Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities


Abstract
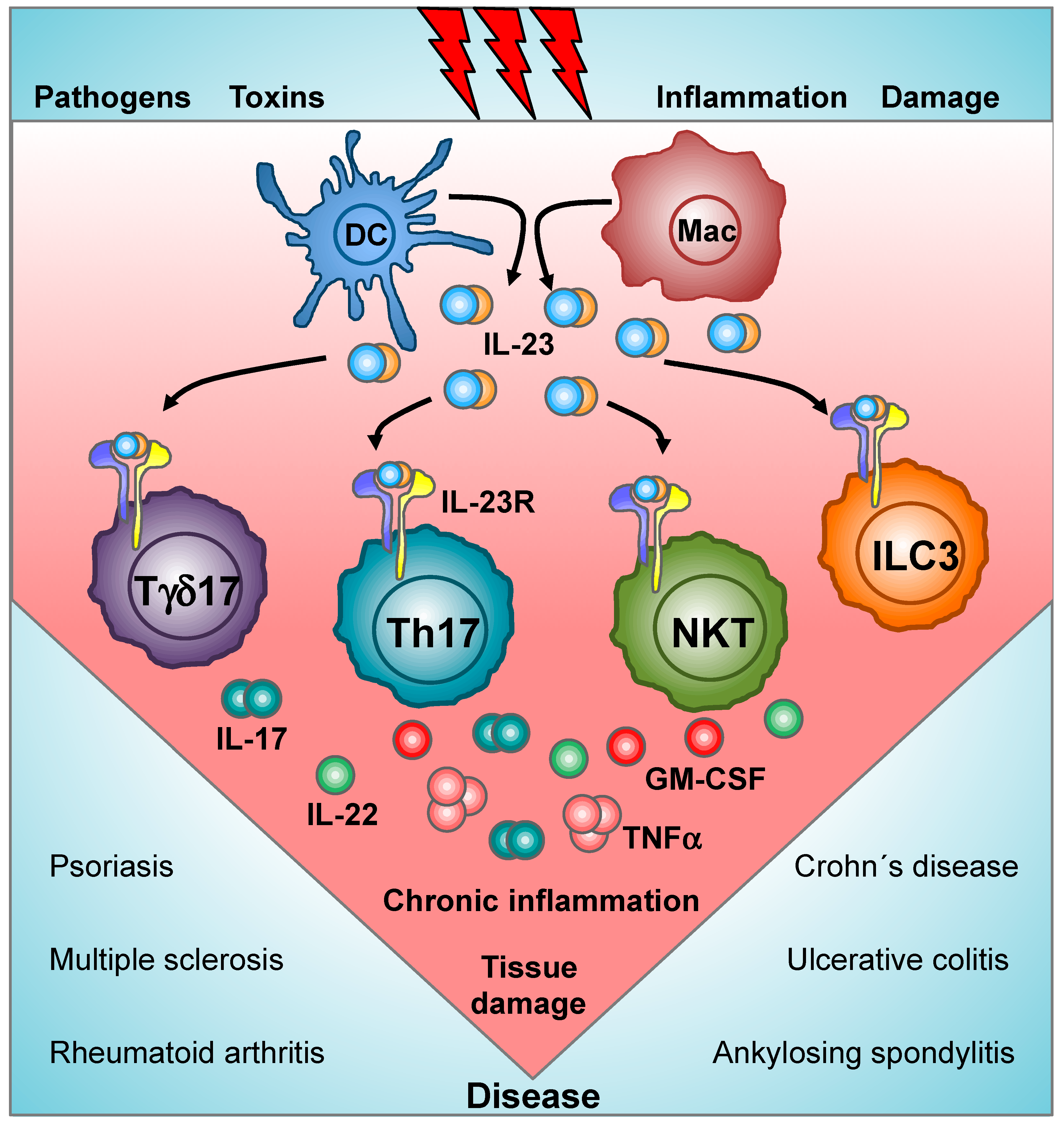
1. Introduction
2. IL-23/IL-23R Structure and Binding
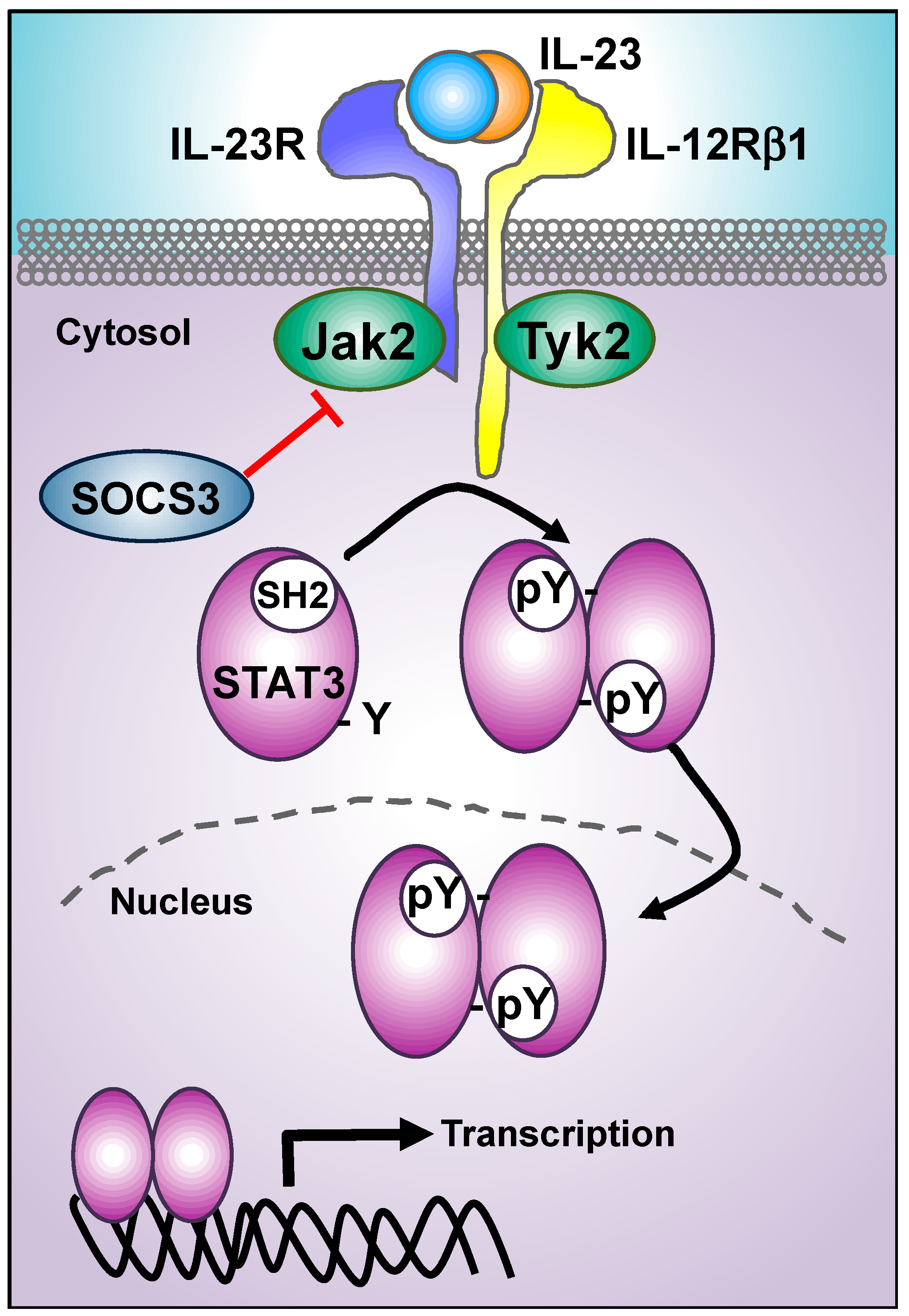
3. IL-23 Proximal Signaling Cascade: JAKs/STATs Module
4. New Players in IL-23 Proximal Signaling Events
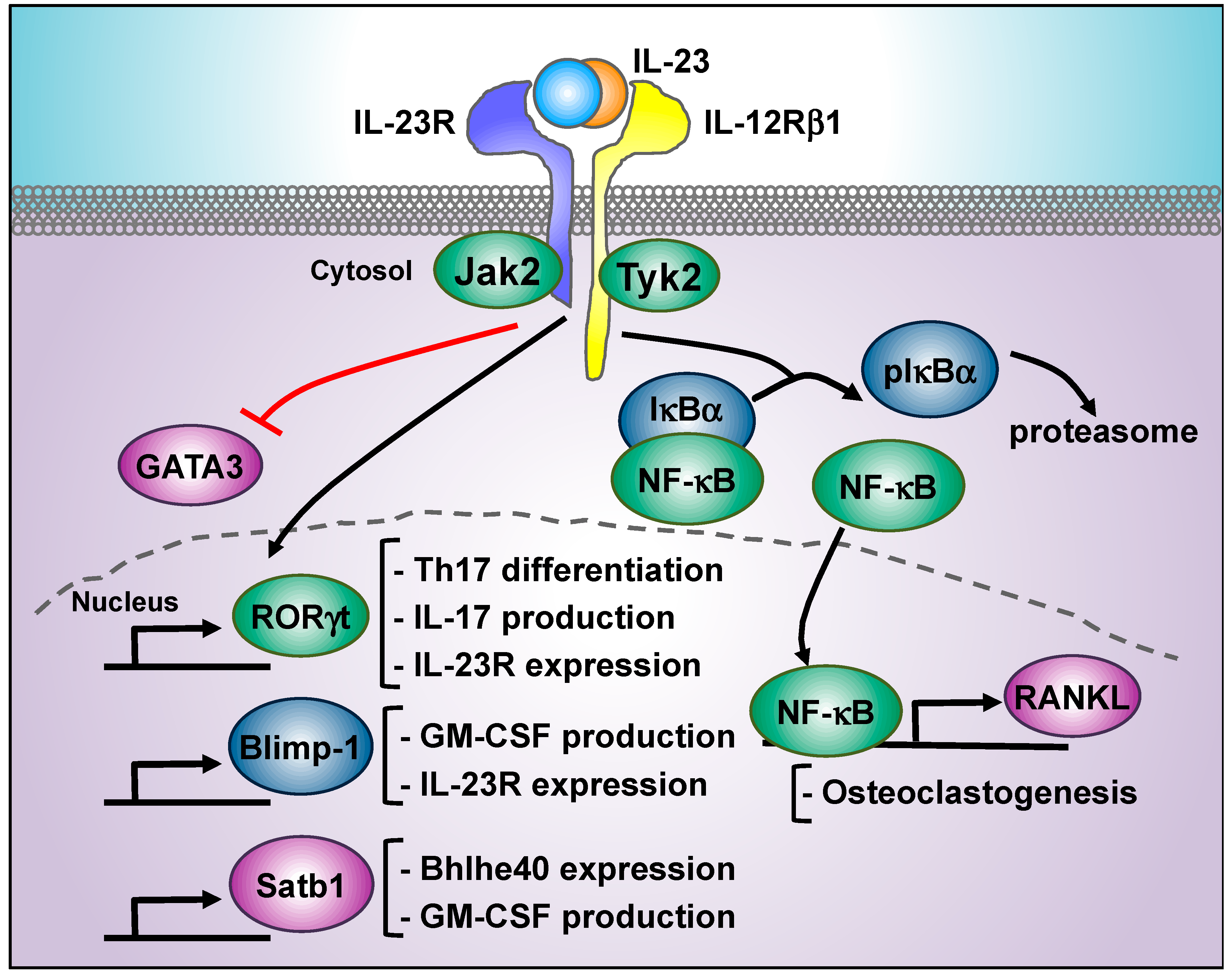
5. IL-23-Regulated Transcription Factors Beyond STAT3: RORγt, Blimp, NF-κB, Tbet, Satb1, and GATA3
5.1. RORγt
5.2. Blimp-1
5.3. NF-κB
5.4. T-bet
5.5. Satb1
5.6. GATA3 Inhibition
6. IL-23 and IL-1β: Signaling Crosstalk

{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Inhibitor | Target | Clinical Phase | Disease/Model | Route | Reference |
|---|---|---|---|---|---|
| IL-23R peptide antagonist | IL-23R | Preclinical | rIL-23-induced skin inflammation model, anti-CD40 systemic inflammation, CIA | i.p.* | [63] |
| IL-23 Alphabodies | IL-23 | Preclinical | rIL-23-induced skin inflammation model | i.d., i.p. | [64] |
| Tofacitinib | Jak1, Jak3, Jak2 | Approved | Rheumatoid and psoriasic arthritis, ulcerative colitis | oral | [78] |
| Phase 3 | Juvenile arthritis, systemic lupus erythematosus | oral | |||
| Phase 2 | Atopic dermatitis | topical | |||
| Ruxolitinib | Jak1, Jak2 | Phase 2 | Rheumatoid arthritis | oral | [78] |
| Phase 2 | Atopic dermatitis | topical | |||
| Baricitinib | Jak1, Jak2 | Approved | Rheumatoid arthritis | oral | [78] |
| Phase 2 | Psoriasis, systemic lupus erythematosus | oral | |||
| Phase 3 | Atopic dermatitis | topical | |||
| BMS-986165 | Tyk2 | Phase 2 | Psoriasis, rheumatoid arthritis, Crohn’s disease, ulcerative colitis, systemic lupus erythematosus | oral | [80] |
| Brepocitinib (PF-06700841) | Tyk2, Jak1 | Phase 2 | Psoriasis, Crohn’s disease, ulcerative colitis | oral | [81] |
| Baicalin | STAT3/SOCS3 | Preclinical | EAE model | i.p. | [98] |
| Fasudil | ROCK | Preclinical | EAE model | oral, i.p. | [116,117] |
| WAR5 compound | ROCK | Preclinical | EAE model | i.p. | [118] |
| Statins | ROCK | Preclinical | EAE model | i.p. | [119] |
| KD025 | ROCK | Phase 2 | Psoriasis vulgaris | oral | [120] |
| Fingolimod (FTY720) | S1PR1 | Approved | Multiple sclerosis | oral | [127] |
| Preclinical | IMQ-induced skin inflammation model | i.p. | [128] | ||
| Natalizumab | VLA4 | Approved | Multiple sclerosis, Crohn’s disease | i.v. | [129] |
| TEPP-46 | PKM2 | Preclinical | EAE model | i.p. | [132] |
| CH-223191 | AhR inhibitor | Preclinical | IL-23-induced skin inflammation model | i.p. | [139] |
| FICZ | AhR agonist | Preclinical | IMQ-induced skin inflammation model | i.p | [141] |
| Nitric oxide carriers | AhR | Preclinical | EAE model | oral | [147] |
| Digoxin | RORγt | Preclinical | EAE model | i.p. | [155] |
| SR1001 | RORαt, RORγt | Preclinical | EAE model | i.p. | [156] |
| TMP778 | RORγt | Preclinical | EAE model | s.c. | [157] |
| GSK805 | RORγt | Preclinical | EAE model | s.c. | [157] |
| JNJ-54271074 | RORγt | Preclinical | CIA and IL-23-induced skin inflammation models | oral | [158] |
| JPH203 | LAT1 | Preclinical | IMQ-induced skin inflammation model | i.p. | [140] |
| Rapamycin | mTOR | Preclinical | IMQ-induced skin inflammation model | i.p. | [140] |
7. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Gaffen, S.L.; Jain, R.; Garg, A.V.; Cua, D.J. The IL-23-IL-17 immune axis: From mechanisms to therapeutic testing. Nat. Rev. Immunol. 2014, 14, 585–600. [Google Scholar] [CrossRef] [PubMed]
- Patel, D.D.; Kuchroo, V.K. Th17 Cell Pathway in Human Immunity: Lessons from Genetics and Therapeutic Interventions. Immunity 2015, 43, 1040–1051. [Google Scholar] [CrossRef] [PubMed]
- Tait Wojno, E.D.; Hunter, C.A.; Stumhofer, J.S. The Immunobiology of the Interleukin-12 Family: Room for Discovery. Immunity 2019, 50, 851–870. [Google Scholar] [CrossRef] [PubMed]
- Oppmann, B.; Lesley, R.; Blom, B.; Timans, J.C.; Xu, Y.; Hunte, B.; Vega, F.; Yu, N.; Wang, J.; Singh, K.; et al. Novel p19 protein engages IL-12p40 to form a cytokine, IL-23, with biological activities similar as well as distinct from IL-12. Immunity 2000, 13, 715–725. [Google Scholar] [CrossRef]
- Langrish, C.L.; Chen, Y.; Blumenschein, W.M.; Mattson, J.; Basham, B.; Sedgwick, J.D.; McClanahan, T.; Kastelein, R.A.; Cua, D.J. IL-23 drives a pathogenic T cell population that induces autoimmune inflammation. J. Exp. Med. 2005, 201, 233–240. [Google Scholar] [CrossRef]
- Zheng, Y.; Danilenko, D.M.; Valdez, P.; Kasman, I.; Eastham-Anderson, J.; Wu, J.; Ouyang, W. Interleukin-22, a T(H)17 cytokine, mediates IL-23-induced dermal inflammation and acanthosis. Nature 2007, 445, 648–651. [Google Scholar] [CrossRef]
- El-Behi, M.; Ciric, B.; Dai, H.; Yan, Y.; Cullimore, M.; Safavi, F.; Zhang, G.-X.; Dittel, B.N.; Rostami, A. The encephalitogenicity of T(H)17 cells is dependent on IL-1- and IL-23-induced production of the cytokine GM-CSF. Nat. Immunol. 2011, 12, 568–575. [Google Scholar] [CrossRef]
- Codarri, L.; Gyülvészi, G.; Tosevski, V.; Hesske, L.; Fontana, A.; Magnenat, L.; Suter, T.; Becher, B. RORγt drives production of the cytokine GM-CSF in helper T cells, which is essential for the effector phase of autoimmune neuroinflammation. Nat. Immunol. 2011, 12, 560–567. [Google Scholar] [CrossRef]
- Chan, J.R.; Blumenschein, W.; Murphy, E.; Diveu, C.; Wiekowski, M.; Abbondanzo, S.; Lucian, L.; Geissler, R.; Brodie, S.; Kimball, A.B.; et al. IL-23 stimulates epidermal hyperplasia via TNF and IL-20R2-dependent mechanisms with implications for psoriasis pathogenesis. J. Exp. Med. 2006, 203, 2577–2587. [Google Scholar] [CrossRef]
- Burkett, P.R.; Meyer zu Horste, G.; Kuchroo, V.K. Pouring fuel on the fire: Th17 cells, the environment, and autoimmunity. J. Clin. Investig. 2015, 125, 2211–2219. [Google Scholar] [CrossRef]
- Zwicky, P.; Unger, S.; Becher, B. Targeting interleukin-17 in chronic inflammatory disease: A clinical perspective. J. Exp. Med. 2020, 217. [Google Scholar] [CrossRef] [PubMed]
- Guttman-Yassky, E.; Lowes, M.A.; Fuentes-Duculan, J.; Zaba, L.C.; Cardinale, I.; Nograles, K.E.; Khatcherian, A.; Novitskaya, I.; Carucci, J.A.; Bergman, R.; et al. Low expression of the IL-23/Th17 pathway in atopic dermatitis compared to psoriasis. J. Immunol. 2008, 181, 7420–7427. [Google Scholar] [CrossRef] [PubMed]
- Perera, G.K.; Di Meglio, P.; Nestle, F.O. Psoriasis. Annu Rev. Pathol 2012, 7, 385–422. [Google Scholar] [CrossRef] [PubMed]
- Acosta-Rodriguez, E.V.; Rivino, L.; Geginat, J.; Jarrossay, D.; Gattorno, M.; Lanzavecchia, A.; Sallusto, F.; Napolitani, G. Surface phenotype and antigenic specificity of human interleukin 17-producing T helper memory cells. Nat. Immunol. 2007, 8, 639–646. [Google Scholar] [CrossRef] [PubMed]
- Onishi, R.M.; Gaffen, S.L. Interleukin-17 and its target genes: Mechanisms of interleukin-17 function in disease. Immunology 2010, 129, 311–321. [Google Scholar] [CrossRef] [PubMed]
- Kim, K.-W.; Kim, H.-R.; Kim, B.-M.; Cho, M.-L.; Lee, S.-H. Th17 cytokines regulate osteoclastogenesis in rheumatoid arthritis. Am. J. Pathol. 2015, 185, 3011–3024. [Google Scholar] [CrossRef]
- Yang, D.; Chertov, O.; Bykovskaia, S.N.; Chen, Q.; Buffo, M.J.; Shogan, J.; Anderson, M.; Schröder, J.M.; Wang, J.M.; Howard, O.M.; et al. Beta-defensins: Linking innate and adaptive immunity through dendritic and T cell CCR. Science 1999, 286, 525–528. [Google Scholar] [CrossRef]
- Goetz, D.H.; Holmes, M.A.; Borregaard, N.; Bluhm, M.E.; Raymond, K.N.; Strong, R.K. The neutrophil lipocalin NGAL is a bacteriostatic agent that interferes with siderophore-mediated iron acquisition. Mol. Cell 2002, 10, 1033–1043. [Google Scholar] [CrossRef]
- Lee, Y.; Awasthi, A.; Yosef, N.; Quintana, F.J.; Xiao, S.; Peters, A.; Wu, C.; Kleinewietfeld, M.; Kunder, S.; Hafler, D.A.; et al. Induction and molecular signature of pathogenic TH17 cells. Nat. Immunol. 2012, 13, 991–999. [Google Scholar] [CrossRef]
- Hueber, W.; Sands, B.E.; Lewitzky, S.; Vandemeulebroecke, M.; Reinisch, W.; Higgins, P.D.R.; Wehkamp, J.; Feagan, B.G.; Yao, M.D.; Karczewski, M.; et al. Secukinumab in Crohn’s Disease Study Group Secukinumab, a human anti-IL-17A monoclonal antibody, for moderate to severe Crohn’s disease: Unexpected results of a randomised, double-blind placebo-controlled trial. Gut 2012, 61, 1693–1700. [Google Scholar] [CrossRef]
