Molecular Genetics and Functional Analysis Implicate CDKN2BAS1-CDKN2B Involvement in POAG Pathogenesis

 , ,
, ,
Abstract
:1. Introduction
2. Materials and Methods
2.1. Enrollment of the Study Subjects
2.2. Molecular Genetic Analysis
2.3. Generation of Luciferase Reporter Constructs and Luciferase Reporter Assay
2.4. Cell Lines and Tissues Used for the Study
2.5. Immunostaining
2.6. CDKN2B-AS1 siRNA Treatment
2.7. β-Galactosidase Assay
2.8. Treatment of Cells with TGF-β1 and TGF-β2
2.9. Semi-Quantitative PCR
3. Results
3.1. Genotype-Phenotype Association Studies for SNP rs4977756
3.2. Potential Regulatory Role of SNP rs4977756
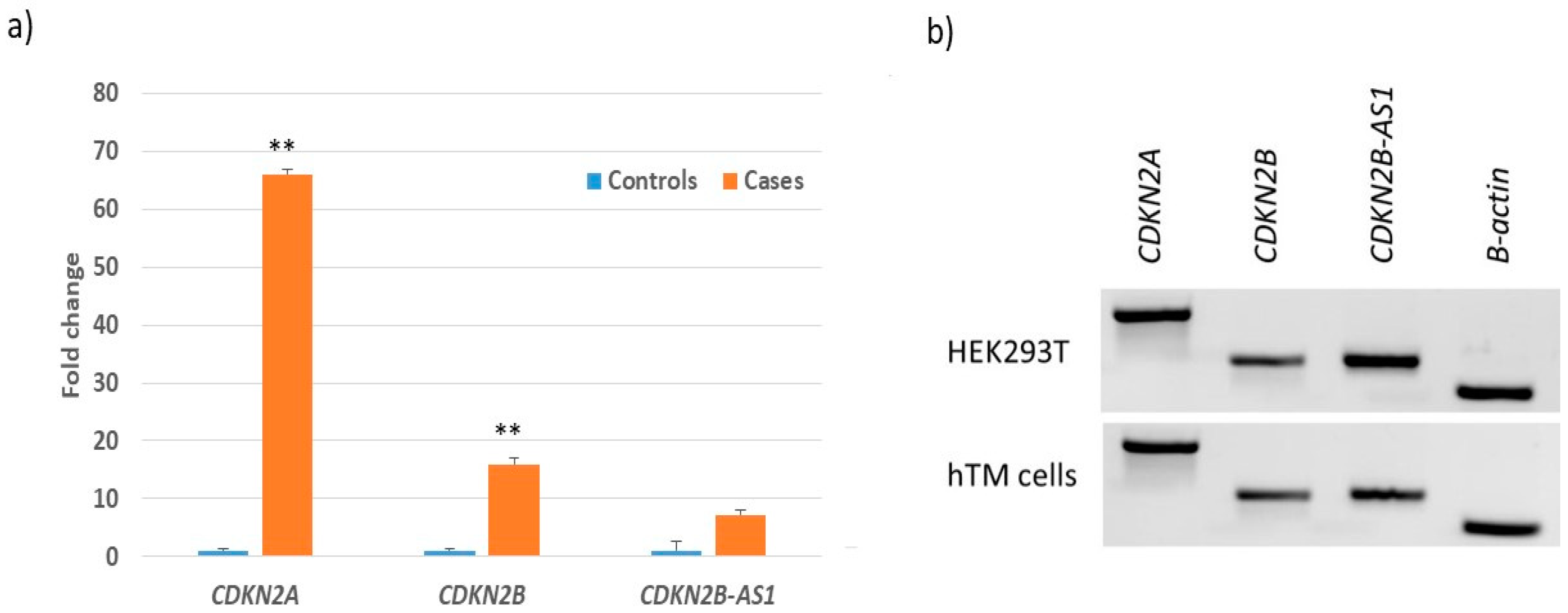
3.3. Transcript Expression of CDKN2B-AS1 Locus in POAG Retina, TM and HEK293T Cells
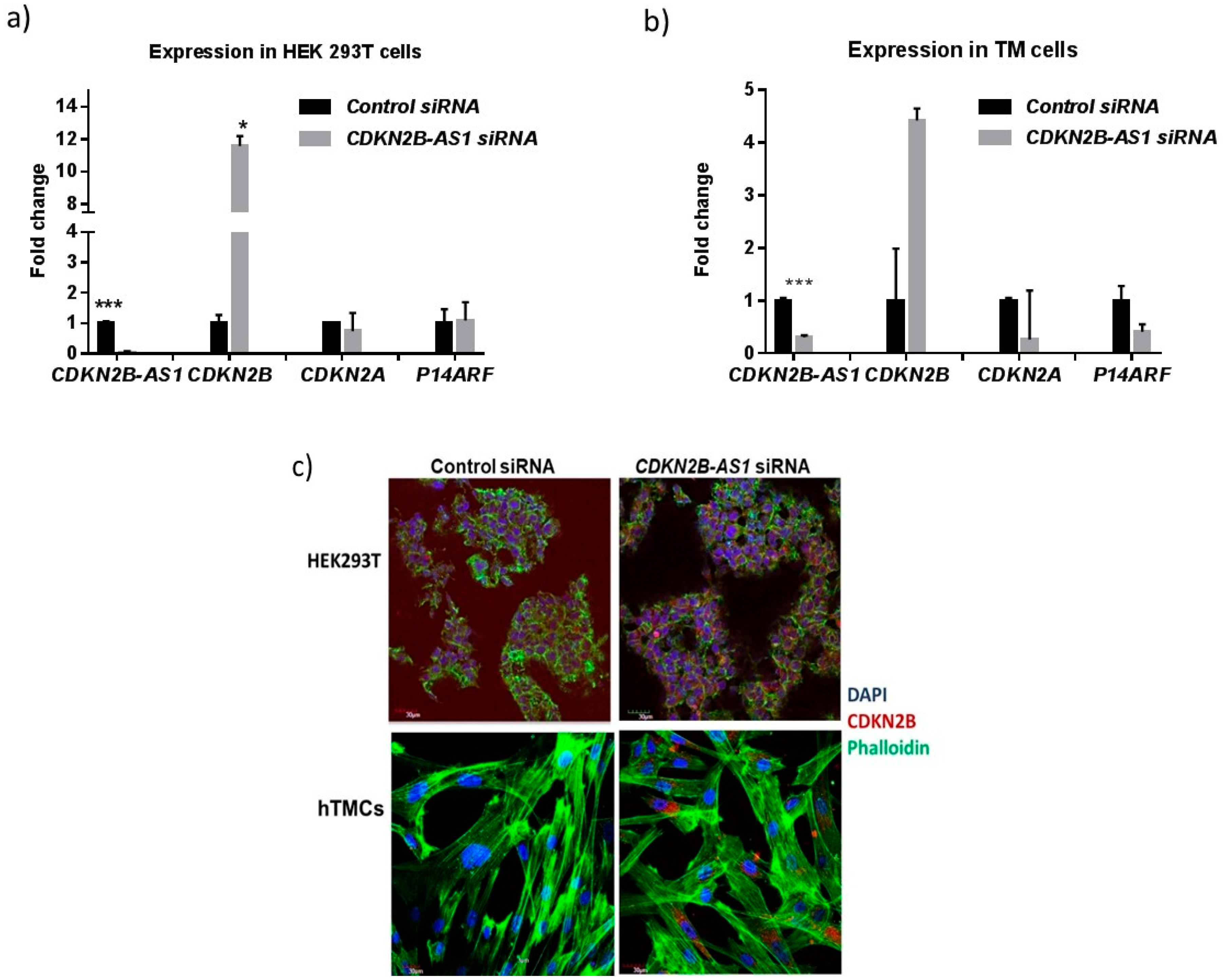
3.4. CDKN2B-AS1 Regulates the Expression of CDKN2B and CDKN2A Transcripts
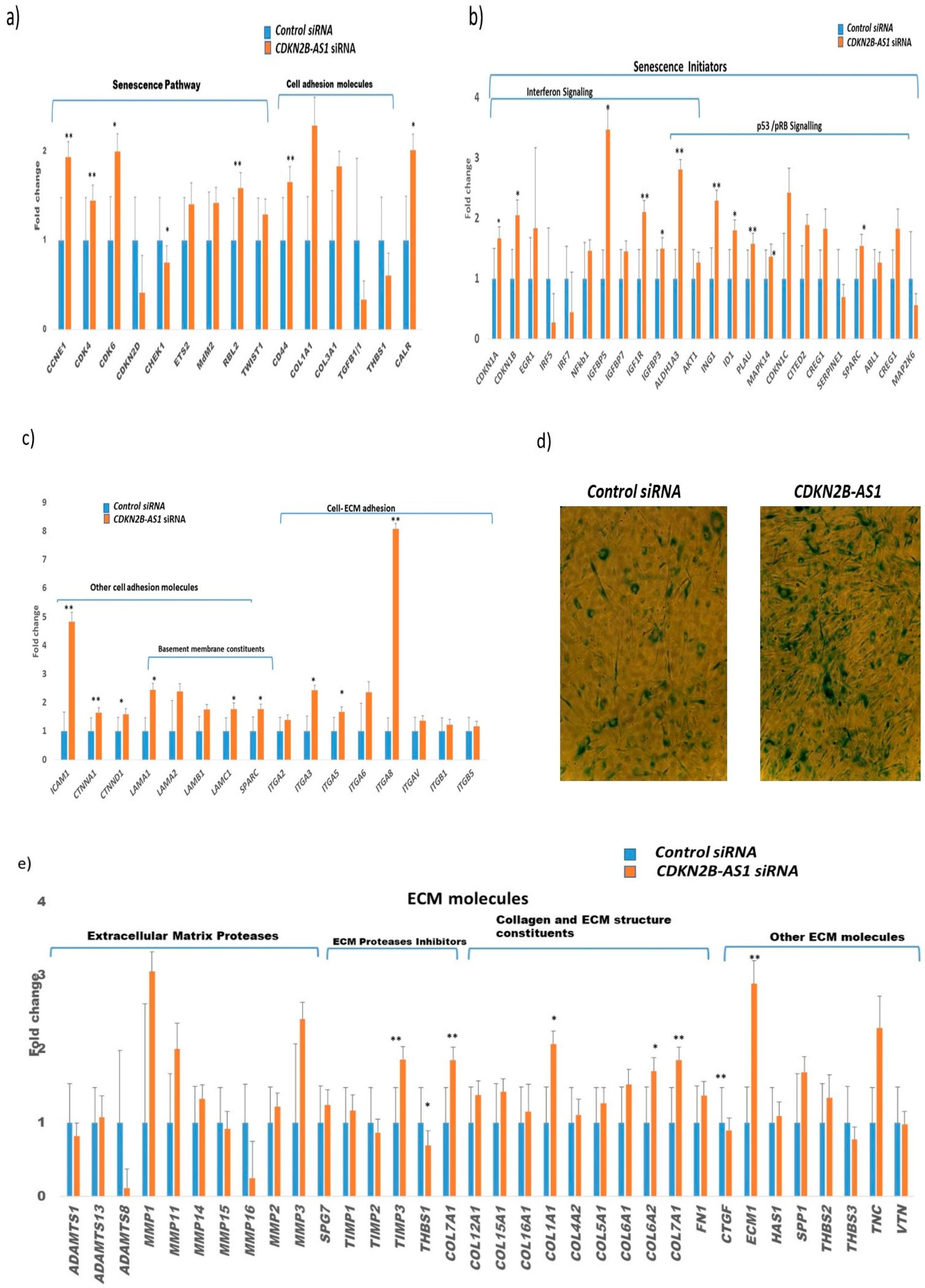
3.5. Role of CDKN2B-AS1 in Causing Cellular Senescence and ECM Homeostasis
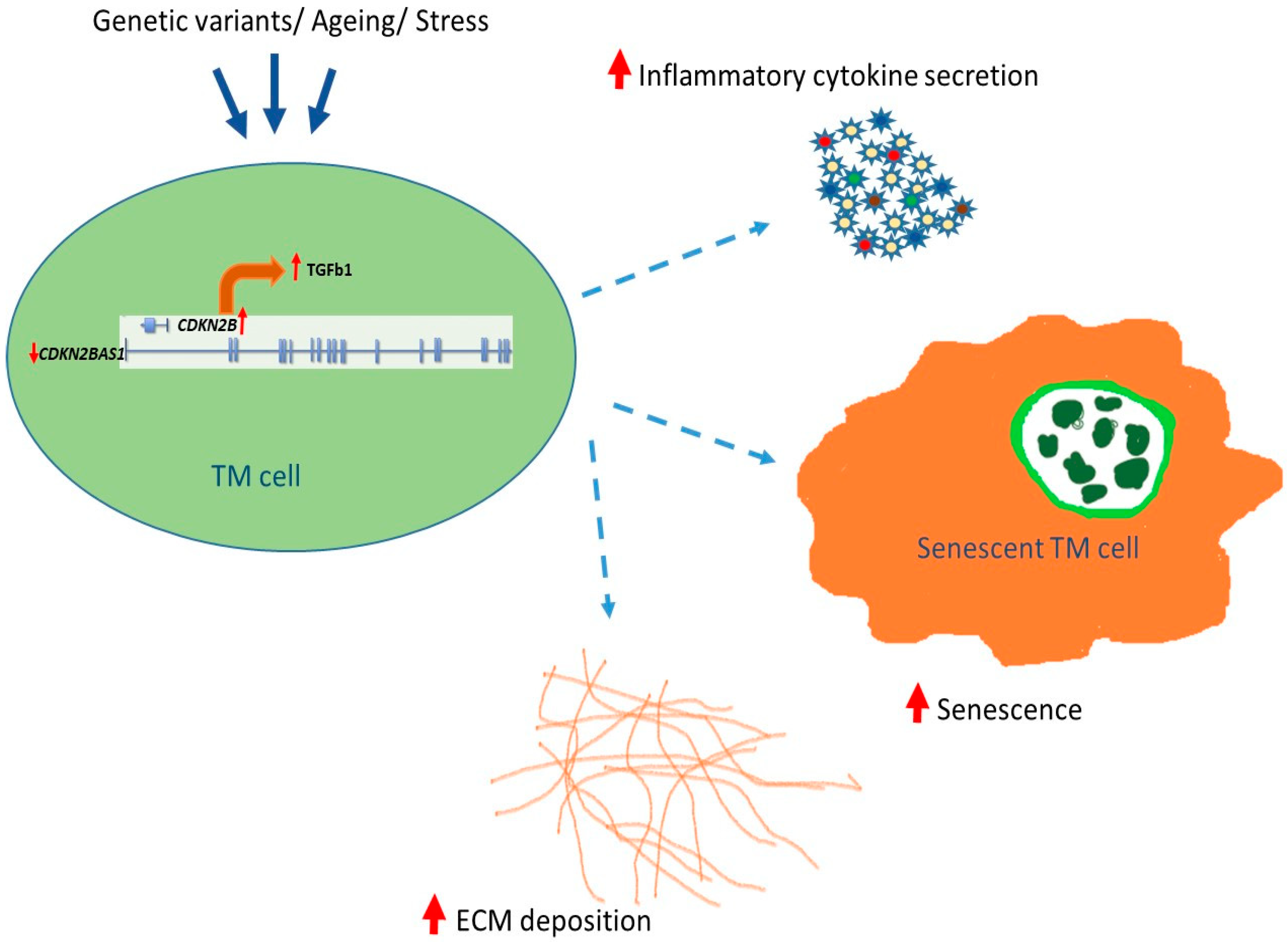
3.6. TGF-β Signaling in Senescent-Inflammatory Phenotype
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Mahabadi, N.; Foris, L.A.; Tripathy, K. Open Angle Glaucoma. In StatPearls; StatPearls Publishing: Treasure Island, FL, USA, 2020. [Google Scholar]
- Beidoe, G.; Mousa, S.A. Current primary open-angle glaucoma treatments and future directions. Clin. Ophthalmol. 2012, 6, 1699–1707. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rudnicka, A.R.; Mt-Isa, S.; Owen, C.G.; Cook, D.G.; Ashby, D. Variations in primary open-angle glaucoma prevalence by age, gender, and race: A Bayesian meta-analysis. Investig. Ophthalmol. Vis. Sci. 2006, 47, 4254–4261. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Friedman, D.S.; Wolfs, R.C.; O’Colmain, B.J.; Klein, B.E.; Taylor, H.R.; West, S.; Leske, M.C.; Mitchell, P.; Congdon, N.; Kempen, J.; et al. Prevalence of open-angle glaucoma among adults in the United States. Arch. Ophthalmol. 2004, 122, 532–538. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Tielsch, J.M.; Sommer, A.; Katz, J.; Royall, R.M.; Quigley, H.A.; Javitt, J. Racial variations in the prevalence of primary open-angle glaucoma. The Baltimore Eye Survey. JAMA 1991, 266, 369–374. [Google Scholar] [CrossRef]
- Vitart, V.; Bencic, G.; Hayward, C.; Skunca Herman, J.; Huffman, J.; Campbell, S.; Bucan, K.; Navarro, P.; Gunjaca, G.; Marin, J.; et al. New loci associated with central cornea thickness include COL5A1, AKAP13 and AVGR8. Hum. Mol. Genet. 2010, 19, 4304–4311. [Google Scholar] [CrossRef] [Green Version]
