Calcium, Bioenergetics, and Parkinson’s Disease


Department of Physiology, Feinberg School of Medicine, Northwestern University, Chicago, IL 60611, USA
*
Author to whom correspondence should be addressed.
Cells 2020, 9(9), 2045; https://doi.org/10.3390/cells9092045
Submission received: 7 August 2020
/
Revised: 4 September 2020
/
Accepted: 7 September 2020
/
Published: 8 September 2020
(This article belongs to the Special Issue Key Signalling Molecules in Aging and Neurodegeneration)
Abstract
:Degeneration of substantia nigra (SN) dopaminergic (DAergic) neurons is responsible for the core motor deficits of Parkinson’s disease (PD). These neurons are autonomous pacemakers that have large cytosolic Ca2+ oscillations that have been linked to basal mitochondrial oxidant stress and turnover. This review explores the origin of Ca2+ oscillations and their role in the control of mitochondrial respiration, bioenergetics, and mitochondrial oxidant stress.
{kind=link}
{kind=link}
{kind=link}
{kind=link}
1. Introduction: The Duality of Intracellular Ca2+ Signaling
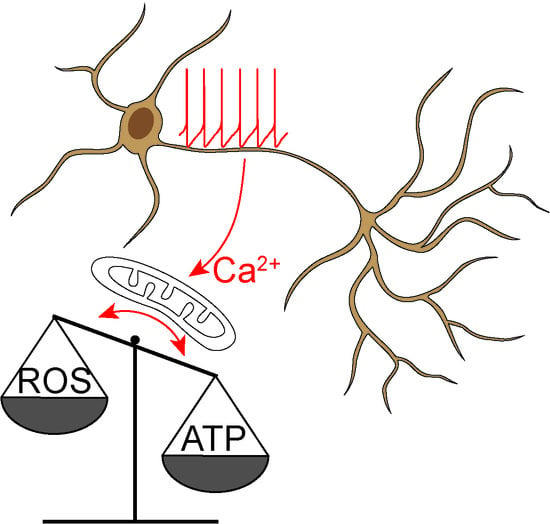
The role of Ca2+ as a second messenger has been explored for decades [1,2,3,4]. One of the most intriguing features of Ca2+ that has emerged from this effort is its duality: Ca2+ signals are necessary for cellular health, but can also trigger dysfunction and death [5]. This duality also manifests itself in substantia nigra (SN) dopaminergic (DAergic) neurons (Figure 1). These neurons—whose degeneration is responsible for the core motor symptoms of Parkinson’s disease (PD) [6,7]—have large cytosolic oscillations in Ca2+ concentration ([Ca2+]). These oscillations play a key role in helping the neurons meet their bioenergetic needs, but they are also linked to cellular stress and vulnerability with aging and PD [8,9,10].
2. Neuronal Ca2+ Homeostasis
Spiking or synaptic activity can trigger transient elevations in cytosolic [Ca2+]. Generally speaking, there are three classes of plasma membrane (PM) proteins that underlie these transients. One class is formed by voltage-dependent Ca2+ permeable ion channels. These channels vary in their voltage-dependence, location, and kinetics, and are accordingly classified as L-type (Cav1.1–1.4), N-type (Cav2.1), P/Q-type (Cav2.2), R-type (Cav2.3), and T-type (Cav3.3) [11]. Voltage-dependent Ca2+ channels provide an elegant means of linking spiking and synaptic activity to intracellular machinery responsible for the control of other channels (e.g., Ca2+ activated K+ channels), transmitter release, metabolism, and gene expression [11]. Another class is formed by ionotropic receptors that are gated by neurotransmitters (e.g., nicotinic acetylcholine receptors) and flux Ca2+. The third class is formed by Gq-linked G-protein coupled receptors (GPCRs) activated by neurotransmitters (e.g., metabotropic glutamate receptors) that do not flux Ca2+ themselves but generate ligands for receptors that release Ca2+ from intracellular stores [12,13]. Other channels that can participate in neuronal Ca2+ signaling include store-operated channels (SOCs) and transient receptor (TRP) channels [12]. The amplitude, kinetics, and spatial distribution of intracellular [Ca2+] transients triggered by these PM proteins are controlled by Ca2+ buffering proteins—proteins endowed with Ca2+ binding sites [14]; Ca2+ binding proteins also can serve as Ca2+ sensors and effectors by interacting with an extraordinary array of other signaling molecules [14].
Unlike most cations, the transmembrane gradient for Ca2+ between the extracellular space and the cytosol is typically several orders of magnitude. As a consequence, a sophisticated collection of molecular mechanisms exists to achieve this end [1,3,15,16]. While extracellular [Ca2+] is 1–2 mM, cytosolic [Ca2+] is generally maintained at nanomolar levels (approximately 100 nM) by pumps and exchangers that extrude Ca2+ across the plasma membrane (PM) or into intracellular stores. The PM is endowed with plasma membrane Ca2+ ATPases (PMCAs) and Na+/Ca2+ exchangers (NCXs) that expel Ca2+. PMCAs pump Ca2+ to the extracellular space by using adenosine triphosphate (ATP), while NCX takes advantage of the Na+ gradient created by the Na+/K+ ATPase to extrude Ca2+ to the extracellular space. Given their differences in affinity for Ca2+, it is likely that basal cytosolic [Ca2+] is largely governed by the PMCA and the NCX is engaged by activity that pushes local [Ca2+] higher, as during repetitive spiking [17].
The endoplasmic reticulum (ER) is the main intracellular Ca2+ store. Elevated ER [Ca2+] is maintained by the sarco/endoplasmic reticulum Ca2+ ATPases (SERCAs) that move Ca2+ from the cytosol. SERCAs serve to terminate cytosolic transients induced by PM processes and to counteract constitutive “leak” of Ca2+ from the ER itself. The ER is richly invested with Ca2+ buffer proteins (e.g., calreticulin) that differ from cytosolic buffers in their affinity and capacity to help stabilize the high (µM) luminal ER [Ca2+]. Ca2+ release from the ER is mediated by the inositol trisphosphate (IP3) receptor (IP3R) and the ryanodine receptor (RyR). IP3R is gated by IP3 generated by phospholipase C in response to the activation of GPCRs. The primary agonist of RyR is Ca2+ itself; thus, cytosolic Ca2+ transients can trigger Ca2+-induced Ca2+ release” (CICR) from the ER [12,13,18]. In this regard, it is important to remember that the ER is a morphologically complex system of cisternae and tubules spread through the neuron, extending into axons, dendrites, and spines [18,19,20]; thus, CICR creates a means of creating propagated Ca2+ waves from one region of a cell to another. In addition to the ER, other organelles—including mitochondria, the Golgi apparatus, lysosomes, and endosomes—act as Ca2+ stores and can contribute to shaping intracellular Ca2+ signaling events [15].
3. Ca2+ and Control of Mitochondria
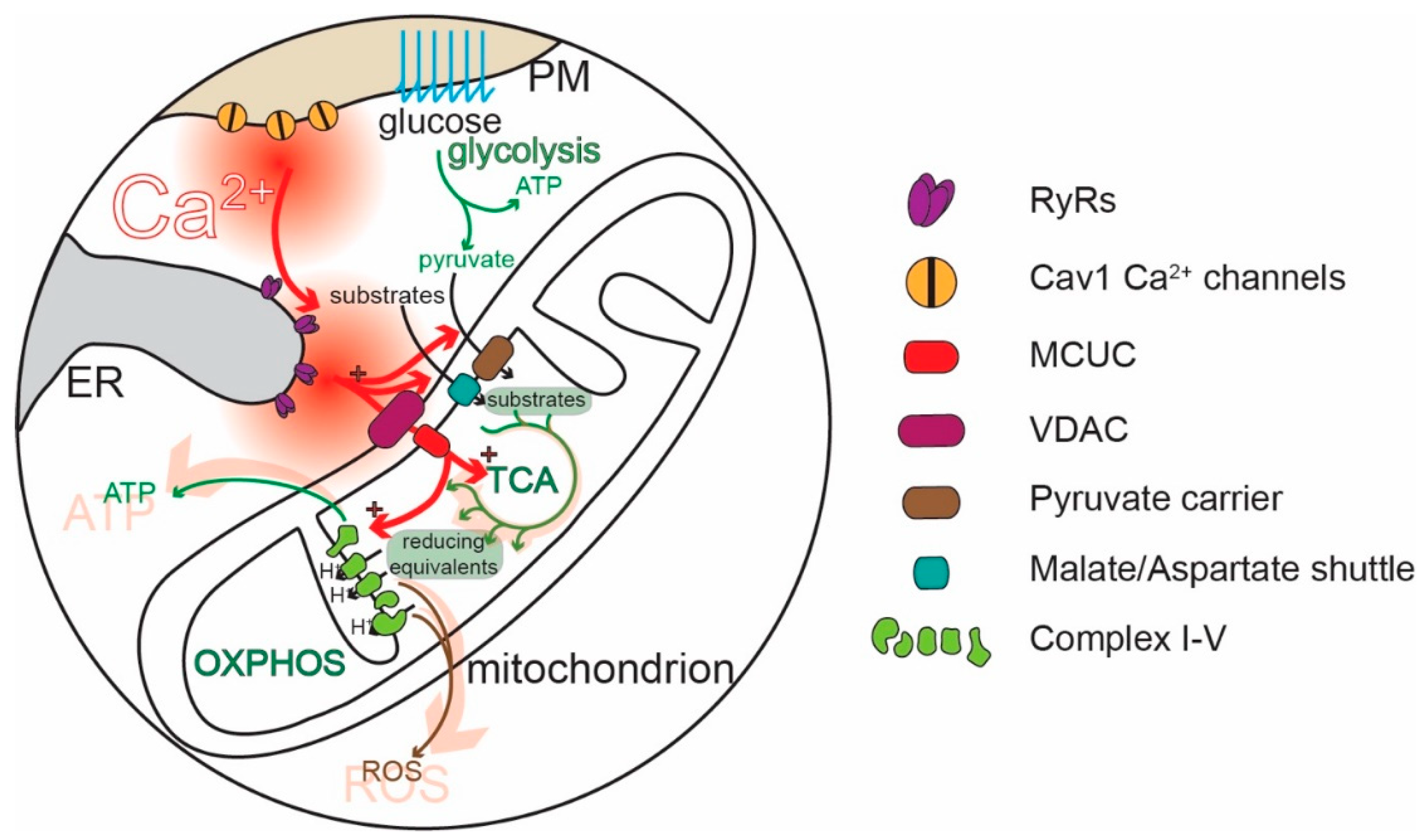
Mitochondria are widely thought to be the “powerhouses” of neurons, meeting the bioenergetic demands of regenerative activity and neurotransmitter release. Hypothesized to be ancient bacterial symbionts, mitochondria have an outer membrane (OMM) perforated by relatively large pores. The OMM surrounds an inner membrane (IMM) with deep invaginations (cristae); in contrast to the OMM, the transit of molecules—and Ca2+—across the IMM is tightly regulated [21,22,23,24]. In healthy mitochondria, the electrochemical gradient across the IMM created by the electron transport chain (ETC) is very steep (~180 mV), providing a strong driving force for Ca2+ entry into the mitochondrial matrix [25,26]. Recently, a great deal of progress has been made in characterizing the molecular machinery responsible for regulating mitochondrial Ca2+ influx [26]. The influx of Ca2+ is controlled by the mitochondrial Ca2+ uniporter complex (MCUC). The MCUC is composed of the channel-forming unit, known as the mitochondrial Ca2+ uniporter (MCU) [27,28], and several accessory proteins that influence MCU gating [29,30]. These subunits limit MCU opening to periods when the intermembrane [Ca2+] is high (~10–20 µM). In physiological situations, this concentration is achieved inside neurons only in microdomains where diffusion is restricted [3,31]. Indeed, this kind of restricted diffusion space is created at specialized junctions between mitochondria and the ER [32,33,34,35,36,37], referred to as “mitochondria-associated membranes” (MAMs) [38,39,40]. There is also evidence that the MCUC is tailored to meet the needs of different subcellular compartments, like the nerve terminal [41].
Ca2+ is extruded from mitochondria by a Ca2+/H+ exchanger and—particularly in excitable cells—a Na+/Ca2+ exchanger (or Na+/Ca2+/Li2+ exchanger, NCLX) [25,42,43]. In contrast to Ca2+ entry through the MCU pore, the extrusion of Ca2+ by exchangers is relatively slow. This difference in dynamics shapes cytosolic Ca2+ signals [44,45,46,47]. Another possible mitochondrial exit pathway for Ca2+ is the mitochondria permeability transition pore (mPTP), which is generally thought to open only in pathological situations when matrix [Ca2+] gets too high [48]; however, the mPTP can also open transiently to modulate mitochondrial Ca2+ levels [26,49,50,51].
Although mitochondria regulate cellular functions in a variety of ways [52], one of their most important roles is the conversion of adenosine diphosphate (ADP) to ATP through oxidative phosphorylation (OXPHOS, Figure 2). OXPHOS complements glycolysis, generating 18 molecules of ATP for each pyruvate molecule produced from the metabolism of glucose [53,54]. Metabolic substrates, like pyruvate (also amino acids or ketones), are taken up by mitochondria and enter the tricarboxylic acid cycle (TCA), which converts them into reducing equivalents for the ETC. Complexes I-IV of the ETC located in the IMM use the reducing equivalents to transfer electrons to molecular oxygen and to pump protons (H+) from the mitochondrial matrix into the intermembrane space (IMS), between the IMM and the OMM. ATP synthase (complex V) then uses the H+ electrochemical gradient to convert ADP to ATP [50,55,56] (Figure 2). The rate of OXPHOS is modulated by cytosolic Ca2+ in several ways [57,58,59,60] (Figure 2). IMS Ca2+ stimulates the transport of metabolites into the matrix [59,61]. Ca2+ entry into the mitochondrial matrix through MCUC stimulates the generation of reducing equivalents by disinhibiting three key TCA dehydrogenases [62]. The mitochondrial matrix Ca2+ stimulates complex V [57]. In this way, Ca2+ signaling links regenerative activity to ATP production [63,64,65,66,67,68,69]. Ablating the MCU and preventing Ca2+ uptake in mitochondria leads to a compensatory upregulation of glycolysis, supporting the critical role of OXPHOS and its stimulation by Ca2+ for neuronal health [70].
4. SN DAergic Neurons and PD
The degeneration of SN DAergic neurons is responsible for the core motor symptoms—bradykinesia and rigidity—of PD [6,71,72]. These neurons are autonomous pacemakers: in the absence of external stimulation, SN DAergic neurons fire broad (~2–3 ms) action potentials (APs) at a regular frequency (1–4 Hz) [73,74,75,76]. SN DAergic neurons are part of the basal ganglia, and dopamine (DA) released from their axons modulates the activity of basal ganglia circuits controlling goal-directed actions and habits. The largest of the basal ganglia nuclei modulated by DA is the striatum. The autonomous pacemaking of SN DAergic neurons is modulated up and down by synaptic inputs [77], allowing bidirectional control of DA release, which in turn bidirectionally modulate basal ganglia circuits [78,79,80]. SN DAergic neurons also release DA from their somatodendritic membrane [81,82,83]. This release is known to modulate synaptic input to neighboring substantia nigra pars reticulata (SNr) GABAergic neurons that form a major portion of the basal ganglia interface with the rest of the brain [84]. The degeneration of SN DAergic neurons distorts cellular and network activity in the basal ganglia, resulting in the core motor symptoms of PD [6,71,72].
5. Ca2+ Signaling in SN DAergic Neurons
It’s our thesis that the vulnerability of SN DAergic neurons to aging and PD [85] is in large measure attributable to their distinctive phenotype [86]. This phenotype not only creates basal metabolic stress in the absence of overt pathology but also increases the impact of genetic mutations and environmental toxins linked to increased risk of developing PD. A key feature of this distinctive phenotype is the way Ca2+ signaling is engaged.
In all neurons, Ca2+ currents through voltage-dependent channels serve to promote and regulate regenerative spiking, as well as to link that activity to a variety of other processes. Specific channel subtypes play specific roles. For example, in presynaptic regions, Cav2 channels control exocytosis of neurotransmitters [11]. Ca2+ flux through somatodendritic Cav1.2 (L-type) channels control processes tied to spiking, as their open probability rises only when neurons spike. This feature allows them to generate Ca2+ signaling that is proportional to spike rate – an important variable not only for ion channels responsible for membrane excitability (e.g., K+ channels) but also the transcriptional machinery involved in processes like homeostatic plasticity [11].
Several types of ion channels—including Ca2+ channels—participate in the initiation and regulation of the autonomous rhythmic activity in SN DAergic neurons [73,87,88,89,90,91,92,93,94,95,96,97,98]. Most voltage-dependent Ca2+ channels (N-, P/Q-, R- and most L-type Ca2+ channels) require relatively depolarized potentials to activate, and they open only at membrane potentials above the spike threshold. For example, high voltage-activated, R-type (Cav2.3) channels contribute to somatic Ca2+ oscillations in SN DAergic neurons during spiking and help regulate spike patterning [99]. However, SN DAergic neurons express two types of voltage-dependent Ca2+ channels (Cav3 (T-type) and Cav1.3 (L-type)) that open at membrane potentials below the spike threshold and thus can help push the membrane potential to the threshold for spiking [100,101]. Indeed, Ca2+ imaging experiments have revealed cytosolic Ca2+ transients in SN DAergic neurons that begin well before the spike and then increase during it [101,102,103,104,105,106,107].
Cav3 channels (classified as low-voltage activated channels) activate at sub-threshold membrane potentials but inactivate with sustained depolarization. Although their activation and inactivation curves partially overlap, creating a “window current” that can destabilize membrane potential [108,109], their gating properties and sub-cellular location makes them particularly well-suited to the regulation of spiking patterns originating in the proximal somatodendritic region and axon initial segment [110]. In fact, recent quantitative Ca2+ imaging experiments have shown that the contribution of Cav3 channels to cytosolic Ca2+ transients is primarily in the proximal dendrites of SN neurons [104]. In this way, Ca2+ entry through Cav3 channels can increase the depolarization needed to trigger spikes (particularly in cases when the membrane is “released” from synaptic hyperpolarization that de-inactivates them [111]), but also help maintain the regularity of pacemaking and the duration of synaptically generated spike “bursts” by activating Ca2+-dependent SK K+ channels that pull the membrane potential in a negative direction [112].
Like Cav3 channels, Cav1 channels (L-type) containing the Cav1.3 pore-forming subunit [113,114,115] open at relatively hyperpolarized potentials and thus can contribute to the depolarization leading to the generation of rhythmic spontaneous spikes [116,117,118]. However, unlike Cav3 channels, Cav1 channels inactivate only modestly with depolarization, making them suitable for modulating more sustained changes in activity. Consistent with these properties, Cav1 channels drive a slow oscillatory potential (SOPs) in SN DAergic neurons when Na+ channels are blocked with tetrodotoxin [93,95,101,102,107,119,120]. However, the opening of Cav1 channels is not necessary for pacemaking, as asserted previously based upon experiments that employed dihydropyridines (DHPs) at concentrations where channel specificity is lost [89,96]. At nanomolar concentrations, where binding is specific to Cav1 channels, DHPs effectively inhibit cytoplasmic Ca2+ transients without changing the pacemaking rate [101]. Rather, the engagement of Cav1 channels increases the robustness of pacemaking that is largely driven by a cation leak channel (NALCN) [101,102,121,122]. Although SN DAergic neurons express both Cav1.2 and Cav1.3 channels, targeted genetic suppression of Cav1.3 channels mimics the effects of DHPs on dendritic Ca2+ transients, pointing to them as primary determinants of this feature of the phenotype [104]. Indeed, because of their gating properties, Cav1.3 channels are open through most of the pacemaking cycle [107].
During pacemaking, the cytosolic Ca2+ transient in the dendrites of SN DAergic neurons is estimated using quantitative Fura2 imaging to rise above 500 nM (near zones of Ca2+ entry or release, the concentration may reach into the microlar range). In part, the magnitude of this transient is attributable to low intrinsic Ca2+ buffering [123]. This allows Ca2+ to diffuse easily through the cytoplasm and control biochemical processes and gating of channels, like SK channels [124,125] and RyRs (triggering CICR) [126]. Indeed, Cav1.3 channels are strongly coupled to ER RYRs [127]. This coupling is responsible for much of the cytosolic Ca2+ transient during pacemaking (unpublished observations and [128]).
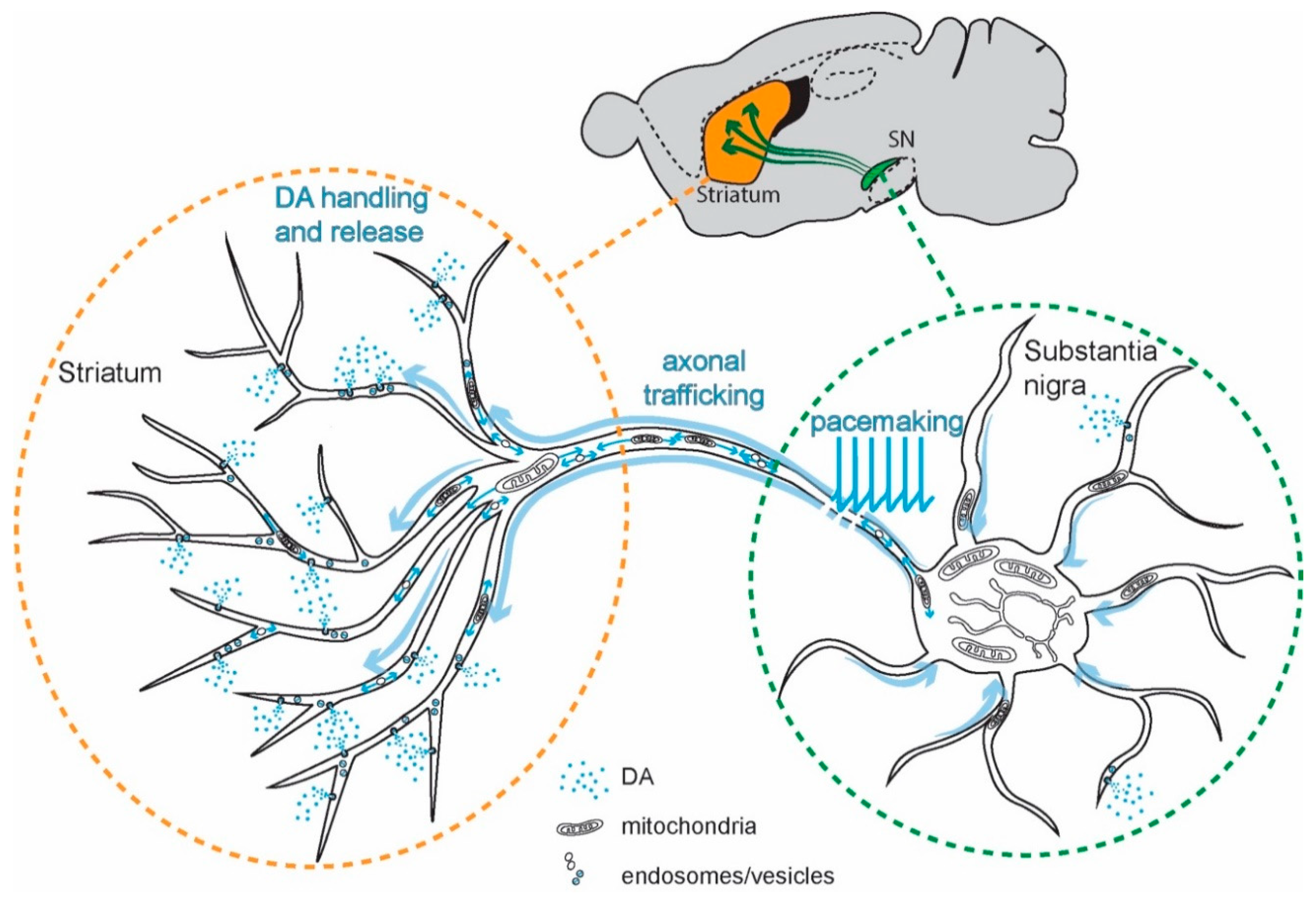
The purpose of Ca2+ signaling triggered by pacemaking in SN DAergic neurons is still being unraveled, but there are some clues. Ca2+ flux through Cav1 channels regulates the expression of genes coding for proteins responsible for the synthesis of DA, linking activity, and anabolic activity [129,130,131] (Figure 3). Another function of Cav1.3 channels in SN DAergic neurons is the control of mitochondrial OXPHOS (Figure 2). Unlike most neurons, SN DAergic neurons appear to have a high basal bioenergetic demand [132,133,134]. This demand may have its roots in several factors. The most important of these is likely to be the neuron’s massive axonal arbor [132,135]. This arbor creates an anabolic demand, as it has to be supplied with release-related proteins and lipids largely delivered by axonal transport from the somatic region [136,137,138,139]. The hundreds of thousands of DA release sites create an independent burden, as the release and recycling of synaptic vesicles is bioenergetically expensive [63,65,68,132]. As proteins and lipids within this arbor become damaged or dysfunctional, they have to be degraded, creating an additional catabolic demand [140]. Moreover, ionic gradients necessary to support regenerative activity throughout the axon poses a significant burden [133,141]. SN DAergic neurons also are constantly active, multiplying the demands associated with the axonal propagation of spikes (Figure 1) [8,133].