- Targan, S.R.; Feagan, B.; Vermeire, S.; Panaccione, R.; Melmed, G.Y.; Landers, C.; Li, D.; Russell, C.; Newmark, R.; Zhang, N.; et al. A Randomized, Double-Blind, Placebo-Controlled Phase 2 Study of Brodalumab in Patients With Moderate-to-Severe Crohn’s Disease. Am. J. Gastroenterol. 2016, 111, 1599–1607. [Google Scholar] [CrossRef] [PubMed]
- Kumar, P.; Monin, L.; Castillo, P.; Elsegeiny, W.; Horne, W.; Eddens, T.; Vikram, A.; Good, M.; Schoenborn, A.A.; Bibby, K.; et al. Intestinal Interleukin-17 Receptor Signaling Mediates Reciprocal Control of the Gut Microbiota and Autoimmune Inflammation. Immunity 2016, 44, 659–671. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.S.; Tato, C.M.; Joyce-Shaikh, B.; Gulen, M.F.; Gulan, F.; Cayatte, C.; Chen, Y.; Blumenschein, W.M.; Judo, M.; Ayanoglu, G.; et al. Interleukin-23-Independent IL-17 Production Regulates Intestinal Epithelial Permeability. Immunity 2015, 43, 727–738. [Google Scholar] [CrossRef] [PubMed]
- Maxwell, J.R.; Zhang, Y.; Brown, W.A.; Smith, C.L.; Byrne, F.R.; Fiorino, M.; Stevens, E.; Bigler, J.; Davis, J.A.; Rottman, J.B.; et al. Differential Roles for Interleukin-23 and Interleukin-17 in Intestinal Immunoregulation. Immunity 2015, 43, 739–750. [Google Scholar] [CrossRef] [PubMed]
- Song, X.; Dai, D.; He, X.; Zhu, S.; Yao, Y.; Gao, H.; Wang, J.; Qu, F.; Qiu, J.; Wang, H.; et al. Growth Factor FGF2 Cooperates with Interleukin-17 to Repair Intestinal Epithelial Damage. Immunity 2015, 43, 488–501. [Google Scholar] [CrossRef] [PubMed]
- Dudakov, J.A.; Hanash, A.M.; van den Brink, M.R.M. Interleukin-22: Immunobiology and pathology. Annu. Rev. Immunol. 2015, 33, 747–785. [Google Scholar] [CrossRef]
- Sonnenberg, G.F.; Fouser, L.A.; Artis, D. Border patrol: Regulation of immunity, inflammation and tissue homeostasis at barrier surfaces by IL-22. Nat. Immunol. 2011, 12, 383–390. [Google Scholar] [CrossRef]
- Mortha, A.; Chudnovskiy, A.; Hashimoto, D.; Bogunovic, M.; Spencer, S.P.; Belkaid, Y.; Merad, M. Microbiota-dependent crosstalk between macrophages and ILC3 promotes intestinal homeostasis. Science 2014, 343, 1249288. [Google Scholar] [CrossRef]
- Sabat, R.; Ouyang, W.; Wolk, K. Therapeutic opportunities of the IL-22-IL-22R1 system. Nat. Rev. Drug Discov 2014, 13, 21–38. [Google Scholar] [CrossRef]
- Wolk, K.; Haugen, H.S.; Xu, W.; Witte, E.; Waggie, K.; Anderson, M.; Baur, V.E.; Witte, K.; Warszawska, K.; Philipp, S.; et al. IL-22 and IL-20 are key mediators of the epidermal alterations in psoriasis while IL-17 and IFN-gamma are not. J. Mol. Med. 2009, 87, 523–536. [Google Scholar] [CrossRef]
- Wolk, K.; Witte, E.; Wallace, E.; Döcke, W.-D.; Kunz, S.; Asadullah, K.; Volk, H.-D.; Sterry, W.; Sabat, R. IL-22 regulates the expression of genes responsible for antimicrobial defense, cellular differentiation, and mobility in keratinocytes: A potential role in psoriasis. Eur. J. Immunol. 2006, 36, 1309–1323. [Google Scholar] [CrossRef] [PubMed]
- Zheng, Y.; Valdez, P.A.; Danilenko, D.M.; Hu, Y.; Sa, S.M.; Gong, Q.; Abbas, A.R.; Modrusan, Z.; Ghilardi, N.; De Sauvage, F.J.; et al. Interleukin-22 mediates early host defense against attaching and effacing bacterial pathogens. Nat. Med. 2008, 14, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.; Heller, J.J.; Guo, X.; Chen, Z.-M.E.; Fish, K.; Fu, Y.-X.; Zhou, L. The aryl hydrocarbon receptor regulates gut immunity through modulation of innate lymphoid cells. Immunity 2012, 36, 92–104. [Google Scholar] [CrossRef] [PubMed]
- Pickert, G.; Neufert, C.; Leppkes, M.; Zheng, Y.; Wittkopf, N.; Warntjen, M.; Lehr, H.-A.; Hirth, S.; Weigmann, B.; Wirtz, S.; et al. STAT3 links IL-22 signaling in intestinal epithelial cells to mucosal wound healing. J. Exp. Med. 2009, 206, 1465–1472. [Google Scholar] [CrossRef]
- Uhlig, H.H.; Powrie, F. Translating Immunology into Therapeutic Concepts for Inflammatory Bowel Disease. Annu. Rev. Immunol. 2018, 36, 755–781. [Google Scholar] [CrossRef]
- Sugimoto, K.; Ogawa, A.; Mizoguchi, E.; Shimomura, Y.; Andoh, A.; Bhan, A.K.; Blumberg, R.S.; Xavier, R.J.; Mizoguchi, A. IL-22 ameliorates intestinal inflammation in a mouse model of ulcerative colitis. J. Clin. Investig. 2008, 118, 534–544. [Google Scholar] [CrossRef]
- Zenewicz, L.A.; Yancopoulos, G.D.; Valenzuela, D.M.; Murphy, A.J.; Stevens, S.; Flavell, R.A. Innate and adaptive interleukin-22 protects mice from inflammatory bowel disease. Immunity 2008, 29, 947–957. [Google Scholar] [CrossRef]
- Stockinger, B.; Omenetti, S. The dichotomous nature of T helper 17 cells. Nat. Rev. Immunol. 2017, 17, 535–544. [Google Scholar] [CrossRef]
- Becher, B.; Tugues, S.; Greter, M. GM-CSF: From Growth Factor to Central Mediator of Tissue Inflammation. Immunity 2016, 45, 963–973. [Google Scholar] [CrossRef]
- Monaghan, K.L.; Wan, E.C.K. The Role of Granulocyte-Macrophage Colony-Stimulating Factor in Murine Models of Multiple Sclerosis. Cells 2020, 9, 611. [Google Scholar] [CrossRef]
- Croxford, A.L.; Lanzinger, M.; Hartmann, F.J.; Schreiner, B.; Mair, F.; Pelczar, P.; Clausen, B.E.; Jung, S.; Greter, M.; Becher, B. The Cytokine GM-CSF Drives the Inflammatory Signature of CCR2+ Monocytes and Licenses Autoimmunity. Immunity 2015, 43, 502–514. [Google Scholar] [CrossRef] [PubMed]
- King, I.L.; Dickendesher, T.L.; Segal, B.M. Circulating Ly-6C+ myeloid precursors migrate to the CNS and play a pathogenic role during autoimmune demyelinating disease. Blood 2009, 113, 3190–3197. [Google Scholar] [CrossRef] [PubMed]
- Ko, H.-J.; Brady, J.L.; Ryg-Cornejo, V.; Hansen, D.S.; Vremec, D.; Shortman, K.; Zhan, Y.; Lew, A.M. GM-CSF-responsive monocyte-derived dendritic cells are pivotal in Th17 pathogenesis. J. Immunol. 2014, 192, 2202–2209. [Google Scholar] [CrossRef] [PubMed]
- Spath, S.; Komuczki, J.; Hermann, M.; Pelczar, P.; Mair, F.; Schreiner, B.; Becher, B. Dysregulation of the Cytokine GM-CSF Induces Spontaneous Phagocyte Invasion and Immunopathology in the Central Nervous System. Immunity 2017, 46, 245–260. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Bowman, E.P.; McElwee, J.J.; Smyth, M.J.; Casanova, J.-L.; Cooper, A.M.; Cua, D.J. IL-12 and IL-23 cytokines: From discovery to targeted therapies for immune-mediated inflammatory diseases. Nat. Med. 2015, 21, 719–729. [Google Scholar] [CrossRef]
- Moschen, A.R.; Tilg, H.; Raine, T. IL-12, IL-23 and IL-17 in IBD: Immunobiology and therapeutic targeting. Nat. Rev. Gastroenterol. Hepatol. 2018, 16, 185–196. [Google Scholar] [CrossRef]
- Ghilardi, N.; Pappu, R.; Arron, J.R.; Chan, A.C. 30 Years of Biotherapeutics Development-What Have We Learned? Annu. Rev. Immunol. 2020, 38, 249–287. [Google Scholar] [CrossRef]
- Cohen, P. Targeting protein kinases for the development of anti-inflammatory drugs. Curr. Opin. Cell Biol. 2009, 21, 317–324. [Google Scholar] [CrossRef]
- Kontzias, A.; Laurence, A.; Gadina, M.; O’Shea, J.J. Kinase inhibitors in the treatment of immune-mediated disease. F1000 Med. Rep. 2012, 4, 5. [Google Scholar] [CrossRef]
- Imai, K.; Takaoka, A. Comparing antibody and small-molecule therapies for cancer. Nat. Rev. Cancer 2006, 6, 714–727. [Google Scholar] [CrossRef]
- Gebauer, M.; Skerra, A. Engineered Protein Scaffolds as Next-Generation Therapeutics. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 391–415. [Google Scholar] [CrossRef] [PubMed]
- Parham, C.; Chirica, M.; Timans, J.; Vaisberg, E.; Travis, M.; Cheung, J.; Pflanz, S.; Zhang, R.; Singh, K.P.; Vega, F.; et al. A receptor for the heterodimeric cytokine IL-23 is composed of IL-12Rbeta1 and a novel cytokine receptor subunit, IL-23R. J. Immunol. 2002, 168, 5699–5708. [Google Scholar] [CrossRef] [PubMed]
- Cua, D.J.; Sherlock, J.; Chen, Y.; Murphy, C.A.; Joyce, B.; Seymour, B.; Lucian, L.; To, W.; Kwan, S.; Churakova, T.; et al. Interleukin-23 rather than interleukin-12 is the critical cytokine for autoimmune inflammation of the brain. Nature 2003, 421, 744–748. [Google Scholar] [CrossRef] [PubMed]
- Yoon, C.; Johnston, S.C.; Tang, J.; Stahl, M.; Tobin, J.F.; Somers, W.S. Charged residues dominate a unique interlocking topography in the heterodimeric cytokine interleukin-12. EMBO J. 2000, 19, 3530–3541. [Google Scholar] [CrossRef]
- Lupardus, P.J.; Garcia, K.C. The structure of interleukin-23 reveals the molecular basis of p40 subunit sharing with interleukin-12. J. Mol. Biol. 2008, 382, 931–941. [Google Scholar] [CrossRef]
