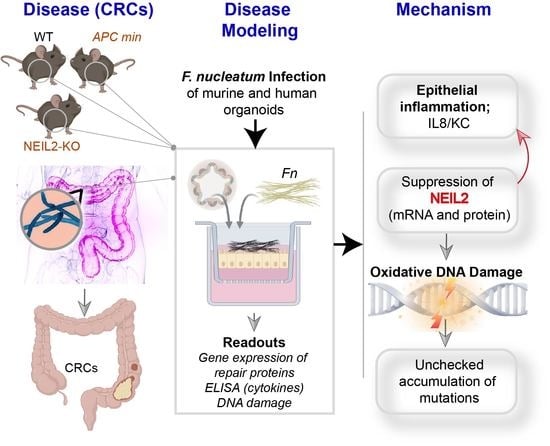
The DNA Glycosylase NEIL2 Suppresses Fusobacterium-Infection-Induced Inflammation and DNA Damage in Colonic Epithelial Cells

 , , , ,
, , , ,
Abstract
:
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Bacterial Cultures
2.3. Isolation and Maintenance of Colonic Organoids
2.4. The Preparation of Enteroid-Derived Monolayers (EDMs)
2.5. RNA Isolation and qPCR for Gene Expression Analysis
2.6. Proteome Profiler Array
2.7. The Measurement of Interlekin (IL)-8 Cytokine by ELISA
2.8. Immunoblot for Detection of Base Excision Repair and Cellular Signaling Proteins
2.9. Assessment of γH2AX Phosphorylation by Immunofluorescence (IF)
2.10. Analysis of Accumulation of DNA Strand Break by Long Amplicon Quantitative PCR (LAqPCR)
2.11. Measurement of Oxidative DNA/RNA Damage
2.12. Lactate Dehydrogenase (LDH) Assay
2.13. Analysis of Datasets with the CRC Patients
2.14. Statistics
3. Results
3.1. NEIL2 is Downregulated in the Human Colonic EDMs Following Fn Infection
3.2. Fn Infection Suppressed the Expression of NEIL2 in the Murine Colonic EDMs
3.3. The Role of NEIL2 in Controlling Inflammation and DNA Strand-Break Accumulation Following Fn Infection in Colonic EDMs
3.4. NEIL2 Knockdown Exacerbated Fn-Induced DNA Double-Strand Breaks (DSBs)
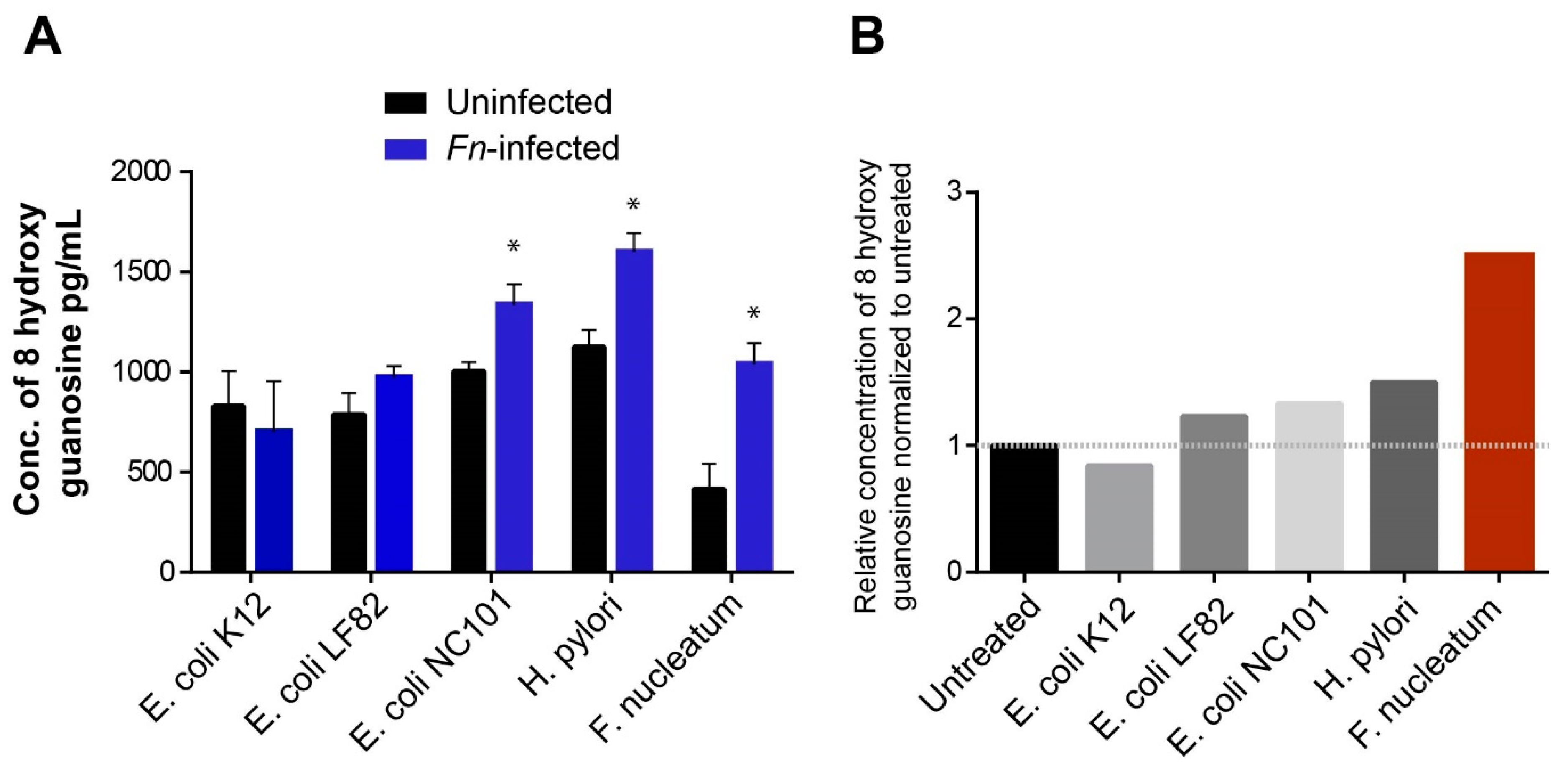
3.5. Fn Infection Downregulated NEIL2 and Increased Oxidative Damage in the Murine CRC Model
3.6. NEIL2 Is Downregulated in the MSS-CRCs Patients
4. Discussion
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
Abbreviations
| EDTA | Ethylenediaminetetraacetic acid |
| TGFb | Transforming Growth Factor b |
| PVDF | Polyvinylidene difluoride |
| NFkB | Nuclear factor kappa-light-chain enhancer of activated B-cells |
| HEPES | 4-(2-hydroxyethyl)-1-piperazineethanesulfonic acid |
References
- Siegel, R.L.; Mph, K.D.M.; Jemal, A. Cancer statistics, 2017. CA Cancer J. Clin. 2017, 67, 7–30. [Google Scholar] [CrossRef] [Green Version]
- Slattery, M.L. Diet, lifestyle, and colon cancer. Semin. Gastrointest. Dis. 2000, 11, 142–146. [Google Scholar]
- Campbell, P.T.; Cotterchio, M.; Dicks, E.; Parfrey, P.; Gallinger, S.; McLaughlin, J.R. Excess body weight and colorectal cancer risk in Canada: Associations in subgroups of clinically defined familial risk of cancer. Cancer Epidemiol. Biomark. Prev. 2007, 16, 1735–1744. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zisman, A.L.; Nickolov, A.; Brand, R.E.; Gorchow, A.; Roy, H.K. Associations between the age at diagnosis and location of colorectal cancer and the use of alcohol and tobacco. Arch. Intern. Med. 2006, 166, 629–634. [Google Scholar] [CrossRef] [Green Version]
