Osteoblast-Osteoclast Communication and Bone Homeostasis


Abstract
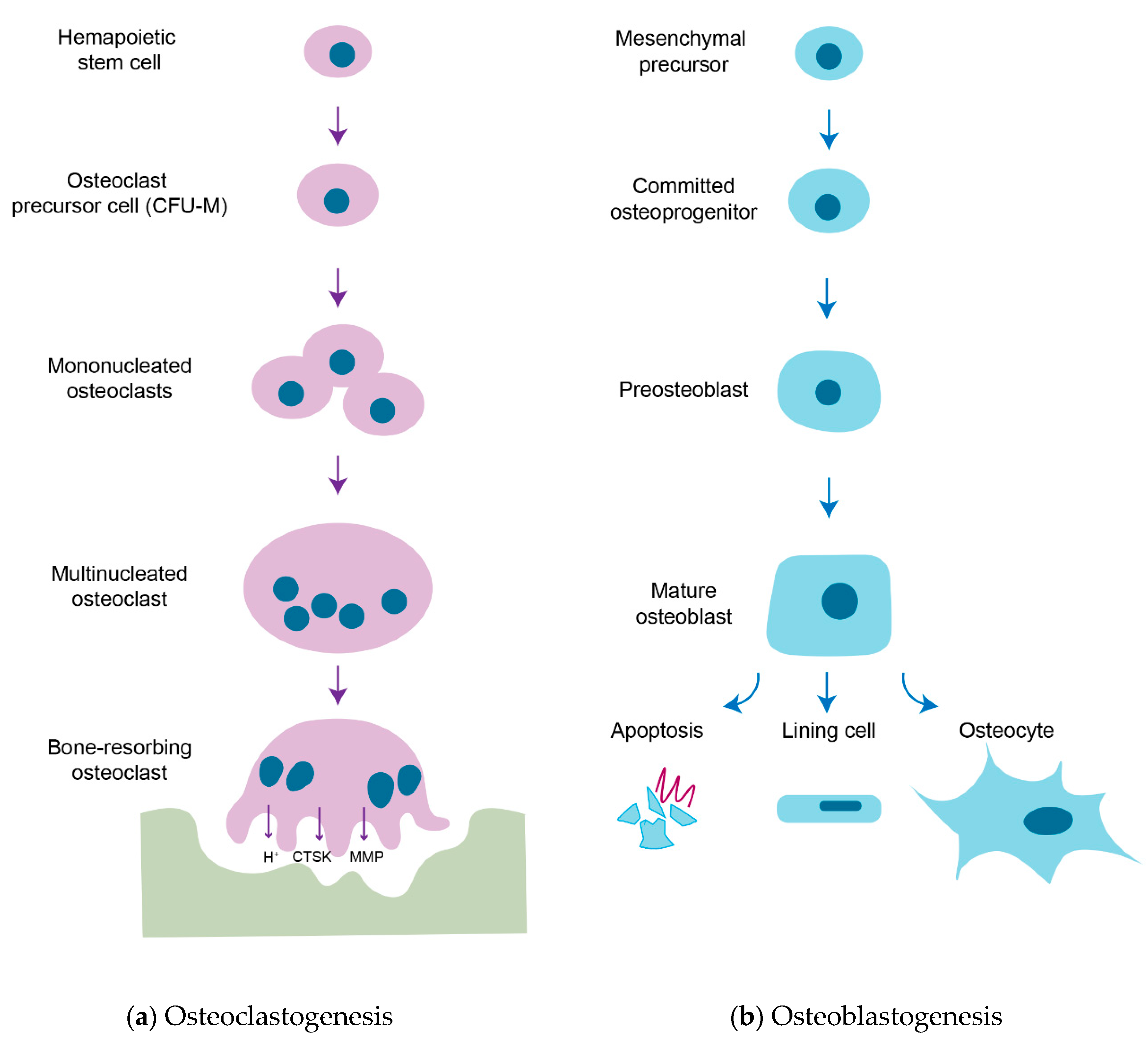
1. Introduction
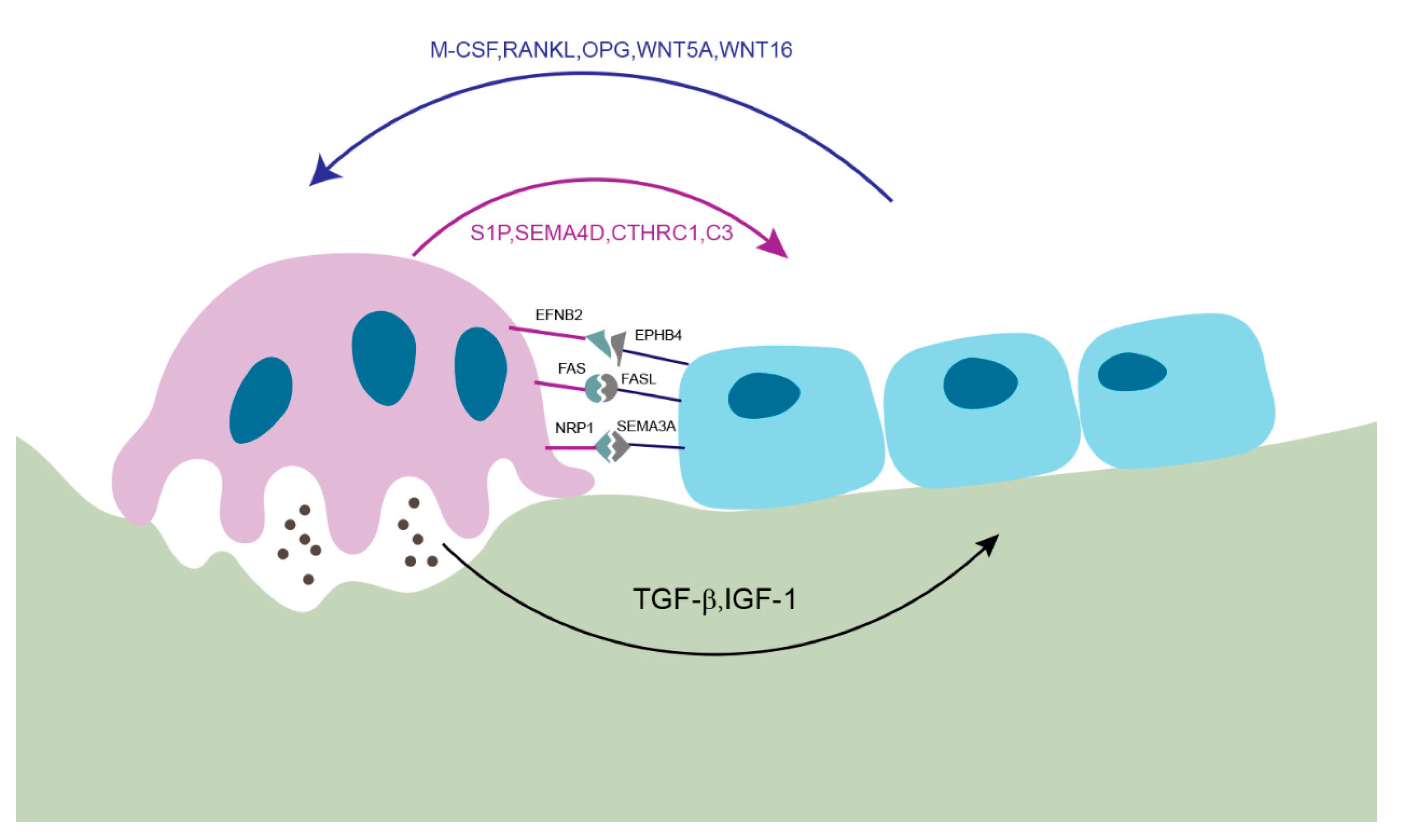
2. Membrane-Bound Mediators of Cell-to-Cell Communication
2.1. EFNB2 (Ephrin B2)-EPHB4
2.2. FAS Ligand (FASL)-FAS
2.3. Semaphorin 3A (SEMA3A)-NRP1
3. Soluble Factors Released from Osteoblasts
3.1. Macrophage Colony-Stimulating Factor (M-CSF)
3.2. Receptor Activator of NF-κB (Nuclear Factor-Kappa B) Ligand (RANKL)
3.3. Osteoprotegerin (OPG)
3.4. WNT5A
3.5. WNT16
4. Soluble Factors Released from Osteoclasts
4.1. Sphingosine 1 Phosphate (S1P)
4.2. Semaphorin 4D (SEMA4D)
4.3. Collagen Triple Helix Repeat Containing 1 (CTHRC1)
4.4. Complement Component C (C3)
4.5. Other Factors
5. Matrix-Derived Coupling Factors by Bone Resorption
5.1. Transforming Growth Factor β1 (TGF-β1)
5.2. Insulin-Like Growth Factor Type 1 (IGF-1)
6. Impact of Therapeutic Agents on Osteoblast-Osteoclast Interactions
7. Conclusions and Future Perspectives
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| RUNX2 | Runt-related transcription factor 2 |
| M-CSF | Macrophage colony-stimulating factor |
| CFU-M | Macrophage colony-forming units |
| RANKL | Receptor activator of NF-κB (nuclear factor-kappa B) ligand |
| RANK | Receptor activator of NF-κB (nuclear factor-kappa B) |
| CTSK | Cathepsin K |
| MMP | Matrix metalloproteinases |
| EFNB2 | Ephrin B2 |
| FASL | Fas ligand |
| SEMA3A | Semaphorin 3A |
| NRP1 | Neuropilin-1 |
| TGF-β1 | Transforming growth factor β1 |
| IGF-1 | Insulin-like growth factor type 1 |
| OPG | Osteoprotegerin |
| S1P | Sphingosine 1 phosphate |
| SEMA4D | Semaphorin 4D |
| CTHRC1 | Collagen triple helix repeat containing 1 |
| C3 | Complement component c |
| PTH | Parathyroid hormone |
| SOST | Sclerostin |
References
- Frost, H.M. Dynamics of bone remodeling. Bone Biodynam. 1964, 14, 315–333. [Google Scholar]
- Feng, X.; McDonald, J.M. Disorders of bone remodeling. Annu. Rev. Pathol. 2011, 6, 121–145. [Google Scholar] [CrossRef] [PubMed]
- Clarke, B. Normal bone anatomy and physiology. Clin. J. Am. 2008, 3, S131–S139. [Google Scholar] [CrossRef] [PubMed]
- Gothlin, G.; Ericsson, J.L. The osteoclast: Review of ultrastructure, origin, and structure-function relationship. Clin. Orthop. Relat. Res. 1976, 1, 201–231. [Google Scholar]
- Feng, X.; Teitelbaum, S.L. Osteoclasts: New Insights. Bone Res. 2013, 1, 11–26. [Google Scholar] [CrossRef]
- Boyle, W.J.; Simonet, W.S.; Lacey, D.L. Osteoclast differentiation and activation. Nature 2003, 423, 337–342. [Google Scholar] [CrossRef]
- Asagiri, M.; Takayanagi, H. The molecular understanding of osteoclast differentiation. Bone 2007, 40, 251–264. [Google Scholar] [CrossRef]
- Zhu, L.; Tang, Y.; Li, X.Y.; Keller, E.T.; Yang, J.; Cho, J.S.; Feinberg, T.Y.; Weiss, S.J. Osteoclast-mediated bone resorption is controlled by a compensatory network of secreted and membrane-tethered metalloproteinases. Sci. Transl. Med. 2020, 12. [Google Scholar] [CrossRef]
- Teitelbaum, S.L. Bone resorption by osteoclasts. Science 2000, 289, 1504–1508. [Google Scholar] [CrossRef]
- Charles, J.F.; Aliprantis, A.O. Osteoclasts: More than ‘bone eaters’. Trends Mol. Med. 2014, 20, 449–459. [Google Scholar] [CrossRef]