- Schröder, J.; Moll, J.M.; Baran, P.; Grötzinger, J.; Scheller, J.; Floss, D.M. Non-canonical interleukin 23 receptor complex assembly: p40 protein recruits interleukin 12 receptor β1 via site II and induces p19/interleukin 23 receptor interaction via site III. J. Biol. Chem. 2015, 290, 359–370. [Google Scholar] [CrossRef]
- Meier, S.; Bohnacker, S.; Klose, C.J.; Lopez, A.; Choe, C.A.; Schmid, P.W.N.; Bloemeke, N.; Rührnößl, F.; Haslbeck, M.; Bieren, J.E.-V.; et al. The molecular basis of chaperone-mediated interleukin 23 assembly control. Nat. Commun 2019, 10, 4121. [Google Scholar] [CrossRef]
- Wang, X.; Liu, X.; Zhang, Y.; Wang, Z.; Zhu, G.; Han, G.; Chen, G.; Hou, C.; Wang, T.; Ma, N.; et al. Interleukin (IL)-39 [IL-23p19/Epstein-Barr virus-induced 3 (Ebi3)] induces differentiation/expansion of neutrophils in lupus-prone mice. Clin. Exp. Immunol. 2016, 186, 144–156. [Google Scholar] [CrossRef]
- Bridgewood, C.; Alase, A.; Watad, A.; Wittmann, M.; Cuthbert, R.; McGonagle, D. The IL-23p19/EBI3 heterodimeric cytokine termed IL-39 remains a theoretical cytokine in man. Inflamm. Res. 2019, 68, 423–426. [Google Scholar] [CrossRef]
- Chua, A.O.; Chizzonite, R.; Desai, B.B.; Truitt, T.P.; Nunes, P.; Minetti, L.J.; Warrier, R.R.; Presky, D.H.; Levine, J.F.; Gately, M.K. Expression cloning of a human IL-12 receptor component. A new member of the cytokine receptor superfamily with strong homology to gp 130. J. Immunol. 1994, 153, 128–136. [Google Scholar]
- Chua, A.O.; Wilkinson, V.L.; Presky, D.H.; Gubler, U. Cloning and characterization of a mouse IL-12 receptor-beta component. J. Immunol. 1995, 155, 4286–4294. [Google Scholar] [PubMed]
- Bloch, Y.; Bouchareychas, L.; Merceron, R.; Składanowska, K.; Van den Bossche, L.; Detry, S.; Govindarajan, S.; Elewaut, D.; Haerynck, F.; Dullaers, M.; et al. Structural Activation of Pro-inflammatory Human Cytokine IL-23 by Cognate IL-23 Receptor Enables Recruitment of the Shared Receptor IL-12Rβ1. Immunity 2018, 48, 45–58.e6. [Google Scholar] [CrossRef] [PubMed]
- Quiniou, C.; Domínguez-Punaro, M.; Cloutier, F.; Erfani, A.; Ennaciri, J.; Sivanesan, D.; Sanchez, M.; Chognard, G.; Hou, X.; Rivera, J.C.; et al. Specific targeting of the IL-23 receptor, using a novel small peptide noncompetitive antagonist, decreases the inflammatory response. Am. J. Physiol. Regul. Integr. Comp. Physiol. 2014, 307, R1216–R1230. [Google Scholar] [CrossRef]
- Desmet, J.; Verstraete, K.; Bloch, Y.; Lorent, E.; Wen, Y.; Devreese, B.; Vandenbroucke, K.; Loverix, S.; Hettmann, T.; Deroo, S.; et al. Structural basis of IL-23 antagonism by an Alphabody protein scaffold. Nat. Commun. 2014, 5, 5237. [Google Scholar] [CrossRef] [PubMed]
- Kuchař, M.; Vaňková, L.; Petroková, H.; Cerný, J.; Osička, R.; Pelák, O.; Sípová, H.; Schneider, B.; Homola, J.; Sebo, P.; et al. Human interleukin-23 receptor antagonists derived from an albumin-binding domain scaffold inhibit IL-23-dependent ex vivo expansion of IL-17-producing T-cells. Proteins 2014, 82, 975–989. [Google Scholar] [CrossRef] [PubMed]
- Křížová, L.; Kuchař, M.; Petroková, H.; Osička, R.; Hlavničková, M.; Pelák, O.; Cerný, J.; Kalina, T.; Malý, P. p19-targeted ABD-derived protein variants inhibit IL-23 binding and exert suppressive control over IL-23-stimulated expansion of primary human IL-17+ T-cells. Autoimmunity 2017, 50, 102–113. [Google Scholar] [CrossRef] [PubMed]
- Hlavničková, M.; Kuchař, M.; Osička, R.; Vaňková, L.; Petroková, H.; Malý, M.; Cerný, J.; Arenberger, P.; Malý, P. ABD-Derived Protein Blockers of Human IL-17 Receptor A as Non-IgG Alternatives for Modulation of IL-17-Dependent Pro-Inflammatory Axis. Int. J. Mol. Sci. 2018, 19, 3089. [Google Scholar] [CrossRef]
- Plavec, T.V.; Kuchař, M.; Benko, A.; Lišková, V.; Cerný, J.; Berlec, A.; Malý, P. Engineered Lactococcus lactis Secreting IL-23 Receptor-Targeted REX Protein Blockers for Modulation of IL-23/Th17-Mediated Inflammation. Microorganisms 2019, 7, 152. [Google Scholar] [CrossRef]
- Škrlec, K.; Zadravec, P.; Hlavničková, M.; Kuchař, M.; Vaňková, L.; Petroková, H.; Křížová, L.; Cerný, J.; Berlec, A.; Malý, P. p19-Targeting ILP Protein Blockers of IL-23/Th-17 Pro-Inflammatory Axis Displayed on Engineered Bacteria of Food Origin. Int. J. Mol. Sci. 2018, 19, 1933. [Google Scholar] [CrossRef]
- Sayago, C.; Gonzalez Valcarcel, I.C.; Qian, Y.; Lee, J.; Alsina-Fernandez, J.; Fite, N.C.; Carrillo, J.J.; Zhang, F.F.; Chalmers, M.J.; Dodge, J.A.; et al. Deciphering Binding Interactions of IL-23R with HDX-MS: Mapping Protein and Macrocyclic Dodecapeptide Ligands. ACS Med. Chem. Lett. 2018, 9, 912–916. [Google Scholar] [CrossRef]
- Stark, G.R.; Darnell, J.E. The JAK-STAT pathway at twenty. Immunity 2012, 36, 503–514. [Google Scholar] [CrossRef] [PubMed]
- Villarino, A.V.; Kanno, Y.; Ferdinand, J.R.; O’Shea, J.J. Mechanisms of Jak/STAT signaling in immunity and disease. J. Immunol. 2015, 194, 21–27. [Google Scholar] [CrossRef] [PubMed]
- Floss, D.M.; Klöcker, T.; Schröder, J.; Lamertz, L.; Mrotzek, S.; Strobl, B.; Hermanns, H.; Scheller, J. Defining the functional binding sites of interleukin 12 receptor β1 and interleukin 23 receptor to Janus kinases. Mol. Biol. Cell 2016, 27, 2301–2316. [Google Scholar] [CrossRef] [PubMed]
- Neubauer, H.; Cumano, A.; Müller, M.; Wu, H.; Huffstadt, U.; Pfeffer, K. Jak2 deficiency defines an essential developmental checkpoint in definitive hematopoiesis. Cell 1998, 93, 397–409. [Google Scholar] [CrossRef]
- Oyamada, A.; Ikebe, H.; Itsumi, M.; Saiwai, H.; Okada, S.; Shimoda, K.; Iwakura, Y.; Nakayama, K.I.; Iwamoto, Y.; Yoshikai, Y.; et al. Tyrosine kinase 2 plays critical roles in the pathogenic CD4 T cell responses for the development of experimental autoimmune encephalomyelitis. J. Immunol. 2009, 183, 7539–7546. [Google Scholar] [CrossRef] [PubMed]
- Ishizaki, M.; Akimoto, T.; Muromoto, R.; Yokoyama, M.; Ohshiro, Y.; Sekine, Y.; Maeda, H.; Shimoda, K.; Oritani, K.; Matsuda, T. Involvement of tyrosine kinase-2 in both the IL-12/Th1 and IL-23/Th17 axes in vivo. J. Immunol. 2011, 187, 181–189. [Google Scholar] [CrossRef] [PubMed]
- Gorman, J.A.; Hundhausen, C.; Kinsman, M.; Arkatkar, T.; Allenspach, E.J.; Clough, C.; West, S.E.; Thomas, K.; Eken, A.; Khim, S.; et al. The TYK2-P1104A Autoimmune Protective Variant Limits Coordinate Signals Required to Generate Specialized T Cell Subsets. Front. Immunol. 2019, 10, 44. [Google Scholar] [CrossRef]
- Gadina, M.; Johnson, C.; Schwartz, D.; Bonelli, M.; Hasni, S.; Kanno, Y.; Changelian, P.; Laurence, A.; O’Shea, J.J. Translational and clinical advances in JAK-STAT biology: The present and future of jakinibs. J. Leukoc. Biol. 2018, 104, 499–514. [Google Scholar] [CrossRef]
- Schwartz, D.M.; Kanno, Y.; Villarino, A.; Ward, M.; Gadina, M.; O’Shea, J.J. JAK inhibition as a therapeutic strategy for immune and inflammatory diseases. Nat. Rev. Drug Discov. 2017, 16, 843–862. [Google Scholar] [CrossRef]
- Papp, K.; Gordon, K.; Thaçi, D.; Morita, A.; Gooderham, M.; Foley, P.; Girgis, I.G.; Kundu, S.; Banerjee, S. Phase 2 Trial of Selective Tyrosine Kinase 2 Inhibition in Psoriasis. N. Engl. J. Med. 2018, 379, 1313–1321. [Google Scholar] [CrossRef]
- Page, K.M.; Suárez-Fariñas, M.; Suprun, M.; Zhang, W.; Garcet, S.; Fuentes-Duculan, J.; Li, X.; Scaramozza, M.; Kieras, E.; Banfield, C.; et al. Molecular and Cellular Responses to the TYK2/JAK1 Inhibitor PF-06700841 Reveal Reduction of Skin Inflammation in Plaque Psoriasis. J. Investig. Dermatol. 2020, 140, 1546–1555. [Google Scholar] [CrossRef] [PubMed]
- Solimani, F.; Meier, K.; Ghoreschi, K. Emerging Topical and Systemic JAK Inhibitors in Dermatology. Front. Immunol. 2019, 10, 2847. [Google Scholar] [CrossRef] [PubMed]
- Floss, D.M.; Mrotzek, S.; Klöcker, T.; Schröder, J.; Grötzinger, J.; Rose-John, S.; Scheller, J. Identification of Canonical Tyrosine-dependent and Non-canonical Tyrosine-independent STAT3 Activation Sites in the Intracellular Domain of the Interleukin 23 Receptor. J. Biol. Chem. 2013, 288, 19386–19400. [Google Scholar] [CrossRef] [PubMed]
- Page, B.D.G.; Croucher, D.C.; Li, Z.H.; Haftchenary, S.; Jimenez-Zepeda, V.H.; Atkinson, J.; Spagnuolo, P.A.; Wong, Y.L.; Colaguori, R.; Lewis, A.M.; et al. Inhibiting aberrant signal transducer and activator of transcription protein activation with tetrapodal, small molecule Src homology 2 domain binders: Promising agents against multiple myeloma. J. Med. Chem. 2013, 56, 7190–7200. [Google Scholar] [CrossRef]