- Botteri, E.; Iodice, S.; Raimondi, S.; Maisonneuve, P.; Lowenfels, A.B. Cigarette smoking and adenomatous polyps: A Meta-analysis. Gastroenterology 2008, 134, 388–395. [Google Scholar] [CrossRef] [PubMed]
- Willett, W.C. Diet and cancer: An evolving picture. JAMA 2005, 293, 233–234. [Google Scholar] [CrossRef] [PubMed]
- Haggar, F.; Boushey, R.P. Colorectal cancer epidemiology: Incidence, mortality, survival, and risk factors. Clin. Colon Rectal Surg. 2009, 22, 191–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Khare, S.; Verma, M. Epigenetics of colon cancer. Adv. Struct. Saf. Stud. 2012, 863, 177–185. [Google Scholar] [CrossRef]
- Bellizzi, A.M.; Frankel, W.L. Colorectal cancer due to deficiency in DNA mismatch repair function. Adv. Anat. Pathol. 2009, 16, 405–417. [Google Scholar] [CrossRef] [PubMed]
- Richman, S.D. Deficient mismatch repair: Read all about it (Review). Int. J. Oncol. 2015, 47, 1189–1202. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Peltomaki, P. Deficient DNA mismatch repair: A common etiologic factor for colon cancer. Hum. Mol. Genet. 2001, 10, 735–740. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- French, A.J.; Sargent, D.J.; Burgart, L.J.; Foster, N.R.; Kabat, B.F.; Goldberg, R.; Shepherd, L.; Windschitl, H.E.; Thibodeau, S.N. Prognostic significance of defective mismatch repair and BRAF V600E in patients with colon cancer. Clin. Cancer Res. 2008, 14, 3408–3415. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Guinney, J.; Dienstmann, R.; Wang, X.; De Reyniès, A.; Schlicker, A.; Soneson, C.; Marisa, L.; Roepman, P.; Nyamundanda, G.; Angelino, P.; et al. The consensus molecular subtypes of colorectal cancer. Nat. Med. 2015, 21, 1350–1356. [Google Scholar] [CrossRef] [PubMed]
- Holmes, E.; Li, J.V.; Athanasiou, T.; Ashrafian, H.; Nicholson, J.K. Understanding the role of gut microbiome–host metabolic signal disruption in health and disease. Trends Microbiol. 2011, 19, 349–359. [Google Scholar] [CrossRef] [PubMed]
- Tsilimigras, M.C.B.; Fodor, A.; Jobin, C. Carcinogenesis and therapeutics: The microbiota perspective. Nat. Microbiol. 2017, 2, 17008. [Google Scholar] [CrossRef]
- Marchesi, J.R.; Dutilh, B.E.; Hall, N.; Peters, W.H.M.; Roelofs, R.; Boleij, A.; Tjalsma, H. Towards the Human Colorectal Cancer Microbiome. PLoS ONE 2011, 6, e20447. [Google Scholar] [CrossRef] [Green Version]
- Sobhani, I.; Tap, J.; Roudot-Thoraval, F.; Roperch, J.P.; Letulle, S.; Langella, P.; Corthier, G.; Van Nhieu, J.T.; Furet, J.P. Microbial Dysbiosis in Colorectal Cancer (CRC) Patients. PLoS ONE 2011, 6, e16393. [Google Scholar] [CrossRef]
- Kostic, A.D.; Gevers, D.; Pedamallu, C.S.; Michaud, M.; Duke, F.; Earl, A.M.; Ojesina, A.I.; Jung, J.; Bass, A.J.; Tabernero, J.; et al. Genomic analysis identifies association of Fusobacterium with colorectal carcinoma. Genome Res. 2011, 22, 292–298. [Google Scholar] [CrossRef] [Green Version]