- Lu, Y.; Dimasi, D.P.; Hysi, P.G.; Hewitt, A.W.; Burdon, K.P.; Toh, T.; Ruddle, J.B.; Li, Y.J.; Mitchell, P.; Healey, P.R.; et al. Common genetic variants near the Brittle Cornea Syndrome locus ZNF469 influence the blinding disease risk factor central corneal thickness. PLoS Genet. 2010, 6, e1000947. [Google Scholar] [CrossRef]
- Macgregor, S.; Hewitt, A.W.; Hysi, P.G.; Ruddle, J.B.; Medland, S.E.; Henders, A.K.; Gordon, S.D.; Andrew, T.; McEvoy, B.; Sanfilippo, P.G.; et al. Genome-wide association identifies ATOH7 as a major gene determining human optic disc size. Hum. Mol. Genet. 2010, 19, 2716–2724. [Google Scholar] [CrossRef] [Green Version]
- Cornes, B.K.; Khor, C.C.; Nongpiur, M.E.; Xu, L.; Tay, W.T.; Zheng, Y.; Lavanya, R.; Li, Y.; Wu, R.; Sim, X.; et al. Identification of four novel variants that influence central corneal thickness in multi-ethnic Asian populations. Hum. Mol. Genet. 2012, 21, 437–445. [Google Scholar] [CrossRef] [Green Version]
- Khor, C.C.; Ramdas, W.D.; Vithana, E.N.; Cornes, B.K.; Sim, X.; Tay, W.T.; Saw, S.M.; Zheng, Y.; Lavanya, R.; Wu, R.; et al. Genome-wide association studies in Asians confirm the involvement of ATOH7 and TGFBR3, and further identify CARD10 as a novel locus influencing optic disc area. Hum. Mol. Genet. 2011, 20, 1864–1872. [Google Scholar] [CrossRef]
- Vithana, E.N.; Aung, T.; Khor, C.C.; Cornes, B.K.; Tay, W.T.; Sim, X.; Lavanya, R.; Wu, R.; Zheng, Y.; Hibberd, M.L.; et al. Collagen-related genes influence the glaucoma risk factor, central corneal thickness. Hum. Mol. Genet. 2011, 20, 649–658. [Google Scholar] [CrossRef]
- Ramdas, W.D.; van Koolwijk, L.M.; Ikram, M.K.; Jansonius, N.M.; de Jong, P.T.; Bergen, A.A.; Isaacs, A.; Amin, N.; Aulchenko, Y.S.; Wolfs, R.C.; et al. A genome-wide association study of optic disc parameters. PLoS Genet. 2010, 6, e1000978. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Danford, I.D.; Verkuil, L.D.; Choi, D.J.; Collins, D.W.; Gudiseva, H.V.; Uyhazi, K.E.; Lau, M.K.; Kanu, L.N.; Grant, G.R.; Chavali, V.R.; et al. Characterizing the “POAGome”: A bioinformatics-driven approach to primary open-angle glaucoma. Prog. Retin. Eye Res. 2017, 58, 89–114. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burdon, K.P.; Macgregor, S.; Hewitt, A.W.; Sharma, S.; Chidlow, G.; Mills, R.A.; Danoy, P.; Casson, R.; Viswanathan, A.C.; Liu, J.Z.; et al. Genome-wide association study identifies susceptibility loci for open angle glaucoma at TMCO1 and CDKN2B-AS1. Nat. Genet. 2011, 43, 574–578. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Takamoto, M.; Kaburaki, T.; Mabuchi, A.; Araie, M.; Amano, S.; Aihara, M.; Tomidokoro, A.; Iwase, A.; Mabuchi, F.; Kashiwagi, K.; et al. Common variants on chromosome 9p21 are associated with normal tension glaucoma. PLoS ONE 2012, 7, e40107. [Google Scholar] [CrossRef] [Green Version]
- Osman, W.; Low, S.K.; Takahashi, A.; Kubo, M.; Nakamura, Y. A genome-wide association study in the Japanese population confirms 9p21 and 14q23 as susceptibility loci for primary open angle glaucoma. Hum. Mol. Genet. 2012, 21, 2836–2842. [Google Scholar] [CrossRef]
- Yoshikawa, M.; Nakanishi, H.; Yamashiro, K.; Miyake, M.; Akagi, T.; Gotoh, N.; Ikeda, H.O.; Suda, K.; Yamada, H.; Hasegawa, T.; et al. Association of Glaucoma-Susceptible Genes to Regional Circumpapillary Retinal Nerve Fiber Layer Thickness and Visual Field Defects. Investig. Ophthalmol. Vis. Sci. 2017, 58, 2510–2519. [Google Scholar] [CrossRef]
- Holdt, L.M.; Teupser, D. Recent studies of the human chromosome 9p21 locus, which is associated with atherosclerosis in human populations. Arterioscler. Thromb. Vasc. Biol. 2012, 32, 196–206. [Google Scholar] [CrossRef] [Green Version]
- Chan, K.; Patel, R.S.; Newcombe, P.; Nelson, C.P.; Qasim, A.; Epstein, S.E.; Burnett, S.; Vaccarino, V.L.; Zafari, A.M.; Shah, S.H.; et al. Association between the chromosome 9p21 locus and angiographic coronary artery disease burden: A collaborative meta-analysis. J. Am. Coll. Cardiol. 2013, 61, 957–970. [Google Scholar] [CrossRef] [Green Version]
- Cheng, X.; Shi, L.; Nie, S.; Wang, F.; Li, X.; Xu, C.; Wang, P.; Yang, B.; Li, Q.; Pan, Z.; et al. The same chromosome 9p21.3 locus is associated with type 2 diabetes and coronary artery disease in a Chinese Han population. Diabetes 2011, 60, 680–684. [Google Scholar] [CrossRef] [Green Version]
- Olsson, S.; Csajbok, L.Z.; Jood, K.; Nylen, K.; Nellgard, B.; Jern, C. Association between genetic variation on chromosome 9p21 and aneurysmal subarachnoid haemorrhage. J. Neurol. Neurosurg. Psychiatry 2011, 82, 384–388. [Google Scholar] [CrossRef] [Green Version]