For those readers interested in the numbers, let us review them. In rodents, the axon of a single SN DAergic neuron can reach a length of over 40 cm and form more than 200,000 synapses, covering a significant portion of the striatum [132,142,143,144,145]. Neighboring ventral tegmental area (VTA) DAergic neurons also have relatively large axonal trees [146,147] but have far fewer transmitter release sites (~12,000–30,000 [132]). Although an order of magnitude less than SN DAergic neurons, this is still substantially greater than many other neurons [148,149]. In the human brain, SN DAergic neurons may have an order of magnitude greater number of release sites than those in the mouse, possibly because of forebrain evolution [132,134,150].
Direct evidence of the bioenergetic burden posed by the axon of SN DAergic neurons comes from a novel in vitro study [151]. The authors not only confirmed that SN DAergic neurons have longer and more branched axons than VTA DAergic neurons, but also that axonal size was directly correlated with oxygen consumption rate (OCR, an index of mitochondria OXPHOS) and mitochondrial oxidant stress. Reducing the size of the axonal arbor decreased OCR, oxidant stress, and vulnerability to parkinsonian toxins. Interestingly, inhibiting Cav1 Ca2+ channels decreased OCR [151]. In a follow-up study in vivo, the authors demonstrated that increasing axonal size in SN DAergic neurons increased their vulnerability to mitochondrial toxins [152]—solidifying the connection between axonal arbor size and mitochondrial stress.
If we accept the proposition that SN DAergic neurons have a high basal bioenergetic demand, how do they satisfy that demand? As noted above, OXPHOS is an efficient means of generating ATP from glucose, fatty acids, and amino acids. It has long been thought that ATP levels were maintained by ATP-mediated feedback control of complex V [153,154,155,156]. The problem with this kind of control mechanism is speed. SN DAergic neurons dynamically regulate basal ganglia circuits controlling escape, attack, and habitual behaviors. If ATP levels fall and DAergic neurons slow their spiking or release of DA because of flagging ATP levels [157,158], the organism’s movement will begin to slow, making it vulnerable. Thus, there must have been strong evolutionary pressure to develop a control strategy that does not depend upon feedback. In muscle, a feedforward, “anticipatory” control mechanism is used to drive OXPHOS [159,160]. A similar mechanism is in place in SN DAergic neurons where the Cav1.3 channel triggered ER release of Ca2+ through RYRs “injects” Ca2+ into mitochondria at MAMs—stimulating OXPHOS in anticipation of need. Indeed, inhibition of mitochondrial OXPHOS or glucose deprivation causes them to hyperpolarize and stop spiking [157,158,161,162]. Interestingly, a recent paper has confirmed in hippocampal neurons that Ca2+ influx through Cav1 channels combined with CICR can regulate mitochondria ATP production, although in these non-pacemaking neurons mitochondrial contribution to cell bioenergetic seems to be relatively small and this mechanism is activated only upon stimulation [163]. At axonal DA release sites, Ca2+ stimulated mitochondrial OXPHOS is complemented by another feedforward system in which DA transiting the cytosol is metabolized by monoamine oxidase (MAO) anchored to the outer membrane of mitochondria; in so doing, MAO generates an electron that is shuttled to the ETC, which supports the electrochemical gradient used by complex V to generate ATP [164]. Thus, feedforward control of mitochondrial OXPHOS is a mechanism by which SN DAergic neurons can maximize their functionality and promote organismal survival.
6. Why are SN DAergic Neurons Preferentially Vulnerable in PD?
Several theories have been advanced to explain the selective vulnerability of SN DAergic neurons in PD. The oldest is that DA is responsible. DA is a reactive molecule that when oxidized or metabolized can damage a variety of cellular proteins and lipids, most importantly α-synuclein (αSYN) [165,166,167,168,169,170,171,172,173,174] (Figure 3). In human mesencephalic DA neurons, cytosolic DA may be particularly high [175], allowing Cav1 channel-driven mitochondrial oxidant stress to significantly increase DA oxidation; the combination of mitochondrially generated reactive oxygen species (ROS) that escape into the cytoplasm and oxidized DA promotes not only misfolding of αSYN but also damage to lysosomal proteins that play a role in clearing misfolded αSYN [175]. Recent work also has shown that monoamine oxidase (MAO) metabolism of cytosolic DA in axons and distal dendrites increases mitochondrial oxidant stress by shuttling electrons to the ETC, creating a novel interaction between DA and mitochondria that could have pathological consequences [164]. That said, while DA might accelerate pathogenesis in PD, it is not the sole culprit. It has become increasingly clear that other transmitter phenotypes, particularly cholinergic and adrenergic neurons, also are highly vulnerable in PD [8,72].
While transmitter phenotype may not be a universally shared trait of vulnerable neurons, other traits are shared [8,72,135]. One cluster of shared traits is modest cytosolic Ca2+ buffering, a slow, broad-spike, autonomous pacemaking, and Cav1.3 channel opening that triggers CICR; together, these traits result in the generation of large Ca2+ transients several times a second. In contrast, VTA DAergic neurons are pacemakers, but do not manifest large [Ca2+] transients and are largely spared in PD [97,105,122,176]. This difference in Ca2+ handling is attributable in part to higher expression of the Ca2+ buffering protein calbindin-D28k (CB-D28k) [90,177,178,179]. CB-D28k expression levels between SN and VTA and within SN itself are correlated with vulnerability in experimental models and in clinical PD [180,181,182,183,184,185,186]. Interestingly, intracellular Ca2+ chelation or over-expression of CB-D28K can protect DAergic neurons against the deleterious effects of Ca2+ entry, including a gain of function mutation in the TRP channel Trp-4 [187].
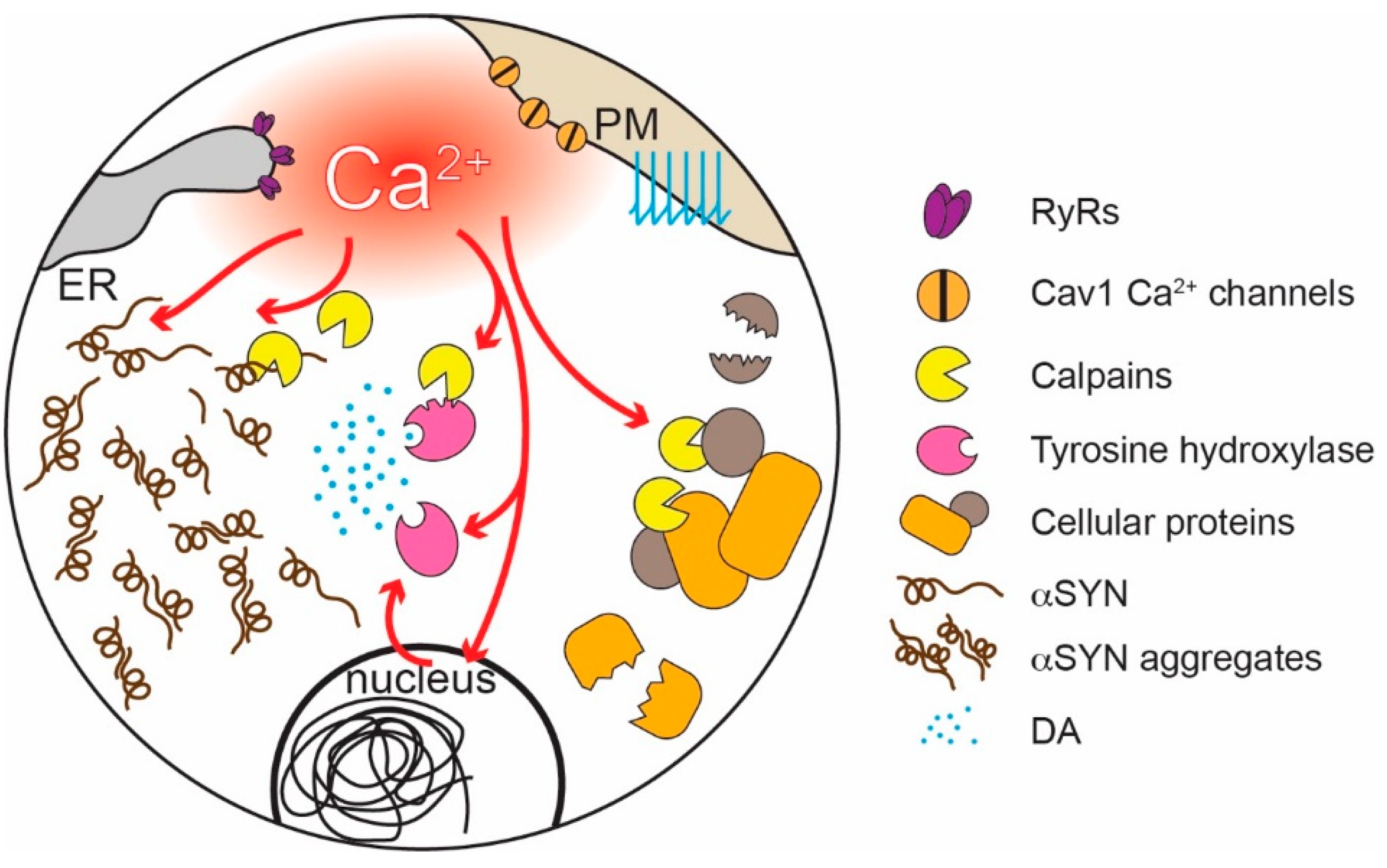
How might physiological levels of Ca2+ experienced by at-risk neurons promote PD pathology? There is growing evidence that Ca2+, directly and indirectly, promotes αSYN pathology—a hallmark of many forms of PD [72,188,189,190]. The negatively charged C-terminal region of αSYN inhibits misfolding and aggregation [191]. Binding of Ca2+ to this region attenuates electrostatic repulsion and promotes the formation of oligomers and higher molecular weight aggregates both in reconstituted preparations and in cells [192,193,194,195,196]. Ca2+ also promotes αSYN aggregation by enhancing calmodulin and membrane binding [197,198]. Conversely, increasing Ca2+ buffering decreases αSYN aggregation [199]. High (low micromolar) cytosolic [Ca2+]—like that achieved in SN DAergic neuron dendrites—also activates proteases known as calpains, which cleave a variety of cellular proteins [200], including αSYN and tyrosine hydroxylase, a key synthetic enzyme for DA (Figure 3) [201,202,203]. Calpain cleaves the C-terminal region of αSYN discussed above, promoting its aggregation [204]. Pharmacological or genetic inhibition of calpains reduces αSYN cleavage, aggregation, and toxicity [205] and is neuroprotective in PD models [206].
Interestingly, Ca2+ signaling also can be shaped by αSYN aggregates. At high concentrations, αSYN can induce the formation of Ca2+ permeable pores in membranes and enhance the activity of SERCA, possibly contributing to Ca2+-induced αSYN aggregation and damage [207,208,209,210,211,212]. Elevated cytosolic [Ca2+] also might promote the spreading of αSYN pathology, as cytosolic Ca2+ enhances αSYN release [213,214]. Moreover, another way in which αSYN affects Ca2+ homeostasis is by modulating ER-mitochondria Ca2+ transfer at the MAMs [215].
Another way in which Ca2+ signaling may increase neuronal vulnerability is through enhancing the production of superoxide and damaging ROS by mitochondria (Figure 2). The movement of electrons along the mitochondrial ETC is inevitably associated with electrons “jumping” to molecular oxygen and the generation of superoxide and ROS, primarily by mitochondrial complex I and III [50,55,216,217,218]. ROS can damage deoxyribonucleic acid (DNA), lipids, and proteins [218]. Although mitochondria are endowed with a variety of antioxidant defenses [219], these systems are imperfect [217,219]. Feedforward ETC stimulation not only results in longer respiratory bouts but also periods of stimulation during which there is little ATP demand and high mitochondrial membrane potential, a situation that is particularly likely to result in superoxide/ROS production [10,220]. Indeed, SN DAergic neurons have high levels of mitochondrial oxidant stress “at rest” and in the absence of pathology, as shown in primary neuronal cultures [106,209] and ex-vivo slices from mice [104,105]. By contrast, mitochondrial oxidant stress in VTA DAergic neurons is much lower [105,106,209]. Suppressing feedforward mitochondrial stimulation by inhibiting Cav1 Ca2+ channels lowers mitochondrial oxidant stress [104,105,106], supporting the connection between normal Ca2+ signaling and oxidant stress.
Mitochondrial ROS can damage mitochondrial proteins and DNA (mtDNA). The accumulation of mtDNA deletions characteristic of sustained oxidant stress is a well-described feature of SN DAergic neurons in aged humans and PD patients, in contrast to other types of neuron [221,222,223,224,225,226]. It is important to mention that mtDNA encodes only 13 proteins, all critical components of the OXPHOS machinery; thus, once a cell accumulates enough mtDNA deletions, the ability of mitochondria to produce ATP will be compromised [24,227]. In addition, because they are proximal to the sites of ROS generation, ETC proteins are prone to damage. Loss of complex I, which is the largest of the ETC complexes and a major source of ROS, is a key feature of SN DAergic neurons in PD patients [228,229,230,231,232].
Interestingly, in the somatodendritic regions of SN DAergic neurons, mitochondrial mass is paradoxically low [233]. Recent work has confirmed this observation and explained why it is this way. It turns out that the high mitochondrial oxidant stress driven by Cav1 Ca2+ channel-dependent stimulation of OXPHOS results in mitochondrial damage and elevated rates of mitophagy in SN DAergic neurons [104]. Systemic administration of DHPs to mice at concentrations that inhibit Cav1 channels decreases mitochondrial oxidant stress, slows mitophagy, and normalizes mitochondrial mass in SN DAergic neurons over the course of about a week [104]. Although it remains to be determined whether macroautophagy or mitochondrial-derived vesicles (MDVs) turnover is engaged by this process [234,235], this challenge is likely to compromise the ability of neurons to deal with other protein degradation tasks, like clearing αSYN aggregates. Moreover, with age the efficiency of macroautophagy declines [236]. This could have particularly dire consequences for SN DAergic neurons as their autophagic capacity may be pushed close to its limit by the combined catabolic demands associated with mitochondria and αSYN aggregation created by the massive axonal arbor. Aging, and any other stressor, like a genetic mutation compromising mitochondrial or autophagic function or an environmental toxin that exacerbates mitochondrial stress, could create a tipping point for degeneration. Interestingly, several studies of non-neuronal cells obtained from sporadic or familial PD patients have revealed bioenergetic and mitochondrial deficits [237,238,239,240,241,242,243,244,245,246], suggesting that there may be a systemic impairment in metabolism in PD, but only in neurons with little spare metabolic capacity (e.g., SN DAergic neurons) does this defect result in degeneration.
7. Is Mitochondrial Dysfunction Sufficient to Cause PD?
Although there are clear signs of mitochondrial pathology in the SN of PD patients, there is a continuing debate about whether mitochondrial dysfunction is a root cause of PD or whether it is merely a tombstone or consequence of pathology. For some time, there was little debate. At high enough doses, mitochondrial toxins, like rotenone and 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine (MPTP), effectively kill SN DAergic neurons in mice and primates and induce a parkinsonian like state within a matter of hours [247,248]. However, drugs that effectively blunt the toxicity of these compounds have consistently failed in human clinical trials [249,250,251]. An influential paper on this topic demonstrated that impairing complex I function in DAergic neurons by deleting one of its subunits (Ndufs4) had little effect on them and did not alter the sensitivity to toxins like rotenone [252,253,254,255]. That said, making complex I insensitive to rotenone or MPTP by knocking down p13 confers neuroprotection in toxin models of PD [256].
More recent attempts to determine the possible impact of mitochondrial dysfunction on PD pathogenesis have turned to genetic strategies. As already mentioned above, it is widely assumed that neurons need mitochondria, particularly in axons [137,257]. Reduced expression of molecular motors associated with axonal transport has been observed in tissue from early-stage PD patients [258]. Exposure to PD toxins (6-Hydroxydopamine, 6-OHDA, or MPTP metabolites) decreased anterograde mitochondrial axonal transport in primary cultures of rodent DAergic neurons [259,260] and in transgenic zebrafish [261].
More compelling evidence of the importance of mitochondrial dynamics in SN DAergic neurons comes from studies based on the manipulation of the molecular machinery responsible for these dynamics. Mitochondria undergo fusion and fission, allowing them to exchange mtDNA and other components (fusion) and to generate smaller isolated organelles (fission) that can be easily transported through the cell or destined for degradation [262,263]. Diminishing mitochondrial fission upon deletion of Drp1 leads to the depletion of mitochondria from the axons of SN DAergic neurons, progressive loss of SN striatal projections, and neuronal loss in SN [264]. Similarly, the deletion of mitofusin 2 (but not mitofusin 1), which is involved in mitochondrial fusion and ER-mitochondria tethering (see below), causes decreased mitochondrial transport and axonal degeneration in SN DAergic neurons [265,266].
Another strategy to test the role mitochondria in SN DAergic neurons is to target mtDNA. The “MitoPark” mouse is based on a DAergic-specific deletion of the mitochondria transcription factor Tfam, compromising mitochondrial transcription and mtDNA maintenance, disrupting the synthesis of critical subunits of the OXPHOS machinery [267]. Within weeks of birth, DAergic neurons in MitoPark mice have dysmorphic mitochondria, impaired spiking; later, SN DAergic neurons degenerate and mice manifest a parkinsonian phenotype [267,268]. Similarly, targeting the endonuclease (PstI) to mitochondria in DAergic neurons, which causes mtDNA damage and OXPHOS dysfunction, results in a slow loss of SN DAergic neurons and motor impairments [269]. These studies demonstrate that mitochondria are necessary for normal functioning and survival of SN DAergic neurons, but they do not resolve the issue about whether the loss of complex I function seen in the SN of PD patients is a driver of pathogenesis.
In an attempt to directly target complex I, a subunit of complex I (Ndufs4) was deleted in DAergic neurons of mice. However, these mice don’t manifest a parkinsonian phenotype [253,254,255,270]. These results need to be interpreted with caution however as Ndufs4 deletion only partially decreases in complex I activity [270,271]. A more complete disruption of complex I activity, like that achieved by deletion of the catalytic subunit (Ndufs2), would be more informative.
The most compelling evidence for the involvement of mitochondria in PD pathogenesis is based upon an examination of the consequences of genetic mutations associated with relatively rare familial forms of the disease [272,273,274]. Many of these mutations modulate mitochondrial homeostasis [275,276,277,278,279,280,281,282,283], dynamics [284,285,286,287,288,289], redox status [105,290], and biogenesis [291,292].
Particularly intriguing is the role of parkin (PARK-2) and PTEN-induced kinase 1 (PINK1, PARK-6) in mitochondria quality control, especially because in SN DAergic neurons mitochondria have elevated oxidant stress, mtDNA damage, and turnover rates (see above). PINK1 (PARK-6) and parkin (PARK-2) cooperate in a pathway that tags damaged mitochondria for mitophagic degradation [293]. Briefly, PINK1 (PARK-6) is constitutively imported and degraded in healthy mitochondria, but upon mitochondrial damage, it accumulates on the OMM, where it recruits and activates parkin (PARK-2) [294]. Parkin (PARK-2), in turn, ubiquitinates OMM proteins and induces the formation of the autophagosome that will engulf the damaged mitochondria, leading to mitophagy [293,295,296]. Another way in which PINK1 (PARK-6) and Parkin (PARK-2) ensure mitochondrial quality control is through the generation of MDVs that contain damaged mitochondrial components targeted for lysosomal degradation [235]. Loss of function mutations in PINK1 (PARK-6) and parkin (PARK-2) mutations observed in familial PD patients suggest that a defect in the elimination (and the consequent accumulation) of dysfunctional mitochondria can increase the already elevated mitochondrial stress of SN DAergic neurons.
These familial mutations can also affect mitochondrial Ca2+ signaling. Deletion of PINK1 (PARK-6) is associated both with either decreased mitochondria Ca2+ uptake due to depolarization [297] or impaired mitochondrial Ca2+ efflux, which facilitates mitochondrial Ca2+ overload [298,299,300]; parkin (PARK-2) regulates the levels and the turnover of the MCUC regulators MICU1/2 [301]. In both cases, the disruption caused by deletion or loss of function mutations in these genes could be attributed to poor quality control. PD-linked mutations in leucine-rich repeat kinase 2 (LRRK2, PARK-8) increase the expression of MCU and MICU1 [302] and decrease mitochondrial Ca2+ efflux [303]. In zebrafish, inhibition of mitochondrial Ca2+ influx protects neurons against the effects of mutations mimicking the functional effects of those seen in PD patients [304,305], just as does inhibition of Cav1 Ca2+ channels responsible for mitochondrial Ca2+ influx in rodent SN DAergic neurons [102,105,306,307,308,309,310].
Another key site that is modulated by genetic mutations associated with PD is the MAM [215,291,311,312,313,314,315,316,317,318,319]. Dysregulation of MAMs has emerged as a key feature of neurodegenerative processes and PD in particular [318,320,321,322,323]. Many of the proteins encoded by genes mutated in familial PD regulate ER-mitochondria junctions, including αSYN (PARK-1/4) [215,311,316,317], Parkin (PARK-2) and PINK1 (PARK-6) [291,312,313,319,324], DJ-1(PARK-7) [314,315,319] and LRRK2 (PARK-8) [313,325]. Given the key role played by mitochondria in SN DAergic neurons, any dysregulation in the Ca2+ signals to the mitochondria could either impair the feed-forward mechanism that maintains the supply of ATP or exacerbate the already high oxidant burden experienced by the organelles.