- Kostic, A.D.; Chun, E.; Robertson, L.; Glickman, J.N.; Gallini, C.A.; Michaud, M.; Clancy, T.E.; Chung, D.C.; Lochhead, P.; Hold, G.L.; et al. Fusobacterium nucleatum potentiates intestinal tumorigenesis and modulates the tumor-immune microenvironment. Cell Host Microbe 2013, 14, 207–215. [Google Scholar] [CrossRef] [Green Version]
- Tahara, T.; Yamamoto, E.; Suzuki, H.; Maruyama, R.; Chung, W.; Garriga, J.; Jelinek, J.; Yamano, H.-O.; Sugai, T.; An, B.; et al. Fusobacterium in colonic flora and molecular features of colorectal carcinoma. Cancer Res. 2014, 74, 1311–1318. [Google Scholar] [CrossRef] [Green Version]
- Mima, K.; Cao, Y.; Chan, A.T.; Qian, Z.R.; Nowak, J.A.; Masugi, Y.; Shi, Y.; Song, M.; Da Silva, A.; Gu, M.; et al. Fusobacterium nucleatum in Colorectal Carcinoma Tissue According to Tumor Location. Clin. Transl. Gastroenterol. 2016, 7, e200. [Google Scholar] [CrossRef] [PubMed]
- Yu, J.; Chen, Y.; Fu, X.; Zhou, X.; Peng, Y.; Shi, L.; Chen, T.; Wu, Y.; And, T.C. InvasiveFusobacterium nucleatummay play a role in the carcinogenesis of proximal colon cancer through the serrated neoplasia pathway. Int. J. Cancer 2016, 139, 1318–1326. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yu, T.; Guo, F.; Yu, Y.; Sun, T.; Ma, D.; Han, J.; Qian, Y.; Kryczek, I.; Sun, D.; Nagarsheth, N.; et al. Fusobacterium nucleatum Promotes Chemoresistance to Colorectal Cancer by Modulating Autophagy. Cell 2017, 170, 548–563. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rubinstein, M.R.; Wang, X.; Liu, W.; Hao, Y.; Cai, G.; Han, Y.W. Fusobacterium nucleatum promotes colorectal carcinogenesis by modulating E-cadherin/β-catenin signaling via its FadA adhesin. Cell Host Microbe 2013, 14, 195–206. [Google Scholar] [CrossRef] [Green Version]
- Rubinstein, M.R.; Baik, J.E.; Lagana, S.M.; Han, R.P.; Raab, W.J.; Sahoo, D.; Dalerba, P.; Wang, T.C.; Han, Y.W. Fusobacterium nucleatum promotes colorectal cancer by inducing Wnt/β-catenin modulator Annexin A1. EMBO Rep. 2019, 20, e47638. [Google Scholar] [CrossRef]
- Geng, F.; Zhang, Y.; Lu, Z.; Zhang, S.; Pan, Y. Fusobacterium nucleatum Caused DNA Damage and Promoted Cell Proliferation by the Ku70/p53 Pathway in Oral Cancer Cells. DNA Cell Biol. 2019, 39, 144–151. [Google Scholar] [CrossRef] [Green Version]
- Kidane, D. Molecular Mechanisms of H. pylori-Induced DNA Double-Strand Breaks. Int. J. Mol. Sci. 2018, 19, 2891. [Google Scholar] [CrossRef] [Green Version]
- Chumduri, C.; Gurumurthy, R.K.; Zietlow, R.; Meyer, T.F. Subversion of host genome integrity by bacterial pathogens. Nat. Rev. Mol. Cell Biol. 2016, 17, 659–673. [Google Scholar] [CrossRef]