- Gu, F.; Pfeiffer, R.M.; Bhattacharjee, S.; Han, S.S.; Taylor, P.R.; Berndt, S.; Yang, H.; Sigurdson, A.J.; Toro, J.; Mirabello, L.; et al. Common genetic variants in the 9p21 region and their associations with multiple tumours. Br. J. Cancer 2013, 108, 1378–1386. [Google Scholar] [CrossRef] [Green Version]
- Aarabi, G.; Zeller, T.; Heydecke, G.; Munz, M.; Schafer, A.; Seedorf, U. Roles of the Chr.9p21.3 ANRIL Locus in Regulating Inflammation and Implications for Anti-Inflammatory Drug Target Identification. Front. Cardiovasc. Med. 2018, 5, 47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zhao, W.; Smith, J.A.; Mao, G.; Fornage, M.; Peyser, P.A.; Sun, Y.V.; Turner, S.T.; Kardia, S.L. The cis and trans effects of the risk variants of coronary artery disease in the Chr9p21 region. BMC Med. Genom. 2015, 8, 21. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Congrains, A.; Kamide, K.; Katsuya, T.; Yasuda, O.; Oguro, R.; Yamamoto, K.; Ohishi, M.; Rakugi, H. CVD-associated non-coding RNA, ANRIL, modulates expression of atherogenic pathways in VSMC. Biochem. Biophys. Res. Commun. 2012, 419, 612–616. [Google Scholar] [CrossRef] [PubMed]
- Qiu, J.J.; Wang, Y.; Liu, Y.L.; Zhang, Y.; Ding, J.X.; Hua, K.Q. The long non-coding RNA ANRIL promotes proliferation and cell cycle progression and inhibits apoptosis and senescence in epithelial ovarian cancer. Oncotarget 2016, 7, 32478–32492. [Google Scholar] [CrossRef] [PubMed]
- Congrains, A.; Kamide, K.; Ohishi, M.; Rakugi, H. ANRIL: Molecular mechanisms and implications in human health. Int. J. Mol. Sci. 2013, 14, 1278–1292. [Google Scholar] [CrossRef] [Green Version]
- Pasquale, L.R.; Loomis, S.J.; Kang, J.H.; Yaspan, B.L.; Abdrabou, W.; Budenz, D.L.; Chen, T.C.; Delbono, E.; Friedman, D.S.; Gaasterland, D.; et al. CDKN2B-AS1 genotype-glaucoma feature correlations in primary open-angle glaucoma patients from the United States. Am. J. Ophthalmol. 2013, 155, 342–353.e5. [Google Scholar] [CrossRef] [Green Version]
- Restrepo, N.A.; Laper, S.M.; Farber-Eger, E.; Crawford, D.C. Local genetic ancestry in CDKN2B-AS1 is associated with primary open-angle glaucoma in an African American cohort extracted from de-identified electronic health records. BMC Med. Genom. 2018, 11, 70. [Google Scholar] [CrossRef] [Green Version]
- Liu, Y.; Hauser, M.A.; Akafo, S.K.; Qin, X.; Miura, S.; Gibson, J.R.; Wheeler, J.; Gaasterland, D.E.; Challa, P.; Herndon, L.W.; et al. Investigation of known genetic risk factors for primary open angle glaucoma in two populations of African ancestry. Investig. Ophthalmol. Vis. Sci. 2013, 54, 6248–6254. [Google Scholar] [CrossRef]