One particularly bothersome aspect of this literature is that mice with PD-linked mutations do not develop a true parkinsonian phenotype. This is true for both the recessive mutations that are tightly linked to mitochondria (PARK-2, 6, 7) [326,327,328] and for the dominant mutations with more complex linkages to mitochondria [327,328]. Why this is the case is unclear, but, likely, human aging (the biggest risk factor for PD) is not faithfully captured in rodents.
8. Other Vulnerable Neuronal Populations
If Ca2+ and feedforward control of mitochondrial OXPHOS are the keys to the vulnerability of SN DAergic neurons in PD, other neuronal populations at-risk in PD should have a similar phenotype. Many other neurons, particularly in the brainstem, are vulnerable in PD [8,72,135]. In the Braak staging model, the earliest signs of Lewy pathology (LP) are in the dorsal motor nucleus of the vagus (DMV) [190,329,330]. As discussed elsewhere, the relationship between LP and neurodegeneration and death is far from clear [72,331]. In the SN, LP trails neurodegeneration [72,331]. Nevertheless, this line of study underscores the fact that several types of neurons distributed along the neuroaxis are vulnerable in PD, warranting a comparative analysis. In general, these other populations have not received the same level of attention as SN DAergic neurons. However, some intriguing similarities have already begun to emerge.
The cholinergic neurons of DMV are among the first neurons affected by LP, according to the Braak staging [330]. They form very long and branched axons that reach many gastro-intestinal-related organs, from the esophagus to the colon [332]. As with SN DA neurons, their firing activity has been described as a slow pacemaker, engaging various Ca2+ channels, including Cav1.2, Cav1.3, and Cav2 [333,334,335,336]. More importantly, DMV neurons manifest cytosolic Ca2+ oscillations and elevated mitochondrial oxidant stress (resembling SN DAergic neurons) [336,337,338]. Another vulnerable population of cholinergic neurons are in the pedunculopontine nucleus (PPN). PPN neurons are heterogeneous, being comprised of glutamatergic, cholinergic, and GABAergic neurons [339,340,341]. Cholinergic neurons are the most vulnerable [342,343,344]. Like SN DAergic and DMV cholinergic neurons, PPN cholinergic neurons are autonomous pacemakers with robust cytosolic Ca2+ oscillations (unpublished observations) and long, highly branched axons [345,346,347,348,349,350,351,352].
Two other PD vulnerable cell types have been studied in some depth. Adrenergic neurons in the locus coeruleus (LC) are among the first to degenerate in PD [190,353,354]. LC neurons show spontaneous rhythmic firing, whose frequency correlates with waking or sleeping states and sensory stimulation [355,356,357,358,359,360,361,362]. As in SN DA neurons, LC neurons engage L-type and T-type Ca2+ channels in pacemaking [128,363,364] and have low intrinsic cytosolic Ca2+ buffering and high levels of mitochondrial oxidant stress [128]. In addition, as other vulnerable neurons studied, LC neurons have long, highly branched axonal arbors [365,366,367,368]. Another vulnerable neuronal population resides in the raphe nuclei (RN). Again, these neurons have highly branched axonal arbors [369,370,371,372,373,374]. RN neurons are active during the waking state but slow down during sleep [375,376,377,378,379,380,381,382]. Spiking of RN neurons is sensitive to inhibition of Cav1 Ca2+ channels [383], but precisely why this is the case is unclear.
Thus, the available data indicates that an extensive axonal branching, autonomous pacemaking, and Cav1 channel-mediated feedforward control of mitochondrial OXPHOS (and the consequent mitochondrial oxidant stress) might be key features determining neuronal vulnerability in PD [8,72,132]. Instead, the neurotransmitter phenotype per se does not seem to represent an intrinsic risk factor in PD: not all DAergic, serotoninergic, adrenergic, and cholinergic neurons are vulnerable in the disease. However, it is noteworthy that the vulnerable neurons exert a widespread neuromodulatory role rather than releasing conventional fast neurotransmitters (i.e., glutamate and GABA) [72].
9. Conclusions and Future Directions
Ca2+ signaling plays a central role in many aspects of neuronal function. One under-appreciated role is in the control of neuronal bioenergetics. In SN DAergic neurons, Ca2+ entry through Cav1 Ca2+ channels couples activity to feedforward control of mitochondrial OXPHOS. This coupling has two apparently unintended consequences. One is a robust oscillation in cytosolic [Ca2+]; another is the excessive production of ROS by mitochondria. Both unintended consequences can have deleterious consequences over time (Figure 2 and Figure 3). This situation may be an example of antagonist pleiotropy [384,385]. Pacemaking-dependent Ca2+-mediated feed-forward stimulation of mitochondria should confer an advantage in the early stages of life when an animal (reprising the example used above) needs to escape predators or hunt for food and ultimately survive to mate. Only later in life, past reproductive age, this design may have negative consequences [141,386]. The average age of diagnosis with PD is about 60 years old [387]. As a consequence, it has only been relatively recently with the extension of the average lifespan that the incidence of PD has risen [388,389,390].
A fundamental question is then whether alleviating mitochondria oxidant stress could safely prevent or alleviate the progression of PD. Many of the early attempts at disease modification in PD have targeted mitochondrial ROS signaling, but all of these have failed to show efficacy [249,250,251]. Recently, epidemiological studies identified a correlation between PD risk and the use of DHPs Cav1 channel inhibitors [391,392,393,394,395], and preclinical studies supported this connection [102,306,307,308,309,310,396,397]. However, a Phase III clinical trial with the DHP isradipine did not show any benefit of the drug versus the placedo in slowing the progression of PD [398]. This trial may have failed for many reasons, but there are two obvious possibilities. One is that even in early-stage PD patients there has been a substantial loss of DAergic neurons and the processes driving the disease forward have changed to ones (e.g., inflammation) that will not be responsive to Cav1 channel inhibition. The epidemiological data supporting a protective role for DHPs invariably comes from presymptomatic patients that may be 5–10 years away from the typical age of PD diagnosis (~60 years of age). Unfortunately, the development of predictive biomarkers of disease onset and progression remains one of the main challenges facing the PD field [399,400]. The other (and to our mind more likely possibility) is that there was inadequate target engagement (Cav1 channel inhibition) with the twice daily, 5 mg immediate-release format isradipine pill that was used in the STEADY-PD III trial. After oral delivery, DHPs like isradipine are cleared within hours and pharmacokinetic modeling suggests that for most of the day plasma (and brain) isradipine concentrations were well below the threshold for protection determined in preclinical studies [306]. In retrospect, the use of a controlled release format of the drug that would have produced a sustained elevation in plasma (and brain) drug concentration, mimicking the preclinical studies, could have resulted in a different outcome.
As outlined above, there may be other Ca2+ channels that could be targeted in PD. For example, DAergic neurons derived from induced pluripotent stem cells from familial PD patients are protected from rotenone toxicity upon Cav3 channel inhibition [401], while knock-out of Cav2.3 channels protects mice from MPTP neurotoxicity [99]. Negative modulators of the MCUC [402] could decrease mitochondrial oxidant stress. Agonists of lysosomal Ca2+ channels could enhance lysosomal exocytosis and diminish αSYN accumulation [403]. However, all of these targets come with caveats given that these channels are widely distributed in the body and brain; as a consequence, it may be difficult to achieve enough biological effect with any one drug to alter disease course without causing intolerable side-effects. In this situation, intersectional approaches may prove worthwhile. That is, to target a combination of proteins in vulnerable neurons to achieve specificity of action, without bringing about unacceptable side-effects.
Funding
This work was supported by the JPB, IDP, and MJF Foundations and NINDS (grant P50NS047085).
Conflicts of Interest
The authors declare no conflict of interest, but D. J. Surmeier is a founding member of Dyad Therapeutics, Inc., a company focused on the development of disease-modifying drug combinations in PD.
References
- Clapham, D.E. Calcium signaling. Cell 2007, 131, 1047–1058. [Google Scholar] [CrossRef] [Green Version]
- Raffaello, A.; Mammucari, C.; Gherardi, G.; Rizzuto, R. Calcium at the Center of Cell Signaling: Interplay between Endoplasmic Reticulum, Mitochondria, and Lysosomes. Trends Biochem. Sci. 2016, 41, 1035–1049. [Google Scholar] [CrossRef] [Green Version]
- Rizzuto, R.; Pozzan, T. Microdomains of intracellular Ca2+: Molecular determinants and functional consequences. Physiol. Rev. 2006, 86, 369–408. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J.; Bootman, M.D.; Roderick, H.L. Calcium signalling: Dynamics, homeostasis and remodelling. Nat. Rev. Mol. Cell Biol. 2003, 4, 517–529. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Danese, A.; Missiroli, S.; Patergnani, S.; Pinton, P. Calcium Dynamics as a Machine for Decoding Signals. Trends Cell Biol. 2018, 28, 258–273. [Google Scholar] [CrossRef] [PubMed]
- Poewe, W.; Seppi, K.; Tanner, C.M.; Halliday, G.M.; Brundin, P.; Volkmann, J.; Schrag, A.E.; Lang, A.E. Parkinson disease. Nat. Rev. Dis. Primers 2017, 3, 17013. [Google Scholar] [CrossRef]
- Obeso, J.A.; Stamelou, M.; Goetz, C.G.; Poewe, W.; Lang, A.E.; Weintraub, D.; Burn, D.; Halliday, G.M.; Bezard, E.; Przedborski, S.; et al. Past, present, and future of Parkinson’s disease: A special essay on the 200th Anniversary of the Shaking Palsy. Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 1264–1310. [Google Scholar] [CrossRef]
- Sulzer, D.; Surmeier, D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2013, 28, 41–50. [Google Scholar] [CrossRef] [Green Version]
- Michel, P.P.; Hirsch, E.C.; Hunot, S. Understanding Dopaminergic Cell Death Pathways in Parkinson Disease. Neuron 2016, 90, 675–691. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Schumacker, P.T.; Guzman, J.D.; Ilijic, E.; Yang, B.; Zampese, E. Calcium and Parkinson’s disease. Biochem. Biophys. Res. Commun. 2017, 483, 1013–1019. [Google Scholar] [CrossRef] [Green Version]
- Catterall, W.A. Voltage-gated calcium channels. Cold Spring Harb. Perspect. Biol. 2011, 3, a003947. [Google Scholar] [CrossRef] [PubMed]
- Brini, M.; Cali, T.; Ottolini, D.; Carafoli, E. Neuronal calcium signaling: Function and dysfunction. Cell. Mol. Life Sci. 2014, 71, 2787–2814. [Google Scholar] [CrossRef] [PubMed]
- Berridge, M.J. Neuronal calcium signaling. Neuron 1998, 21, P13–P26. [Google Scholar] [CrossRef] [Green Version]
- Schwaller, B. Cytosolic Ca2+ buffers. Cold Spring Harb. Perspect. Biol. 2010, 2, a004051. [Google Scholar] [CrossRef] [PubMed]
- Zampese, E.; Pizzo, P. Intracellular organelles in the saga of Ca2+ homeostasis: Different molecules for different purposes? Cell. Mol. Life Sci. 2012, 69, 1077–1104. [Google Scholar] [CrossRef]
- Bagur, R.; Hajnóczky, G. Intracellular Ca2+ Sensing: Its Role in Calcium Homeostasis and Signaling. Mol. Cell 2017, 66, 780–788. [Google Scholar] [CrossRef] [Green Version]
- Brini, M.; Carafoli, E. The plasma membrane Ca2+ ATPase and the plasma membrane sodium calcium exchanger cooperate in the regulation of cell calcium. Cold Spring Harb. Perspect. Biol. 2011, 3. [Google Scholar] [CrossRef]
- Karagas, N.E.; Venkatachalam, K. Roles for the Endoplasmic Reticulum in Regulation of Neuronal Calcium Homeostasis. Cells 2019, 8, 1232. [Google Scholar] [CrossRef] [Green Version]
- Choi, Y.M.; Kim, S.H.; Chung, S.; Uhm, D.Y.; Park, M.K. Regional interaction of endoplasmic reticulum Ca2+ signals between soma and dendrites through rapid luminal Ca2+ diffusion. J. Neurosci. 2006, 26, 12127–12136. [Google Scholar] [CrossRef] [Green Version]
- Myoung Kyu, P.; Yu Mi, C.; Yun Kyung, K.; Petersen, O.H. The Endoplasmic Reticulum as an Integrator of Multiple Dendritic Events. Neuroscientist 2007, 14, 68–77. [Google Scholar] [CrossRef]
- Pizzo, P.; Pozzan, T. Mitochondria-endoplasmic reticulum choreography: Structure and signaling dynamics. Trends Cell Biol. 2007, 17, 511–517. [Google Scholar] [CrossRef] [PubMed]
- Nunnari, J.; Suomalainen, A. Mitochondria: In sickness and in health. Cell 2012, 148, 1145–1159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giorgi, C.; Marchi, S.; Pinton, P. The machineries, regulation and cellular functions of mitochondrial calcium. Nat. Rev. Mol. Cell Biol. 2018, 19, 713–730. [Google Scholar] [CrossRef] [PubMed]
- Elfawy, H.A.; Das, B. Crosstalk between mitochondrial dysfunction, oxidative stress, and age related neurodegenerative disease: Etiologies and therapeutic strategies. Life Sci. 2019, 218, 165–184. [Google Scholar] [CrossRef] [PubMed]
- Pizzo, P.; Drago, I.; Filadi, R.; Pozzan, T. Mitochondrial Ca2+ homeostasis: Mechanism, role, and tissue specificities. Pflug. Arch. 2012, 464, 3–17. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; De Stefani, D.; Raffaello, A.; Mammucari, C. Mitochondria as sensors and regulators of calcium signalling. Nat. Rev. Mol. Cell Biol. 2012, 13, 566–578. [Google Scholar] [CrossRef]
- Baughman, J.M.; Perocchi, F.; Girgis, H.S.; Plovanich, M.; Belcher-Timme, C.A.; Sancak, Y.; Bao, X.R.; Strittmatter, L.; Goldberger, O.; Bogorad, R.L.; et al. Integrative genomics identifies MCU as an essential component of the mitochondrial calcium uniporter. Nature 2011, 476, 341–345. [Google Scholar] [CrossRef] [Green Version]
- De Stefani, D.; Raffaello, A.; Teardo, E.; Szabo, I.; Rizzuto, R. A forty-kilodalton protein of the inner membrane is the mitochondrial calcium uniporter. Nature 2011, 476, 336–340. [Google Scholar] [CrossRef]
- Penna, E.; Espino, J.; De Stefani, D.; Rizzuto, R. The MCU complex in cell death. Cell Calcium 2018, 69, 73–80. [Google Scholar] [CrossRef]
- Kamer, K.J.; Mootha, V.K. The molecular era of the mitochondrial calcium uniporter. Nat. Rev. Mol. Cell Biol. 2015, 16, 545–553. [Google Scholar] [CrossRef]
- De Stefani, D.; Rizzuto, R.; Pozzan, T. Enjoy the Trip: Calcium in Mitochondria Back and Forth. Annu. Rev. Biochem. 2016, 85, 161–192. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Brini, M.; Murgia, M.; Pozzan, T. Microdomains with high Ca2+ close to IP3-sensitive channels that are sensed by neighboring mitochondria. Science 1993, 262, 744–747. [Google Scholar] [CrossRef] [PubMed]
- Rizzuto, R.; Pinton, P.; Carrington, W.; Fay, F.S.; Fogarty, K.E.; Lifshitz, L.M.; Tuft, R.A.; Pozzan, T. Close contacts with the endoplasmic reticulum as determinants of mitochondrial Ca2+ responses. Science 1998, 280, 1763–1766. [Google Scholar] [CrossRef] [PubMed]
- Csordas, G.; Renken, C.; Varnai, P.; Walter, L.; Weaver, D.; Buttle, K.F.; Balla, T.; Mannella, C.A.; Hajnoczky, G. Structural and functional features and significance of the physical linkage between ER and mitochondria. J. Cell Biol. 2006, 174, 915–921. [Google Scholar] [CrossRef] [Green Version]
- Csordas, G.; Varnai, P.; Golenar, T.; Roy, S.; Purkins, G.; Schneider, T.G.; Balla, T.; Hajnoczky, G. Imaging interorganelle contacts and local calcium dynamics at the ER-mitochondrial interface. Mol. Cell 2010, 39, 121–132. [Google Scholar] [CrossRef]
- Giacomello, M.; Drago, I.; Bortolozzi, M.; Scorzeto, M.; Gianelle, A.; Pizzo, P.; Pozzan, T. Ca2+ hot spots on the mitochondrial surface are generated by Ca2+ mobilization from stores, but not by activation of store-operated Ca2+ channels. Mol. Cell 2010, 38, 280–290. [Google Scholar] [CrossRef]
- Rizzuto, R.; Duchen, M.R.; Pozzan, T. Flirting in little space: The ER/mitochondria Ca2+ liaison. Sci. STKE Signal Transduct. Knowl. Environ. 2004, 2004, re1. [Google Scholar] [CrossRef]
- Hayashi, T.; Rizzuto, R.; Hajnoczky, G.; Su, T.-P. MAM: More than just a housekeeper. Trends Cell Biol. 2009, 19, 81–88. [Google Scholar] [CrossRef] [Green Version]
- Rowland, A.A.; Voeltz, G.K. Endoplasmic reticulum-mitochondria contacts: Function of the junction. Nat. Rev. Mol. Cell Biol. 2012, 13, 607–625. [Google Scholar] [CrossRef] [Green Version]
- Csordas, G.; Weaver, D.; Hajnoczky, G. Endoplasmic Reticulum-Mitochondrial Contactology: Structure and Signaling Functions. Trends Cell Biol. 2018, 28, 523–540. [Google Scholar] [CrossRef]
- Ashrafi, G.; de Juan-Sanz, J.; Farrell, R.J.; Ryan, T.A. Molecular Tuning of the Axonal Mitochondrial Ca2+ Uniporter Ensures Metabolic Flexibility of Neurotransmission. Neuron 2020, 105, 678–687.e5. [Google Scholar] [CrossRef] [PubMed]
- Boyman, L.; Williams, G.S.; Khananshvili, D.; Sekler, I.; Lederer, W.J. NCLX: The mitochondrial sodium calcium exchanger. J. Mol. Cell. Cardiol. 2013, 59, 205–213. [Google Scholar] [CrossRef] [Green Version]
- Palty, R.; Silverman, W.F.; Hershfinkel, M.; Caporale, T.; Sensi, S.L.; Parnis, J.; Nolte, C.; Fishman, D.; Shoshan-Barmatz, V.; Herrmann, S.; et al. NCLX is an essential component of mitochondrial Na+/Ca2+ exchange. Proc. Natl. Acad. Sci. USA 2010, 107, 436–441. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Young, K.W.; Bampton, E.T.; Pinon, L.; Bano, D.; Nicotera, P. Mitochondrial Ca2+ signalling in hippocampal neurons. Cell Calcium 2008, 43, 296–306. [Google Scholar] [CrossRef] [PubMed]
- Devine, M.J.; Kittler, J.T. Mitochondria at the neuronal presynapse in health and disease. Nat. Rev. Neurosci. 2018, 19, 63–80. [Google Scholar] [CrossRef] [PubMed]
- Friel, D.D. Mitochondria as regulators of stimulus-evoked calcium signals in neurons. Cell Calcium 2000, 28, 307–316. [Google Scholar] [CrossRef] [PubMed]