- Karsenty, G.; Kronenberg, H.M.; Settembre, C. Genetic control of bone formation. Annu. Rev. Cell Dev. Biol. 2009, 25, 629–648. [Google Scholar] [CrossRef] [PubMed]
- Long, F. Building strong bones: Molecular regulation of the osteoblast lineage. Nat. Rev. Mol. Cell Biol. 2011, 13, 27–38. [Google Scholar] [CrossRef] [PubMed]
- Harada, S.; Rodan, G.A. Control of osteoblast function and regulation of bone mass. Nature 2003, 423, 349–355. [Google Scholar] [CrossRef] [PubMed]
- Plotkin, L.I.; Bellido, T. Osteocytic signalling pathways as therapeutic targets for bone fragility. Nat. Rev. Endocrinol. 2016, 12, 593–605. [Google Scholar] [CrossRef] [PubMed]
- Blair, H.C.; Larrouture, Q.C.; Li, Y.; Lin, H.; Beer-Stoltz, D.; Liu, L.; Tuan, R.S.; Robinson, L.J.; Schlesinger, P.H.; Nelson, D.J. Osteoblast Differentiation and Bone Matrix Formation In Vivo and In Vitro. Tissue Eng. Part. B Rev. 2017, 23, 268–280. [Google Scholar] [CrossRef]
- Hattner, R.; Epker, B.N.; Frost, H.M. Suggested sequential mode of control of changes in cell behaviour in adult bone remodelling. Nature 1965, 206, 489–490. [Google Scholar] [CrossRef]
- Langdahl, B.; Ferrari, S.; Dempster, D.W. Bone modeling and remodeling: Potential as therapeutic targets for the treatment of osteoporosis. Ther. Adv. Musculoskelet. Dis. 2016, 8, 225–235. [Google Scholar] [CrossRef]
- Crockett, J.C.; Rogers, M.J.; Coxon, F.P.; Hocking, L.J.; Helfrich, M.H. Bone remodelling at a glance. J. Cell Sci. 2011, 124, 991–998. [Google Scholar] [CrossRef]
- Parfitt, A.M. The coupling of bone formation to bone resorption: A critical analysis of the concept and of its relevance to the pathogenesis of osteoporosis. Metab. Bone Dis. Relat. Res. 1982, 4, 1–6. [Google Scholar] [CrossRef]
- Khosla, S.; Oursler, M.J.; Monroe, D.G. Estrogen and the skeleton. Trends Endocrinol. Metab. 2012, 23, 576–581. [Google Scholar] [CrossRef]
- Sobacchi, C.; Schulz, A.; Coxon, F.P.; Villa, A.; Helfrich, M.H. Osteopetrosis: Genetics, treatment and new insights into osteoclast function. Nat. Rev. Endocrinol. 2013, 9, 522–536. [Google Scholar] [CrossRef] [PubMed]
- Mirza, F.; Canalis, E. Management of endocrine disease: Secondary osteoporosis: Pathophysiology and management. Eur. J. Endocrinol. 2015, 173, R131–R151. [Google Scholar] [CrossRef] [PubMed]
- Andersen, T.L.; Abdelgawad, M.E.; Kristensen, H.B.; Hauge, E.M.; Rolighed, L.; Bollerslev, J.; Kjaersgaard-Andersen, P.; Delaisse, J.M. Understanding coupling between bone resorption and formation: Are reversal cells the missing link? Am. J. Pathol. 2013, 183, 235–246. [Google Scholar] [CrossRef] [PubMed]
- Baron, R. Importance of the intermediate phases between resorption and formation in the measurement and understanding of the bone remodeling sequence. In Bone Histomorphometry; Lab Armour Montagu: Paris, France, 1977; pp. 179–183. [Google Scholar]
- Everts, V.; Delaisse, J.M.; Korper, W.; Jansen, D.C.; Tigchelaar-Gutter, W.; Saftig, P.; Beertsen, W. The bone lining cell: Its role in cleaning Howship’s lacunae and initiating bone formation. J. Bone Miner. Res. 2002, 17, 77–90. [Google Scholar] [CrossRef]
- Zhao, C.; Irie, N.; Takada, Y.; Shimoda, K.; Miyamoto, T.; Nishiwaki, T.; Suda, T.; Matsuo, K. Bidirectional ephrinB2-EphB4 signaling controls bone homeostasis. Cell Metab. 2006, 4, 111–121. [Google Scholar] [CrossRef]