- Hanahan, D.; Weinberg, R.A. Hallmarks of Cancer: The Next Generation. Cell 2011, 144, 646–674. [Google Scholar] [CrossRef] [Green Version]
- Krokan, H.E.; Bjørås, M. Base Excision Repair. Cold Spring Harb. Perspect. Biol. 2013, 5, a012583. [Google Scholar] [CrossRef]
- Chakraborty, A.; Wakamiya, M.; Venkova-Canova, T.; Pandita, R.K.; Aguilera-Aguirre, L.; Sarker, A.H.; Singh, D.K.; Hosoki, K.; Wood, T.G.; Sharma, G.; et al. Neil2-null Mice Accumulate Oxidized DNA Bases in the Transcriptionally Active Sequences of the Genome and Are Susceptible to Innate Inflammation. J. Biol. Chem. 2015, 290, 24636–24648. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sahan, A.Z.; Hazra, T.K.; Das, S. The Pivotal Role of DNA Repair in Infection Mediated-Inflammation and Cancer. Front. Microbiol. 2018, 9, 663. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schärer, O.D.; Jiricny, J. Recent progress in the biology, chemistry and structural biology of DNA glycosylases. BioEssays 2001, 23, 270–281. [Google Scholar] [CrossRef]
- Hegde, M.L.; Hazra, T.K.; Mitra, S. Early steps in the DNA base excision/single-strand interruption repair pathway in mammalian cells. Cell Res. 2008, 18, 27–47. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Hazra, T.K.; Das, A.; Das, S.; Choudhury, S.; Kow, Y.W.; Roy, R. Oxidative DNA damage repair in mammalian cells: A new perspective. DNA Repair 2007, 6, 470–480. [Google Scholar] [CrossRef] [Green Version]
- Ba, X.; Aguilera-Aguirre, L.; Rashid, Q.T.A.N.; Bacsi, A.; Radák, Z.; Sur, S.; Hosoki, K.; Hegde, M.L.; Brasier, A.R. The Role of 8-Oxoguanine DNA Glycosylase-1 in Inflammation. Int. J. Mol. Sci. 2014, 15, 16975–16997. [Google Scholar] [CrossRef] [Green Version]
- Shinmura, K.; Kato, H.; Kawanishi, Y.; Igarashi, H.; Goto, M.; Tao, H.; Inoue, Y.; Nakamura, S.; Misawa, K.; Mineta, H.; et al. Abnormal Expressions of DNA Glycosylase Genes NEIL1, NEIL2, and NEIL3 Are Associated with Somatic Mutation Loads in Human Cancer. Oxidat. Med. Cell. Longev. 2016, 2016, 1–10. [Google Scholar] [CrossRef] [Green Version]
- Dou, H.; Mitra, S.; Hazra, T.K. Repair of Oxidized Bases in DNA Bubble Structures by Human DNA Glycosylases NEIL1 and NEIL2. J. Biol. Chem. 2003, 278, 49679–49684. [Google Scholar] [CrossRef] [Green Version]
- Dey, S.; Maiti, A.K.; Hegde, M.L.; Hegde, P.M.; Boldogh, I.; Sarkar, P.S.; Abdel-Rahman, S.; Sarker, A.H.; Hang, B.; Xie, J.; et al. Increased risk of lung cancer associated with a functionally impaired polymorphic variant of the human DNA glycosylase NEIL2. DNA Repair 2012, 11, 570–578. [Google Scholar] [CrossRef] [Green Version]