- Cao, D.; Jiao, X.; Liu, X.; Hennis, A.; Leske, M.C.; Nemesure, B.; Hejtmancik, J.F. CDKN2B polymorphism is associated with primary open-angle glaucoma (POAG) in the Afro-Caribbean population of Barbados, West Indies. PLoS ONE 2012, 7, e39278. [Google Scholar] [CrossRef]
- Gudiseva, H.V.; Verma, S.S.; Chavali, V.R.M.; Salowe, R.J.; Lucas, A.; Collins, D.W.; Rathi, S.; He, J.; Lee, R.; Merriam, S.; et al. Genome wide-association study identifies novel loci in the Primary Open-Angle African American Glaucoma Genetics (POAAGG) study. BioRxiv 2020. [Google Scholar] [CrossRef]
- Gudiseva, H.V.; Hansen, M.; Gutierrez, L.; Collins, D.W.; He, J.; Verkuil, L.D.; Danford, I.D.; Sagaser, A.; Bowman, A.S.; Salowe, R.; et al. Saliva DNA quality and genotyping efficiency in a predominantly elderly population. BMC Med. Genom. 2016, 9, 17. [Google Scholar] [CrossRef] [Green Version]
- Verkuil, L.; Danford, I.; Pistilli, M.; Collins, D.W.; Gudiseva, H.V.; Trachtman, B.T.; He, J.; Rathi, S.; Haider, N.; Ying, G.S.; et al. SNP located in an AluJb repeat downstream of TMCO1, rs4657473, is protective for POAG in African Americans. Br. J. Ophthalmol. 2019, 103, 1530–1536. [Google Scholar] [CrossRef] [Green Version]
- Chavali, V.R.M.; Haider, N.; Rathi, S.; Vrathasha, V.; Alapati, T.; He, J.; Gill, K.; Nikonov, R.; Duong, T.T.; McDougald, D.S.; et al. Dual SMAD inhibition and Wnt inhibition enable efficient and reproducible differentiations of induced pluripotent stem cells into retinal ganglion cells. Sci. Rep. 2020, 10, 11828. [Google Scholar] [CrossRef] [PubMed]
- Sibin, M.K.; Dhananjaya, I.B.; Narasingarao, K.V.; Harshitha, S.M.; Jeru-Manoj, M.; Chetan, G.K. Two gene polymorphisms (rs4977756 and rs11515) in CDKN2A/B and glioma risk in South Indian population. Meta Gene 2016, 9, 215–218. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Cunnington, M.S.; Santibanez Koref, M.; Mayosi, B.M.; Burn, J.; Keavney, B. Chromosome 9p21 SNPs Associated with Multiple Disease Phenotypes Correlate with ANRIL Expression. PLoS Genet. 2010, 6, e1000899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Liton, P.B.; Challa, P.; Stinnett, S.; Luna, C.; Epstein, D.L.; Gonzalez, P. Cellular senescence in the glaucomatous outflow pathway. Exp. Gerontol. 2005, 40, 745–748. [Google Scholar] [CrossRef] [Green Version]