- Babcock, D.F.; Hille, B. Mitochondrial oversight of cellular Ca2+ signaling. Curr. Opin. Neurobiol. 1998, 8, 398–404. [Google Scholar] [CrossRef]
- Szalai, G.; Krishnamurthy, R.; Hajnóczky, G. Apoptosis driven by IP(3)-linked mitochondrial calcium signals. EMBO J. 1999, 18, 6349–6361. [Google Scholar] [CrossRef]
- Brenner, C.; Moulin, M. Physiological roles of the permeability transition pore. Circ. Res. 2012, 111, 1237–1247. [Google Scholar] [CrossRef] [Green Version]
- Nicholls, D.G.; Budd, S.L. Mitochondria and neuronal survival. Physiol. Rev. 2000, 80, 315–360. [Google Scholar] [CrossRef] [Green Version]
- Agarwal, A.; Wu, P.H.; Hughes, E.G.; Fukaya, M.; Tischfield, M.A.; Langseth, A.J.; Wirtz, D.; Bergles, D.E. Transient Opening of the Mitochondrial Permeability Transition Pore Induces Microdomain Calcium Transients in Astrocyte Processes. Neuron 2017, 93, 587–605.e7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Martínez-Reyes, I.; Chandel, N.S. Mitochondrial TCA cycle metabolites control physiology and disease. Nat. Commun. 2020, 11, 102. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Magistretti, P.J.; Allaman, I. A cellular perspective on brain energy metabolism and functional imaging. Neuron 2015, 86, 883–901. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J.; Guzman, J.N.; Sanchez, J.; Schumacker, P.T. Physiological phenotype and vulnerability in Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2012, 2, a009290. [Google Scholar] [CrossRef] [Green Version]
- Brookes, P.S.; Yoon, Y.; Robotham, J.L.; Anders, M.W.; Sheu, S.S. Calcium, ATP, and ROS: A mitochondrial love-hate triangle. Am. J. Physiol. Cell Physiol. 2004, 287, C817–C833. [Google Scholar] [CrossRef] [PubMed]
- Kann, O.; Kovacs, R. Mitochondria and neuronal activity. Am. J. Physiol. Cell Physiol. 2007, 292, C641–C657. [Google Scholar] [CrossRef]
- Griffiths, E.J.; Rutter, G.A. Mitochondrial calcium as a key regulator of mitochondrial ATP production in mammalian cells. Biochim. Biophys. Acta 2009, 1787, 1324–1333. [Google Scholar] [CrossRef] [Green Version]
- Rossi, A.; Pizzo, P.; Filadi, R. Calcium, mitochondria and cell metabolism: A functional triangle in bioenergetics. Biochim. Biophys. Acta Mol. Cell Res. 2019, 1866, 1068–1078. [Google Scholar] [CrossRef]
- Gellerich, F.N.; Gizatullina, Z.; Gainutdinov, T.; Muth, K.; Seppet, E.; Orynbayeva, Z.; Vielhaber, S. The control of brain mitochondrial energization by cytosolic calcium: The mitochondrial gas pedal. IUBMB Life 2013, 65, 180–190. [Google Scholar] [CrossRef]
- Llorente-Folch, I.; Rueda, C.B.; Pardo, B.; Szabadkai, G.; Duchen, M.R.; Satrustegui, J. The regulation of neuronal mitochondrial metabolism by calcium. J. Physiol. 2015, 593, 3447–3462. [Google Scholar] [CrossRef] [Green Version]
- Szibor, M.; Gizatullina, Z.; Gainutdinov, T.; Endres, T.; Debska-Vielhaber, G.; Kunz, M.; Karavasili, N.; Hallmann, K.; Schreiber, F.; Bamberger, A.; et al. Cytosolic, but not matrix, calcium is essential for adjustment of mitochondrial pyruvate supply. J. Biol. Chem. 2020, 295, 4383–4397. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Denton, R.M. Regulation of mitochondrial dehydrogenases by calcium ions. Biochim. Biophys. Acta 2009, 1787, 1309–1316. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rangaraju, V.; Calloway, N.; Ryan, T.A. Activity-driven local ATP synthesis is required for synaptic function. Cell 2014, 156, 825–835. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Verstreken, P.; Ly, C.V.; Venken, K.J.; Koh, T.W.; Zhou, Y.; Bellen, H.J. Synaptic mitochondria are critical for mobilization of reserve pool vesicles at Drosophila neuromuscular junctions. Neuron 2005, 47, 365–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Harris, J.J.; Jolivet, R.; Attwell, D. Synaptic energy use and supply. Neuron 2012, 75, 762–777. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hall, C.N.; Klein-Flugge, M.C.; Howarth, C.; Attwell, D. Oxidative phosphorylation, not glycolysis, powers presynaptic and postsynaptic mechanisms underlying brain information processing. J. Neurosci. 2012, 32, 8940–8951. [Google Scholar] [CrossRef]
- Chouhan, A.K.; Ivannikov, M.V.; Lu, Z.; Sugimori, M.; Llinas, R.R.; Macleod, G.T. Cytosolic calcium coordinates mitochondrial energy metabolism with presynaptic activity. J. Neurosci. 2012, 32, 1233–1243. [Google Scholar] [CrossRef] [Green Version]
- Pathak, D.; Shields, L.Y.; Mendelsohn, B.A.; Haddad, D.; Lin, W.; Gerencser, A.A.; Kim, H.; Brand, M.D.; Edwards, R.H.; Nakamura, K. The role of mitochondrially derived ATP in synaptic vesicle recycling. J. Biol. Chem. 2015, 290, 22325–22336. [Google Scholar] [CrossRef] [Green Version]
- Sheng, Z.H. The Interplay of Axonal Energy Homeostasis and Mitochondrial Trafficking and Anchoring. Trends Cell Biol. 2017, 27, 403–416. [Google Scholar] [CrossRef]
- Nichols, M.; Elustondo, P.A.; Warford, J.; Thirumaran, A.; Pavlov, E.V.; Robertson, G.S. Global ablation of the mitochondrial calcium uniporter increases glycolysis in cortical neurons subjected to energetic stressors. J. Cereb. Blood Flow Metab. 2017, 37, 3027–3041. [Google Scholar] [CrossRef] [Green Version]
- Raza, C.; Anjum, R.; Shakeel, N.U.A. Parkinson’s disease: Mechanisms, translational models and management strategies. Life Sci. 2019, 226, 77–90. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017, 18, 101–113. [Google Scholar] [CrossRef] [PubMed]
- Grace, A.A.; Onn, S.P. Morphology and electrophysiological properties of immunocytochemically identified rat dopamine neurons recorded in vitro. J. Neurosci. 1989, 9, 3463–3481. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sanghera, M.K.; Trulson, M.E.; German, D.C. Electrophysiological properties of mouse dopamine neurons: In vivo and in vitro studies. Neuroscience 1984, 12, 793–801. [Google Scholar] [CrossRef]
- Grace, A.A.; Bunney, B.S. Intracellular and extracellular electrophysiology of nigral dopaminergic neurons—1. Identification and characterization. Neuroscience 1983, 10, 301–315. [Google Scholar] [CrossRef]
- Hainsworth, A.H.; Röper, J.; Kapoor, R.; Ashcroft, F.M. Identification and electrophysiology of isolated pars-compacta neurons from guinea-pig substantia nigra. Neuroscience 1991, 43, 81–93. [Google Scholar] [CrossRef]
- Paladini, C.A.; Roeper, J. Generating bursts (and pauses) in the dopamine midbrain neurons. Neuroscience 2014, 282, 109–121. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Graves, S.M.; Shen, W. Dopaminergic modulation of striatal networks in health and Parkinson’s disease. Curr. Opin. Neurobiol. 2014, 29, 109–117. [Google Scholar] [CrossRef] [Green Version]
- Gerfen, C.R.; Surmeier, D.J. Modulation of striatal projection systems by dopamine. Annu. Rev. Neurosci. 2011, 34, 441–466. [Google Scholar] [CrossRef] [Green Version]
- Howe, M.W.; Dombeck, D.A. Rapid signalling in distinct dopaminergic axons during locomotion and reward. Nature 2016, 535, 505–510. [Google Scholar] [CrossRef]
- Yee, A.G.; Forbes, B.; Cheung, P.Y.; Martini, A.; Burrell, M.H.; Freestone, P.S.; Lipski, J. Action potential and calcium dependence of tonic somatodendritic dopamine release in the Substantia Nigra pars compacta. J. Neurochem. 2019, 148, 462–479. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Geffen, L.B.; Jessell, T.M.; Cuello, A.C.; Iversen, L.L. Release of dopamine from dendrites in rat substantia nigra. Nature 1976, 260, 258–260. [Google Scholar] [CrossRef] [PubMed]
- Chen, B.T.; Rice, M.E. Novel Ca2+ dependence and time course of somatodendritic dopamine release: Substantia nigra versus striatum. J. Neurosci. 2001, 21, 7841–7847. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Mallet, N.; Delgado, L.; Chazalon, M.; Miguelez, C.; Baufreton, J. Cellular and Synaptic Dysfunctions in Parkinson’s Disease: Stepping out of the Striatum. Cells 2019, 8, 1005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Collier, T.J.; Kanaan, N.M.; Kordower, J.H. Aging and Parkinson’s disease: Different sides of the same coin? Mov. Disord. Off. J. Mov. Disord. Soc. 2017, 32, 983–990. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Surmeier, D.J.; Halliday, G.M.; Simuni, T. Calcium, mitochondrial dysfunction and slowing the progression of Parkinson’s disease. Exp. Neurol. 2017, 298, 202–209. [Google Scholar] [CrossRef]
- Ping, H.X.; Shepard, P.D. Apamin-sensitive Ca(2+)-activated K+ channels regulate pacemaker activity in nigral dopamine neurons. Neuroreport 1996, 7, 809–814. [Google Scholar] [CrossRef]
- Shepard, P.D.; Stump, D. Nifedipine blocks apamin-induced bursting activity in nigral dopamine-containing neurons. Brain Res. 1999, 817, 104–109. [Google Scholar] [CrossRef]
- Puopolo, M.; Raviola, E.; Bean, B.P. Roles of subthreshold calcium current and sodium current in spontaneous firing of mouse midbrain dopamine neurons. J. Neurosci. 2007, 27, 645–656. [Google Scholar] [CrossRef] [Green Version]
- Neuhoff, H.; Neu, A.; Liss, B.; Roeper, J. I(h) channels contribute to the different functional properties of identified dopaminergic subpopulations in the midbrain. J. Neurosci. 2002, 22, 1290–1302. [Google Scholar] [CrossRef] [Green Version]
- Iyer, R.; Ungless, M.A.; Faisal, A.A. Calcium-activated SK channels control firing regularity by modulating sodium channel availability in midbrain dopamine neurons. Sci. Rep. 2017, 7. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shi, W.X. Electrophysiological characteristics of dopamine neurons: A 35-year update. J. Neural Transm. Suppl. 2009, 73, 103–119. [Google Scholar] [CrossRef]
- Yung, W.H.; Häusser, M.A.; Jack, J.J. Electrophysiology of dopaminergic and non-dopaminergic neurones of the guinea-pig substantia nigra pars compacta in vitro. J. Physiol. 1991, 436, 643–667. [Google Scholar] [CrossRef] [PubMed]
- Gantz, S.C.; Ford, C.P.; Morikawa, H.; Williams, J.T. The Evolving Understanding of Dopamine Neurons in the Substantia Nigra and Ventral Tegmental Area. Annu. Rev. Physiol. 2018, 80, 219–241. [Google Scholar] [CrossRef] [PubMed]
- Nedergaard, S.; Flatman, J.A.; Engberg, I. Nifedipine- and omega-conotoxin-sensitive Ca2+ conductances in guinea-pig substantia nigra pars compacta neurones. J. Physiol. 1993, 466, 727–747. [Google Scholar]
- Mercuri, N.B.; Bonci, A.; Calabresi, P.; Stratta, F.; Stefani, A.; Bernardi, G. Effects of dihydropyridine calcium antagonists on rat midbrain dopaminergic neurones. Br. J. Pharmacol. 1994, 113, 831–838. [Google Scholar] [CrossRef] [Green Version]
- Philippart, F.; Destreel, G.; Merino-Sepúlveda, P.; Henny, P.; Engel, D.; Seutin, V. Differential Somatic Ca2+ Channel Profile in Midbrain Dopaminergic Neurons. J. Neurosci. Off. J. Soc. Neurosci. 2016, 36, 7234–7245. [Google Scholar] [CrossRef] [Green Version]
- Ortner, N.J.; Bock, G.; Dougalis, A.; Kharitonova, M.; Duda, J.; Hess, S.; Tuluc, P.; Pomberger, T.; Stefanova, N.; Pitterl, F.; et al. Lower Affinity of Isradipine for L-Type Ca2+ Channels during Substantia Nigra Dopamine Neuron-Like Activity: Implications for Neuroprotection in Parkinson’s Disease. J. Neurosci. 2017, 37, 6761–6777. [Google Scholar] [CrossRef] [Green Version]
- Benkert, J.; Hess, S.; Roy, S.; Beccano-Kelly, D.; Wiederspohn, N.; Duda, J.; Simons, C.; Patil, K.; Gaifullina, A.; Mannal, N.; et al. Cav2.3 channels contribute to dopaminergic neuron loss in a model of Parkinson’s disease. Nat. Commun. 2019, 10. [Google Scholar] [CrossRef]
- Dufour, M.A.; Woodhouse, A.; Goaillard, J.-M. Somatodendritic ion channel expression in substantia nigra pars compacta dopaminergic neurons across postnatal development. J. Neurosci. Res. 2014, 92, 981–999. [Google Scholar] [CrossRef]
- Guzman, J.N.; Sanchez-Padilla, J.; Chan, C.S.; Surmeier, D.J. Robust pacemaking in substantia nigra dopaminergic neurons. J. Neurosci. 2009, 29, 11011–11019. [Google Scholar] [CrossRef] [PubMed]
- Chan, C.S.; Guzman, J.N.; Ilijic, E.; Mercer, J.N.; Rick, C.; Tkatch, T.; Meredith, G.E.; Surmeier, D.J. ‘Rejuvenation’ protects neurons in mouse models of Parkinson’s disease. Nature 2007, 447, 1081–1086. [Google Scholar] [CrossRef] [PubMed]
- Hage, T.A.; Khaliq, Z.M. Tonic firing rate controls dendritic Ca2+ signaling and synaptic gain in substantia nigra dopamine neurons. J. Neurosci. 2015, 35, 5823–5836. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guzman, J.N.; Ilijic, E.; Yang, B.; Sanchez-Padilla, J.; Wokosin, D.; Galtieri, D.; Kondapalli, J.; Schumacker, P.T.; Surmeier, D.J. Systemic isradipine treatment diminishes calcium-dependent mitochondrial oxidant stress. J. Clin. Investig. 2018, 128, 2266–2280. [Google Scholar] [CrossRef] [Green Version]
- Guzman, J.N.; Sanchez-Padilla, J.; Wokosin, D.; Kondapalli, J.; Ilijic, E.; Schumacker, P.T.; Surmeier, D.J. Oxidant stress evoked by pacemaking in dopaminergic neurons is attenuated by DJ-1. Nature 2010, 468, 696–700. [Google Scholar] [CrossRef] [Green Version]
- Dryanovski, D.I.; Guzman, J.N.; Xie, Z.; Galteri, D.J.; Volpicelli-Daley, L.A.; Lee, V.M.; Miller, R.J.; Schumacker, P.T.; Surmeier, D.J. Calcium entry and alpha-synuclein inclusions elevate dendritic mitochondrial oxidant stress in dopaminergic neurons. J. Neurosci. 2013, 33, 10154–10164. [Google Scholar] [CrossRef] [Green Version]
- Wilson, C.J.; Callaway, J.C. Coupled oscillator model of the dopaminergic neuron of the substantia nigra. J. Neurophysiol. 2000, 83, 3084–3100. [Google Scholar] [CrossRef]
- Weiss, N.; Zamponi, G.W. T-type calcium channels: From molecule to therapeutic opportunities. Int. J. Biochem. Cell Biol. 2019, 108, 34–39. [Google Scholar] [CrossRef]
- Perez-Reyes, E. Molecular Physiology of Low-Voltage-Activated T-type Calcium Channels. Physiol. Rev. 2003, 83, 117–161. [Google Scholar] [CrossRef] [Green Version]
- Galtieri, D.J.; Estep, C.M.; Wokosin, D.L.; Traynelis, S.; Surmeier, D.J. Pedunculopontine glutamatergic neurons control spike patterning in substantia nigra dopaminergic neurons. Elife 2017, 6. [Google Scholar] [CrossRef]
- Evans, R.C.; Zhu, M.; Khaliq, Z.M. Dopamine Inhibition Differentially Controls Excitability of Substantia Nigra Dopamine Neuron Subpopulations through T-Type Calcium Channels. J. Neurosci. 2017, 37, 3704–3720. [Google Scholar] [CrossRef] [PubMed]
- Wolfart, J.; Roeper, J. Selective coupling of T-type calcium channels to SK potassium channels prevents intrinsic bursting in dopaminergic midbrain neurons. J. Neurosci. 2002, 22, 3403–3413. [Google Scholar] [CrossRef] [Green Version]
- Lipscombe, D.; Helton, T.D.; Xu, W. L-type calcium channels: The low down. J. Neurophysiol. 2004, 92, 2633–2641. [Google Scholar] [CrossRef] [PubMed]
- Calin-Jageman, I.; Lee, A. Ca(v)1 L-type Ca2+ channel signaling complexes in neurons. J. Neurochem. 2008, 105, 573–583. [Google Scholar] [CrossRef] [PubMed]
- Takada, M.; Kang, Y.; Imanishi, M. Immunohistochemical localization of voltage-gated calcium channels in substantia nigra dopamine neurons. Eur. J. Neurosci. 2001, 13, 757–762. [Google Scholar] [CrossRef]
- Koschak, A.; Reimer, D.; Huber, I.; Grabner, M.; Glossmann, H.; Engel, J.; Striessnig, J. alpha 1D (Cav1.3) subunits can form l-type Ca2+ channels activating at negative voltages. J. Biol. Chem. 2001, 276, 22100–22106. [Google Scholar] [CrossRef] [Green Version]
- Scholze, A.; Plant, T.D.; Dolphin, A.C.; Nürnberg, B. Functional expression and characterization of a voltage-gated CaV1.3 (alpha1D) calcium channel subunit from an insulin-secreting cell line. Mol. Endocrinol. 2001, 15, 1211–1221. [Google Scholar] [CrossRef] [Green Version]
- Putzier, I.; Kullmann, P.H.; Horn, J.P.; Levitan, E.S. Cav1.3 channel voltage dependence, not Ca2+ selectivity, drives pacemaker activity and amplifies bursts in nigral dopamine neurons. J. Neurosci. 2009, 29, 15414–15419. [Google Scholar] [CrossRef]
- Fujimura, K.; Matsuda, Y. Autogenous oscillatory potentials in neurons of the guinea pig substantia nigra pars compacta in vitro. Neurosci. Lett. 1989, 104, 53–57. [Google Scholar] [CrossRef]
- Kang, Y.; Kitai, S.T. Calcium spike underlying rhytmic firing in dopaminergic neurons of the rat substantia nigra. Neurosci. Res. 1993, 18, 195–207. [Google Scholar] [CrossRef]
- Dragicevic, E.; Poetschke, C.; Duda, J.; Schlaudraff, F.; Lammel, S.; Schiemann, J.; Fauler, M.; Hetzel, A.; Watanabe, M.; Lujan, R.; et al. Cav1.3 channels control D2-autoreceptor responses via NCS-1 in substantia nigra dopamine neurons. Brain J. Neurol. 2014, 137, 2287–2302. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khaliq, Z.M.; Bean, B.P. Pacemaking in Dopaminergic Ventral Tegmental Area Neurons: Depolarizing Drive from Background and Voltage-Dependent Sodium Conductances. J. Neurosci. 2010, 30, 7401–7413. [Google Scholar] [CrossRef] [PubMed]
- Foehring, R.C.; Zhang, X.F.; Lee, J.C.; Callaway, J.C. Endogenous calcium buffering capacity of substantia nigral dopamine neurons. J. Neurophysiol. 2009, 102, 2326–2333. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shepard, P.D.; Bunney, B.S. Repetitive firing properties of putative dopamine-containing neurons in vitro: Regulation by an apamin-sensitive Ca2+-activated K+ conductance. Exp. Brain Res. 1991, 86, 141–150. [Google Scholar] [CrossRef]
- de Vrind, V.; Scuvee-Moreau, J.; Drion, G.; Hmaied, C.; Philippart, F.; Engel, D.; Seutin, V. Interactions between calcium channels and SK channels in midbrain dopamine neurons and their impact on pacemaker regularity: Contrasting roles of N- and L-type channels. Eur. J. Pharmacol. 2016, 788, 274–279. [Google Scholar] [CrossRef]
- Verkhratsky, A.; Shmigol, A. Calcium-induced calcium release in neurones. Cell Calcium 1996, 19, 1–14. [Google Scholar] [CrossRef]
- Kim, S.; Yun, H.-M.; Baik, J.-H.; Chung, K.C.; Nah, S.-Y.; Rhim, H. Functional Interaction of Neuronal Cav1.3 L-type Calcium Channel with Ryanodine Receptor Type 2 in the Rat Hippocampus. J. Biol. Chem. 2007, 282, 32877–32889. [Google Scholar] [CrossRef] [Green Version]
- Sanchez-Padilla, J.; Guzman, J.N.; Ilijic, E.; Kondapalli, J.; Galtieri, D.J.; Yang, B.; Schieber, S.; Oertel, W.; Wokosin, D.; Schumacker, P.T.; et al. Mitochondrial oxidant stress in locus coeruleus is regulated by activity and nitric oxide synthase. Nat. Neurosci. 2014, 17, 832–840. [Google Scholar] [CrossRef] [Green Version]
