0055086 | Nucleobase-containing small molecule metabolic process |
| W0-W6 down | BP | GO:0006091 | Generation of precursor metabolites and energy |
| W0-WB down | BP | GO:0006091 | Generation of precursor metabolites and energy |
| W4-W6 down | BP | GO:0006091 | Generation of precursor metabolites and energy |
| W4-WB down | BP | GO:0006812 | Cation transport |
| W6-WB down | BP | GO:0006091 | Generation of precursor metabolites and energy |
| W0-W4 up | MF | GO:0003700 | DNA-binding transcription factor activity |
| W0-W6 up | MF | GO:0003700 | DNA-binding transcription factor activity |
| W0-WB up | MF | GO:0003700 | DNA-binding transcription factor activity |
| W4-W6 up | MF | GO:0005198 | Structural molecule activity |
| W4-WB up | MF | GO:0005198 | Structural molecule activity |
| W6-WB up | MF | GO:0003700 | DNA-binding transcription factor activity |
| W0-W4 down | MF | GO:0005198 | Structural molecule activity |
| W0-W6 down | MF | GO:0005198 | Structural molecule activity |
| W0-WB down | MF | GO:0005198 | Structural molecule activity |
| W4-W6 down | MF | GO:0003700 | DNA-binding transcription factor activity |
| W4-WB down | MF | GO:0015075 | Ion transmembrane transporter activity |
| W6-WB down | MF | GO:0015075 | Ion transmembrane transporter activity |
| W0-W4 up | CC | GO:0031974 | Membrane-enclosed lumen |
| GO:0043233 | Organelle lumen | ||
| GO:0070013 | Intracellular organelle lumen | ||
| W0-W6 up | CC | GO:0005829 | Cytosol |
| W0-WB up | CC | GO:0031974 | Membrane-enclosed lumen |
| GO:0043233 | Organelle lumen | ||
| GO:0070013 | Intracellular organelle lumen | ||
| W4-W6 up | CC | GO:0005840 | Ribosome |
| W4-WB up | CC | GO:0005840 | Ribosome |
| W6-WB up | CC | GO:0031974 | Membrane-enclosed lumen |
| GO:0043233 | Organelle lumen | ||
| GO:0070013 | Intracellular organelle lumen | ||
| W0-W4 down | CC | GO:0005840 | Ribosome |
| W0-W6 down | CC | GO:0005840 | Ribosome |
| W0-WB down | CC | GO:0098796 | Membrane protein complex |
| W4-W6 down | CC | GO:0009579 | Thylakoid |
| GO:0044434 | Chloroplast part | ||
| GO:0044435 | Plastid part | ||
| W4-WB down | CC | GO:0044435 | Plastid part |
| W6-WB down | CC | GO:0098796 | Membrane protein complex |
| Flowering Time Genes | Cluster Number |
|---|---|
| AqTFL | C0 |
| FRIGIDA LIKE 3 | C0 |
| FRIGIDA LIKE 4A | C0 |
| AqGI | C0 |
| AqVIN3A | C1 |
| AqCLF | C1 |
| AqCO | C2 |
| AqFT3 | C3 |
| AqMSI | C3 |
| AqFT2 | C5 |
| AqLFY | C6 |
| AqUFO2 | C6 |
| AqUFO2A | C6 |
| AqSWN | C7 |
| AqFT | C8 |
| AqMFT | C8 |
| FRIGIDA LIKE | C9 |
| FRIGIDA LIKE 1 | C9 |
| AqVIN3B | C9 |
| AqVRN5 | C10 |
| AqUFO1 | C11 |
| MADS-Box TFs | Cluster Number |
|---|---|
| AqAGL61 | C2 |
| AqAGL63 | C2 |
| AaAGL63 LIKE | C2 |
| AqAGL64 | C2 |
| AqAGL86 LIKE | C2 |
| AqAGL86 LIKE A | C2 |
| AqAGl12 | C2 |
| AqSOC1.3 | C2 |
| AqSOC 1.4 | C2 |
| AqAG1 | C2 |
| AqBS | C2 |
| AqAGL24.1 | C3 |
| AqAGL80 LIKE | C3 |
| AqAG2 | C4 |
| AqAGL84 | C5 |
| AqAGL85 | C5 |
| AqAGL24.2 | C5 |
| AqAGL65 | C5 |
| TM8 LIKE | C6 |
| AqSOC1.2 | C6 |
| AqAGL15 | C8 |
| AqAP3-1 | C8 |
| AqAP3-2 | C8 |
| AqAP3-3 | C8 |
| AqAG1B | C8 |
| AqSEP2B | C8 |
| AqSEP3 | C8 |
| AqAGL63 LIKE A | C8 |
| AqFL3 | C9 |
| SRF TYPE-TF | C10 |
| AqAGL6 | C11 |
| AqFL1A | C11 |
| AqFL1B | C11 |
| AqAP3-3b | C11 |
| AqPI | C11 |
| AqSEP1 | C11 |
| AqSEP2A | C11 |
© 2019 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sharma, B.; Batz, T.A.; Kaundal, R.; Kramer, E.M.; Sanders, U.R.; Mellano, V.J.; Duhan, N.; Larson, R.B. Developmental and Molecular Changes Underlying the Vernalization-Induced Transition to Flowering in Aquilegia coerulea (James). Genes 2019, 10, 734. https://doi.org/10.3390/genes10100734
Sharma B, Batz TA, Kaundal R, Kramer EM, Sanders UR, Mellano VJ, Duhan N, Larson RB. Developmental and Molecular Changes Underlying the Vernalization-Induced Transition to Flowering in Aquilegia coerulea (James). Genes. 2019; 10(10):734. https://doi.org/10.3390/genes10100734
Chicago/Turabian StyleSharma, Bharti, Timothy A. Batz, Rakesh Kaundal, Elena M. Kramer, Uriah R. Sanders, Valerie J. Mellano, Naveen Duhan, and Rousselene B. Larson. 2019. "Developmental and Molecular Changes Underlying the Vernalization-Induced Transition to Flowering in Aquilegia coerulea (James)" Genes 10, no. 10: 734. https://doi.org/10.3390/genes10100734
APA StyleSharma, B., Batz, T. A., Kaundal, R., Kramer, E. M., Sanders, U. R., Mellano, V. J., Duhan, N., & Larson, R. B. (2019). Developmental and Molecular Changes Underlying the Vernalization-Induced Transition to Flowering in Aquilegia coerulea (James). Genes, 10(10), 734. https://doi.org/10.3390/genes10100734