- Fletcher, S.; Singh, J.; Zhang, X.; Yue, P.; Page, B.D.G.; Sharmeen, S.; Shahani, V.M.; Zhao, W.; Schimmer, A.D.; Turkson, J.; et al. Disruption of transcriptionally active Stat3 dimers with non-phosphorylated, salicylic acid-based small molecules: Potent in vitro and tumor cell activities. Chembiochem 2009, 10, 1959–1964. [Google Scholar] [CrossRef]
- Orlova, A.; Wagner, C.; de Araujo, E.D.; Bajusz, D.; Neubauer, H.A.; Herling, M.; Gunning, P.T.; Keserű, G.M.; Moriggl, R. Direct Targeting Options for STAT3 and STAT5 in Cancer. Cancers 2019, 11, 1930. [Google Scholar] [CrossRef]
- Takeda, K.; Noguchi, K.; Shi, W.; Tanaka, T.; Matsumoto, M.; Yoshida, N.; Kishimoto, T.; Akira, S. Targeted disruption of the mouse Stat3 gene leads to early embryonic lethality. Proc. Natl. Acad. Sci. USA 1997, 94, 3801–3804. [Google Scholar] [CrossRef]
- Bharadwaj, U.; Kasembeli, M.M.; Robinson, P.; Tweardy, D.J. Targeting Janus Kinases and Signal Transducer and Activator of Transcription 3 to Treat Inflammation, Fibrosis, and Cancer: Rationale, Progress, and Caution. Pharmacol. Rev. 2020, 72, 486–526. [Google Scholar] [CrossRef]
- Minegishi, Y.; Saito, M.; Tsuchiya, S.; Tsuge, I.; Takada, H.; Hara, T.; Kawamura, N.; Ariga, T.; Pasic, S.; Stojkovic, O.; et al. Dominant-negative mutations in the DNA-binding domain of STAT3 cause hyper-IgE syndrome. Nature 2007, 448, 1058–1062. [Google Scholar] [CrossRef]
- Milner, J.D.; Brenchley, J.M.; Laurence, A.; Freeman, A.F.; Hill, B.J.; Elias, K.M.; Kanno, Y.; Spalding, C.; Elloumi, H.Z.; Paulson, M.L.; et al. Impaired T(H)17 cell differentiation in subjects with autosomal dominant hyper-IgE syndrome. Nature 2008, 452, 773–776. [Google Scholar] [CrossRef]
- Ma, C.S.; Chew, G.Y.J.; Simpson, N.; Priyadarshi, A.; Wong, M.; Grimbacher, B.; Fulcher, D.A.; Tangye, S.G.; Cook, M.C. Deficiency of Th17 cells in hyper IgE syndrome due to mutations in STAT. J. Exp. Med. 2008, 205, 1551–1557. [Google Scholar] [CrossRef] [PubMed]
- Ma, C.S.; Avery, D.T.; Chan, A.; Batten, M.; Bustamante, J.; Boisson-Dupuis, S.; Arkwright, P.D.; Kreins, A.Y.; Averbuch, D.; Engelhard, D.; et al. Functional STAT3 deficiency compromises the generation of human T follicular helper cells. Blood 2012, 119, 3997–4008. [Google Scholar] [CrossRef] [PubMed]
- Deenick, E.K.; Avery, D.T.; Chan, A.; Berglund, L.J.; Ives, M.L.; Moens, L.; Stoddard, J.L.; Bustamante, J.; Boisson-Dupuis, S.; Tsumura, M.; et al. Naive and memory human B cells have distinct requirements for STAT3 activation to differentiate into antibody-secreting plasma cells. J. Exp. Med. 2013, 210, 2739–2753. [Google Scholar] [CrossRef] [PubMed]
- Wu, C.; Yosef, N.; Thalhamer, T.; Zhu, C.; Xiao, S.; Kishi, Y.; Regev, A.; Kuchroo, V.K. Induction of pathogenic TH17 cells by inducible salt-sensing kinase SGK 1. Nature 2013, 496, 513–517. [Google Scholar] [CrossRef] [PubMed]
- Cao, W.; Yang, Y.; Wang, Z.; Liu, A.; Fang, L.; Wu, F.; Hong, J.; Shi, Y.; Leung, S.; Dong, C.; et al. Leukemia inhibitory factor inhibits T helper 17 cell differentiation and confers treatment effects of neural progenitor cell therapy in autoimmune disease. Immunity 2011, 35, 273–284. [Google Scholar] [CrossRef]
- Eriksen, K.W.; Woetmann, A.; Skov, L.; Krejsgaard, T.; Bovin, L.F.; Hansen, M.L.; Grønbaek, K.; Billestrup, N.; Nissen, M.H.; Geisler, C.; et al. Deficient SOCS3 and SHP-1 expression in psoriatic T cells. J. Investig. Dermatol. 2010, 130, 1590–1597. [Google Scholar] [CrossRef]
- Frisullo, G.; Mirabella, M.; Angelucci, F.; Caggiula, M.; Morosetti, R.; Sancricca, C.; Patanella, A.K.; Nociti, V.; Iorio, R.; Bianco, A.; et al. The effect of disease activity on leptin, leptin receptor and suppressor of cytokine signalling-3 expression in relapsing-remitting multiple sclerosis. J. Neuroimmunol. 2007, 192, 174–183. [Google Scholar] [CrossRef]
- Zhang, Y.; Li, X.; Ciric, B.; Ma, C.-G.; Gran, B.; Rostami, A.; Zhang, G.-X. Therapeutic effect of baicalin on experimental autoimmune encephalomyelitis is mediated by SOCS3 regulatory pathway. Sci. Rep. 2015, 5, 17407. [Google Scholar] [CrossRef]
- Qu, X.; Han, J.; Zhang, Y.; Wang, Y.; Zhou, J.; Fan, H.; Yao, R. MiR-384 Regulates the Th17/Treg Ratio during Experimental Autoimmune Encephalomyelitis Pathogenesis. Front. Cell Neurosci. 2017, 11, 88. [Google Scholar] [CrossRef]
- Liu, X.; Zhou, F.; Yang, Y.; Wang, W.; Niu, L.; Zuo, D.; Li, X.; Hua, H.; Zhang, B.; Kou, Y.; et al. MiR-409-3p and MiR-1896 co-operatively participate in IL-17-induced inflammatory cytokine production in astrocytes and pathogenesis of EAE mice via targeting SOCS3/STAT3 signaling. Glia 2019, 67, 101–112. [Google Scholar] [CrossRef]
- Mujtaba, M.G.; Flowers, L.O.; Patel, C.B.; Patel, R.A.; Haider, M.I.; Johnson, H.M. Treatment of mice with the suppressor of cytokine signaling-1 mimetic peptide, tyrosine kinase inhibitor peptide, prevents development of the acute form of experimental allergic encephalomyelitis and induces stable remission in the chronic relapsing/remitting form. J. Immunol. 2005, 175, 5077–5086. [Google Scholar] [PubMed]
- Madonna, S.; Scarponi, C.; Doti, N.; Carbone, T.; Cavani, A.; Scognamiglio, P.L.; Marasco, D.; Albanesi, C. Therapeutical potential of a peptide mimicking the SOCS1 kinase inhibitory region in skin immune responses. Eur. J. Immunol. 2013, 43, 1883–1895. [Google Scholar] [CrossRef] [PubMed]
- Park, H.; Li, Z.; Yang, X.O.; Chang, S.H.; Nurieva, R.; Wang, Y.-H.; Wang, Y.; Hood, L.; Zhu, Z.; Tian, Q.; et al. A distinct lineage of CD4 T cells regulates tissue inflammation by producing interleukin. Nat. Immunol. 2005, 6, 1133–1141. [Google Scholar]
- Harrington, L.E.; Hatton, R.D.; Mangan, P.R.; Turner, H.; Murphy, T.L.; Murphy, K.M.; Weaver, C.T. Interleukin 17-producing CD4+ effector T cells develop via a lineage distinct from the T helper type 1 and 2 lineages. Nat. Immunol. 2005, 6, 1123–1132. [Google Scholar] [CrossRef]
- Mathur, A.N.; Chang, H.-C.; Zisoulis, D.G.; Stritesky, G.L.; Yu, Q.; O’Malley, J.T.; Kapur, R.; Levy, D.E.; Kansas, G.S.; Kaplan, M.H. Stat3 and Stat4 direct development of IL-17-secreting Th cells. J. Immunol. 2007, 178, 4901–4907. [Google Scholar] [CrossRef]
- Lee, P.W.; Smith, A.J.; Yang, Y.; Selhorst, A.J.; Liu, Y.; Racke, M.K.; Lovett-Racke, A.E. IL-23R-activated STAT3/STAT4 is essential for Th1/Th17-mediated CNS autoimmunity. JCI Insight 2017, 2. [Google Scholar] [CrossRef]
- Agerholm, R.; Rizk, J.; Viñals, M.T.; Bekiaris, V. STAT3 but not STAT4 is critical for γδT17 cell responses and skin inflammation. EMBO Rep. 2019, 20, e48647. [Google Scholar] [CrossRef]
- Lee, Y.K.; Turner, H.; Maynard, C.L.; Oliver, J.R.; Chen, D.; Elson, C.O.; Weaver, C.T. Late developmental plasticity in the T helper 17 lineage. Immunity 2009, 30, 92–107. [Google Scholar] [CrossRef]
- Sun, R.; Hedl, M.; Abraham, C. IL23 induces IL23R recycling and amplifies innate receptor-induced signalling and cytokines in human macrophages, and the IBD-protective IL23R R381Q variant modulates these outcomes. Gut 2020, 69, 264–273. [Google Scholar] [CrossRef]
- Duerr, R.H.; Taylor, K.D.; Brant, S.R.; Rioux, J.D.; Silverberg, M.S.; Daly, M.J.; Steinhart, A.H.; Abraham, C.; Regueiro, M.; Griffiths, A.; et al. A genome-wide association study identifies IL23R as an inflammatory bowel disease gene. Science 2006, 314, 1461–1463. [Google Scholar] [CrossRef]
- Capon, F.; Di Meglio, P.; Szaub, J.; Prescott, N.J.; Dunster, C.; Baumber, L.; Timms, K.; Gutin, A.; Abkevic, V.; Burden, A.D.; et al. Sequence variants in the genes for the interleukin-23 receptor (IL23R) and its ligand (IL12B) confer protection against psoriasis. Hum. Genet. 2007, 122, 201–206. [Google Scholar] [CrossRef] [PubMed]
- Di Meglio, P.; Di Cesare, A.; Laggner, U.; Chu, C.-C.; Napolitano, L.; Villanova, F.; Tosi, I.; Capon, F.; Trembath, R.C.; Peris, K.; et al. The IL23R R381Q gene variant protects against immune-mediated diseases by impairing IL-23-induced Th17 effector response in humans. PLoS ONE 2011, 6, e17160. [Google Scholar] [CrossRef] [PubMed]
- Álvarez-Salamero, C.; Castillo-González, R.; Pastor-Fernández, G.; Mariblanca, I.R.; Pino, J.; Cibrian, D.; Navarro, M.N. IL-23 signaling regulation of pro-inflammatory T-cell migration uncovered by phosphoproteomics. PLoS Biol. 2020, 18, e3000646. [Google Scholar] [CrossRef]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef]