- Aumann, T.; Horne, M. Activity-dependent regulation of the dopamine phenotype in substantia nigra neurons. J. Neurochem. 2012, 121, 497–515. [Google Scholar] [CrossRef]
- Aumann, T.D.; Egan, K.; Lim, J.; Boon, W.C.; Bye, C.R.; Chua, H.K.; Baban, N.; Parish, C.L.; Bobrovskaya, L.; Dickson, P.; et al. Neuronal activity regulates expression of tyrosine hydroxylase in adult mouse substantia nigra pars compacta neurons. J. Neurochem. 2011, 116, 646–658. [Google Scholar] [CrossRef]
- Menezes, A.; Zeman, R.; Sabban, E. Involvement of intracellular or extracellular calcium in activation of tyrosine hydroxylase gene expression in PC12 cells. J. Neurochem. 1996, 67, 2316–2324. [Google Scholar] [CrossRef] [PubMed]
- Bolam, J.P.; Pissadaki, E.K. Living on the edge with too many mouths to feed: Why dopamine neurons die. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 1478–1483. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pissadaki, E.K.; Bolam, J.P. The energy cost of action potential propagation in dopamine neurons: Clues to susceptibility in Parkinson’s disease. Front. Comput. Neurosci. 2013, 7, 13. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Diederich, N.J.; James Surmeier, D.; Uchihara, T.; Grillner, S.; Goetz, C.G. Parkinson’s disease: Is it a consequence of human brain evolution? Mov. Disord. Off. J. Mov. Disord. Soc. 2019, 34, 453–459. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giguère, N.; Burke Nanni, S.; Trudeau, L.-E. On Cell Loss and Selective Vulnerability of Neuronal Populations in Parkinson’s Disease. Front. Neurol. 2018, 9. [Google Scholar] [CrossRef]
- De Vos, K.J.; Grierson, A.J.; Ackerley, S.; Miller, C.C. Role of axonal transport in neurodegenerative diseases. Annu. Rev. Neurosci. 2008, 31, 151–173. [Google Scholar] [CrossRef]
- Millecamps, S.; Julien, J.P. Axonal transport deficits and neurodegenerative diseases. Nat. Rev. Neurosci. 2013, 14, 161–176. [Google Scholar] [CrossRef]
- Gennerich, A.; Vale, R.D. Walking the walk: How kinesin and dynein coordinate their steps. Curr. Opin. Cell Biol. 2009, 21, 59–67. [Google Scholar] [CrossRef] [Green Version]
- Maday, S.; Twelvetrees, A.E.; Moughamian, A.J.; Holzbaur, E.L.F. Axonal transport: Cargo-specific mechanisms of motility and regulation. Neuron 2014, 84, 292–309. [Google Scholar] [CrossRef] [Green Version]
- Pease, S.E.; Segal, R.A. Preserve and protect: Maintaining axons within functional circuits. Trends Neurosci. 2014, 37, 572–582. [Google Scholar] [CrossRef] [Green Version]
- Surmeier, D.J.; Schumacker, P.T. Calcium, bioenergetics, and neuronal vulnerability in Parkinson’s disease. J. Biol. Chem. 2013, 288, 10736–10741. [Google Scholar] [CrossRef] [Green Version]
- Andén, N.E.; Hfuxe, K.; Hamberger, B.; Hökfelt, T. A quantitative study on the nigro-neostriatal dopamine neuron system in the rat. Acta Physiol. Scand. 1966, 67, 306–312. [Google Scholar] [CrossRef]
- Matsuda, W.; Furuta, T.; Nakamura, K.C.; Hioki, H.; Fujiyama, F.; Arai, R.; Kaneko, T. Single nigrostriatal dopaminergic neurons form widely spread and highly dense axonal arborizations in the neostriatum. J. Neurosci. 2009, 29, 444–453. [Google Scholar] [CrossRef] [Green Version]
- Arbuthnott, G.W.; Wickens, J. Space, time and dopamine. Trends Neurosci. 2007, 30, 62–69. [Google Scholar] [CrossRef]
- Gauthier, J.; Parent, M.; Lévesque, M.; Parent, A. The axonal arborization of single nigrostriatal neurons in rats. Brain Res. 1999, 834, 228–232. [Google Scholar] [CrossRef]
- Fallon, J.H. Collateralization of monoamine neurons: Mesotelencephalic dopamine projections to caudate, septum, and frontal cortex. J. Neurosci. 1981, 1, 1361–1368. [Google Scholar] [CrossRef]
- Loughlin, S.E.; Fallon, J.H. Substantia nigra and ventral tegmental area projections to cortex: Topography and collateralization. Neuroscience 1984, 11, 425–435. [Google Scholar] [CrossRef]
- Kawaguchi, Y.; Wilson, C.J.; Emson, P.C. Projection subtypes of rat neostriatal matrix cells revealed by intracellular injection of biocytin. J. Neurosci. 1990, 10, 3421–3428. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wu, Y.; Richard, S.; Parent, A. The organization of the striatal output system: A single-cell juxtacellular labeling study in the rat. Neurosci. Res. 2000, 38, 49–62. [Google Scholar] [CrossRef]
- Garcia-Ruiz, P.J.; Espay, A.J. Parkinson Disease: An Evolutionary Perspective. Front. Neurol. 2017, 8, 157. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pacelli, C.; Giguere, N.; Bourque, M.J.; Levesque, M.; Slack, R.S.; Trudeau, L.E. Elevated Mitochondrial Bioenergetics and Axonal Arborization Size Are Key Contributors to the Vulnerability of Dopamine Neurons. Curr. Biol. 2015, 25, 2349–2360. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Giguere, N.; Delignat-Lavaud, B.; Herborg, F.; Voisin, A.; Li, Y.; Jacquemet, V.; Anand-Srivastava, M.; Gether, U.; Giros, B.; Trudeau, L.E. Increased vulnerability of nigral dopamine neurons after expansion of their axonal arborization size through D2 dopamine receptor conditional knockout. PLoS Genet. 2019, 15, e1008352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Balaban, R.S. Regulation of oxidative phosphorylation in the mammalian cell. Am. J. Physiol. Cell Physiol. 1990, 258, C377–C389. [Google Scholar] [CrossRef] [PubMed]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. I. Kinetics of oxygen utilization. J. Biol. Chem. 1955, 217, 383–393. [Google Scholar] [PubMed]
- Chance, B.; Williams, G.R. Respiratory enzymes in oxidative phosphorylation. VI. The effects of adenosine diphosphate on azide-treated mitochondria. J. Biol. Chem. 1956, 221, 477–489. [Google Scholar]
- Lardy, H.A.; Wellman, H. Oxidative phosphorylations; rôle of inorganic phosphate and acceptor systems in control of metabolic rates. J. Biol. Chem. 1952, 195, 215–224. [Google Scholar]
- Röper, J.; Ashcroft, F.M. Metabolic inhibition and low internal ATP activate K-ATP channels in rat dopaminergic substantia nigra neurones. Pflug. Arch. 1995, 430, 44–54. [Google Scholar] [CrossRef]
- Marinelli, S.; Bernardi, G.; Giacomini, P.; Mercuri, N.B. Pharmacological identification of the K+ currents mediating the hypoglycemic hyperpolarization of rat midbrain dopaminergic neurones. Neuropharmacology 2000, 39, 1021–1028. [Google Scholar] [CrossRef]
- Viola, H.M.; Arthur, P.G.; Hool, L.C. Evidence for regulation of mitochondrial function by the L-type Ca2+ channel in ventricular myocytes. J. Mol. Cell. Cardiol. 2009, 46, 1016–1026. [Google Scholar] [CrossRef]
- Díaz-Vegas, A.R.; Cordova, A.; Valladares, D.; Llanos, P.; Hidalgo, C.; Gherardi, G.; De Stefani, D.; Mammucari, C.; Rizzuto, R.; Contreras-Ferrat, A.; et al. Mitochondrial Calcium Increase Induced by RyR1 and IP3R Channel Activation After Membrane Depolarization Regulates Skeletal Muscle Metabolism. Front. Physiol. 2018, 9, 791. [Google Scholar] [CrossRef] [Green Version]
- Liss, B.; Haeckel, O.; Wildmann, J.; Miki, T.; Seino, S.; Roeper, J. K-ATP channels promote the differential degeneration of dopaminergic midbrain neurons. Nat. Neurosci. 2005, 8, 1742–1751. [Google Scholar] [CrossRef]
- Guatteo, E.; Marinelli, S.; Geracitano, R.; Tozzi, A.; Federici, M.; Bernardi, G.; Mercuri, N.B. Dopamine-containing neurons are silenced by energy deprivation: A defensive response or beginning of cell death? Neurotoxicology 2005, 26, 857–868. [Google Scholar] [CrossRef]
- Hotka, M.; Cagalinec, M.; Hilber, K.; Hool, L.; Boehm, S.; Kubista, H. L-type Ca2+ channel-mediated Ca2+ influx adjusts neuronal mitochondrial function to physiological and pathophysiological conditions. Sci. Signal. 2020, 13. [Google Scholar] [CrossRef]
- Graves, S.M.; Xie, Z.; Stout, K.A.; Zampese, E.; Burbulla, L.F.; Shih, J.C.; Kondapalli, J.; Patriarchi, T.; Tian, L.; Brichta, L.; et al. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat. Neurosci. 2020, 23, 15–20. [Google Scholar] [CrossRef]
- Bisaglia, M.; Filograna, R.; Beltramini, M.; Bubacco, L. Are dopamine derivatives implicated in the pathogenesis of Parkinson’s disease? Ageing Res. Rev. 2014, 13, 107–114. [Google Scholar] [CrossRef]
- Asanuma, M.; Miyazaki, I.; Diaz-Corrales, F.J.; Ogawa, N. Quinone formation as dopaminergic neuron-specific oxidative stress in the pathogenesis of sporadic Parkinson’s disease and neurotoxin-induced parkinsonism. Acta Med. Okayama 2004, 58, 221–233. [Google Scholar]
- Burke, W.J.; Li, S.W.; Williams, E.A.; Nonneman, R.; Zahm, D.S. 3,4-Dihydroxyphenylacetaldehyde is the toxic dopamine metabolite in vivo: Implications for Parkinson’s disease pathogenesis. Brain Res. 2003, 989, 205–213. [Google Scholar] [CrossRef]
- Mosharov, E.V.; Larsen, K.E.; Kanter, E.; Phillips, K.A.; Wilson, K.; Schmitz, Y.; Krantz, D.E.; Kobayashi, K.; Edwards, R.H.; Sulzer, D. Interplay between cytosolic dopamine, calcium, and alpha-synuclein causes selective death of substantia nigra neurons. Neuron 2009, 62, 218–229. [Google Scholar] [CrossRef] [Green Version]
- Post, M.R.; Lieberman, O.J.; Mosharov, E.V. Can Interactions Between alpha-Synuclein, Dopamine and Calcium Explain Selective Neurodegeneration in Parkinson’s Disease? Front. Neurosci. 2018, 12, 161. [Google Scholar] [CrossRef]
- Segura-Aguilar, J.; Paris, I.; Munoz, P.; Ferrari, E.; Zecca, L.; Zucca, F.A. Protective and toxic roles of dopamine in Parkinson’s disease. J. Neurochem. 2014, 129, 898–915. [Google Scholar] [CrossRef]
- Zahid, M.; Saeed, M.; Yang, L.; Beseler, C.; Rogan, E.; Cavalieri, E.L. Formation of dopamine quinone-DNA adducts and their potential role in the etiology of Parkinson’s disease. IUBMB Life 2011, 63, 1087–1093. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Jain, M.K.; Bhat, R. Modulation of human alpha-synuclein aggregation by a combined effect of calcium and dopamine. Neurobiol. Dis. 2014, 63, 115–128. [Google Scholar] [CrossRef] [PubMed]
- Burke, W.J.; Kumar, V.B.; Pandey, N.; Panneton, W.M.; Gan, Q.; Franko, M.W.; O’Dell, M.; Li, S.W.; Pan, Y.; Chung, H.D.; et al. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008, 115, 193–203. [Google Scholar] [CrossRef] [PubMed]
- Conway, K.A.; Rochet, J.C.; Bieganski, R.M.; Lansbury, P.T.J. Kinetic stabilization of the alpha-synuclein protofibril by a dopamine-alpha-synuclein adduct. Science 2001, 294, 1346–1349. [Google Scholar] [CrossRef]
- Burbulla, L.F.; Song, P.; Mazzulli, J.R.; Zampese, E.; Wong, Y.C.; Jeon, S.; Santos, D.P.; Blanz, J.; Obermaier, C.D.; Strojny, C.; et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science 2017, 357, 1255–1261. [Google Scholar] [CrossRef] [Green Version]
- Hirsch, E.; Graybiel, A.M.; Agid, Y.A. Melanized dopaminergic neurons are differentially susceptible to degeneration in Parkinson’s disease. Nature 1988, 334, 345–348. [Google Scholar] [CrossRef]
- Liang, C.L.; Sinton, C.M.; German, D.C. Midbrain dopaminergic neurons in the mouse: Co-localization with Calbindin-D28K and calretinin. Neuroscience 1996, 75, 523–533. [Google Scholar] [CrossRef]
- Fu, Y.; Yuan, Y.; Halliday, G.; Rusznák, Z.; Watson, C.; Paxinos, G. A cytoarchitectonic and chemoarchitectonic analysis of the dopamine cell groups in the substantia nigra, ventral tegmental area, and retrorubral field in the mouse. Brain Struct. Funct. 2012, 217, 591–612. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. I. Nigrosomes and the nigral matrix, a compartmental organization based on calbindin D(28K) immunohistochemistry. Brain J. Neurol. 1999, 122 Pt 8, 1421–1436. [Google Scholar] [CrossRef] [Green Version]
- Yamada, T.; McGeer, P.L.; Baimbridge, K.G.; McGeer, E.G. Relative sparing in Parkinson’s disease of substantia nigra dopamine neurons containing calbindin-D28K. Brain Res. 1990, 526, 303–307. [Google Scholar] [CrossRef]
- Liang, C.L.; Sinton, C.M.; Sonsalla, P.K.; German, D.C. Midbrain dopaminergic neurons in the mouse that contain calbindin-D28k exhibit reduced vulnerability to MPTP-induced neurodegeneration. Neurodegeneration 1996, 5, 313–318. [Google Scholar] [CrossRef] [PubMed]
- Gaspar, P.; Ben Jelloun, N.; Febvret, A. Sparing of the dopaminergic neurons containing calbindin-D28k and of the dopaminergic mesocortical projections in weaver mutant mice. Neuroscience 1994, 61, 293–305. [Google Scholar] [CrossRef]
- Mouatt-Prigent, A.; Agid, Y.; Hirsch, E.C. Does the calcium binding protein calretinin protect dopaminergic neurons against degeneration in Parkinson’s disease? Brain Res. 1994, 668, 62–70. [Google Scholar] [CrossRef]
- Tsuboi, K.; Kimber, T.A.; Shults, C.W. Calretinin-containing axons and neurons are resistant to an intrastriatal 6-hydroxydopamine lesion. Brain Res. 2000, 866, 55–64. [Google Scholar] [CrossRef]
- Damier, P.; Hirsch, E.C.; Agid, Y.; Graybiel, A.M. The substantia nigra of the human brain. II. Patterns of loss of dopamine-containing neurons in Parkinson’s disease. Brain J. Neurol. 1999, 122 Pt 8, 1437–1448. [Google Scholar] [CrossRef]
- German, D.C.; Manaye, K.F.; Sonsalla, P.K.; Brooks, B.A. Midbrain dopaminergic cell loss in Parkinson’s disease and MPTP-induced parkinsonism: Sparing of calbindin-D28k-containing cells. Ann. N. Y. Acad. Sci. 1992, 648, 42–62. [Google Scholar] [CrossRef]
- Nagarajan, A.; Ning, Y.; Reisner, K.; Buraei, Z.; Larsen, J.P.; Hobert, O.; Doitsidou, M. Progressive degeneration of dopaminergic neurons through TRP channel-induced cell death. J. Neurosci. 2014, 34, 5738–5746. [Google Scholar] [CrossRef]
- Spillantini, M.G.; Crowther, R.A.; Jakes, R.; Hasegawa, M.; Goedert, M. alpha-Synuclein in filamentous inclusions of Lewy bodies from Parkinson’s disease and dementia with lewy bodies. Proc. Natl. Acad. Sci. USA 1998, 95, 6469–6473. [Google Scholar] [CrossRef] [Green Version]
- Spillantini, M.G.; Schmidt, M.L.; Lee, V.M.; Trojanowski, J.Q.; Jakes, R.; Goedert, M. Alpha-synuclein in Lewy bodies. Nature 1997, 388, 839–840. [Google Scholar] [CrossRef]
- Braak, H.; Del Tredici, K.; Rüb, U.; de Vos, R.A.; Jansen Steur, E.N.; Braak, E. Staging of brain pathology related to sporadic Parkinson’s disease. Neurobiol. Aging 2003, 24, 197–211. [Google Scholar] [CrossRef]
- Park, S.M.; Jung, H.Y.; Kim, T.D.; Park, J.H.; Yang, C.H.; Kim, J. Distinct roles of the N-terminal-binding domain and the C-terminal-solubilizing domain of alpha-synuclein, a molecular chaperone. J. Biol. Chem. 2002, 277, 28512–28520. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nath, S.; Goodwin, J.; Engelborghs, Y.; Pountney, D.L. Raised calcium promotes alpha-synuclein aggregate formation. Mol. Cell. Neurosci. 2011, 46, 516–526. [Google Scholar] [CrossRef] [PubMed]
- Lowe, R.; Pountney, D.L.; Jensen, P.H.; Gai, W.P.; Voelcker, N.H. Calcium(II) selectively induces alpha-synuclein annular oligomers via interaction with the C-terminal domain. Protein Sci. 2004, 13, 3245–3252. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Follett, J.; Darlow, B.; Wong, M.B.; Goodwin, J.; Pountney, D.L. Potassium depolarization and raised calcium induces alpha-synuclein aggregates. Neurotox. Res. 2013, 23, 378–392. [Google Scholar] [CrossRef]
- Nielsen, M.S.; Vorum, H.; Lindersson, E.; Jensen, P.H. Ca2+ binding to alpha-synuclein regulates ligand binding and oligomerization. J. Biol. Chem. 2001, 276, 22680–22684. [Google Scholar] [CrossRef] [Green Version]
- Han, J.Y.; Choi, T.S.; Kim, H.I. Molecular Role of Ca2+ and Hard Divalent Metal Cations on Accelerated Fibrillation and Interfibrillar Aggregation of alpha-Synuclein. Sci. Rep. 2018, 8, 1895. [Google Scholar] [CrossRef] [Green Version]
- Martinez, J.; Moeller, I.; Erdjument-Bromage, H.; Tempst, P.; Lauring, B. Parkinson’s disease-associated alpha-synuclein is a calmodulin substrate. J. Biol. Chem. 2003, 278, 17379–17387. [Google Scholar] [CrossRef] [Green Version]
- Tamamizu-Kato, S.; Kosaraju, M.G.; Kato, H.; Raussens, V.; Ruysschaert, J.M.; Narayanaswami, V. Calcium-triggered membrane interaction of the alpha-synuclein acidic tail. Biochemistry 2006, 45, 10947–10956. [Google Scholar] [CrossRef] [Green Version]
- Rcom-H’cheo-Gauthier, A.N.; Meedeniya, A.C.; Pountney, D.L. Calcipotriol inhibits alpha-synuclein aggregation in SH-SY5Y neuroblastoma cells by a Calbindin-D28k-dependent mechanism. J. Neurochem. 2017, 141, 263–274. [Google Scholar] [CrossRef] [Green Version]
- Liu, J.; Liu, M.C.; Wang, K.K. Calpain in the CNS: From synaptic function to neurotoxicity. Sci. Signal. 2008, 1, re1. [Google Scholar] [CrossRef]
- Togari, A.; Ichikawa, S.; Nagatsu, T. Activation of tyrosine hydroxylase by Ca2+-dependent neutral protease, calpain. Biochem. Biophys. Res. Commun. 1986, 134, 749–754. [Google Scholar] [CrossRef]
- Kiuchi, K.; Kiuchi, K.; Titani, K.; Fujita, K.; Suzuki, K.; Nagatsu, T. Limited proteolysis of tyrosine hydroxylase by Ca(2+)-activated neutral protease (calpain). Biochemistry 1991, 30, 10416–10419. [Google Scholar] [CrossRef] [PubMed]
- Mishizen-Eberz, A.J.; Guttmann, R.P.; Giasson, B.I.; Day, G.A., 3rd; Hodara, R.; Ischiropoulos, H.; Lee, V.M.; Trojanowski, J.Q.; Lynch, D.R. Distinct cleavage patterns of normal and pathologic forms of alpha-synuclein by calpain I in vitro. J. Neurochem. 2003, 86, 836–847. [Google Scholar] [CrossRef] [PubMed]
- Dufty, B.M.; Warner, L.R.; Hou, S.T.; Jiang, S.X.; Gomez-Isla, T.; Leenhouts, K.M.; Oxford, J.T.; Feany, M.B.; Masliah, E.; Rohn, T.T. Calpain-cleavage of alpha-synuclein: Connecting proteolytic processing to disease-linked aggregation. Am. J. Pathol. 2007, 170, 1725–1738. [Google Scholar] [CrossRef] [Green Version]
- Diepenbroek, M.; Casadei, N.; Esmer, H.; Saido, T.C.; Takano, J.; Kahle, P.J.; Nixon, R.A.; Rao, M.V.; Melki, R.; Pieri, L.; et al. Overexpression of the calpain-specific inhibitor calpastatin reduces human alpha-Synuclein processing, aggregation and synaptic impairment in [A30P]alphaSyn transgenic mice. Hum. Mol. Genet. 2014, 23, 3975–3989. [Google Scholar] [CrossRef]
- Crocker, S.J.; Smith, P.D.; Jackson-Lewis, V.; Lamba, W.R.; Hayley, S.P.; Grimm, E.; Callaghan, S.M.; Slack, R.S.; Melloni, E.; Przedborski, S.; et al. Inhibition of calpains prevents neuronal and behavioral deficits in an MPTP mouse model of Parkinson’s disease. J. Neurosci. 2003, 23, 4081–4091. [Google Scholar] [CrossRef]