- Zhu, L.; Zhang, Q.; Li, S.; Jiang, S.; Cui, J.; Dang, G. Interference of the long noncoding RNA CDKN2B-AS1 upregulates miR-181a-5p/TGFbetaI axis to restrain the metastasis and promote apoptosis and senescence of cervical cancer cells. Cancer Med. 2019, 8, 1721–1730. [Google Scholar] [CrossRef] [Green Version]
- Takai, Y.; Tanito, M.; Ohira, A. Multiplex cytokine analysis of aqueous humor in eyes with primary open-angle glaucoma, exfoliation glaucoma, and cataract. Investig. Ophthalmol. Vis. Sci. 2012, 53, 241–247. [Google Scholar] [CrossRef] [Green Version]
- Wallace, D.M.; Pokrovskaya, O.; O’Brien, C.J. The Function of Matricellular Proteins in the Lamina Cribrosa and Trabecular Meshwork in Glaucoma. J. Ocul. Pharmacol. Ther. 2015, 31, 386–395. [Google Scholar] [CrossRef]
- Chen, D.; Zhang, Z.; Mao, C.; Zhou, Y.; Yu, L.; Yin, Y.; Wu, S.; Mou, X.; Zhu, Y. ANRIL inhibits p15(INK4b) through the TGFbeta1 signaling pathway in human esophageal squamous cell carcinoma. Cell. Immunol. 2014, 289, 91–96. [Google Scholar] [CrossRef] [PubMed]
- Wiggs, J.L.; Yaspan, B.L.; Hauser, M.A.; Kang, J.H.; Allingham, R.R.; Olson, L.M.; Abdrabou, W.; Fan, B.J.; Wang, D.Y.; Brodeur, W.; et al. Common variants at 9p21 and 8q22 are associated with increased susceptibility to optic nerve degeneration in glaucoma. PLoS Genet. 2012, 8, e1002654. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Wang, C.; Kane, M.A.; Napoli, J.L. Multiple retinol and retinal dehydrogenases catalyze all-trans-retinoic acid biosynthesis in astrocytes. J. Biol. Chem. 2011, 286, 6542–6553. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kel, A.E.; Gossling, E.; Reuter, I.; Cheremushkin, E.; Kel-Margoulis, O.V.; Wingender, E. MATCH: A tool for searching transcription factor binding sites in DNA sequences. Nucleic Acids Res. 2003, 31, 3576–3579. [Google Scholar] [CrossRef] [Green Version]
- Folkersen, L.; Kyriakou, T.; Goel, A.; Peden, J.; Malarstig, A.; Paulsson-Berne, G.; Hamsten, A.; Hugh, W.; Franco-Cereceda, A.; Gabrielsen, A.; et al. Relationship between CAD risk genotype in the chromosome 9p21 locus and gene expression. Identification of eight new ANRIL splice variants. PLoS ONE 2009, 4, e7677. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Burd, C.E.; Jeck, W.R.; Liu, Y.; Sanoff, H.K.; Wang, Z.; Sharpless, N.E. Expression of linear and novel circular forms of an INK4/ARF-associated non-coding RNA correlates with atherosclerosis risk. PLoS Genet. 2010, 6, e1001233. [Google Scholar] [CrossRef]