- Pernis, A.B.; Ricker, E.; Weng, C.-H.; Rozo, C.; Yi, W. Rho Kinases in Autoimmune Diseases. Annu. Rev. Med. 2016, 67, 355–374. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Minohara, M.; Kikuchi, H.; Ishizu, T.; Tanaka, M.; Piao, H.; Osoegawa, M.; Ohyagi, Y.; Shimokawa, H.; Kira, J.-I. The selective Rho-kinase inhibitor Fasudil is protective and therapeutic in experimental autoimmune encephalomyelitis. J. Neuroimmunol. 2006, 180, 126–134. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.-Z.; Ding, J.; Ma, C.-G.; Sun, C.-H.; Sun, Y.-F.; Lu, C.-Z.; Xiao, B.-G. Therapeutic potential of experimental autoimmune encephalomyelitis by Fasudil, a Rho kinase inhibitor. J. Neurosci. Res. 2010, 88, 1664–1672. [Google Scholar] [CrossRef]
- Li, Y.-H.; Yu, J.-Z.; Xin, Y.-L.; Feng, L.; Chai, Z.; Liu, J.-C.; Zhang, H.-Z.; Zhang, G.-X.; Xiao, B.-G.; Ma, C.-G. Protective effect of a novel Rho kinase inhibitor WAR-5 in experimental autoimmune encephalomyelitis by modulating inflammatory response and neurotrophic factors. Exp. Mol. Pathol. 2015, 99, 220–228. [Google Scholar] [CrossRef]
- Paintlia, A.S.; Paintlia, M.K.; Singh, A.K.; Singh, I. Inhibition of rho family functions by lovastatin promotes myelin repair in ameliorating experimental autoimmune encephalomyelitis. Mol. Pharmacol. 2008, 73, 1381–1393. [Google Scholar] [CrossRef]
- Zanin-Zhorov, A.; Weiss, J.M.; Trzeciak, A.; Chen, W.; Zhang, J.; Nyuydzefe, M.S.; Arencibia, C.; Polimera, S.; Schueller, O.; Fuentes-Duculan, J.; et al. Cutting Edge: Selective Oral ROCK2 Inhibitor Reduces Clinical Scores in Patients with Psoriasis Vulgaris and Normalizes Skin Pathology via Concurrent Regulation of IL-17 and IL-10. J. Immunol. 2017, 198, 3809–3814. [Google Scholar] [CrossRef]
- Singh, A.K.; Eken, A.; Fry, M.; Bettelli, E.; Oukka, M. DOCK8 regulates protective immunity by controlling the function and survival of RORγt+ ILCs. Nat. Commun. 2014, 5, 4603. [Google Scholar] [CrossRef]
- Mangan, P.R.; Harrington, L.E.; O’Quinn, D.B.; Helms, W.S.; Bullard, D.C.; Elson, C.O.; Hatton, R.D.; Wahl, S.M.; Schoeb, T.R.; Weaver, C.T. Transforming growth factor-beta induces development of the T(H)17 lineage. Nature 2006, 441, 231–234. [Google Scholar] [CrossRef] [PubMed]
- Eken, A.; Cansever, M.; Okus, F.Z.; Erdem, S.; Nain, E.; Azizoglu, Z.B.; Haliloglu, Y.; Karakukcu, M.; Ozcan, A.; Devecioglu, O.; et al. ILC3 deficiency and generalized ILC abnormalities in DOCK8-deficient patients. Allergy 2020, 75, 921–932. [Google Scholar] [CrossRef] [PubMed]
- Keles, S.; Charbonnier, L.M.; Kabaleeswaran, V.; Reisli, I.; Genel, F.; Gulez, N.; Al-Herz, W.; Ramesh, N.; Perez-Atayde, A.; Karaca, N.E.; et al. Dedicator of cytokinesis 8 regulates signal transducer and activator of transcription 3 activation and promotes TH17 cell differentiation. J. Allergy Clin. Immunol. 2016, 138, 1384–1394.e2. [Google Scholar] [CrossRef] [PubMed]
- Razawy, W.; Asmawidjaja, P.S.; Mus, A.-M.; Salioska, N.; Davelaar, N.; Kops, N.; Oukka, M.; Alves, C.H.; Lubberts, E. CD4+ CCR6+ T cells, but not γδ T cells, are important for the IL-23R-dependent progression of antigen-induced inflammatory arthritis in mice. Eur. J. Immunol. 2020, 50, 245–255. [Google Scholar] [CrossRef]
- Matloubian, M.; Lo, C.G.; Cinamon, G.; Lesneski, M.J.; Xu, Y.; Brinkmann, V.; Allende, M.L.; Proia, R.L.; Cyster, J.G. Lymphocyte egress from thymus and peripheral lymphoid organs is dependent on S1P receptor. Nature 2004, 427, 355–360. [Google Scholar] [CrossRef]
- Derfuss, T.; Mehling, M.; Papadopoulou, A.; Bar-Or, A.; Cohen, J.A.; Kappos, L. Advances in oral immunomodulating therapies in relapsing multiple sclerosis. Lancet Neurol. 2020, 19, 336–347. [Google Scholar] [CrossRef]
- Ramírez-Valle, F.; Gray, E.E.; Cyster, J.G. Inflammation induces dermal Vγ4+ γδT17 memory-like cells that travel to distant skin and accelerate secondary IL-17-driven responses. Proc. Natl. Acad. Sci. USA 2015, 112, 8046–8051. [Google Scholar] [CrossRef]
- Tintore, M.; Vidal-Jordana, A.; Sastre-Garriga, J. Treatment of multiple sclerosis - success from bench to bedside. Nat. Rev. Neurol 2019, 15, 53–58. [Google Scholar] [CrossRef]
- Lochmatter, C.; Fischer, R.; Charles, P.D.; Yu, Z.; Powrie, F.; Kessler, B.M. Integrative Phosphoproteomics Links IL-23R Signaling with Metabolic Adaptation in Lymphocytes. Sci. Rep. 2016, 6, 24491. [Google Scholar] [CrossRef]
- Kono, M.; Maeda, K.; Stocton-Gavanescu, I.; Pan, W.; Umeda, M.; Katsuyama, E.; Burbano, C.; Orite, S.Y.K.; Vukelic, M.; Tsokos, M.G.; et al. Pyruvate kinase M2 is requisite for Th1 and Th17 differentiation. JCI Insight 2019, 4. [Google Scholar] [CrossRef] [PubMed]
- Angiari, S.; Runtsch, M.C.; Sutton, C.E.; Palsson-McDermott, E.M.; Kelly, B.; Rana, N.; Kane, H.; Papadopoulou, G.; Pearce, E.L.; Mills, K.H.G.; et al. Pharmacological Activation of Pyruvate Kinase M2 Inhibits CD4+ T Cell Pathogenicity and Suppresses Autoimmunity. Cell Metab. 2020, 31, 391–405.e8. [Google Scholar] [CrossRef]
- Patel, C.H.; Leone, R.D.; Horton, M.R.; Powell, J.D. Targeting metabolism to regulate immune responses in autoimmunity and cancer. Nat. Rev. Drug Discov. 2019, 18, 669–688. [Google Scholar] [CrossRef]
- Stockinger, B.; Di Meglio, P.; Gialitakis, M.; Duarte, J.H. The aryl hydrocarbon receptor: Multitasking in the immune system. Annu. Rev. Immunol. 2014, 32, 403–432. [Google Scholar] [CrossRef] [PubMed]
- Gutiérrez-Vázquez, C.; Quintana, F.J. Regulation of the Immune Response by the Aryl Hydrocarbon Receptor. Immunity 2018, 48, 19–33. [Google Scholar] [CrossRef] [PubMed]
- Veldhoen, M.; Hocking, R.J.; Flavell, R.A.; Stockinger, B. Signals mediated by transforming growth factor-beta initiate autoimmune encephalomyelitis, but chronic inflammation is needed to sustain disease. Nat. Immunol. 2006, 7, 1151–1156. [Google Scholar] [CrossRef]
- Veldhoen, M.; Hirota, K.; Westendorf, A.M.; Buer, J.; Dumoutier, L.; Renauld, J.-C.; Stockinger, B. The aryl hydrocarbon receptor links TH17-cell-mediated autoimmunity to environmental toxins. Nature 2008, 453, 106–109. [Google Scholar] [CrossRef]
- Martin, B.; Hirota, K.; Cua, D.J.; Stockinger, B.; Veldhoen, M. Interleukin-17-producing gammadelta T cells selectively expand in response to pathogen products and environmental signals. Immunity 2009, 31, 321–330. [Google Scholar] [CrossRef]
- Cibrian, D.; Saiz, M.L.; de la Fuente, H.; Sánchez-Díaz, R.; Moreno-Gonzalo, O.; Jorge, I.; Ferrarini, A.; Vázquez, J.; Punzón, C.; Fresno, M.; et al. CD69 controls the uptake of L-tryptophan through LAT1-CD98 and AhR-dependent secretion of IL-22 in psoriasis. Nat. Immunol. 2016, 17, 985–996. [Google Scholar] [CrossRef]
- Cibrian, D.; Castillo-González, R.; Fernández-Gallego, N.; de la Fuente, H.; Jorge, I.; Saiz, M.L.; Punzón, C.; Ramírez-Huesca, M.; Vicente-Manzanares, M.; Fresno, M.; et al. Targeting L-type amino acid transporter 1 in innate and adaptive T cells efficiently controls skin inflammation. J. Allergy Clin. Immunol. 2020, 145, 199–214.e11. [Google Scholar] [CrossRef]
- Di Meglio, P.; Duarte, J.H.; Ahlfors, H.; Owens, N.D.L.; Li, Y.; Villanova, F.; Tosi, I.; Hirota, K.; Nestle, F.O.; Mrowietz, U.; et al. Activation of the Aryl Hydrocarbon Receptor Dampens the Severity of Inflammatory Skin Conditions. Immunity 2014, 40, 989–1001. [Google Scholar] [CrossRef] [PubMed]
- Zhu, Z.; Chen, J.; Lin, Y.; Zhang, C.; Li, W.; Qiao, H.; Fu, M.; Dang, E.; Wang, G. Aryl Hydrocarbon Receptor in Cutaneous Vascular Endothelial Cells Restricts Psoriasis Development by Negatively Regulating Neutrophil Recruitment. J. Investig. Dermatol. 2020, 140, 1233–1243.e9. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Innocentin, S.; Withers, D.R.; Roberts, N.A.; Gallagher, A.R.; Grigorieva, E.F.; Wilhelm, C.; Veldhoen, M. Exogenous stimuli maintain intraepithelial lymphocytes via aryl hydrocarbon receptor activation. Cell 2011, 147, 629–640. [Google Scholar] [CrossRef] [PubMed]
- Basu, R.; O’Quinn, D.B.; Silberger, D.J.; Schoeb, T.R.; Fouser, L.; Ouyang, W.; Hatton, R.D.; Weaver, C.T. Th22 cells are an important source of IL-22 for host protection against enteropathogenic bacteria. Immunity 2012, 37, 1061–1075. [Google Scholar] [CrossRef]
- Rothhammer, V.; Quintana, F.J. The aryl hydrocarbon receptor: An environmental sensor integrating immune responses in health and disease. Nat. Rev. Immunol. 2019, 19, 184–197. [Google Scholar] [CrossRef]
- Niedbala, W.; Alves-Filho, J.C.; Fukada, S.Y.; Vieira, S.M.; Mitani, A.; Sonego, F.; Mirchandani, A.; Nascimento, D.C.; Cunha, F.Q.; Liew, F.Y. Regulation of type 17 helper T-cell function by nitric oxide during inflammation. Proc. Natl. Acad. Sci. USA 2011, 108, 9220–9225. [Google Scholar] [CrossRef]