- Sarker, A.H.; Chatterjee, A.; Williams, M.; Lin, S.; Havel, C.; Iii, P.J.; Boldogh, I.; Hazra, T.K.; Talbot, P.; Hang, B. NEIL2 Protects against Oxidative DNA Damage Induced by Sidestream Smoke in Human Cells. PLoS ONE 2014, 9, e90261. [Google Scholar] [CrossRef]
- Zhai, X.; Zhao, H.; Liu, Z.; Wang, L.-E.; El-Naggar, A.K.; Sturgis, E.M.; Wei, Q. Functional variants of the NEIL1 and NEIL2 genes and risk and progression of squamous cell carcinoma of the oral cavity and oropharynx. Clin. Cancer Res. 2008, 14, 4345–4352. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Anurag, M.; Punturi, N.; Hoog, J.; Bainbridge, M.N.; Ellis, M.J.; Haricharan, S. Comprehensive Profiling of DNA Repair Defects in Breast Cancer Identifies a Novel Class of Endocrine Therapy Resistance Drivers. Clin. Cancer Res. 2018, 24, 4887–4899. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Meira, L.; Bugni, J.M.; Green, S.L.; Lee, C.-W.; Pang, B.; Borenshtein, D.; Rickman, B.H.; Rogers, A.B.; Moroski-Erkul, C.A.; McFaline, J.L.; et al. DNA damage induced by chronic inflammation contributes to colon carcinogenesis in mice. J. Clin. Investig. 2008, 118, 2516–2525. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Sayed, I.; Sahan, A.Z.; Venkova, T.; Chakraborty, A.; Mukhopadhyay, D.; Bimczok, D.; Beswick, E.J.; Reyes, V.E.; Pinchuk, I.; Sahoo, D.; et al. Helicobacter pylori infection downregulates the DNA glycosylase NEIL2, resulting in increased genome damage and inflammation in gastric epithelial cells. J. Biol. Chem. 2020, 295, 11082–11098. [Google Scholar] [CrossRef] [PubMed]
- Sayed, I.; Suarez, K.; Lim, E.; Singh, S.; Pereira, M.; Ibeawuchi, S.; Katkar, G.; Dunkel, Y.; Mittal, Y.; Chattopadhyay, R.; et al. Host engulfment pathway controls inflammation in inflammatory bowel disease. FEBS J. 2020. [Google Scholar] [CrossRef]
- Mahe, M.M.; Sundaram, N.; Watson, C.L.; Shroyer, N.F.; Helmrath, M.A. Establishment of human epithelial enteroids and colonoids from whole tissue and biopsy. J. Vis. Exp. 2015, e52483. [Google Scholar] [CrossRef] [Green Version]
- Sato, T.; Vries, R.G.; Snippert, H.J.; Van De Wetering, M.; Barker, N.; Stange, D.E.; Van Es, J.H.; Abo, A.; Kujala, P.; Peters, P.J.; et al. Single Lgr5 stem cells build crypt-villus structures in vitro without a mesenchymal niche. Nature 2009, 459, 262–265. [Google Scholar] [CrossRef]
- Miyoshi, H.; Stappenbeck, T.S. In vitro expansion and genetic modification of gastrointestinal stem cells in spheroid culture. Nat. Protoc. 2013, 8, 2471–2482. [Google Scholar] [CrossRef]
- Ghosh, P.; Swanson, L.; Sayed, I.; Mittal, Y.; Lim, B.B.; Ibeawuchi, S.-R.; Foretz, M.; Viollet, B.; Sahoo, D.; Das, S. The stress polarity signaling (SPS) pathway serves as a marker and a target in the leaky gut barrier: Implications in aging and cancer. Life Sci. Alliance 2020, 3, e201900481. [Google Scholar] [CrossRef] [Green Version]