- Broadbent, H.M.; Peden, J.F.; Lorkowski, S.; Goel, A.; Ongen, H.; Green, F.; Clarke, R.; Collins, R.; Franzosi, M.G.; Tognoni, G.; et al. Susceptibility to coronary artery disease and diabetes is encoded by distinct, tightly linked SNPs in the ANRIL locus on chromosome 9p. Hum. Mol. Genet. 2008, 17, 806–814. [Google Scholar] [CrossRef]
- Congrains, A.; Kamide, K.; Oguro, R.; Yasuda, O.; Miyata, K.; Yamamoto, E.; Kawai, T.; Kusunoki, H.; Yamamoto, H.; Takeya, Y.; et al. Genetic variants at the 9p21 locus contribute to atherosclerosis through modulation of ANRIL and CDKN2A/B. Atherosclerosis 2012, 220, 449–455. [Google Scholar] [CrossRef] [Green Version]
- Kotake, Y.; Nakagawa, T.; Kitagawa, K.; Suzuki, S.; Liu, N.; Kitagawa, M.; Xiong, Y. Long non-coding RNA ANRIL is required for the PRC2 recruitment to and silencing of p15(INK4B) tumor suppressor gene. Oncogene 2011, 30, 1956–1962. [Google Scholar] [CrossRef] [Green Version]
- Yap, K.L.; Li, S.; Munoz-Cabello, A.M.; Raguz, S.; Zeng, L.; Mujtaba, S.; Gil, J.; Walsh, M.J.; Zhou, M.M. Molecular interplay of the noncoding RNA ANRIL and methylated histone H3 lysine 27 by polycomb CBX7 in transcriptional silencing of INK4a. Mol. Cell 2010, 38, 662–674. [Google Scholar] [CrossRef] [Green Version]
- Puvvula, P.K. LncRNAs Regulatory Networks in Cellular Senescence. Int. J. Mol. Sci. 2019, 20, 2615. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Montes, M.; Lund, A.H. Emerging roles of lncRNAs in senescence. FEBS J. 2016, 283, 2414–2426. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Alexander, P.B.; Wang, X.F. TGF-beta Family Signaling in the Control of Cell Proliferation and Survival. Cold Spring Harb. Perspect. Biol. 2017, 9. [Google Scholar] [CrossRef] [PubMed] [Green Version]




{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Locus | Genotype | Case (Frequency) | Control (Frequency) | Total (Frequency) | Age and Sex Adjusted Odds Ratio (95% CI) | |
|---|---|---|---|---|---|
| rs4977756 | GG | 181 (11.6%) | 199 (12.4%) | 380 (12.0%) | 1 |
| AG | 691 (44.1%) | 753 (47.1%) | 1444 (45.6%) | 1.02 (0.80, 1.30) | |
| AA | 695 (44.4%) | 648 (40.5%) | 1343 (42.4%) | 1.21 (0.94, 1.55) | |
| Total | 1567 (49.5%) | 1600 (50.5%) | 3167 | ||
| Locus | # of Risk Alleles | Case (Frequency) | Control (Frequency) | Total (Frequency) | Age and Sex Adjusted Odds Ratio (95% CI) | |
|---|---|---|---|---|---|
| rs4977756 | 0-1 Risk Alleles (GG or AG) | 872 (55.7%) | 952 (59.5%) | 1824 (57.6%) | 1 |