- Angelova, P.R.; Ludtmann, M.H.; Horrocks, M.H.; Negoda, A.; Cremades, N.; Klenerman, D.; Dobson, C.M.; Wood, N.W.; Pavlov, E.V.; Gandhi, S.; et al. Ca2+ is a key factor in alpha-synuclein-induced neurotoxicity. J. Cell Sci. 2016, 129, 1792–1801. [Google Scholar] [CrossRef] [Green Version]
- Hettiarachchi, N.T.; Parker, A.; Dallas, M.L.; Pennington, K.; Hung, C.C.; Pearson, H.A.; Boyle, J.P.; Robinson, P.; Peers, C. alpha-Synuclein modulation of Ca2+ signaling in human neuroblastoma (SH-SY5Y) cells. J. Neurochem. 2009, 111, 1192–1201. [Google Scholar] [CrossRef]
- Lieberman, O.J.; Choi, S.J.; Kanter, E.; Saverchenko, A.; Frier, M.D.; Fiore, G.M.; Wu, M.; Kondapalli, J.; Zampese, E.; Surmeier, D.J.; et al. alpha-Synuclein-Dependent Calcium Entry Underlies Differential Sensitivity of Cultured SN and VTA Dopaminergic Neurons to a Parkinsonian Neurotoxin. eNeuro 2017, 4. [Google Scholar] [CrossRef] [Green Version]
- Mironov, S.L. alpha-Synuclein forms non-selective cation channels and stimulates ATP-sensitive potassium channels in hippocampal neurons. J. Physiol. 2015, 593, 145–159. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Caraveo, G.; Auluck, P.K.; Whitesell, L.; Chung, C.Y.; Baru, V.; Mosharov, E.V.; Yan, X.; Ben-Johny, M.; Soste, M.; Picotti, P.; et al. Calcineurin determines toxic versus beneficial responses to α-synuclein. Proc. Natl. Acad. Sci. USA 2014, 111, E3544–E3552. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Betzer, C.; Lassen, L.B.; Olsen, A.; Kofoed, R.H.; Reimer, L.; Gregersen, E.; Zheng, J.; Cali, T.; Gai, W.P.; Chen, T.; et al. Alpha-synuclein aggregates activate calcium pump SERCA leading to calcium dysregulation. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef] [PubMed]
- Yamada, K.; Iwatsubo, T. Extracellular alpha-synuclein levels are regulated by neuronal activity. Mol. Neurodegener 2018, 13, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Emmanouilidou, E.; Melachroinou, K.; Roumeliotis, T.; Garbis, S.D.; Ntzouni, M.; Margaritis, L.H.; Stefanis, L.; Vekrellis, K. Cell-produced alpha-synuclein is secreted in a calcium-dependent manner by exosomes and impacts neuronal survival. J. Neurosci. 2010, 30, 6838–6851. [Google Scholar] [CrossRef] [Green Version]
- Cali, T.; Ottolini, D.; Negro, A.; Brini, M. alpha-Synuclein controls mitochondrial calcium homeostasis by enhancing endoplasmic reticulum-mitochondria interactions. J. Biol. Chem. 2012, 287, 17914–17929. [Google Scholar] [CrossRef] [Green Version]
- Murphy, M.P. How mitochondria produce reactive oxygen species. Biochem. J. 2009, 417, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Nickel, A.; Kohlhaas, M.; Maack, C. Mitochondrial reactive oxygen species production and elimination. J. Mol. Cell. Cardiol. 2014, 73, 26–33. [Google Scholar] [CrossRef]
- Stefanatos, R.; Sanz, A. The role of mitochondrial ROS in the aging brain. FEBS Lett. 2018, 592, 743–758. [Google Scholar] [CrossRef] [Green Version]
- Scheibye-Knudsen, M.; Fang, E.F.; Croteau, D.L.; Wilson, D.M., 3rd; Bohr, V.A. Protecting the mitochondrial powerhouse. Trends Cell Biol. 2014. [Google Scholar] [CrossRef] [Green Version]
- Votyakova, T.V.; Reynolds, I.J. DeltaPsi(m)-Dependent and -independent production of reactive oxygen species by rat brain mitochondria. J. Neurochem. 2001, 79, 266–277. [Google Scholar] [CrossRef]
- Dolle, C.; Flones, I.; Nido, G.S.; Miletic, H.; Osuagwu, N.; Kristoffersen, S.; Lilleng, P.K.; Larsen, J.P.; Tysnes, O.B.; Haugarvoll, K.; et al. Defective mitochondrial DNA homeostasis in the substantia nigra in Parkinson disease. Nat. Commun. 2016, 7, 13548. [Google Scholar] [CrossRef] [PubMed]
- Muller, S.K.; Bender, A.; Laub, C.; Hogen, T.; Schlaudraff, F.; Liss, B.; Klopstock, T.; Elstner, M. Lewy body pathology is associated with mitochondrial DNA damage in Parkinson’s disease. Neurobiol. Aging 2013, 34, 2231–2233. [Google Scholar] [CrossRef] [PubMed]
- Sanders, L.H.; McCoy, J.; Hu, X.; Mastroberardino, P.G.; Dickinson, B.C.; Chang, C.J.; Chu, C.T.; Van Houten, B.; Greenamyre, J.T. Mitochondrial DNA damage: Molecular marker of vulnerable nigral neurons in Parkinson’s disease. Neurobiol. Dis. 2014, 70, 214–223. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bender, A.; Krishnan, K.J.; Morris, C.M.; Taylor, G.A.; Reeve, A.K.; Perry, R.H.; Jaros, E.; Hersheson, J.S.; Betts, J.; Klopstock, T.; et al. High levels of mitochondrial DNA deletions in substantia nigra neurons in aging and Parkinson disease. Nat. Genet. 2006, 38, 515–517. [Google Scholar] [CrossRef] [PubMed]
- Kraytsberg, Y.; Kudryavtseva, E.; McKee, A.C.; Geula, C.; Kowall, N.W.; Khrapko, K. Mitochondrial DNA deletions are abundant and cause functional impairment in aged human substantia nigra neurons. Nat. Genet. 2006, 38, 518–520. [Google Scholar] [CrossRef] [PubMed]
- Elstner, M.; Müller, S.K.; Leidolt, L.; Laub, C.; Krieg, L.; Schlaudraff, F.; Liss, B.; Morris, C.; Turnbull, D.M.; Masliah, E.; et al. Neuromelanin, neurotransmitter status and brainstem location determine the differential vulnerability of catecholaminergic neurons to mitochondrial DNA deletions. Mol. Brain 2011, 4, 1–10. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Greaves, L.C.; Reeve, A.K.; Taylor, R.W.; Turnbull, D.M. Mitochondrial DNA and disease. J. Pathol. 2012, 226, 274–286. [Google Scholar] [CrossRef]
- Keeney, P.M.; Xie, J.; Capaldi, R.A.; Bennett, J.P., Jr. Parkinson’s disease brain mitochondrial complex I has oxidatively damaged subunits and is functionally impaired and misassembled. J. Neurosci. 2006, 26, 5256–5264. [Google Scholar] [CrossRef]
- Mann, V.M.; Cooper, J.M.; Daniel, S.E.; Srai, K.; Jenner, P.; Marsden, C.D.; Schapira, A.H. Complex I, iron, and ferritin in Parkinson’s disease substantia nigra. Ann. Neurol. 1994, 36, 876–881. [Google Scholar] [CrossRef]
- Schapira, A.H.; Cooper, J.M.; Dexter, D.; Clark, J.B.; Jenner, P.; Marsden, C.D. Mitochondrial complex I deficiency in Parkinson’s disease. J. Neurochem. 1990, 54, 823–827. [Google Scholar] [CrossRef]
- Schapira, A.H.; Mann, V.M.; Cooper, J.M.; Dexter, D.; Daniel, S.E.; Jenner, P.; Clark, J.B.; Marsden, C.D. Anatomic and disease specificity of NADH CoQ1 reductase (complex I) deficiency in Parkinson’s disease. J. Neurochem. 1990, 55, 2142–2145. [Google Scholar] [CrossRef] [PubMed]
- Grunewald, A.; Rygiel, K.A.; Hepplewhite, P.D.; Morris, C.M.; Picard, M.; Turnbull, D.M. Mitochondrial DNA Depletion in Respiratory Chain-Deficient Parkinson Disease Neurons. Ann. Neurol. 2016, 79, 366–378. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liang, C.L.; Wang, T.T.; Luby-Phelps, K.; German, D.C. Mitochondria mass is low in mouse substantia nigra dopamine neurons: Implications for Parkinson’s disease. Exp. Neurol. 2007, 203, 370–380. [Google Scholar] [CrossRef] [PubMed]
- Ashrafi, G.; Schwarz, T.L. The pathways of mitophagy for quality control and clearance of mitochondria. Cell Death Differ. 2013, 20, 31–42. [Google Scholar] [CrossRef] [Green Version]
- McLelland, G.L.; Soubannier, V.; Chen, C.X.; McBride, H.M.; Fon, E.A. Parkin and PINK1 function in a vesicular trafficking pathway regulating mitochondrial quality control. EMBO J. 2014, 33, 282–295. [Google Scholar] [CrossRef]
- Hipp, M.S.; Kasturi, P.; Hartl, F.U. The proteostasis network and its decline in ageing. Nat. Rev. Mol. Cell Biol. 2019, 20, 421–435. [Google Scholar] [CrossRef]
- Haas, R.H.; Nasirian, F.; Nakano, K.; Ward, D.; Pay, M.; Hill, R.; Shults, C.W. Low platelet mitochondrial complex I and complex II/III activity in early untreated Parkinson’s disease. Ann. Neurol. 1995, 37, 714–722. [Google Scholar] [CrossRef]
- Ambrosi, G.; Ghezzi, C.; Sepe, S.; Milanese, C.; Payan-Gomez, C.; Bombardieri, C.R.; Armentero, M.T.; Zangaglia, R.; Pacchetti, C.; Mastroberardino, P.G.; et al. Bioenergetic and proteolytic defects in fibroblasts from patients with sporadic Parkinson’s disease. Biochim. Biophys. Acta 2014, 1842, 1385–1394. [Google Scholar] [CrossRef] [Green Version]
- Deus, C.M.; Pereira, S.P.; Cunha-Oliveira, T.; Pereira, F.B.; Raimundo, N.; Oliveira, P.J. Mitochondrial remodeling in human skin fibroblasts from sporadic PD male patients uncovers metabolic and mitochondrial bioenergetics defects. Biochim. Biophys. Acta Mol. Basis Dis. 2019, 165615. [Google Scholar] [CrossRef]
- Moon, H.E.; Yoon, S.H.; Hur, Y.S.; Park, H.W.; Ha, J.Y.; Kim, K.H.; Shim, J.H.; Yoo, S.H.; Son, J.H.; Paek, S.L.; et al. Mitochondrial dysfunction of immortalized human adipose tissue-derived mesenchymal stromal cells from patients with Parkinson’s disease. Exp. Neurobiol. 2013, 22, 283–300. [Google Scholar] [CrossRef] [Green Version]
- Muftuoglu, M.; Elibol, B.; Dalmizrak, O.; Ercan, A.; Kulaksiz, G.; Ogus, H.; Dalkara, T.; Ozer, N. Mitochondrial complex I and IV activities in leukocytes from patients with parkin mutations. Mov. Disord. Off. J. Mov. Disord. Soc. 2004, 19, 544–548. [Google Scholar] [CrossRef] [PubMed]
- Smith, A.M.; Depp, C.; Ryan, B.J.; Johnston, G.I.; Alegre-Abarrategui, J.; Evetts, S.; Rolinski, M.; Baig, F.; Ruffmann, C.; Simon, A.K.; et al. Mitochondrial dysfunction and increased glycolysis in prodromal and early Parkinson’s blood cells. Mov. Disord. Off. J. Mov. Disord. Soc. 2018, 33, 1580–1590. [Google Scholar] [CrossRef] [PubMed]
- Mak, S.K.; Tewari, D.; Tetrud, J.W.; Langston, J.W.; Schule, B. Mitochondrial dysfunction in skin fibroblasts from a Parkinson’s disease patient with an alpha-synuclein triplication. J. Parkinsons Dis. 2011, 1, 175–183. [Google Scholar] [CrossRef] [PubMed]
- Mortiboys, H.; Thomas, K.J.; Koopman, W.J.; Klaffke, S.; Abou-Sleiman, P.; Olpin, S.; Wood, N.W.; Willems, P.H.; Smeitink, J.A.; Cookson, M.R.; et al. Mitochondrial function and morphology are impaired in parkin-mutant fibroblasts. Ann. Neurol. 2008, 64, 555–565. [Google Scholar] [CrossRef] [PubMed]
- Teves, J.M.Y.; Bhargava, V.; Kirwan, K.R.; Corenblum, M.J.; Justiniano, R.; Wondrak, G.T.; Anandhan, A.; Flores, A.J.; Schipper, D.A.; Khalpey, Z.; et al. Parkinson’s Disease Skin Fibroblasts Display Signature Alterations in Growth, Redox Homeostasis, Mitochondrial Function, and Autophagy. Front. Neurosci. 2017, 11, 737. [Google Scholar] [CrossRef] [Green Version]
- Van der Merwe, C.; Loos, B.; Swart, C.; Kinnear, C.; Henning, F.; van der Merwe, L.; Pillay, K.; Muller, N.; Zaharie, D.; Engelbrecht, L.; et al. Mitochondrial impairment observed in fibroblasts from South African Parkinson’s disease patients with parkin mutations. Biochem. Biophys. Res. Commun. 2014, 447, 334–340. [Google Scholar] [CrossRef] [Green Version]
- Martinez, T.N.; Greenamyre, J.T. Toxin models of mitochondrial dysfunction in Parkinson’s disease. Antioxid. Redox Signal. 2012, 16, 920–934. [Google Scholar] [CrossRef] [Green Version]
- Tieu, K. A guide to neurotoxic animal models of Parkinson’s disease. Cold Spring Harb. Perspect. Med. 2011, 1, a009316. [Google Scholar] [CrossRef]
- Beal, M.F.; Oakes, D.; Shoulson, I.; Henchcliffe, C.; Galpern, W.R.; Haas, R.; Juncos, J.L.; Nutt, J.G.; Voss, T.S.; Ravina, B.; et al. A randomized clinical trial of high-dosage coenzyme Q10 in early Parkinson disease: No evidence of benefit. JAMA Neurol. 2014, 71, 543–552. [Google Scholar] [CrossRef]
- Kieburtz, K.; Tilley, B.C.; Elm, J.J.; Babcock, D.; Hauser, R.; Ross, G.W.; Augustine, A.H.; Augustine, E.U.; Aminoff, M.J.; Bodis-Wollner, I.G.; et al. Effect of creatine monohydrate on clinical progression in patients with Parkinson disease: A randomized clinical trial. JAMA 2015, 313, 584–593. [Google Scholar] [CrossRef]
- Snow, B.J.; Rolfe, F.L.; Lockhart, M.M.; Frampton, C.M.; O’Sullivan, J.D.; Fung, V.; Smith, R.A.; Murphy, M.P.; Taylor, K.M. A double-blind, placebo-controlled study to assess the mitochondria-targeted antioxidant MitoQ as a disease-modifying therapy in Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2010, 25, 1670–1674. [Google Scholar] [CrossRef] [PubMed]
- Choi, W.S.; Kruse, S.E.; Palmiter, R.D.; Xia, Z. Mitochondrial complex I inhibition is not required for dopaminergic neuron death induced by rotenone, MPP+, or paraquat. Proc. Natl. Acad. Sci. USA 2008, 105, 15136–15141. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, H.W.; Choi, W.S.; Sorscher, N.; Park, H.J.; Tronche, F.; Palmiter, R.D.; Xia, Z. Genetic reduction of mitochondrial complex I function does not lead to loss of dopamine neurons in vivo. Neurobiol. Aging 2015, 36, 2617–2627. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Choi, W.S.; Palmiter, R.D.; Xia, Z. Loss of mitochondrial complex I activity potentiates dopamine neuron death induced by microtubule dysfunction in a Parkinson’s disease model. J. Cell Biol. 2011, 192, 873–882. [Google Scholar] [CrossRef]
- Choi, W.-S.; Kim, H.-W.; Tronche, F.; Palmiter, R.D.; Storm, D.R.; Xia, Z. Conditional deletion of Ndufs4 in dopaminergic neurons promotes Parkinson’s disease-like non-motor symptoms without loss of dopamine neurons. Sci. Rep. 2017, 7, 44989. [Google Scholar] [CrossRef]
- Inoue, N.; Ogura, S.; Kasai, A.; Nakazawa, T.; Ikeda, K.; Higashi, S.; Isotani, A.; Baba, K.; Mochizuki, H.; Fujimura, H.; et al. Knockdown of the mitochondria-localized protein p13 protects against experimental parkinsonism. EMBO Rep. 2018, 19. [Google Scholar] [CrossRef]
- Misgeld, T.; Schwarz, T.L. Mitostasis in Neurons: Maintaining Mitochondria in an Extended Cellular Architecture. Neuron 2017, 96, 651–666. [Google Scholar] [CrossRef] [Green Version]
- Chu, Y.; Morfini, G.A.; Langhamer, L.B.; He, Y.; Brady, S.T.; Kordower, J.H. Alterations in axonal transport motor proteins in sporadic and experimental Parkinson’s disease. Brain J. Neurol. 2012, 135, 2058–2073. [Google Scholar] [CrossRef] [Green Version]
- Kim-Han, J.S.; Antenor-Dorsey, J.A.; O’Malley, K.L. The parkinsonian mimetic, MPP+, specifically impairs mitochondrial transport in dopamine axons. J. Neurosci. 2011, 31, 7212–7221. [Google Scholar] [CrossRef] [Green Version]
- Lu, X.; Kim-Han, J.S.; Harmon, S.; Sakiyama-Elbert, S.E.; O’Malley, K.L. The Parkinsonian mimetic, 6-OHDA, impairs axonal transport in dopaminergic axons. Mol. Neurodegener 2014, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Dukes, A.A.; Bai, Q.; Van Laar, V.S.; Zhou, Y.; Ilin, V.; David, C.N.; Agim, Z.S.; Bonkowsky, J.L.; Cannon, J.R.; Watkins, S.C.; et al. Live imaging of mitochondrial dynamics in CNS dopaminergic neurons in vivo demonstrates early reversal of mitochondrial transport following MPP(+) exposure. Neurobiol. Dis. 2016, 95, 238–249. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gottlieb, R.A.; Bernstein, D. Mitochondrial remodeling: Rearranging, recycling, and reprogramming. Cell Calcium 2016, 60, 88–101. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, J.R.; Nunnari, J. Mitochondrial form and function. Nature 2014, 505, 335–343. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Berthet, A.; Margolis, E.B.; Zhang, J.; Hsieh, I.; Zhang, J.; Hnasko, T.S.; Ahmad, J.; Edwards, R.H.; Sesaki, H.; Huang, E.J.; et al. Loss of mitochondrial fission depletes axonal mitochondria in midbrain dopamine neurons. J. Neurosci. 2014, 34, 14304–14317. [Google Scholar] [CrossRef] [Green Version]
- Pham, A.H.; Meng, S.; Chu, Q.N.; Chan, D.C. Loss of Mfn2 results in progressive, retrograde degeneration of dopaminergic neurons in the nigrostriatal circuit. Hum. Mol. Genet. 2012, 21, 4817–4826. [Google Scholar] [CrossRef]
- Lee, S.; Sterky, F.H.; Mourier, A.; Terzioglu, M.; Cullheim, S.; Olson, L.; Larsson, N.G. Mitofusin 2 is necessary for striatal axonal projections of midbrain dopamine neurons. Hum. Mol. Genet. 2012, 21, 4827–4835. [Google Scholar] [CrossRef] [Green Version]
- Ekstrand, M.I.; Terzioglu, M.; Galter, D.; Zhu, S.; Hofstetter, C.; Lindqvist, E.; Thams, S.; Bergstrand, A.; Hansson, F.S.; Trifunovic, A.; et al. Progressive parkinsonism in mice with respiratory-chain-deficient dopamine neurons. Proc. Natl. Acad. Sci. USA 2007, 104, 1325–1330. [Google Scholar] [CrossRef] [Green Version]
- Ekstrand, M.I.; Galter, D. The MitoPark Mouse—An animal model of Parkinson’s disease with impaired respiratory chain function in dopamine neurons. Parkinsonism Relat. Disord. 2009, 15, S185–S188. [Google Scholar] [CrossRef]
- Pickrell, A.M.; Pinto, M.; Hida, A.; Moraes, C.T. Striatal dysfunctions associated with mitochondrial DNA damage in dopaminergic neurons in a mouse model of Parkinson’s disease. J. Neurosci. 2011, 31, 17649–17658. [Google Scholar] [CrossRef] [Green Version]
- Sterky, F.H.; Hoffman, A.F.; Milenkovic, D.; Bao, B.; Paganelli, A.; Edgar, D.; Wibom, R.; Lupica, C.R.; Olson, L.; Larsson, N.G. Altered dopamine metabolism and increased vulnerability to MPTP in mice with partial deficiency of mitochondrial complex I in dopamine neurons. Hum. Mol. Genet. 2012, 21, 1078–1089. [Google Scholar] [CrossRef] [Green Version]
- Kruse, S.E.; Watt, W.C.; Marcinek, D.J.; Kapur, R.P.; Schenkman, K.A.; Palmiter, R.D. Mice with mitochondrial complex I deficiency develop a fatal encephalomyopathy. Cell Metab. 2008, 7, 312–320. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lin, M.K.; Farrer, M.J. Genetics and genomics of Parkinson’s disease. Genome Med. 2014, 6, 48. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Larsen, S.B.; Hanss, Z.; Krüger, R. The genetic architecture of mitochondrial dysfunction in Parkinson’s disease. Cell Tissue Res. 2018, 373, 21–37. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cieri, D.; Brini, M.; Calì, T. Emerging (and converging) pathways in Parkinson’s disease: Keeping mitochondrial wellness. Biochem. Biophys. Res. Commun. 2017, 483, 1020–1030. [Google Scholar] [CrossRef]
- Ludtmann, M.H.; Angelova, P.R.; Ninkina, N.N.; Gandhi, S.; Buchman, V.L.; Abramov, A.Y. Monomeric Alpha-Synuclein Exerts a Physiological Role on Brain ATP Synthase. J. Neurosci. 2016, 36, 10510–10521. [Google Scholar] [CrossRef]