- Nath, N.; Morinaga, O.; Singh, I. S-nitrosoglutathione a physiologic nitric oxide carrier attenuates experimental autoimmune encephalomyelitis. J. Neuroimmune Pharmacol. 2010, 5, 240–251. [Google Scholar] [CrossRef]
- García-Ortiz, A.; Serrador, J.M. Nitric Oxide Signaling in T Cell-Mediated Immunity. Trends Mol. Med. 2018, 24, 412–427. [Google Scholar] [CrossRef]
- Ivanov, I.I.; McKenzie, B.S.; Zhou, L.; Tadokoro, C.E.; Lepelley, A.; Lafaille, J.J.; Cua, D.J.; Littman, D.R. The orphan nuclear receptor RORgammat directs the differentiation program of proinflammatory IL-17+ T helper cells. Cell 2006, 126, 1121–1133. [Google Scholar] [CrossRef]
- Zhou, L.; Ivanov, I.I.; Spolski, R.; Min, R.; Shenderov, K.; Egawa, T.; Levy, D.E.; Leonard, W.J.; Littman, D.R. IL-6 programs T(H)-17 cell differentiation by promoting sequential engagement of the IL-21 and IL-23 pathways. Nat. Immunol. 2007, 8, 967–974. [Google Scholar] [CrossRef]
- Schmolka, N.; Serre, K.; Grosso, A.R.; Rei, M.; Pennington, D.J.; Gomes, A.Q.; Silva-Santos, B. Epigenetic and transcriptional signatures of stable versus plastic differentiation of proinflammatory γδ T cell subsets. Nat. Immunol. 2013, 14, 1093–1100. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.O.; Pappu, B.P.; Nurieva, R.; Akimzhanov, A.; Kang, H.S.; Chung, Y.; Ma, L.; Shah, B.; Panopoulos, A.D.; Schluns, K.S.; et al. T helper 17 lineage differentiation is programmed by orphan nuclear receptors ROR alpha and ROR gamma. Immunity 2008, 28, 29–39. [Google Scholar] [CrossRef]
- Heng, T.S.P.; Painter, M.W. Immunological Genome Project Consortium The Immunological Genome Project: Networks of gene expression in immune cells. Nat. Immunol. 2008, 9, 1091–1094. [Google Scholar] [CrossRef] [PubMed]
- Tang, L.; Yang, X.; Liang, Y.; Xie, H.; Dai, Z.; Zheng, G. Transcription Factor Retinoid-Related Orphan Receptor γt: A Promising Target for the Treatment of Psoriasis. Front. Immunol 2018, 9, 1210. [Google Scholar] [CrossRef] [PubMed]
- Huh, J.R.; Leung, M.W.L.; Huang, P.; Ryan, D.A.; Krout, M.R.; Malapaka, R.R.V.; Chow, J.; Manel, N.; Ciofani, M.; Kim, S.V.; et al. Digoxin and its derivatives suppress TH17 cell differentiation by antagonizing RORγt activity. Nature 2011, 472, 486–490. [Google Scholar] [CrossRef]
- Solt, L.A.; Kumar, N.; Nuhant, P.; Wang, Y.; Lauer, J.L.; Liu, J.; Istrate, M.A.; Kamenecka, T.M.; Roush, W.R.; Vidović, D.; et al. Suppression of TH17 differentiation and autoimmunity by a synthetic ROR ligand. Nature 2011, 472, 491–494. [Google Scholar] [CrossRef] [PubMed]
- Xiao, S.; Yosef, N.; Yang, J.; Wang, Y.; Zhou, L.; Zhu, C.; Wu, C.; Baloglu, E.; Schmidt, D.; Ramesh, R.; et al. Small-molecule RORγt antagonists inhibit T helper 17 cell transcriptional network by divergent mechanisms. Immunity 2014, 40, 477–489. [Google Scholar] [CrossRef] [PubMed]
- Xue, X.; Soroosh, P.; De Leon-Tabaldo, A.; Luna-Roman, R.; Sablad, M.; Rozenkrants, N.; Yu, J.; Castro, G.; Banie, H.; Fung-Leung, W.-P.; et al. Pharmacologic modulation of RORγt translates to efficacy in preclinical and translational models of psoriasis and inflammatory arthritis. Sci. Rep. 2016, 6, 37977. [Google Scholar] [CrossRef]
- Venken, K.; Jacques, P.; Mortier, C.; Labadia, M.E.; Decruy, T.; Coudenys, J.; Hoyt, K.; Wayne, A.L.; Hughes, R.; Turner, M.; et al. RORγt inhibition selectively targets IL-17 producing iNKT and γδ-T cells enriched in Spondyloarthritis patients. Nat. Commun 2019, 10, 9. [Google Scholar] [CrossRef]
- Hang, S.; Paik, D.; Yao, L.; Kim, E.; Trinath, J.; Lu, J.; Ha, S.; Nelson, B.N.; Kelly, S.P.; Wu, L.; et al. Bile acid metabolites control TH17 and Treg cell differentiation. Nature 2019, 576, 143–148. [Google Scholar] [CrossRef]
- Jetten, A.M.; Cook, D.N. (Inverse) Agonists of Retinoic Acid-Related Orphan Receptor γ: Regulation of Immune Responses, Inflammation, and Autoimmune Disease. Annu. Rev. Pharmacol. Toxicol. 2020, 60, 371–390. [Google Scholar] [CrossRef] [PubMed]
- Jain, R.; Chen, Y.; Kanno, Y.; Joyce-Shaikh, B.; Vahedi, G.; Hirahara, K.; Blumenschein, W.M.; Sukumar, S.; Haines, C.J.; Sadekova, S.; et al. Interleukin-23-Induced Transcription Factor Blimp-1 Promotes Pathogenicity of T Helper 17 Cells. Immunity 2016, 44, 131–142. [Google Scholar] [CrossRef] [PubMed]
- Shapiro-Shelef, M.; Lin, K.-I.; McHeyzer-Williams, L.J.; Liao, J.; McHeyzer-Williams, M.G.; Calame, K. Blimp-1 is required for the formation of immunoglobulin secreting plasma cells and pre-plasma memory B cells. Immunity 2003, 19, 607–620. [Google Scholar] [CrossRef]
- Kallies, A.; Xin, A.; Belz, G.T.; Nutt, S.L. Blimp-1 transcription factor is required for the differentiation of effector CD8(+) T cells and memory responses. Immunity 2009, 31, 283–295. [Google Scholar] [CrossRef]
- Heinemann, C.; Heink, S.; Petermann, F.; Vasanthakumar, A.; Rothhammer, V.; Doorduijn, E.; Mitsdoerffer, M.; Sie, C.; Prazeres da Costa, O.; Buch, T.; et al. IL-27 and IL-12 oppose pro-inflammatory IL-23 in CD4+ T cells by inducing Blimp1. Nat. Commun. 2014, 5, 3770. [Google Scholar] [CrossRef]
- Aqel, S.I.; Granitto, M.C.; Nuro-Gyina, P.K.; Pei, W.; Liu, Y.; Lovett-Racke, A.E.; Racke, M.K.; Yang, Y. Distinct roles for Blimp-1 in autoreactive CD4 T cells during priming and effector phase of autoimmune encephalomyelitis. J. Neuroimmunol. 2018, 325, 20–28. [Google Scholar] [CrossRef]
- Theill, L.E.; Boyle, W.J.; Penninger, J.M. RANK-L and RANK: T cells, bone loss, and mammalian evolution. Annu. Rev. Immunol. 2002, 20, 795–823. [Google Scholar] [CrossRef]
- Sato, K.; Suematsu, A.; Okamoto, K.; Yamaguchi, A.; Morishita, Y.; Kadono, Y.; Tanaka, S.; Kodama, T.; Akira, S.; Iwakura, Y.; et al. Th17 functions as an osteoclastogenic helper T cell subset that links T cell activation and bone destruction. J. Exp. Med. 2006, 203, 2673–2682. [Google Scholar] [CrossRef]
- Cho, M.-L.; Kang, J.-W.; Moon, Y.-M.; Nam, H.-J.; Jhun, J.-Y.; Heo, S.-B.; Jin, H.-T.; Min, S.-Y.; Ju, J.-H.; Park, K.-S.; et al. STAT3 and NF-kappaB signal pathway is required for IL-23-mediated IL-17 production in spontaneous arthritis animal model IL-1 receptor antagonist-deficient mice. J. Immunol. 2006, 176, 5652–5661. [Google Scholar] [CrossRef]
- Ju, J.-H.; Cho, M.-L.; Moon, Y.-M.; Oh, H.-J.; Park, J.-S.; Jhun, J.-Y.; Min, S.-Y.; Cho, Y.-G.; Park, K.-S.; Yoon, C.-H.; et al. IL-23 induces receptor activator of NF-kappaB ligand expression on CD4+ T cells and promotes osteoclastogenesis in an autoimmune arthritis model. J. Immunol. 2008, 181, 1507–1518. [Google Scholar] [CrossRef]
- Li, X.; Kim, K.-W.; Cho, M.-L.; Ju, J.-H.; Kang, C.-M.; Oh, H.-J.; Min, J.-K.; Lee, S.-H.; Park, S.-H.; Kim, H.-Y. IL-23 induces receptor activator of NF-kappaB ligand expression in fibroblast-like synoviocytes via STAT3 and NF-kappaB signal pathways. Immunol. Lett. 2010, 127, 100–107. [Google Scholar] [CrossRef]
- Sutton, C.; Brereton, C.; Keogh, B.; Mills, K.H.G.; Lavelle, E.C. A crucial role for interleukin (IL)-1 in the induction of IL-17-producing T cells that mediate autoimmune encephalomyelitis. J. Exp. Med. 2006, 203, 1685–1691. [Google Scholar] [CrossRef] [PubMed]
- Vallabhapurapu, S.; Karin, M. Regulation and function of NF-kappaB transcription factors in the immune system. Annu. Rev. Immunol. 2009, 27, 693–733. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Zhang, L.; Joo, D.; Sun, S.-C. NF-κB signaling in inflammation. Signal. Transduct Target. Ther 2017, 2. [Google Scholar] [CrossRef]
- Zhang, Q.; Lenardo, M.J.; Baltimore, D. 30 Years of NF-κB: A Blossoming of Relevance to Human Pathobiology. Cell 2017, 168, 37–57. [Google Scholar] [CrossRef] [PubMed]
- Yin, M.J.; Yamamoto, Y.; Gaynor, R.B. The anti-inflammatory agents aspirin and salicylate inhibit the activity of I(kappa)B kinase-beta. Nature 1998, 396, 77–80. [Google Scholar] [CrossRef]
- Kim, H.J.; Hawke, N.; Baldwin, A.S. NF-kappaB and IKK as therapeutic targets in cancer. Cell Death Differ. 2006, 13, 738–747. [Google Scholar] [CrossRef] [PubMed]
- Hayden, M.S.; Ghosh, S. NF-κB, the first quarter-century: Remarkable progress and outstanding questions. Genes Dev. 2012, 26, 203–234. [Google Scholar] [CrossRef]