- Tonna, S.; Poulton, I.J.; Taykar, F.; Ho, P.W.; Tonkin, B.; Crimeen-Irwin, B.; Tatarczuch, L.; McGregor, N.E.; Mackie, E.J.; Martin, T.J.; et al. Chondrocytic ephrin B2 promotes cartilage destruction by osteoclasts in endochondral ossification. Development 2016, 143, 648–657. [Google Scholar] [CrossRef]
- Takyar, F.M.; Tonna, S.; Ho, P.W.; Crimeen-Irwin, B.; Baker, E.K.; Martin, T.J.; Sims, N.A. EphrinB2/EphB4 inhibition in the osteoblast lineage modifies the anabolic response to parathyroid hormone. J. Bone Miner. Res. 2013, 28, 912–925. [Google Scholar] [CrossRef]
- Kullander, K.; Klein, R. Mechanisms and functions of Eph and ephrin signalling. Nat. Rev. Mol. Cell Biol. 2002, 3, 475–486. [Google Scholar] [CrossRef]
- Tonna, S.; Takyar, F.M.; Vrahnas, C.; Crimeen-Irwin, B.; Ho, P.W.; Poulton, I.J.; Brennan, H.J.; McGregor, N.E.; Allan, E.H.; Nguyen, H.; et al. EphrinB2 signaling in osteoblasts promotes bone mineralization by preventing apoptosis. FASEB J. 2014, 28, 4482–4496. [Google Scholar] [CrossRef]
- Green, D.R.; Ferguson, T.A. The role of Fas ligand in immune privilege. Nat. Rev. Mol. Cell Biol. 2001, 2, 917–924. [Google Scholar] [CrossRef]
- Wajant, H. The Fas signaling pathway: More than a paradigm. Science 2002, 296, 1635–1636. [Google Scholar] [CrossRef]
- Sambrook, P.; Cooper, C. Osteoporosis. Lancet 2006, 367, 2010–2018. [Google Scholar] [CrossRef]
- Kameda, T.; Mano, H.; Yuasa, T.; Mori, Y.; Miyazawa, K.; Shiokawa, M.; Nakamaru, Y.; Hiroi, E.; Hiura, K.; Kameda, A.; et al. Estrogen inhibits bone resorption by directly inducing apoptosis of the bone-resorbing osteoclasts. J. Exp. Med. 1997, 186, 489–495. [Google Scholar] [CrossRef] [PubMed]
- Krum, S.A.; Miranda-Carboni, G.A.; Hauschka, P.V.; Carroll, J.S.; Lane, T.F.; Freedman, L.P.; Brown, M. Estrogen protects bone by inducing Fas ligand in osteoblasts to regulate osteoclast survival. EMBO J. 2008, 27, 535–545. [Google Scholar] [CrossRef] [PubMed]
- Wang, L.; Liu, S.; Zhao, Y.; Liu, D.; Liu, Y.; Chen, C.; Karray, S.; Shi, S.; Jin, Y. Osteoblast-induced osteoclast apoptosis by fas ligand/FAS pathway is required for maintenance of bone mass. Cell Death Differ. 2015, 22, 1654–1664. [Google Scholar] [CrossRef] [PubMed]
- Kolodkin, A.L.; Matthes, D.J.; Goodman, C.S. The semaphorin genes encode a family of transmembrane and secreted growth cone guidance molecules. Cell 1993, 75, 1389–1399. [Google Scholar] [CrossRef]
- Hayashi, M.; Nakashima, T.; Taniguchi, M.; Kodama, T.; Kumanogoh, A.; Takayanagi, H. Osteoprotection by semaphorin 3A. Nature 2012, 485, 69–74. [Google Scholar] [CrossRef]
- Negishi-Koga, T.; Shinohara, M.; Komatsu, N.; Bito, H.; Kodama, T.; Friedel, R.H.; Takayanagi, H. Suppression of bone formation by osteoclastic expression of semaphorin 4D. Nat. Med. 2011, 17, 1473–1480. [Google Scholar] [CrossRef]
- Xu, R. Semaphorin 3A: A new player in bone remodeling. Cell Adh. Migr. 2014, 8, 5–10. [Google Scholar] [CrossRef]
- Behar, O.; Golden, J.A.; Mashimo, H.; Schoen, F.J.; Fishman, M.C. Semaphorin III is needed for normal patterning and growth of nerves, bones and heart. Nature 1996, 383, 525–528. [Google Scholar] [CrossRef]
- Fukuda, T.; Takeda, S.; Xu, R.; Ochi, H.; Sunamura, S.; Sato, T.; Shibata, S.; Yoshida, Y.; Gu, Z.; Kimura, A.; et al. Sema3A regulates bone-mass accrual through sensory innervations. Nature 2013, 497, 490–493. [Google Scholar] [CrossRef] [PubMed]