| 2 Risk Alleles (AA) | 695 (44.3%) | 648 (40.5%) | 1343 (42.4%) | 1.19 (1.02, 1.39) | |
| Total | 1567 (49.5%) | 1600 (50.5%) | 3167 | ||
| rs4977756 | |||||
|---|---|---|---|---|---|
| POAG Risk Factor | ANOVA p-Value | GG Mean (Std Dev) | AG Mean (Std Dev) | AA Mean (Std Dev) | All Genotypes |
| Mean CCT | 0.03 * | 535.80 (39.50) | 535.39 (41.52) | 530.28 (38.32) | 533.22 (39.98) |
| Mean IOP | 0.65 | 15.83 (4.57) | 16.03 (3.82) | 16.06 (4.07) | 16.02 (4.02) |
| Mean VCDR | 0.1 | 0.54 (0.25) | 0.54 (0.25) | 0.56 (0.25) | 0.55 (0.25) |
| Mean RNFL | 0.69 | 77.47 (13.37) | 76.42 (15.19) | 76.21 (15.33) | 76.45 (15.04) |
| rs4977756 (0-1 vs. 2 Risk Alleles) | ||||
|---|---|---|---|---|
| POAG Risk Factor | ANOVA p-Value | 0-1 Risk Alleles Mean (std dev) | 2 Risk Alleles Mean (std dev) | All Genotypes Mean (std dev) |
| Mean CCT | 0.008 * | 535.48 (41) | 530.28 (38) | 533.22 (39) |
| Mean IOP | 0.65 | 15.99 (3.99) | 16.06 (4.07) | 16.02 (4.02) |
| Mean VCDR | 0.033 * | 0.54 (0.25) | 0.56 (0.25) | 0.55 (0.25) |
| Mean RNFL | 0.64 | 76.64 (14.8) | 76.21 (15.3) | 76.45 (15.) |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Rathi, S.; Danford, I.; Gudiseva, H.V.; Verkuil, L.; Pistilli, M.; Vishwakarma, S.; Kaur, I.; Dave, T.V.; O’Brien, J.M.; Chavali, V.R.M. Molecular Genetics and Functional Analysis Implicate CDKN2BAS1-CDKN2B Involvement in POAG Pathogenesis. Cells 2020, 9, 1934. https://doi.org/10.3390/cells9091934
Rathi S, Danford I, Gudiseva HV, Verkuil L, Pistilli M, Vishwakarma S, Kaur I, Dave TV, O’Brien JM, Chavali VRM. Molecular Genetics and Functional Analysis Implicate CDKN2BAS1-CDKN2B Involvement in POAG Pathogenesis. Cells. 2020; 9(9):1934. https://doi.org/10.3390/cells9091934
Chicago/Turabian StyleRathi, Sonika, Ian Danford, Harini V. Gudiseva, Lana Verkuil, Maxwell Pistilli, Sushma Vishwakarma, Inderjeet Kaur, Tarjani Vivek Dave, Joan M. O’Brien, and Venkata R. M. Chavali. 2020. "Molecular Genetics and Functional Analysis Implicate CDKN2BAS1-CDKN2B Involvement in POAG Pathogenesis" Cells 9, no. 9: 1934. https://doi.org/10.3390/cells9091934
APA StyleRathi, S., Danford, I., Gudiseva, H. V., Verkuil, L., Pistilli, M., Vishwakarma, S., Kaur, I., Dave, T. V., O’Brien, J. M., & Chavali, V. R. M. (2020). Molecular Genetics and Functional Analysis Implicate CDKN2BAS1-CDKN2B Involvement in POAG Pathogenesis. Cells, 9(9), 1934. https://doi.org/10.3390/cells9091934