- Ludtmann, M.H.R.; Angelova, P.R.; Horrocks, M.H.; Choi, M.L.; Rodrigues, M.; Baev, A.Y.; Berezhnov, A.V.; Yao, Z.; Little, D.; Banushi, B.; et al. alpha-synuclein oligomers interact with ATP synthase and open the permeability transition pore in Parkinson’s disease. Nat. Commun. 2018, 9, 2293. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cali, T.; Ottolini, D.; Soriano, M.E.; Brini, M. A new split-GFP-based probe reveals DJ-1 translocation into the mitochondrial matrix to sustain ATP synthesis upon nutrient deprivation. Hum. Mol. Genet. 2015, 24, 1045–1060. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Heo, J.Y.; Park, J.H.; Kim, S.J.; Seo, K.S.; Han, J.S.; Lee, S.H.; Kim, J.M.; Park, J.I.; Park, S.K.; Lim, K.; et al. DJ-1 null dopaminergic neuronal cells exhibit defects in mitochondrial function and structure: Involvement of mitochondrial complex I assembly. PLoS ONE 2012, 7, e32629. [Google Scholar] [CrossRef] [PubMed]
- Morais, V.A.; Verstreken, P.; Roethig, A.; Smet, J.; Snellinx, A.; Vanbrabant, M.; Haddad, D.; Frezza, C.; Mandemakers, W.; Vogt-Weisenhorn, D.; et al. Parkinson’s disease mutations in PINK1 result in decreased Complex I activity and deficient synaptic function. EMBO Mol. Med. 2009, 1, 99–111. [Google Scholar] [CrossRef] [PubMed]
- Chen, R.; Park, H.A.; Mnatsakanyan, N.; Niu, Y.; Licznerski, P.; Wu, J.; Miranda, P.; Graham, M.; Tang, J.; Boon, A.J.W.; et al. Parkinson’s disease protein DJ-1 regulates ATP synthase protein components to increase neuronal process outgrowth. Cell Death Dis. 2019, 10, 469. [Google Scholar] [CrossRef] [Green Version]
- Lopez-Fabuel, I.; Martin-Martin, L.; Resch-Beusher, M.; Azkona, G.; Sanchez-Pernaute, R.; Bolanos, J.P. Mitochondrial respiratory chain disorganization in Parkinson’s disease-relevant PINK1 and DJ1 mutants. Neurochem. Int. 2017, 109, 101–105. [Google Scholar] [CrossRef] [PubMed]
- Zambon, F.; Cherubini, M.; Fernandes, H.J.R.; Lang, C.; Ryan, B.J.; Volpato, V.; Bengoa-Vergniory, N.; Vingill, S.; Attar, M.; Booth, H.D.E.; et al. Cellular alpha-synuclein pathology is associated with bioenergetic dysfunction in Parkinson’s iPSC-derived dopamine neurons. Hum. Mol. Genet. 2019, 28, 2001–2013. [Google Scholar] [CrossRef] [PubMed]
- Giguere, N.; Pacelli, C.; Saumure, C.; Bourque, M.J.; Matheoud, D.; Levesque, D.; Slack, R.S.; Park, D.S.; Trudeau, L.E. Comparative analysis of Parkinson’s disease-associated genes in mice reveals altered survival and bioenergetics of Parkin-deficient dopamine neurons. J. Biol. Chem. 2018, 293, 9580–9593. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, X.; Winter, D.; Ashrafi, G.; Schlehe, J.; Wong, Y.L.; Selkoe, D.; Rice, S.; Steen, J.; LaVoie, M.J.; Schwarz, T.L. PINK1 and Parkin target Miro for phosphorylation and degradation to arrest mitochondrial motility. Cell 2011, 147, 893–906. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Saha, A.R.; Hill, J.; Utton, M.A.; Asuni, A.A.; Ackerley, S.; Grierson, A.J.; Miller, C.C.; Davies, A.M.; Buchman, V.L.; Anderton, B.H.; et al. Parkinson’s disease alpha-synuclein mutations exhibit defective axonal transport in cultured neurons. J. Cell Sci. 2004, 117, 1017–1024. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Sawada, T.; Lee, S.; Yu, W.; Silverio, G.; Alapatt, P.; Millan, I.; Shen, A.; Saxton, W.; Kanao, T.; et al. Parkinson’s disease-associated kinase PINK1 regulates Miro protein level and axonal transport of mitochondria. PLoS Genet. 2012, 8, e1002537. [Google Scholar] [CrossRef]
- Irrcher, I.; Aleyasin, H.; Seifert, E.L.; Hewitt, S.J.; Chhabra, S.; Phillips, M.; Lutz, A.K.; Rousseaux, M.W.; Bevilacqua, L.; Jahani-Asl, A.; et al. Loss of the Parkinson’s disease-linked gene DJ-1 perturbs mitochondrial dynamics. Hum. Mol. Genet. 2010, 19, 3734–3746. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.; Petrie, T.G.; Liu, Y.; Liu, J.; Fujioka, H.; Zhu, X. Parkinson’s disease-associated DJ-1 mutations impair mitochondrial dynamics and cause mitochondrial dysfunction. J. Neurochem. 2012, 121, 830–839. [Google Scholar] [CrossRef]
- Pozo Devoto, V.M.; Dimopoulos, N.; Alloatti, M.; Pardi, M.B.; Saez, T.M.; Otero, M.G.; Cromberg, L.E.; Marín-Burgin, A.; Scassa, M.E.; Stokin, G.B.; et al. αSynuclein control of mitochondrial homeostasis in human-derived neurons is disrupted by mutations associated with Parkinson’s disease. Sci. Rep. 2017, 7, 5042. [Google Scholar] [CrossRef]
- Giaime, E.; Yamaguchi, H.; Gautier, C.A.; Kitada, T.; Shen, J. Loss of DJ-1 does not affect mitochondrial respiration but increases ROS production and mitochondrial permeability transition pore opening. PLoS ONE 2012, 7, e40501. [Google Scholar] [CrossRef] [Green Version]
- Zheng, L.; Bernard-Marissal, N.; Moullan, N.; D’Amico, D.; Auwerx, J.; Moore, D.J.; Knott, G.; Aebischer, P.; Schneider, B.L. Parkin functionally interacts with PGC-1α to preserve mitochondria and protect dopaminergic neurons. Hum. Mol. Genet. 2017, 26, 582–598. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Shin, J.H.; Ko, H.S.; Kang, H.; Lee, Y.; Lee, Y.I.; Pletinkova, O.; Troconso, J.C.; Dawson, V.L.; Dawson, T.M. PARIS (ZNF746) repression of PGC-1alpha contributes to neurodegeneration in Parkinson’s disease. Cell 2011, 144, 689–702. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pickrell, A.M.; Youle, R.J. The roles of PINK1, parkin, and mitochondrial fidelity in Parkinson’s disease. Neuron 2015, 85, 257–273. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Narendra, D.P.; Jin, S.M.; Tanaka, A.; Suen, D.F.; Gautier, C.A.; Shen, J.; Cookson, M.R.; Youle, R.J. PINK1 is selectively stabilized on impaired mitochondria to activate Parkin. PLoS Biol. 2010, 8, e1000298. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nguyen, T.N.; Padman, B.S.; Lazarou, M. Deciphering the Molecular Signals of PINK1/Parkin Mitophagy. Trends Cell Biol. 2016, 26, 733–744. [Google Scholar] [CrossRef]
- Narendra, D.; Tanaka, A.; Suen, D.F.; Youle, R.J. Parkin is recruited selectively to impaired mitochondria and promotes their autophagy. J. Cell Biol. 2008, 183, 795–803. [Google Scholar] [CrossRef] [Green Version]
- Heeman, B.; Van den Haute, C.; Aelvoet, S.A.; Valsecchi, F.; Rodenburg, R.J.; Reumers, V.; Debyser, Z.; Callewaert, G.; Koopman, W.J.; Willems, P.H.; et al. Depletion of PINK1 affects mitochondrial metabolism, calcium homeostasis and energy maintenance. J. Cell Sci. 2011, 124, 1115–1125. [Google Scholar] [CrossRef] [Green Version]
- Gandhi, S.; Wood-Kaczmar, A.; Yao, Z.; Plun-Favreau, H.; Deas, E.; Klupsch, K.; Downward, J.; Latchman, D.S.; Tabrizi, S.J.; Wood, N.W.; et al. PINK1-associated Parkinson’s disease is caused by neuronal vulnerability to calcium-induced cell death. Mol. Cell 2009, 33, 627–638. [Google Scholar] [CrossRef] [Green Version]
- Kostic, M.; Ludtmann, M.H.; Bading, H.; Hershfinkel, M.; Steer, E.; Chu, C.T.; Abramov, A.Y.; Sekler, I. PKA Phosphorylation of NCLX Reverses Mitochondrial Calcium Overload and Depolarization, Promoting Survival of PINK1-Deficient Dopaminergic Neurons. Cell Rep. 2015, 13, 376–386. [Google Scholar] [CrossRef] [Green Version]
- Huang, E.; Qu, D.; Huang, T.; Rizzi, N.; Boonying, W.; Krolak, D.; Ciana, P.; Woulfe, J.; Klein, C.; Slack, R.S.; et al. PINK1-mediated phosphorylation of LETM1 regulates mitochondrial calcium transport and protects neurons against mitochondrial stress. Nat. Commun. 2017, 8, 1399. [Google Scholar] [CrossRef]
- Matteucci, A.; Patron, M.; Vecellio Reane, D.; Gastaldello, S.; Amoroso, S.; Rizzuto, R.; Brini, M.; Raffaello, A.; Calì, T. Parkin-dependent regulation of the MCU complex component MICU1. Sci. Rep. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Verma, M.; Callio, J.; Otero, P.A.; Sekler, I.; Wills, Z.P.; Chu, C.T. Mitochondrial Calcium Dysregulation Contributes to Dendrite Degeneration Mediated by PD/LBD-Associated LRRK2 Mutants. J. Neurosci. 2017, 37, 11151–11165. [Google Scholar] [CrossRef] [PubMed]
- Ludtmann, M.H.R.; Kostic, M.; Horne, A.; Gandhi, S.; Sekler, I.; Abramov, A.Y. LRRK2 deficiency induced mitochondrial Ca2+ efflux inhibition can be rescued by Na+/Ca2+/Li+ exchanger upregulation. Cell Death Dis. 2019, 10, 265. [Google Scholar] [CrossRef] [PubMed]
- Soman, S.K.; Bazała, M.; Keatinge, M.; Bandmann, O.; Kuznicki, J. Restriction of mitochondrial calcium overload by mcu inactivation renders a neuroprotective effect in zebrafish models of Parkinson’s disease. Biol. Open 2019, 8, bio044347. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Soman, S.; Keatinge, M.; Moein, M.; Da Costa, M.; Mortiboys, H.; Skupin, A.; Sugunan, S.; Bazala, M.; Kuznicki, J.; Bandmann, O. Inhibition of the mitochondrial calcium uniporter rescues dopaminergic neurons in pink1(-/-) zebrafish. Eur. J. Neurosci. 2017, 45, 528–535. [Google Scholar] [CrossRef]
- Ilijic, E.; Guzman, J.N.; Surmeier, D.J. The L-type channel antagonist isradipine is neuroprotective in a mouse model of Parkinson’s disease. Neurobiol. Dis. 2011, 43, 364–371. [Google Scholar] [CrossRef] [Green Version]
- Kupsch, A.; Gerlach, M.; Pupeter, S.C.; Sautter, J.; Dirr, A.; Arnold, G.; Opitz, W.; Przuntek, H.; Riederer, P.; Oertel, W.H. Pretreatment with nimodipine prevents MPTP-induced neurotoxicity at the nigral, but not at the striatal level in mice. Neuroreport 1995, 6, 621–625. [Google Scholar] [CrossRef]
- Wang, Q.-M.; Xu, Y.-Y.; Liu, S.; Ma, Z.-G. Isradipine attenuates MPTP-induced dopamine neuron degeneration by inhibiting up-regulation of L-type calcium channels and iron accumulation in the substantia nigra of mice. Oncotarget 2017, 8, 47284–47295. [Google Scholar] [CrossRef] [Green Version]
- Wang, R.; Ma, Z.; Wang, J.; Xie, J. L-type Cav1.2 calcium channel is involved in 6-hydroxydopamine-induced neurotoxicity in rats. Neurotox. Res. 2012, 21, 266–270. [Google Scholar] [CrossRef]
- Singh, A.; Verma, P.; Balaji, G.; Samantaray, S.; Mohanakumar, K.P. Nimodipine, an L-type calcium channel blocker attenuates mitochondrial dysfunctions to protect against 1-methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced Parkinsonism in mice. Neurochem. Int. 2016, 99, 221–232. [Google Scholar] [CrossRef]
- Cali, T.; Ottolini, D.; Vicario, M.; Catoni, C.; Vallese, F.; Cieri, D.; Barazzuol, L.; Brini, M. splitGFP Technology Reveals Dose-Dependent ER-Mitochondria Interface Modulation by alpha-Synuclein A53T and A30P Mutants. Cells 2019, 8, 1072. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gautier, C.A.; Erpapazoglou, Z.; Mouton-Liger, F.; Muriel, M.P.; Cormier, F.; Bigou, S.; Duffaure, S.; Girard, M.; Foret, B.; Iannielli, A.; et al. The endoplasmic reticulum-mitochondria interface is perturbed in PARK2 knockout mice and patients with PARK2 mutations. Hum. Mol. Genet. 2016, 25, 2972–2984. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lee, K.S.; Huh, S.; Lee, S.; Wu, Z.; Kim, A.K.; Kang, H.Y.; Lu, B. Altered ER-mitochondria contact impacts mitochondria calcium homeostasis and contributes to neurodegeneration in vivo in disease models. Proc. Natl. Acad. Sci. USA 2018, 115, E8844–E8853. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ottolini, D.; Calì, T.; Negro, A.; Brini, M. The Parkinson disease-related protein DJ-1 counteracts mitochondrial impairment induced by the tumour suppressor protein p53 by enhancing endoplasmic reticulum-mitochondria tethering. Hum. Mol. Genet. 2013, 22, 2152–2168. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Ma, X.; Fujioka, H.; Liu, J.; Chen, S.; Zhu, X. DJ-1 regulates the integrity and function of ER-mitochondria association through interaction with IP3R3-Grp75-VDAC1. Proc. Natl. Acad. Sci. USA 2019, 116, 25322–25328. [Google Scholar] [CrossRef] [PubMed]
- Guardia-Laguarta, C.; Area-Gomez, E.; Rub, C.; Liu, Y.; Magrane, J.; Becker, D.; Voos, W.; Schon, E.A.; Przedborski, S. alpha-Synuclein is localized to mitochondria-associated ER membranes. J. Neurosci. 2014, 34, 249–259. [Google Scholar] [CrossRef]
- Paillusson, S.; Gomez-Suaga, P.; Stoica, R.; Little, D.; Gissen, P.; Devine, M.J.; Noble, W.; Hanger, D.P.; Miller, C.C.J. alpha-Synuclein binds to the ER-mitochondria tethering protein VAPB to disrupt Ca2+ homeostasis and mitochondrial ATP production. Acta Neuropathol. 2017, 134, 129–149. [Google Scholar] [CrossRef] [Green Version]
- Gomez-Suaga, P.; Bravo-San Pedro, J.M.; Gonzalez-Polo, R.A.; Fuentes, J.M.; Niso-Santano, M. ER-mitochondria signaling in Parkinson’s disease. Cell Death Dis. 2018, 9, 337. [Google Scholar] [CrossRef] [Green Version]
- Parrado-Fernández, C.; Schneider, B.; Ankarcrona, M.; Conti, M.M.; Cookson, M.R.; Kivipelto, M.; Cedazo-Mínguez, Á.; Sandebring-Matton, A. Reduction of PINK1 or DJ-1 impair mitochondrial motility in neurites and alter ER-mitochondria contacts. J. Cell. Mol. Med. 2018, 22, 5439–5449. [Google Scholar] [CrossRef]
- Janikiewicz, J.; Szymanski, J.; Malinska, D.; Patalas-Krawczyk, P.; Michalska, B.; Duszynski, J.; Giorgi, C.; Bonora, M.; Dobrzyn, A.; Wieckowski, M.R. Mitochondria-associated membranes in aging and senescence: Structure, function, and dynamics. Cell Death Dis. 2018, 9, 332. [Google Scholar] [CrossRef]
- Erpapazoglou, Z.; Mouton-Liger, F.; Corti, O. From dysfunctional endoplasmic reticulum-mitochondria coupling to neurodegeneration. Neurochem. Int. 2017, 109, 171–183. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Paillusson, S.; Stoica, R.; Gomez-Suaga, P.; Lau, D.H.W.; Mueller, S.; Miller, T.; Miller, C.C.J. There’s Something Wrong with my MAM; the ER-Mitochondria Axis and Neurodegenerative Diseases. Trends Neurosci. 2016, 39, 146–157. [Google Scholar] [CrossRef] [Green Version]
- Sironi, L.; Restelli, L.M.; Tolnay, M.; Neutzner, A.; Frank, S. Dysregulated Interorganellar Crosstalk of Mitochondria in the Pathogenesis of Parkinson’s Disease. Cells 2020, 9, 233. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Celardo, I.; Costa, A.C.; Lehmann, S.; Jones, C.; Wood, N.; Mencacci, N.E.; Mallucci, G.R.; Loh, S.H.; Martins, L.M. Mitofusin-mediated ER stress triggers neurodegeneration in pink1/parkin models of Parkinson’s disease. Cell Death Dis. 2016, 7, e2271. [Google Scholar] [CrossRef] [Green Version]
- Toyofuku, T.; Okamoto, Y.; Ishikawa, T.; Sasawatari, S.; Kumanogoh, A. LRRK2 regulates endoplasmic reticulum-mitochondrial tethering through the PERK-mediated ubiquitination pathway. Embo J. 2020, 39, e100875. [Google Scholar] [CrossRef] [PubMed]
- Bezard, E.; Przedborski, S. A tale on animal models of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2011, 26, 993–1002. [Google Scholar] [CrossRef]
- Dawson, T.M.; Ko, H.S.; Dawson, V.L. Genetic animal models of Parkinson’s disease. Neuron 2010, 66, 646–661. [Google Scholar] [CrossRef] [Green Version]
- Chia, S.J.; Tan, E.K.; Chao, Y.X. Historical Perspective: Models of Parkinson’s Disease. Int. J. Mol. Sci. 2020, 21, 2464. [Google Scholar] [CrossRef] [Green Version]
- Braak, H.; Del Tredici, K.; Bratzke, H.; Hamm-Clement, J.; Sandmann-Keil, D.; Rub, U. Staging of the intracerebral inclusion body pathology associated with idiopathic Parkinson’s disease (preclinical and clinical stages). J. Neurol. 2002, 249 (Suppl. S3), iii1–iii5. [Google Scholar] [CrossRef]
- Braak, H.; Ghebremedhin, E.; Rub, U.; Bratzke, H.; Del Tredici, K. Stages in the development of Parkinson’s disease-related pathology. Cell Tissue Res. 2004, 318, 121–134. [Google Scholar] [CrossRef]
- Surmeier, D.J.; Obeso, J.A.; Halliday, G.M. Parkinson’s Disease Is Not Simply a Prion Disorder. J. Neurosci. 2017, 37, 9799–9807. [Google Scholar] [CrossRef] [Green Version]
- Greene, J.G. Causes and consequences of degeneration of the dorsal motor nucleus of the vagus nerve in Parkinson’s disease. Antioxid. Redox Signal. 2014, 21, 649–667. [Google Scholar] [CrossRef]
- Mo, Z.L.; Katafuchi, T.; Muratani, H.; Hori, T. Effects of vasopressin and angiotensin II on neurones in the rat dorsal motor nucleus of the vagus, in vitro. J. Physiol. 1992, 458, 561–577. [Google Scholar] [CrossRef]
- Travagli, R.A.; Gillis, R.A.; Rossiter, C.D.; Vicini, S. Glutamate and GABA-mediated synaptic currents in neurons of the rat dorsal motor nucleus of the vagus. Am. J. Physiol. 1991, 260, G531–G536. [Google Scholar] [CrossRef]
- Marks, J.D.; Donnelly, D.F.; Haddad, G.G. Adenosine-induced inhibition of vagal motoneuron excitability: Receptor subtype and mechanisms. Am. J. Physiol. 1993, 264, L124–L132. [Google Scholar] [CrossRef]
- Cooper, G.; Lasser-Katz, E.; Simchovitz, A.; Sharon, R.; Soreq, H.; Surmeier, D.J.; Goldberg, J.A. Functional segregation of voltage-activated calcium channels in motoneurons of the dorsal motor nucleus of the vagus. J. Neurophysiol. 2015, 114, 1513–1520. [Google Scholar] [CrossRef] [Green Version]
- Lasser-Katz, E.; Simchovitz, A.; Chiu, W.H.; Oertel, W.H.; Sharon, R.; Soreq, H.; Roeper, J.; Goldberg, J.A. Mutant alpha-Synuclein Overexpression Induces Stressless Pacemaking in Vagal Motoneurons at Risk in Parkinson’s Disease. J. Neurosci. 2017, 37, 47–57. [Google Scholar] [CrossRef] [Green Version]
- Goldberg, J.A.; Guzman, J.N.; Estep, C.M.; Ilijic, E.; Kondapalli, J.; Sanchez-Padilla, J.; Surmeier, D.J. Calcium entry induces mitochondrial oxidant stress in vagal neurons at risk in Parkinson’s disease. Nat. Neurosci. 2012, 15, 1414–1421. [Google Scholar] [CrossRef]
- Pienaar, I.S.; Vernon, A.; Winn, P. The Cellular Diversity of the Pedunculopontine Nucleus: Relevance to Behavior in Health and Aspects of Parkinson’s Disease. Neuroscientist 2017, 23, 415–431. [Google Scholar] [CrossRef] [Green Version]
- Tubert, C.; Galtieri, D.; Surmeier, D.J. The pedunclopontine nucleus and Parkinson’s disease. Neurobiol. Dis. 2019, 128, 3–8. [Google Scholar] [CrossRef]
- Wang, H.L.; Morales, M. Pedunculopontine and laterodorsal tegmental nuclei contain distinct populations of cholinergic, glutamatergic and GABAergic neurons in the rat. Eur. J. Neurosci. 2009, 29, 340–358. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hirsch, E.C.; Graybiel, A.M.; Duyckaerts, C.; Javoy-Agid, F. Neuronal loss in the pedunculopontine tegmental nucleus in Parkinson disease and in progressive supranuclear palsy. Proc. Natl. Acad. Sci. USA 1987, 84, 5976–5980. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Pienaar, I.S.; Elson, J.L.; Racca, C.; Nelson, G.; Turnbull, D.M.; Morris, C.M. Mitochondrial abnormality associates with type-specific neuronal loss and cell morphology changes in the pedunculopontine nucleus in Parkinson disease. Am. J. Pathol. 2013, 183, 1826–1840. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rinne, J.O.; Ma, S.Y.; Lee, M.S.; Collan, Y.; Roytta, M. Loss of cholinergic neurons in the pedunculopontine nucleus in Parkinson’s disease is related to disability of the patients. Parkinsonism Relat. Disord. 2008, 14, 553–557. [Google Scholar] [CrossRef] [PubMed]