- Fixe, P.; Praloran, V. M-CSF: Haematopoietic growth factor or inflammatory cytokine? Cytokine 1998, 10, 32–37. [Google Scholar] [CrossRef] [PubMed]
- Stanley, E.R.; Cifone, M.; Heard, P.M.; Defendi, V. Factors regulating macrophage production and growth: Identity of colony-stimulating factor and macrophage growth factor. J. Exp. Med. 1976, 143, 631–647. [Google Scholar] [CrossRef]
- Lacey, D.L.; Erdmann, J.M.; Shima, M.; Kling, S.; Matayoshi, A.; Ohara, J.; Perkins, S.L. Interleukin 4 enhances osteoblast macrophage colony-stimulating factor, but not interleukin 6, production. Calcif. Tissue Int. 1994, 55, 21–28. [Google Scholar] [CrossRef] [PubMed]
- Lacey, D.L.; Timms, E.; Tan, H.L.; Kelley, M.J.; Dunstan, C.R.; Burgess, T.; Elliott, R.; Colombero, A.; Elliott, G.; Scully, S.; et al. Osteoprotegerin ligand is a cytokine that regulates osteoclast differentiation and activation. Cell 1998, 93, 165–176. [Google Scholar] [CrossRef]
- Croft, M.; Benedict, C.A.; Ware, C.F. Clinical targeting of the TNF and TNFR superfamilies. Nat. Rev. Drug Discov. 2013, 12, 147–168. [Google Scholar] [CrossRef]
- Kong, Y.Y.; Yoshida, H.; Sarosi, I.; Tan, H.L.; Timms, E.; Capparelli, C.; Morony, S.; Oliveira-dos-Santos, A.J.; Van, G.; Itie, A.; et al. OPGL is a key regulator of osteoclastogenesis, lymphocyte development and lymph-node organogenesis. Nature 1999, 397, 315–323. [Google Scholar] [CrossRef]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Yamaguchi, K.; Kinosaki, M.; Mochizuki, S.; Tomoyasu, A.; Yano, K.; Goto, M.; Murakami, A.; et al. Osteoclast differentiation factor is a ligand for osteoprotegerin/osteoclastogenesis-inhibitory factor and is identical to TRANCE/RANKL. Proc. Natl. Acad. Sci. USA 1998, 95, 3597–3602. [Google Scholar] [CrossRef]
- Xiong, J.; Onal, M.; Jilka, R.L.; Weinstein, R.S.; Manolagas, S.C.; O’Brien, C.A. Matrix-embedded cells control osteoclast formation. Nat. Med. 2011, 17, 1235–1241. [Google Scholar] [CrossRef]
- Li, J.; Sarosi, I.; Yan, X.Q.; Morony, S.; Capparelli, C.; Tan, H.L.; McCabe, S.; Elliott, R.; Scully, S.; Van, G.; et al. RANK is the intrinsic hematopoietic cell surface receptor that controls osteoclastogenesis and regulation of bone mass and calcium metabolism. Proc. Natl. Acad. Sci. USA 2000, 97, 1566–1571. [Google Scholar] [CrossRef]
- Mizuno, A.; Kanno, T.; Hoshi, M.; Shibata, O.; Yano, K.; Fujise, N.; Kinosaki, M.; Yamaguchi, K.; Tsuda, E.; Murakami, A.; et al. Transgenic mice overexpressing soluble osteoclast differentiation factor (sODF) exhibit severe osteoporosis. J. Bone Miner. Metab. 2002, 20, 337–344. [Google Scholar] [CrossRef] [PubMed]
- Simonet, W.S.; Lacey, D.L.; Dunstan, C.R.; Kelley, M.; Chang, M.S.; Luthy, R.; Nguyen, H.Q.; Wooden, S.; Bennett, L.; Boone, T.; et al. Osteoprotegerin: A novel secreted protein involved in the regulation of bone density. Cell 1997, 89, 309–319. [Google Scholar] [CrossRef]
- Yasuda, H.; Shima, N.; Nakagawa, N.; Mochizuki, S.I.; Yano, K.; Fujise, N.; Sato, Y.; Goto, M.; Yamaguchi, K.; Kuriyama, M.; et al. Identity of osteoclastogenesis inhibitory factor (OCIF) and osteoprotegerin (OPG): A mechanism by which OPG/OCIF inhibits osteoclastogenesis in vitro. Endocrinology 1998, 139, 1329–1337. [Google Scholar] [CrossRef]