- Courtois, G.; Smahi, A.; Reichenbach, J.; Döffinger, R.; Cancrini, C.; Bonnet, M.; Puel, A.; Chable-Bessia, C.; Yamaoka, S.; Feinberg, J.; et al. A hypermorphic IkappaBalpha mutation is associated with autosomal dominant anhidrotic ectodermal dysplasia and T cell immunodeficiency. J. Clin. Investig. 2003, 112, 1108–1115. [Google Scholar] [CrossRef]
- Smahi, A.; Courtois, G.; Vabres, P.; Yamaoka, S.; Heuertz, S.; Munnich, A.; Israël, A.; Heiss, N.S.; Klauck, S.M.; Kioschis, P.; et al. Genomic rearrangement in NEMO impairs NF-kappaB activation and is a cause of incontinentia pigmenti. The International Incontinentia Pigmenti (IP) Consortium. Nature 2000, 405, 466–472. [Google Scholar]
- Döffinger, R.; Smahi, A.; Bessia, C.; Geissmann, F.; Feinberg, J.; Durandy, A.; Bodemer, C.; Kenwrick, S.; Dupuis-Girod, S.; Blanche, S.; et al. X-linked anhidrotic ectodermal dysplasia with immunodeficiency is caused by impaired NF-kappaB signaling. Nat. Genet. 2001, 27, 277–285. [Google Scholar] [CrossRef] [PubMed]
- Nenci, A.; Becker, C.; Wullaert, A.; Gareus, R.; van Loo, G.; Danese, S.; Huth, M.; Nikolaev, A.; Neufert, C.; Madison, B.; et al. Epithelial NEMO links innate immunity to chronic intestinal inflammation. Nature 2007, 446, 557–561. [Google Scholar] [CrossRef] [PubMed]
- Zhou, Q.; Wang, H.; Schwartz, D.M.; Stoffels, M.; Park, Y.H.; Zhang, Y.; Yang, D.; Demirkaya, E.; Takeuchi, M.; Tsai, W.L.; et al. Loss-of-function mutations in TNFAIP3 leading to A20 haploinsufficiency cause an early-onset autoinflammatory disease. Nat. Genet. 2016, 48, 67–73. [Google Scholar] [CrossRef] [PubMed]
- Badran, Y.R.; Dedeoglu, F.; Leyva Castillo, J.M.; Bainter, W.; Ohsumi, T.K.; Bousvaros, A.; Goldsmith, J.D.; Geha, R.S.; Chou, J. Human RELA haploinsufficiency results in autosomal-dominant chronic mucocutaneous ulceration. J. Exp. Med. 2017, 214, 1937–1947. [Google Scholar] [CrossRef]
- Hirota, K.; Duarte, J.H.; Veldhoen, M.; Hornsby, E.; Li, Y.; Cua, D.J.; Ahlfors, H.; Wilhelm, C.; Tolaini, M.; Menzel, U.; et al. Fate mapping of IL-17-producing T cells in inflammatory responses. Nat. Immunol. 2011, 12, 255–263. [Google Scholar] [CrossRef]
- Duhen, R.; Glatigny, S.; Arbelaez, C.A.; Blair, T.C.; Oukka, M.; Bettelli, E. Cutting edge: The pathogenicity of IFN-γ-producing Th17 cells is independent of T-bet. J. Immunol. 2013, 190, 4478–4482. [Google Scholar] [CrossRef]
- Spath, S.; Becher, B. T-bet or not T-bet: Taking the last bow on the autoimmunity stage. Eur. J. Immunol. 2013, 43, 2810–2813. [Google Scholar] [CrossRef]
- Chen, Y.; Langrish, C.L.; McKenzie, B.; Joyce-Shaikh, B.; Stumhofer, J.S.; McClanahan, T.; Blumenschein, W.; Churakovsa, T.; Low, J.; Presta, L.; et al. Anti-IL-23 therapy inhibits multiple inflammatory pathways and ameliorates autoimmune encephalomyelitis. J. Clin. Investig. 2006, 116, 1317–1326. [Google Scholar] [CrossRef]
- Krausgruber, T.; Schiering, C.; Adelmann, K.; Harrison, O.J.; Chomka, A.; Pearson, C.; Ahern, P.P.; Shale, M.; Oukka, M.; Powrie, F. T-bet is a key modulator of IL-23-driven pathogenic CD4(+) T cell responses in the intestine. Nat. Commun 2016, 7, 11627. [Google Scholar] [CrossRef]
- Yasuda, K.; Kitagawa, Y.; Kawakami, R.; Isaka, Y.; Watanabe, H.; Kondoh, G.; Kohwi-Shigematsu, T.; Sakaguchi, S.; Hirota, K. Satb1 regulates the effector program of encephalitogenic tissue Th17 cells in chronic inflammation. Nat. Commun 2019, 10, 549. [Google Scholar] [CrossRef]
- Alvarez, J.D.; Yasui, D.H.; Niida, H.; Joh, T.; Loh, D.Y.; Kohwi-Shigematsu, T. The MAR-binding protein SATB1 orchestrates temporal and spatial expression of multiple genes during T-cell development. Genes Dev. 2000, 14, 521–535. [Google Scholar] [PubMed]
- Lin, C.-C.; Bradstreet, T.R.; Schwarzkopf, E.A.; Sim, J.; Carrero, J.A.; Chou, C.; Cook, L.E.; Egawa, T.; Taneja, R.; Murphy, T.L.; et al. Bhlhe40 controls cytokine production by T cells and is essential for pathogenicity in autoimmune neuroinflammation. Nat. Commun. 2014, 5, 3551. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.-C.; Bradstreet, T.R.; Schwarzkopf, E.A.; Jarjour, N.N.; Chou, C.; Archambault, A.S.; Sim, J.; Zinselmeyer, B.H.; Carrero, J.A.; Wu, G.F.; et al. IL-1-induced Bhlhe40 identifies pathogenic T helper cells in a model of autoimmune neuroinflammation. J. Exp. Med. 2016, 213, 251–271. [Google Scholar] [CrossRef] [PubMed]
- Pavan Kumar, P.; Purbey, P.K.; Sinha, C.K.; Notani, D.; Limaye, A.; Jayani, R.S.; Galande, S. Phosphorylation of SATB1, a global gene regulator, acts as a molecular switch regulating its transcriptional activity in vivo. Mol. Cell 2006, 22, 231–243. [Google Scholar] [CrossRef]
- Izcue, A.; Hue, S.; Buonocore, S.; Arancibia-Cárcamo, C.V.; Ahern, P.P.; Iwakura, Y.; Maloy, K.J.; Powrie, F. Interleukin-23 restrains regulatory T cell activity to drive T cell-dependent colitis. Immunity 2008, 28, 559–570. [Google Scholar] [CrossRef]
- Ahern, P.P.; Schiering, C.; Buonocore, S.; McGeachy, M.J.; Cua, D.J.; Maloy, K.J.; Powrie, F. Interleukin-23 drives intestinal inflammation through direct activity on T cells. Immunity 2010, 33, 279–288. [Google Scholar] [CrossRef]
- Schiering, C.; Krausgruber, T.; Chomka, A.; Fröhlich, A.; Adelmann, K.; Wohlfert, E.A.; Pott, J.; Griseri, T.; Bollrath, J.; Hegazy, A.N.; et al. The alarmin IL-33 promotes regulatory T-cell function in the intestine. Nature 2014, 513, 564–568. [Google Scholar] [CrossRef]
- Lopetuso, L.R.; De Salvo, C.; Pastorelli, L.; Rana, N.; Senkfor, H.N.; Petito, V.; Di Martino, L.; Scaldaferri, F.; Gasbarrini, A.; Cominelli, F.; et al. IL-33 promotes recovery from acute colitis by inducing miR-320 to stimulate epithelial restitution and repair. Proc. Natl. Acad. Sci. USA 2018, 115, E9362–E9370. [Google Scholar] [CrossRef]
- Chen, Z.; Hu, Y.; Gong, Y.; Zhang, X.; Cui, L.; Chen, R.; Yu, Y.; Yu, Q.; Chen, Y.; Diao, H.; et al. Interleukin-33 alleviates psoriatic inflammation by suppressing the T helper type 17 immune response. Immunology 2020, 160, 382–392. [Google Scholar] [CrossRef]
- Cohen, P. The TLR and IL-1 signalling network at a glance. J. Cell. Sci. 2014, 127, 2383–2390. [Google Scholar] [CrossRef]
- Mantovani, A.; Dinarello, C.A.; Molgora, M.; Garlanda, C. Interleukin-1 and Related Cytokines in the Regulation of Inflammation and Immunity. Immunity 2019, 50, 778–795. [Google Scholar] [CrossRef] [PubMed]
- Sutton, C.E.; Lalor, S.J.; Sweeney, C.M.; Brereton, C.F.; Lavelle, E.C.; Mills, K.H.G. Interleukin-1 and IL-23 induce innate IL-17 production from gammadelta T cells, amplifying Th17 responses and autoimmunity. Immunity 2009, 31, 331–341. [Google Scholar] [CrossRef] [PubMed]
- Coccia, M.; Harrison, O.J.; Schiering, C.; Asquith, M.J.; Becher, B.; Powrie, F.; Maloy, K.J. IL-1β mediates chronic intestinal inflammation by promoting the accumulation of IL-17A secreting innate lymphoid cells and CD4(+) Th17 cells. J. Exp. Med. 2012, 209, 1595–1609. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Xue, F.; Fleming, C.; Yang, J.; Ding, C.; Ma, Y.; Liu, M.; Zhang, H.-G.; Zheng, J.; Xiong, N.; et al. Differential developmental requirement and peripheral regulation for dermal Vγ4 and Vγ6T17 cells in health and inflammation. Nat. Commun. 2014, 5, 3986. [Google Scholar] [CrossRef]
- Komuczki, J.; Tuzlak, S.; Friebel, E.; Hartwig, T.; Spath, S.; Rosenstiel, P.; Waisman, A.; Opitz, L.; Oukka, M.; Schreiner, B.; et al. Fate-Mapping of GM-CSF Expression Identifies a Discrete Subset of Inflammation-Driving T Helper Cells Regulated by Cytokines IL-23 and IL-1β. Immunity 2019, 50, 1289–1304.e6. [Google Scholar] [CrossRef]
- Chang, J.; Burkett, P.R.; Borges, C.M.; Kuchroo, V.K.; Turka, L.A.; Chang, C.-H. MyD88 is essential to sustain mTOR activation necessary to promote T helper 17 cell proliferation by linking IL-1 and IL-23 signaling. Proc. Natl. Acad. Sci. USA 2013, 110, 2270–2275. [Google Scholar] [CrossRef]
- McElroy, W.T. Interleukin-1 receptor-associated kinase 4 (IRAK4) inhibitors: An updated patent review (2016–2018). Expert Opin. Ther. Pat. 2019, 29, 243–259. [Google Scholar] [CrossRef]
- Gulen, M.F.; Kang, Z.; Bulek, K.; Youzhong, W.; Kim, T.W.; Chen, Y.; Altuntas, C.Z.; Sass Bak-Jensen, K.; McGeachy, M.J.; Do, J.-S.; et al. The receptor SIGIRR suppresses Th17 cell proliferation via inhibition of the interleukin-1 receptor pathway and mTOR kinase activation. Immunity 2010, 32, 54–66. [Google Scholar] [CrossRef]
- Chung, Y.; Chang, S.H.; Martinez, G.J.; Yang, X.O.; Nurieva, R.; Kang, H.S.; Ma, L.; Watowich, S.S.; Jetten, A.M.; Tian, Q.; et al. Critical regulation of early Th17 cell differentiation by interleukin-1 signaling. Immunity 2009, 30, 576–587. [Google Scholar] [CrossRef]