- Dautan, D.; Hacioglu Bay, H.; Bolam, J.P.; Gerdjikov, T.V.; Mena-Segovia, J. Extrinsic Sources of Cholinergic Innervation of the Striatal Complex: A Whole-Brain Mapping Analysis. Front. Neuroanat 2016, 10, 1. [Google Scholar] [CrossRef] [Green Version]
- Dautan, D.; Huerta-Ocampo, I.; Witten, I.B.; Deisseroth, K.; Bolam, J.P.; Gerdjikov, T.; Mena-Segovia, J. A major external source of cholinergic innervation of the striatum and nucleus accumbens originates in the brainstem. J. Neurosci. 2014, 34, 4509–4518. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Lavoie, B.; Parent, A. Pedunculopontine nucleus in the squirrel monkey: Projections to the basal ganglia as revealed by anterograde tract-tracing methods. J. Comp. Neurol. 1994, 344, 210–231. [Google Scholar] [CrossRef]
- Mena-Segovia, J.; Sims, H.M.; Magill, P.J.; Bolam, J.P. Cholinergic brainstem neurons modulate cortical gamma activity during slow oscillations. J. Physiol. 2008, 586, 2947–2960. [Google Scholar] [CrossRef]
- Ros, H.; Magill, P.J.; Moss, J.; Bolam, J.P.; Mena-Segovia, J. Distinct types of non-cholinergic pedunculopontine neurons are differentially modulated during global brain states. Neuroscience 2010, 170, 78–91. [Google Scholar] [CrossRef] [Green Version]
- Takakusaki, K.; Kitai, S.T. Ionic mechanisms involved in the spontaneous firing of tegmental pedunculopontine nucleus neurons of the rat. Neuroscience 1997, 78, 771–794. [Google Scholar] [CrossRef]
- Petzold, A.; Valencia, M.; Pal, B.; Mena-Segovia, J. Decoding brain state transitions in the pedunculopontine nucleus: Cooperative phasic and tonic mechanisms. Front. Neural Circuits 2015, 9, 68. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Scarnati, E.; Proia, A.; Di Loreto, S.; Pacitti, C. The reciprocal electrophysiological influence between the nucleus tegmenti pedunculopontinus and the substantia nigra in normal and decorticated rats. Brain Res. 1987, 423, 116–124. [Google Scholar] [CrossRef]
- Buchman, A.S.; Nag, S.; Shulman, J.M.; Lim, A.S.P.; VanderHorst, V.G.J.M.; Leurgans, S.E.; Schneider, J.A.; Bennett, D.A. Locus coeruleus neuron density and parkinsonism in older adults without Parkinson’s disease. Mov. Disord. 2012, 27, 1625–1631. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Del Tredici, K.; Rüb, U.; De Vos, R.A.; Bohl, J.R.; Braak, H. Where does parkinson disease pathology begin in the brain? J. Neuropathol. Exp. Neurol. 2002, 61, 413–426. [Google Scholar] [CrossRef]
- Alreja, M.; Aghajanian, G.K. Pacemaker activity of locus coeruleaus neurons: Whole-cell recordings in brain slices show dependence on cAMP and protein kinase A. Brain Res. 1991, 556, 339–343. [Google Scholar] [CrossRef]
- Graham, A.W.; Aghajanian, G.K. Effects of amphetamine on single cell activity in a catecholamine nucleus, the locus coeruleus. Nature 1971, 234, 100–102. [Google Scholar] [CrossRef]
- Masuko, S.; Nakajima, Y.; Nakajima, S.; Yamaguchi, K. Noradrenergic neurons from the locus ceruleus in dissociated cell culture: Culture methods, morphology, and electrophysiology. J. Neurosci. 1986, 6, 3229–3241. [Google Scholar] [CrossRef] [Green Version]
- Williams, J.T.; North, R.A.; Shefner, S.A.; Nishi, S.; Egan, T.M. Membrane properties of rat locus coeruleus neurones. Neuroscience 1984, 13, 137–156. [Google Scholar] [CrossRef]
- Aston-Jones, G.; Bloom, F.E. Activity of norepinephrine-containing locus coeruleus neurons in behaving rats anticipates fluctuations in the sleep-waking cycle. J. Neurosci. 1981, 1, 876–886. [Google Scholar] [CrossRef]
- Chu, N.; Bloom, F.E. Norepinephrine-containing neurons: Changes in spontaneous discharge patterns during sleeping and waking. Science 1973, 179, 908–910. [Google Scholar] [CrossRef]
- Foote, S.L.; Aston-Jones, G.; Bloom, F.E. Impulse activity of locus coeruleus neurons in awake rats and monkeys is a function of sensory stimulation and arousal. Proc. Natl. Acad. Sci. USA 1980, 77, 3033–3037. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Gervasoni, D.; Darracq, L.; Fort, P.; Soulière, F.; Chouvet, G.; Luppi, P.-H. Electrophysiological evidence that noradrenergic neurons of the rat locus coeruleus are tonically inhibited by GABA during sleep. Eur. J. Neurosci. 1998, 10, 964–970. [Google Scholar] [CrossRef] [PubMed]
- Imber, A.N.; Putnam, R.W. Postnatal development and activation of L-type Ca2+ currents in locus ceruleus neurons: Implications for a role for Ca2+ in central chemosensitivity. J. Appl. Physiol. (1985) 2012, 112, 1715–1726. [Google Scholar] [CrossRef] [Green Version]
- Matschke, L.A.; Bertoune, M.; Roeper, J.; Snutch, T.P.; Oertel, W.H.; Rinne, S.; Decher, N. A concerted action of L- and T-type Ca2+ channels regulates locus coeruleus pacemaking. Mol. Cell. Neurosci. 2015, 68, 293–302. [Google Scholar] [CrossRef] [PubMed]
- Aston-Jones, G.; Waterhouse, B. Locus coeruleus: From global projection system to adaptive regulation of behavior. Brain Res. 2016, 1645, 75–78. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schwarz, L.A.; Luo, L. Organization of the locus coeruleus-norepinephrine system. Curr. Biol. 2015, 25, R1051–R1056. [Google Scholar] [CrossRef] [Green Version]
- Ungerstedt, U. Stereotaxic Mapping of the Monoamine Pathways in the Rat Brain. Acta Physiol. Scand. 1971, 82, 1–48. [Google Scholar] [CrossRef]
- Jones, B.E.; Moore, R.Y. Ascending projections of the locus coeruleus in the rat. II. Autoradiographic study. Brain Res. 1977, 127, 23–53. [Google Scholar] [CrossRef]
- Hornung, J.-P. The human raphe nuclei and the serotonergic system. J. Chem. Neuroanat. 2003, 26, 331–343. [Google Scholar] [CrossRef]
- Jacobs, B.L.; Azmitia, E.C. Structure and function of the brain serotonin system. Physiol. Rev. 1992, 72, 165–229. [Google Scholar] [CrossRef] [Green Version]
- Maeda, T.; Fujimiya, M.; Kitahama, K.; Imai, H.; Kimura, H. Serotonin neurons and their physiological roles. Arch. Histol. Cytol. 1989, 52, 113–120. [Google Scholar] [CrossRef] [Green Version]
- Michelsen, K.A.; Schmitz, C.; Steinbusch, H.W. The dorsal raphe nucleus—From silver stainings to a role in depression. Brain Res. Rev. 2007, 55, 329–342. [Google Scholar] [CrossRef]
- Muzerelle, A.; Scotto-Lomassese, S.; Bernard, J.F.; Soiza-Reilly, M.; Gaspar, P. Conditional anterograde tracing reveals distinct targeting of individual serotonin cell groups (B5-B9) to the forebrain and brainstem. Brain Struct. Funct. 2016, 221, 535–561. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Vertes, R.P.; Linley, S.B. Comparison of projections of the dorsal and median raphe nuclei, with some functional considerations. Int. Congr. Ser. 2007, 1304, 98–120. [Google Scholar] [CrossRef]
- Crunelli, V.; Segal, M. An electrophysiological study of neurones in the rat median raphe and their projections to septum and hippocampus. Neuroscience 1985, 15, 47–60. [Google Scholar] [CrossRef]
- Heym, J.; Steinfels, G.F.; Jacobs, B.L. Activity of serotonin-containing neurons in the nucleus raphe pallidus of freely moving cats. Brain Res. 1982, 251, 259–276. [Google Scholar] [CrossRef]
- McGinty, D.J.; Harper, R.M. Dorsal raphe neurons: Depression of firing during sleep in cats. Brain Res. 1976, 101, 569–575. [Google Scholar] [CrossRef]
- Sakai, K. Sleep-waking discharge profiles of dorsal raphe nucleus neurons in mice. Neuroscience 2011, 197, 200–224. [Google Scholar] [CrossRef]
- Trulson, M.E.; Crisp, T.; Trulson, V.M. Activity of serotonin-containing nucleus centralis superior (Raphe medianus) neurons in freely moving cats. Exp. Brain Res. 1984, 54, 33–44. [Google Scholar] [CrossRef]
- Trulson, M.E.; Frederickson, C.J. A comparison of the electrophysiological and pharmacological properties of serotonin-containing neurons in the nucleus raphe dorsalis, raphe medianus and raphe pallidus recorded from mouse brain slices in vitro: Role of autoreceptors. Brain Res. Bull. 1987, 18, 179–190. [Google Scholar] [CrossRef]
- Trulson, M.E.; Jacobs, B.L. Raphe unit activity in freely moving cats: Correlation with level of behavioral arousal. Brain Res. 1979, 163, 135–150. [Google Scholar] [CrossRef]
- Urbain, N.; Creamer, K.; Debonnel, G. Electrophysiological diversity of the dorsal raphe cells across the sleep-wake cycle of the rat. J. Physiol. 2006, 573, 679–695. [Google Scholar] [CrossRef] [PubMed]
- Asaoka, N.; Nishitani, N.; Kinoshita, H.; Kawai, H.; Shibui, N.; Nagayasu, K.; Shirakawa, H.; Nakagawa, T.; Kaneko, S. Chronic antidepressant potentiates spontaneous activity of dorsal raphe serotonergic neurons by decreasing GABAB receptor-mediated inhibition of L-type calcium channels. Sci. Rep. 2017, 7, 13609. [Google Scholar] [CrossRef] [PubMed]
- Kirkwood, T.B.; Austad, S.N. Why do we age? Nature 2000, 408, 233–238. [Google Scholar] [CrossRef] [PubMed]
- Byars, S.G.; Voskarides, K. Antagonistic Pleiotropy in Human Disease. J. Mol. Evol. 2020, 88, 12–25. [Google Scholar] [CrossRef] [PubMed]
- Surmeier, D.J.; Guzman, J.N.; Sanchez-Padilla, J. Calcium, cellular aging, and selective neuronal vulnerability in Parkinson’s disease. Cell Calcium 2010, 47, 175–182. [Google Scholar] [CrossRef] [Green Version]
- Lees, A.J.; Hardy, J.; Revesz, T. Parkinson’s disease. Lancet 2009, 373, 2055–2066. [Google Scholar] [CrossRef]
- Dorsey, E.R.; Constantinescu, R.; Thompson, J.P.; Biglan, K.M.; Holloway, R.G.; Kieburtz, K.; Marshall, F.J.; Ravina, B.M.; Schifitto, G.; Siderowf, A.; et al. Projected number of people with Parkinson disease in the most populous nations, 2005 through 2030. Neurology 2007, 68, 384–386. [Google Scholar] [CrossRef]
- Global, regional, and national burden of Parkinson’s disease, 1990-2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Neurol. 2018, 17, 939–953. [CrossRef] [Green Version]
- Rodriguez, M.; Rodriguez-Sabate, C.; Morales, I.; Sanchez, A.; Sabate, M. Parkinson’s disease as a result of aging. Aging Cell 2015, 14, 293–308. [Google Scholar] [CrossRef]
- Becker, C.; Jick, S.S.; Meier, C.R. Use of antihypertensives and the risk of Parkinson disease. Neurology 2008, 70, 1438–1444. [Google Scholar] [CrossRef] [PubMed]
- Lee, Y.C.; Lin, C.H.; Wu, R.M.; Lin, J.W.; Chang, C.H.; Lai, M.S. Antihypertensive agents and risk of Parkinson’s disease: A nationwide cohort study. PLoS ONE 2014, 9, e98961. [Google Scholar] [CrossRef] [PubMed]
- Marras, C.; Gruneir, A.; Rochon, P.; Wang, X.; Anderson, G.; Brotchie, J.; Bell, C.M.; Fox, S.; Austin, P.C. Dihydropyridine calcium channel blockers and the progression of parkinsonism. Ann. Neurol. 2012, 71, 362–369. [Google Scholar] [CrossRef] [PubMed]
- Pasternak, B.; Svanstrom, H.; Nielsen, N.M.; Fugger, L.; Melbye, M.; Hviid, A. Use of calcium channel blockers and Parkinson’s disease. Am. J. Epidemiol. 2012, 175, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Ritz, B.; Rhodes, S.L.; Qian, L.; Schernhammer, E.; Olsen, J.H.; Friis, S. L-type calcium channel blockers and Parkinson disease in Denmark. Ann. Neurol. 2010, 67, 600–606. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kupsch, A.; Sautter, J.; Schwartz, J.; Rieder, P.; Gerlach, M.; Oertel, W.H. 1-Methyl-4-phenyl-1,2,3,6-tetrahydropyridine-induced neurotoxicity in non-human primates is antagonized by pretreatment with nimodipine at the nigral, but not at the striatal level. Brain Res. 1996, 741, 185–196. [Google Scholar] [CrossRef]
- Liss, B.; Striessnig, J. The Potential of L-Type Calcium Channels as a Drug Target for Neuroprotective Therapy in Parkinson’s Disease. Annu. Rev. Pharmacol. Toxicol. 2019, 59, 263–289. [Google Scholar] [CrossRef]
- Parkinson Study Group STEADY-PD III Investigators. Isradipine Versus Placebo in Early Parkinson Disease. Ann. Internal Med. 2020, 172, 591–598. [Google Scholar] [CrossRef] [PubMed]
- Stern, M.B.; Lang, A.; Poewe, W. Toward a redefinition of Parkinson’s disease. Mov. Disord. Off. J. Mov. Disord. Soc. 2012, 27, 54–60. [Google Scholar] [CrossRef]
- Mantri, S.; Morley, J.F.; Siderowf, A.D. The importance of preclinical diagnostics in Parkinson disease. Parkinsonism Relat. Disord. 2019, 64, 20–28. [Google Scholar] [CrossRef]
- Tabata, Y.; Imaizumi, Y.; Sugawara, M.; Andoh-Noda, T.; Banno, S.; Chai, M.; Sone, T.; Yamazaki, K.; Ito, M.; Tsukahara, K.; et al. T-type Calcium Channels Determine the Vulnerability of Dopaminergic Neurons to Mitochondrial Stress in Familial Parkinson Disease. Stem Cell Rep. 2018, 11, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Dey, K.; Bazala, M.A.; Kuznicki, J. Targeting mitochondrial calcium pathways as a potential treatment against Parkinson’s disease. Cell Calcium 2020, 89, 102216. [Google Scholar] [CrossRef] [PubMed]
- Tsunemi, T.; Perez-Rosello, T.; Ishiguro, Y.; Yoroisaka, A.; Jeon, S.; Hamada, K.; Rammonhan, M.; Wong, Y.C.; Xie, Z.; Akamatsu, W.; et al. Increased Lysosomal Exocytosis Induced by Lysosomal Ca2+ Channel Agonists Protects Human Dopaminergic Neurons from α-Synuclein Toxicity. J. Neurosci. 2019, 39, 5760–5772. [Google Scholar] [CrossRef] [PubMed] [Green Version]
Figure 1.
Schematic representation of a substantia nigra (SN) dopaminergic (DAergic) neuron, its axonal projections, and the factors contributing to its high bioenergetic demands. Inset: the positions of substantia nigra (SN, green) and the striatum (orange) and nigral projections to the striatum. Main: sources of high energy demand in SN DAergic neurons. In neurons, three main processes are responsible for the bioenergetic burden: the maintenance of the axonal tree through anterograde/retrograde transport, the propagation of action potentials (APs) followed by the re-establishment of ionic gradients, and the synaptic vesicles cycling. SN DAergic neurons have particularly high energetic demands because of the huge size of their axon, the autonomous pacemaking activity coupled to broad APs, and the high number of synapses or dopamine (DA) release sites. Note that DA release occurs not only from the axonal domain but also in the somatodendritic region.
Figure 1.
Schematic representation of a substantia nigra (SN) dopaminergic (DAergic) neuron, its axonal projections, and the factors contributing to its high bioenergetic demands. Inset: the positions of substantia nigra (SN, green) and the striatum (orange) and nigral projections to the striatum. Main: sources of high energy demand in SN DAergic neurons. In neurons, three main processes are responsible for the bioenergetic burden: the maintenance of the axonal tree through anterograde/retrograde transport, the propagation of action potentials (APs) followed by the re-establishment of ionic gradients, and the synaptic vesicles cycling. SN DAergic neurons have particularly high energetic demands because of the huge size of their axon, the autonomous pacemaking activity coupled to broad APs, and the high number of synapses or dopamine (DA) release sites. Note that DA release occurs not only from the axonal domain but also in the somatodendritic region.
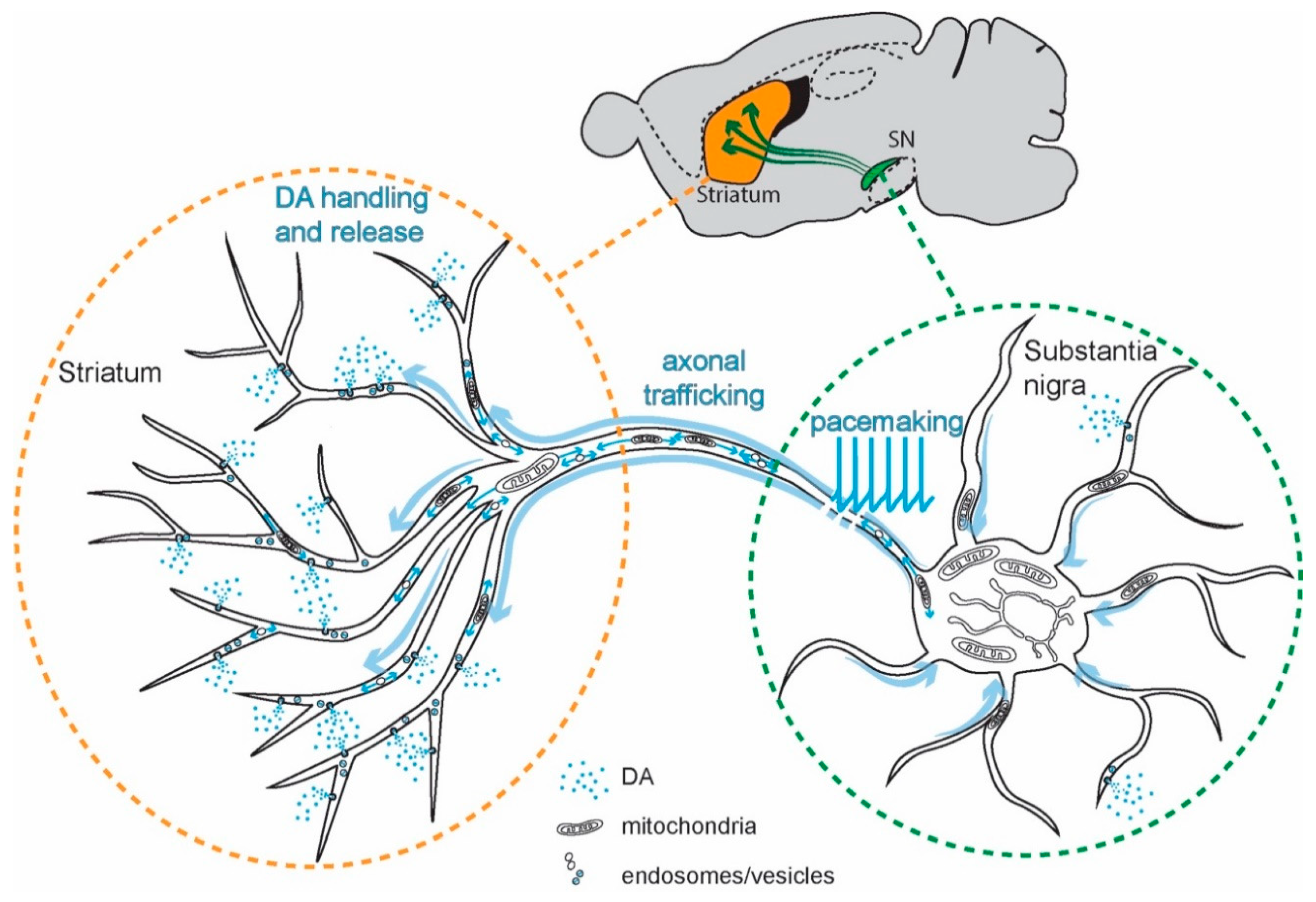
Figure 2.
Effects of Ca2+ on mitochondrial bioenergetic metabolism and reactive oxygen species (ROS) production. The two primary sources of ATP in neurons are glycolysis and mitochondrial OXPHOS. During pacemaking, Ca2+ entry through Cav1 channels, coupled with CICR through RyRs, generates an elevation in [Ca2+]. Elevated [Ca2+] reaching the mitochondria favors the uptake of substrates by the mitochondria by acting on transporters and carriers on the IMM; Ca2+ taken up by mitochondria through the voltage-dependent anion channel (VDAC) and MCUC increases OXPHOS by enhancing the activity of the TCA cycle and by stimulating ATP synthesis by Complex V. The downside of this stimulation is an increase in the generation of ROS.
Figure 2.
Effects of Ca2+ on mitochondrial bioenergetic metabolism and reactive oxygen species (ROS) production. The two primary sources of ATP in neurons are glycolysis and mitochondrial OXPHOS. During pacemaking, Ca2+ entry through Cav1 channels, coupled with CICR through RyRs, generates an elevation in [Ca2+]. Elevated [Ca2+] reaching the mitochondria favors the uptake of substrates by the mitochondria by acting on transporters and carriers on the IMM; Ca2+ taken up by mitochondria through the voltage-dependent anion channel (VDAC) and MCUC increases OXPHOS by enhancing the activity of the TCA cycle and by stimulating ATP synthesis by Complex V. The downside of this stimulation is an increase in the generation of ROS.
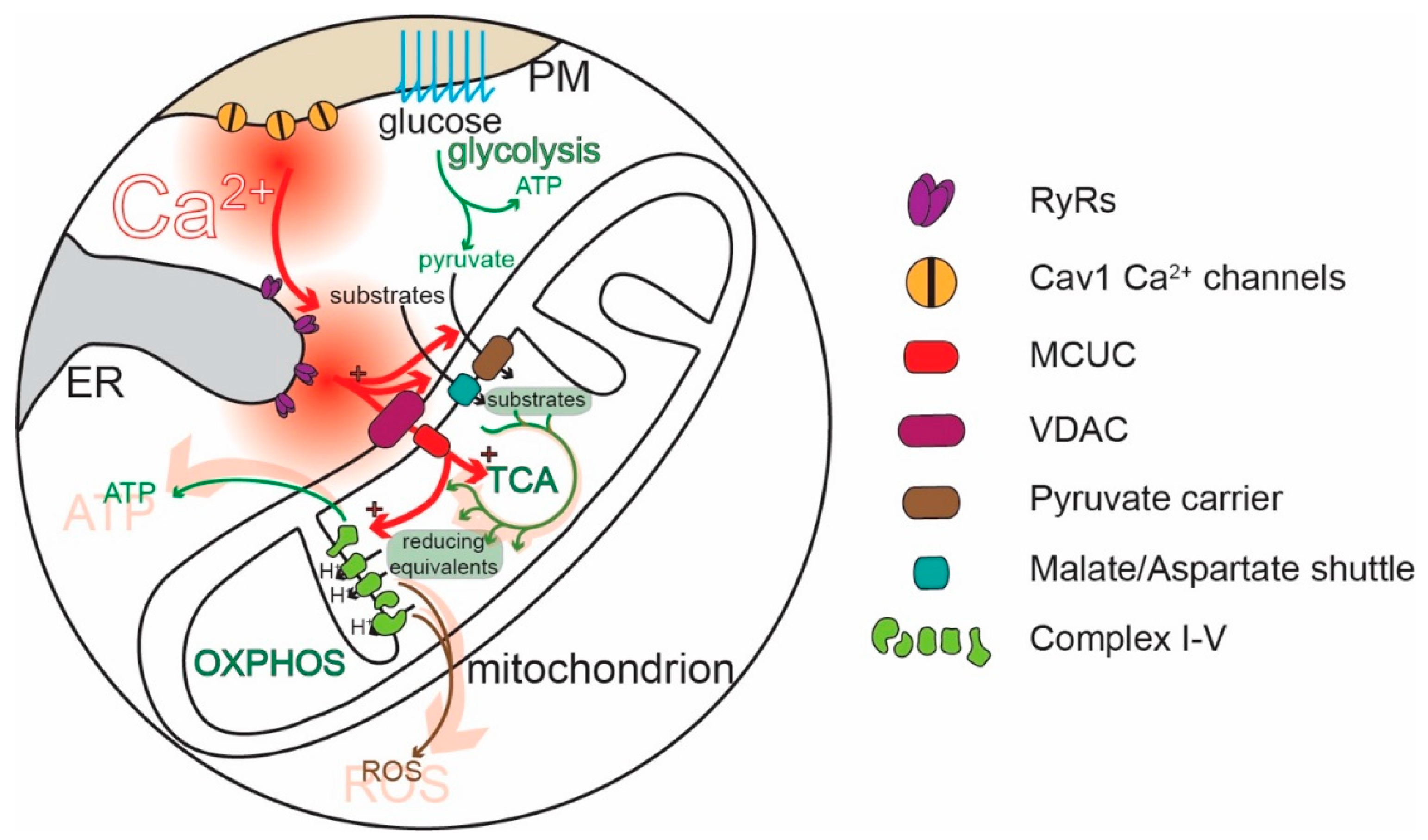
Figure 3.
Toxic effects of elevated [Ca2+] in SN DAergic neurons. The engagement of Cav1 channels in pacemaking and the release of Ca2+ through RyRs generate elevated cytosolic Ca2+ levels that increase α-synuclein (αSYN) aggregation, directly or through the activation of calpains; Ca2+-activated calpains can also damage several intracellular proteins. Ca2+ and calpain cleavage can increase or dysregulate tyrosine hydroxylase activity and DA production, and DA oxidation can favor the aggregation of αSYN.
Figure 3.
Toxic effects of elevated [Ca2+] in SN DAergic neurons. The engagement of Cav1 channels in pacemaking and the release of Ca2+ through RyRs generate elevated cytosolic Ca2+ levels that increase α-synuclein (αSYN) aggregation, directly or through the activation of calpains; Ca2+-activated calpains can also damage several intracellular proteins. Ca2+ and calpain cleavage can increase or dysregulate tyrosine hydroxylase activity and DA production, and DA oxidation can favor the aggregation of αSYN.
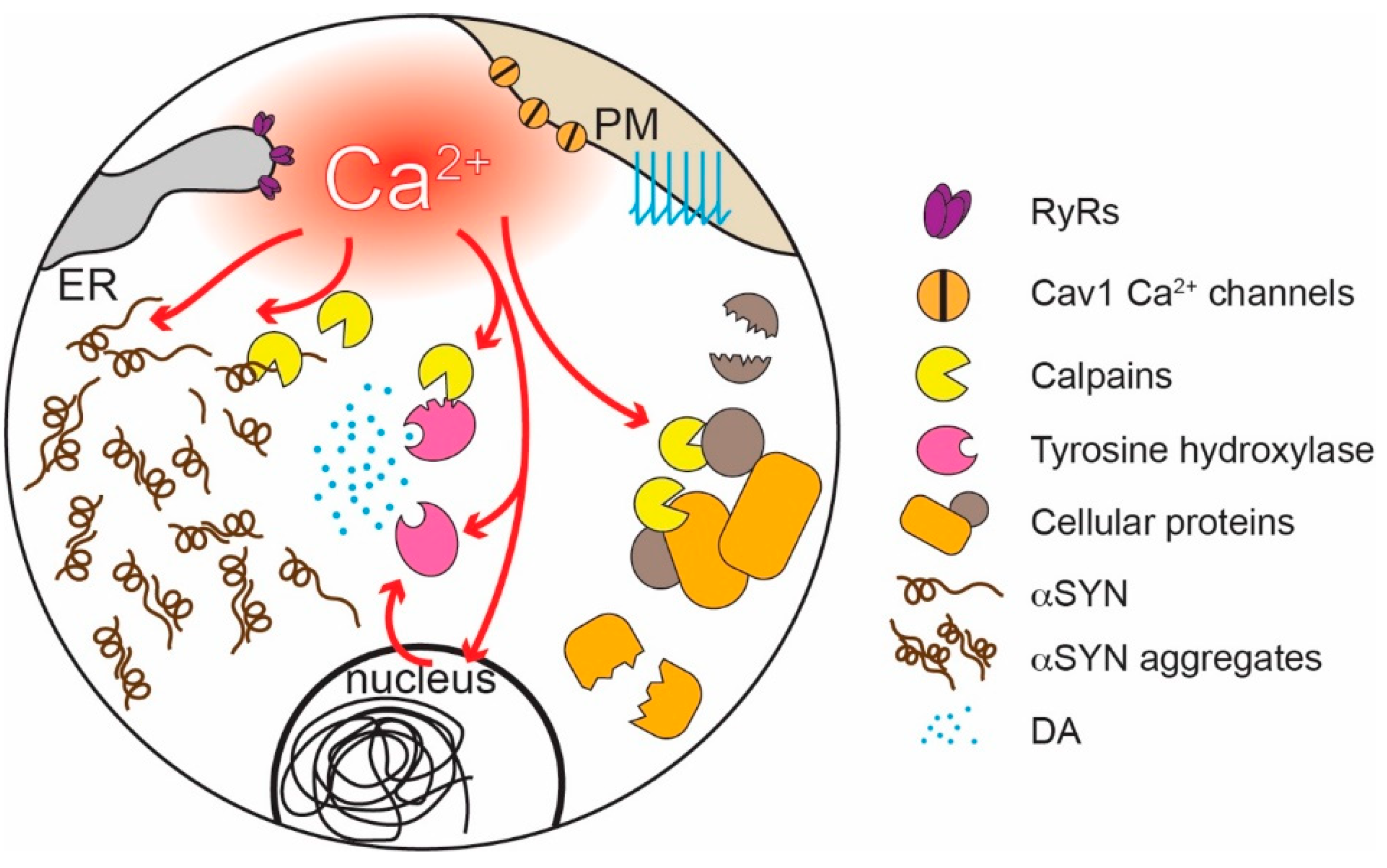
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
MDPI and ACS Style
Zampese, E.; Surmeier, D.J. Calcium, Bioenergetics, and Parkinson’s Disease. Cells 2020, 9, 2045. https://doi.org/10.3390/cells9092045
AMA Style
Zampese E, Surmeier DJ. Calcium, Bioenergetics, and Parkinson’s Disease. Cells. 2020; 9(9):2045. https://doi.org/10.3390/cells9092045
Chicago/Turabian StyleZampese, Enrico, and D. James Surmeier. 2020. "Calcium, Bioenergetics, and Parkinson’s Disease" Cells 9, no. 9: 2045. https://doi.org/10.3390/cells9092045
Note that from the first issue of 2016, this journal uses article numbers instead of page numbers. See further details here.