- Li, Y.; Toraldo, G.; Li, A.; Yang, X.; Zhang, H.; Qian, W.P.; Weitzmann, M.N. B cells and T cells are critical for the preservation of bone homeostasis and attainment of peak bone mass in vivo. Blood 2007, 109, 3839–3848. [Google Scholar] [CrossRef] [PubMed]
- Bucay, N.; Sarosi, I.; Dunstan, C.R.; Morony, S.; Tarpley, J.; Capparelli, C.; Scully, S.; Tan, H.L.; Xu, W.; Lacey, D.L.; et al. osteoprotegerin-deficient mice develop early onset osteoporosis and arterial calcification. Genes Dev. 1998, 12, 1260–1268. [Google Scholar] [CrossRef] [PubMed]
- Theoleyre, S.; Wittrant, Y.; Tat, S.K.; Fortun, Y.; Redini, F.; Heymann, D. The molecular triad OPG/RANK/RANKL: Involvement in the orchestration of pathophysiological bone remodeling. Cytokine Growth Factor Rev. 2004, 15, 457–475. [Google Scholar] [CrossRef]
- Logan, C.Y.; Nusse, R. The Wnt signaling pathway in development and disease. Annu Rev. Cell Dev. Biol. 2004, 20, 781–810. [Google Scholar] [CrossRef]
- Maeda, K.; Kobayashi, Y.; Udagawa, N.; Uehara, S.; Ishihara, A.; Mizoguchi, T.; Kikuchi, Y.; Takada, I.; Kato, S.; Kani, S.; et al. Wnt5a-Ror2 signaling between osteoblast-lineage cells and osteoclast precursors enhances osteoclastogenesis. Nat. Med. 2012, 18, 405–412. [Google Scholar] [CrossRef]
- Medina-Gomez, C.; Kemp, J.P.; Estrada, K.; Eriksson, J.; Liu, J.; Reppe, S.; Evans, D.M.; Heppe, D.H.; Vandenput, L.; Herrera, L.; et al. Meta-analysis of genome-wide scans for total body BMD in children and adults reveals allelic heterogeneity and age-specific effects at the WNT16 locus. PLoS Genet. 2012, 8, e1002718. [Google Scholar] [CrossRef]
- Estrada, K.; Styrkarsdottir, U.; Evangelou, E.; Hsu, Y.H.; Duncan, E.L.; Ntzani, E.E.; Oei, L.; Albagha, O.M.; Amin, N.; Kemp, J.P.; et al. Genome-wide meta-analysis identifies 56 bone mineral density loci and reveals 14 loci associated with risk of fracture. Nat. Genet. 2012, 44, 491–501. [Google Scholar] [CrossRef]
- Koller, D.L.; Zheng, H.F.; Karasik, D.; Yerges-Armstrong, L.; Liu, C.T.; McGuigan, F.; Kemp, J.P.; Giroux, S.; Lai, D.; Edenberg, H.J.; et al. Meta-analysis of genome-wide studies identifies WNT16 and ESR1 SNPs associated with bone mineral density in premenopausal women. J. Bone Miner. Res. 2013, 28, 547–558. [Google Scholar] [CrossRef] [PubMed]
- Moverare-Skrtic, S.; Henning, P.; Liu, X.; Nagano, K.; Saito, H.; Borjesson, A.E.; Sjogren, K.; Windahl, S.H.; Farman, H.; Kindlund, B.; et al. Osteoblast-derived WNT16 represses osteoclastogenesis and prevents cortical bone fragility fractures. Nat. Med. 2014, 20, 1279–1288. [Google Scholar] [CrossRef] [PubMed]
- Ishii, M.; Egen, J.G.; Klauschen, F.; Meier-Schellersheim, M.; Saeki, Y.; Vacher, J.; Proia, R.L.; Germain, R.N. Sphingosine-1-phosphate mobilizes osteoclast precursors and regulates bone homeostasis. Nature 2009, 458, 524–528. [Google Scholar] [CrossRef] [PubMed]
- Pederson, L.; Ruan, M.; Westendorf, J.J.; Khosla, S.; Oursler, M.J. Regulation of bone formation by osteoclasts involves Wnt/BMP signaling and the chemokine sphingosine-1-phosphate. Proc. Natl. Acad. Sci. USA 2008, 105, 20764–20769. [Google Scholar] [CrossRef]
- Ryu, J.; Kim, H.J.; Chang, E.J.; Huang, H.; Banno, Y.; Kim, H.H. Sphingosine 1-phosphate as a regulator of osteoclast differentiation and osteoclast-osteoblast coupling. EMBO J. 2006, 25, 5840–5851. [Google Scholar] [CrossRef]