- Basu, R.; Whitley, S.K.; Bhaumik, S.; Zindl, C.L.; Schoeb, T.R.; Benveniste, E.N.; Pear, W.S.; Hatton, R.D.; Weaver, C.T. IL-1 signaling modulates activation of STAT transcription factors to antagonize retinoic acid signaling and control the TH17 cell-iTreg cell balance. Nat. Immunol. 2015, 16, 286–295. [Google Scholar] [CrossRef]
- Whitley, S.K.; Balasubramani, A.; Zindl, C.L.; Sen, R.; Shibata, Y.; Crawford, G.E.; Weathington, N.M.; Hatton, R.D.; Weaver, C.T. IL-1R signaling promotes STAT3 and NF-κB factor recruitment to distal cis-regulatory elements that regulate Il17a/f transcription. J. Biol. Chem. 2018, 293, 15790–15800. [Google Scholar] [CrossRef] [PubMed]
- Taylor, P.R.; Roy, S.; Leal, S.M.; Sun, Y.; Howell, S.J.; Cobb, B.A.; Li, X.; Pearlman, E. Activation of neutrophils by autocrine IL-17A-IL-17RC interactions during fungal infection is regulated by IL-6, IL-23, RORγt and dectin. Nat. Immunol. 2014, 15, 143–151. [Google Scholar] [CrossRef] [PubMed]
- Chen, F.; Cao, A.; Yao, S.; Evans-Marin, H.L.; Liu, H.; Wu, W.; Carlsen, E.D.; Dann, S.M.; Soong, L.; Sun, J.; et al. mTOR Mediates IL-23 Induction of Neutrophil IL-17 and IL-22 Production. J. Immunol. 2016, 196, 4390–4399. [Google Scholar] [CrossRef] [PubMed]
- Bain, J.; Plater, L.; Elliott, M.; Shpiro, N.; Hastie, C.J.; McLauchlan, H.; Klevernic, I.; Arthur, J.S.C.; Alessi, D.R.; Cohen, P. The selectivity of protein kinase inhibitors: A further update. Biochem. J. 2007, 408, 297–315. [Google Scholar] [CrossRef] [PubMed]
- Cai, Y.; Xue, F.; Qin, H.; Chen, X.; Liu, N.; Fleming, C.; Hu, X.; Zhang, H.-G.; Chen, F.; Zheng, J.; et al. Differential Roles of the mTOR-STAT3 Signaling in Dermal γδ T Cell Effector Function in Skin Inflammation. Cell Rep. 2019, 27, 3034–3048.e5. [Google Scholar] [CrossRef] [PubMed]
- Delgoffe, G.M.; Pollizzi, K.N.; Waickman, A.T.; Heikamp, E.; Meyers, D.J.; Horton, M.R.; Xiao, B.; Worley, P.F.; Powell, J.D. The kinase mTOR regulates the differentiation of helper T cells through the selective activation of signaling by mTORC1 and mTORC2. Nat. Immunol. 2011, 12, 295–303. [Google Scholar] [CrossRef]
- Sarbassov, D.D.; Ali, S.M.; Sengupta, S.; Sheen, J.-H.; Hsu, P.P.; Bagley, A.F.; Markhard, A.L.; Sabatini, D.M. Prolonged rapamycin treatment inhibits mTORC2 assembly and Akt/PKB. Mol. Cell 2006, 22, 159–168. [Google Scholar] [CrossRef]
- Ciofani, M.; Madar, A.; Galan, C.; Sellars, M.; Mace, K.; Pauli, F.; Agarwal, A.; Huang, W.; Parkurst, C.N.; Muratet, M.; et al. A validated regulatory network for Th17 cell specification. Cell 2012, 151, 289–303. [Google Scholar] [CrossRef]
- Hayashi, K.; Anzai, N. Novel therapeutic approaches targeting L-type amino acid transporters for cancer treatment. World J. Gastrointest. Oncol. 2017, 9, 21–29. [Google Scholar] [CrossRef]
- Silva-Santos, B.; Mensurado, S.; Coffelt, S.B. γδ T cells: Pleiotropic immune effectors with therapeutic potential in cancer. Nat. Rev. Cancer 2019, 19, 392–404. [Google Scholar] [CrossRef]
- Langowski, J.L.; Zhang, X.; Wu, L.; Mattson, J.D.; Chen, T.; Smith, K.; Basham, B.; McClanahan, T.; Kastelein, R.A.; Oft, M. IL-23 promotes tumour incidence and growth. Nature 2006, 442, 461–465. [Google Scholar] [CrossRef] [PubMed]
- Teng, M.W.L.; Andrews, D.M.; McLaughlin, N.; von Scheidt, B.; Ngiow, S.F.; Möller, A.; Hill, G.R.; Iwakura, Y.; Oft, M.; Smyth, M.J. IL-23 suppresses innate immune response independently of IL-17A during carcinogenesis and metastasis. Proc. Natl. Acad. Sci. USA 2010, 107, 8328–8333. [Google Scholar] [CrossRef] [PubMed]
- Grivennikov, S.I.; Wang, K.; Mucida, D.; Stewart, C.A.; Schnabl, B.; Jauch, D.; Taniguchi, K.; Yu, G.-Y.; Osterreicher, C.H.; Hung, K.E.; et al. Adenoma-linked barrier defects and microbial products drive IL-23/IL-17-mediated tumour growth. Nature 2012, 491, 254–258. [Google Scholar] [CrossRef] [PubMed]
- Chan, I.H.; Jain, R.; Tessmer, M.S.; Gorman, D.; Mangadu, R.; Sathe, M.; Vives, F.; Moon, C.; Penaflor, E.; Turner, S.; et al. Interleukin-23 is sufficient to induce rapid de novo gut tumorigenesis, independent of carcinogens, through activation of innate lymphoid cells. Mucosal Immunol. 2014, 7, 842–856. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Allen, S.; Vijayan, D.; Li, X.-Y.; Harjunpää, H.; Takeda, K.; Liu, J.; Cua, D.J.; Smyth, M.J.; Teng, M.W.L. Experimental Lung Metastases in Mice Are More Effectively Inhibited by Blockade of IL23R than IL23. Cancer Immunol. Res. 2018, 6, 978–987. [Google Scholar] [CrossRef] [PubMed]
- Yan, J.; Cua, D.J.; Teng, M.W. IL-23 promotes the development of castration-resistant prostate cancer. Immunol. Cell Biol. 2018, 96, 883–885. [Google Scholar] [CrossRef] [PubMed]
- Vom Berg, J.; Prokop, S.; Miller, K.R.; Obst, J.; Kälin, R.E.; Lopategui-Cabezas, I.; Wegner, A.; Mair, F.; Schipke, C.G.; Peters, O.; et al. Inhibition of IL-12/IL-23 signaling reduces Alzheimer’s disease-like pathology and cognitive decline. Nat. Med. 2012, 18, 1812–1819. [Google Scholar] [CrossRef]
- Muschaweckh, A.; Petermann, F.; Korn, T. IL-1β and IL-23 Promote Extrathymic Commitment of CD27+CD122-γδ T Cells to γδT17 Cells. J. Immunol. 2017, 199, 2668–2679. [Google Scholar] [CrossRef]
- Papotto, P.H.; Gonçalves-Sousa, N.; Schmolka, N.; Iseppon, A.; Mensurado, S.; Stockinger, B.; Ribot, J.C.; Silva-Santos, B. IL-23 drives differentiation of peripheral γδ17 T cells from adult bone marrow-derived precursors. EMBO Rep. 2017, 18, 1957–1967. [Google Scholar] [CrossRef]
- Riol-Blanco, L.; Lazarevic, V.; Awasthi, A.; Mitsdoerffer, M.; Wilson, B.S.; Croxford, A.; Waisman, A.; Kuchroo, V.K.; Glimcher, L.H.; Oukka, M. IL-23 receptor regulates unconventional IL-17-producing T cells that control bacterial infections. J. Immunol. 2010, 184, 1710–1720. [Google Scholar] [CrossRef]
- Happel, K.I.; Dubin, P.J.; Zheng, M.; Ghilardi, N.; Lockhart, C.; Quinton, L.J.; Odden, A.R.; Shellito, J.E.; Bagby, G.J.; Nelson, S.; et al. Divergent roles of IL-23 and IL-12 in host defense against Klebsiella pneumoniae. J. Exp. Med. 2005, 202, 761–769. [Google Scholar] [CrossRef] [PubMed]
- Huang, W.; Na, L.; Fidel, P.L.; Schwarzenberger, P. Requirement of interleukin-17A for systemic anti-Candida albicans host defense in mice. J. Infect. Dis. 2004, 190, 624–631. [Google Scholar] [CrossRef] [PubMed]
- Maher, C.O.; Dunne, K.; Comerford, R.; O’Dea, S.; Loy, A.; Woo, J.; Rogers, T.R.; Mulcahy, F.; Dunne, P.J.; Doherty, D.G. Candida albicans stimulates IL-23 release by human dendritic cells and downstream IL-17 secretion by Vδ1 T cells. J. Immunol. 2015, 194, 5953–5960. [Google Scholar] [CrossRef] [PubMed]
- Cho, J.S.; Pietras, E.M.; Garcia, N.C.; Ramos, R.I.; Farzam, D.M.; Monroe, H.R.; Magorien, J.E.; Blauvelt, A.; Kolls, J.K.; Cheung, A.L.; et al. IL-17 is essential for host defense against cutaneous Staphylococcus aureus infection in mice. J. Clin. Investig. 2010, 120, 1762–1773. [Google Scholar] [CrossRef] [PubMed]
- Allen, A.C.; Wilk, M.M.; Misiak, A.; Borkner, L.; Murphy, D.; Mills, K.H.G. Sustained protective immunity against Bordetella pertussis nasal colonization by intranasal immunization with a vaccine-adjuvant combination that induces IL-17-secreting TRM cells. Mucosal Immunol. 2018, 11, 1763–1776. [Google Scholar] [CrossRef]
- Yi, P.; Liang, Y.; Yuan, D.M.K.; Jie, Z.; Kwota, Z.; Chen, Y.; Cong, Y.; Fan, X.; Sun, J. A tightly regulated IL-22 response maintains immune functions and homeostasis in systemic viral infection. Sci. Rep. 2017, 7, 3857. [Google Scholar] [CrossRef]





© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Pastor-Fernández, G.; Mariblanca, I.R.; Navarro, M.N. Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities. Cells 2020, 9, 2044. https://doi.org/10.3390/cells9092044
Pastor-Fernández G, Mariblanca IR, Navarro MN. Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities. Cells. 2020; 9(9):2044. https://doi.org/10.3390/cells9092044
Chicago/Turabian StylePastor-Fernández, Gloria, Isabel R. Mariblanca, and María N. Navarro. 2020. "Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities" Cells 9, no. 9: 2044. https://doi.org/10.3390/cells9092044
APA StylePastor-Fernández, G., Mariblanca, I. R., & Navarro, M. N. (2020). Decoding IL-23 Signaling Cascade for New Therapeutic Opportunities. Cells, 9(9), 2044. https://doi.org/10.3390/cells9092044