- Lotinun, S.; Kiviranta, R.; Matsubara, T.; Alzate, J.A.; Neff, L.; Luth, A.; Koskivirta, I.; Kleuser, B.; Vacher, J.; Vuorio, E.; et al. Osteoclast-specific cathepsin K deletion stimulates S1P-dependent bone formation. J. Clin. Investig. 2013, 123, 666–681. [Google Scholar] [CrossRef]
- Cao, X. Targeting osteoclast-osteoblast communication. Nat. Med. 2011, 17, 1344–1346. [Google Scholar] [CrossRef]
- Takeshita, S.; Fumoto, T.; Matsuoka, K.; Park, K.A.; Aburatani, H.; Kato, S.; Ito, M.; Ikeda, K. Osteoclast-secreted CTHRC1 in the coupling of bone resorption to formation. J. Clin. Investig. 2013, 123, 3914–3924. [Google Scholar] [CrossRef]
- Matsuoka, K.; Park, K.A.; Ito, M.; Ikeda, K.; Takeshita, S. Osteoclast-derived complement component 3a stimulates osteoblast differentiation. J. Bone Miner. Res. 2014, 29, 1522–1530. [Google Scholar] [CrossRef]
- Bennett, C.N.; Longo, K.A.; Wright, W.S.; Suva, L.J.; Lane, T.F.; Hankenson, K.D.; MacDougald, O.A. Regulation of osteoblastogenesis and bone mass by Wnt10b. Proc. Natl. Acad. Sci. USA 2005, 102, 3324–3329. [Google Scholar] [CrossRef]
- Bennett, C.N.; Ouyang, H.; Ma, Y.L.; Zeng, Q.; Gerin, I.; Sousa, K.M.; Lane, T.F.; Krishnan, V.; Hankenson, K.D.; MacDougald, O.A. Wnt10b increases postnatal bone formation by enhancing osteoblast differentiation. J. Bone Miner. Res. 2007, 22, 1924–1932. [Google Scholar] [CrossRef] [PubMed]
- Ikebuchi, Y.; Aoki, S.; Honma, M.; Hayashi, M.; Sugamori, Y.; Khan, M.; Kariya, Y.; Kato, G.; Tabata, Y.; Penninger, J.M.; et al. Coupling of bone resorption and formation by RANKL reverse signalling. Nature 2018, 561, 195–200. [Google Scholar] [CrossRef] [PubMed]
- Xiong, J.; Cawley, K.; Piemontese, M.; Fujiwara, Y.; Zhao, H.; Goellner, J.J.; O’Brien, C.A. Soluble RANKL contributes to osteoclast formation in adult mice but not ovariectomy-induced bone loss. Nat. Commun. 2018, 9, 2909. [Google Scholar] [CrossRef]
- Hinz, B. The extracellular matrix and transforming growth factor-beta1: Tale of a strained relationship. Matrix Biol. 2015, 47, 54–65. [Google Scholar] [CrossRef] [PubMed]
- Dallas, S.L.; Park-Snyder, S.; Miyazono, K.; Twardzik, D.; Mundy, G.R.; Bonewald, L.F. Characterization and autoregulation of latent transforming growth factor beta (TGF beta) complexes in osteoblast-like cell lines. Production of a latent complex lacking the latent TGF beta-binding protein. J. Biol. Chem. 1994, 269, 6815–6821. [Google Scholar]
- Tang, Y.; Wu, X.; Lei, W.; Pang, L.; Wan, C.; Shi, Z.; Zhao, L.; Nagy, T.R.; Peng, X.; Hu, J.; et al. TGF-beta1-induced migration of bone mesenchymal stem cells couples bone resorption with formation. Nat. Med. 2009, 15, 757–765. [Google Scholar] [CrossRef]
- Mohan, S.; Jennings, J.C.; Linkhart, T.A.; Baylink, D.J. Primary structure of human skeletal growth factor: Homology with human insulin-like growth factor-II. Biochim. Biophys. Acta 1988, 966, 44–55. [Google Scholar] [CrossRef]
- Bautista, C.M.; Mohan, S.; Baylink, D.J. Insulin-like growth factors I and II are present in the skeletal tissues of ten vertebrates. Metabolism 1990, 39, 96–100. [Google Scholar] [CrossRef]
- Howard, G.A.; Bottemiller, B.L.; Turner, R.T.; Rader, J.I.; Baylink, D.J. Parathyroid hormone stimulates bone formation and resorption in organ culture: Evidence for a coupling mechanism. Proc. Natl. Acad. Sci. USA 1981, 78, 3204–3208. [Google Scholar] [CrossRef]
- Xian, L.; Wu, X.; Pang, L.; Lou, M.; Rosen, C.J.; Qiu, T.; Crane, J.; Frassica, F.; Zhang, L.; Rodriguez, J.P.; et al. Matrix IGF-1 maintains bone mass by activation of mTOR in mesenchymal stem cells. Nat. Med. 2012, 18, 1095–1101. [Google Scholar] [CrossRef]
- Cummings, S.R.; San Martin, J.; McClung, M.R.; Siris, E.S.; Eastell, R.; Reid, I.R.; Delmas, P.; Zoog, H.B.; Austin, M.; Wang, A.; et al. Denosumab for prevention of fractures in postmenopausal women with osteoporosis. N. Engl. J. Med. 2009, 361, 756–765. [Google Scholar] [CrossRef] [PubMed]
- Bekker, P.J.; Holloway, D.L.; Rasmussen, A.S.; Murphy, R.; Martin, S.W.; Leese, P.T.; Holmes, G.B.; Dunstan, C.R.; DePaoli, A.M. A single-dose placebo-controlled study of AMG 162, a fully human monoclonal antibody to RANKL, in postmenopausal women. J. Bone Miner. Res. 2004, 19, 1059–1066. [Google Scholar] [CrossRef] [PubMed]
- McClung, M.R.; Lewiecki, E.M.; Cohen, S.B.; Bolognese, M.A.; Woodson, G.C.; Moffett, A.H.; Peacock, M.; Miller, P.D.; Lederman, S.N.; Chesnut, C.H.; et al. Denosumab in postmenopausal women with low bone mineral density. N. Engl. J. Med. 2006, 354, 821–831. [Google Scholar] [CrossRef]
- Sims, N.A.; Ng, K.W. Implications of osteoblast-osteoclast interactions in the management of osteoporosis by antiresorptive agents denosumab and odanacatib. Curr. Osteoporos. Rep. 2014, 12, 98–106. [Google Scholar] [CrossRef] [PubMed]
- Drake, M.T.; Clarke, B.L.; Oursler, M.J.; Khosla, S. Cathepsin K Inhibitors for Osteoporosis: Biology, Potential Clinical Utility, and Lessons Learned. Endocr. Rev. 2017, 38, 325–350. [Google Scholar] [CrossRef]
- Costa, A.G.; Cusano, N.E.; Silva, B.C.; Cremers, S.; Bilezikian, J.P. Cathepsin K: Its skeletal actions and role as a therapeutic target in osteoporosis. Nat. Rev. Rheumatol. 2011, 7, 447–456. [Google Scholar] [CrossRef]
- Xue, Y.; Cai, T.; Shi, S.; Wang, W.; Zhang, Y.; Mao, T.; Duan, X. Clinical and animal research findings in pycnodysostosis and gene mutations of cathepsin K from 1996 to 2011. Orphanet. J. Rare Dis. 2011, 6, 20. [Google Scholar] [CrossRef]
- Fratzl-Zelman, N.; Valenta, A.; Roschger, P.; Nader, A.; Gelb, B.D.; Fratzl, P.; Klaushofer, K. Decreased bone turnover and deterioration of bone structure in two cases of pycnodysostosis. J. Clin. Endocrinol. Metab. 2004, 89, 1538–1547. [Google Scholar] [CrossRef]
- Debnath, S.; Yallowitz, A.R.; McCormick, J.; Lalani, S.; Zhang, T.; Xu, R.; Li, N.; Liu, Y.; Yang, Y.S.; Eiseman, M.; et al. Discovery of a periosteal stem cell mediating intramembranous bone formation. Nature 2018, 562, 133–139. [Google Scholar] [CrossRef]
- Recker, R.; Dempster, D.; Langdahl, B.; Giezek, H.; Clark, S.; Ellis, G.; de Villiers, T.; Valter, I.; Zerbini, C.A.; Cohn, D.; et al. Effects of Odanacatib on Bone Structure and Quality in Postmenopausal Women With Osteoporosis: 5-Year Data From the Phase 3 Long-Term Odanacatib Fracture Trial (LOFT) and its Extension. J. Bone Miner. Res. 2020, 35, 1289–1299. [Google Scholar] [CrossRef]
- Mullard, A. Merck &Co. drops osteoporosis drug odanacatib. Nat. Rev. Drug Discov. 2016, 15, 669. [Google Scholar] [CrossRef] [PubMed]
- Fan, Y.; Hanai, J.I.; Le, P.T.; Bi, R.; Maridas, D.; DeMambro, V.; Figueroa, C.A.; Kir, S.; Zhou, X.; Mannstadt, M.; et al. Parathyroid Hormone Directs Bone Marrow Mesenchymal Cell Fate. Cell Metab. 2017, 25, 661–672. [Google Scholar] [CrossRef] [PubMed]
- Ishizuya, T.; Yokose, S.; Hori, M.; Noda, T.; Suda, T.; Yoshiki, S.; Yamaguchi, A. Parathyroid hormone exerts disparate effects on osteoblast differentiation depending on exposure time in rat osteoblastic cells. J. Clin. Investig. 1997, 99, 2961–2970. [Google Scholar] [CrossRef] [PubMed]
- Silva, B.C.; Bilezikian, J.P. Parathyroid hormone: Anabolic and catabolic actions on the skeleton. Curr. Opin. Pharmacol. 2015, 22, 41–50. [Google Scholar] [CrossRef]
- Keller, H.; Kneissel, M. SOST is a target gene for PTH in bone. Bone 2005, 37, 148–158. [Google Scholar] [CrossRef]
- O’Brien, C.A.; Plotkin, L.I.; Galli, C.; Goellner, J.J.; Gortazar, A.R.; Allen, M.R.; Robling, A.G.; Bouxsein, M.; Schipani, E.; Turner, C.H.; et al. Control of bone mass and remodeling by PTH receptor signaling in osteocytes. PLoS ONE. 2008, 3, e2942. [Google Scholar] [CrossRef]
- Sims, N.A.; Martin, T.J. Osteoclasts Provide Coupling Signals to Osteoblast Lineage Cells Through Multiple Mechanisms. Annu. Rev. Physiol. 2020, 82, 507–529. [Google Scholar] [CrossRef]



{kind=link}
{kind=link}
| Osteoblast-Derived Factor | Mode of Action | Influences on Osteoclasts | References |
|---|---|---|---|
| EFNB2 | Membrane-bound | Inhibits osteoclastogenesis | [26] |
| FASL | Membrane-bound | Induces osteoclast apoptosis | [36] |
| SEMA3A | Membrane-bound | Inhibits RANKL-induced osteoclastogenesis | [38] |
| M-CSF | Secreted | Promotes proliferation and survival of osteoclast precursor | [45] |
| RANKL | Membrane-bound and secreted | Promotes osteoclast differentiation and activation | [6,51] |
| OPG | Secreted | Inhibits osteoclastogenesis | [6,57] |
| WNT5A | Secreted | Promotes osteoclastogenesis | [58] |
| WNT16 | Secreted | Inhibits osteoclastogenesis | [63] |
| Osteoclast-Derived Factor | Mode of Action | Influence on Osteoblasts | References |
|---|---|---|---|
| EPHB4 | Membrane-bound | Promotes osteoblastogenesis and suppresses osteoblast apoptosis | [26,30] |
| S1P | Secreted | Promotes osteoblast migration and survival | [64,65] |
| SEMA4D | Secreted | Suppresses osteoblastogenesis | [39] |
| CTHRC1 | Secreted | Recruits stromal cells and induces osteoblastogenesis | [69] |
| C3 | Secreted | Promotes osteoblastogenesis | [70] |
| WNT10B | Secreted | Promotes osteoblastogenesis | [71,72] |
| Vesicular RANK | Secreted | Promotes osteoblastogenesis | [73] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kim, J.-M.; Lin, C.; Stavre, Z.; Greenblatt, M.B.; Shim, J.-H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells 2020, 9, 2073. https://doi.org/10.3390/cells9092073
Kim J-M, Lin C, Stavre Z, Greenblatt MB, Shim J-H. Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells. 2020; 9(9):2073. https://doi.org/10.3390/cells9092073
Chicago/Turabian StyleKim, Jung-Min, Chujiao Lin, Zheni Stavre, Matthew B. Greenblatt, and Jae-Hyuck Shim. 2020. "Osteoblast-Osteoclast Communication and Bone Homeostasis" Cells 9, no. 9: 2073. https://doi.org/10.3390/cells9092073
APA StyleKim, J.-M., Lin, C., Stavre, Z., Greenblatt, M. B., & Shim, J.-H. (2020). Osteoblast-Osteoclast Communication and Bone Homeostasis. Cells, 9(9), 2073. https://doi.org/10.3390/cells9092073