- Den Hartog, G.; Chattopadhyay, R.; Ablack, A.; Hall, E.H.; Butcher, L.D.; Bhattacharyya, A.; Eckmann, L.; Harris, P.R.; Das, S.; Ernst, P.B.; et al. Regulation of rac1 and reactive oxygen species production in response to infection of gastrointestinal epithelia. PLoS Pathog. 2016, 12, e1005382. [Google Scholar] [CrossRef]
- Gào, X.; Holleczek, B.; Cuk, K.; Zhang, Y.; Anusruti, A.; Xuan, Y.; Xu, Y.; Brenner, H.; Schöttker, B. Investigation on potential associations of oxidatively generated DNA/RNA damage with lung, colorectal, breast, prostate and total cancer incidence. Sci. Rep. 2019, 9, 7109. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Rodrigues, D.G.B.; Coelho, D.D.M.; Sitta, Â.; Jacques, C.E.D.; Hauschild, T.; Manfredini, V.; Bakkali, A.; Struys, E.A.; Jakobs, C.; Wajner, M.; et al. Experimental evidence of oxidative stress in patients with l-2-hydroxyglutaric aciduria and that l-carnitine attenuates in vitro DNA damage caused by d-2-hydroxyglutaric and l-2-hydroxyglutaric acids. Toxicol. Vitr. 2017, 42, 47–53. [Google Scholar] [CrossRef]
- Hsieh, P.; Yamane, K. DNA mismatch repair: Molecular mechanism, cancer, and ageing. Mech. Ageing Dev. 2008, 129, 391–407. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Fukui, K. DNA Mismatch Repair in Eukaryotes and Bacteria. J. Nucleic Acids 2010, 2010, 260512. [Google Scholar] [CrossRef] [Green Version]
- Moser, A.R.; Pitot, H.C.; Dove, W.F. A dominant mutation that predisposes to multiple intestinal neoplasia in the mouse. Science 1990, 247, 322–324. [Google Scholar] [CrossRef]
- De Smedt, L.; Lemahieu, J.; Palmans, S.; Govaere, O.; Tousseyn, T.; Van Cutsem, E.; Prenen, H.; Tejpar, S.; Spaepen, M.; Matthijs, G.; et al. Microsatellite instable vs. stable colon carcinomas: Analysis of tumour heterogeneity, inflammation and angiogenesis. Br. J. Cancer 2015, 113, 500–509. [Google Scholar] [CrossRef] [Green Version]
- Brenner, H.; Kloor, M.; Pox, C.P. Colorectal cancer. Lancet 2014, 383, 1490–1502. [Google Scholar] [CrossRef]
- Cancer Genome Atlas Network. Comprehensive molecular characterization of human colon and rectal cancer. Nature 2012, 487, 330–337. [Google Scholar] [CrossRef] [Green Version]
- D’Errico, M.; De Rinaldis, E.; Blasi, M.F.; Viti, V.; Falchetti, M.; Calcagnile, A.; Sera, F.; Saieva, C.; Ottini, L.; Palli, D.; et al. Genome-wide expression profile of sporadic gastric cancers with microsatellite instability. Eur. J. Cancer 2009, 45, 461–469. [Google Scholar] [CrossRef]
- Jorissen, R.N.; Lipton, L.; Gibbs, P.; Chapman, M.; Desai, J.; Jones, I.T.; Yeatman, T.J.; East, P.; Tomlinson, I.P.; Verspaget, H.W.; et al. DNA Copy-Number Alterations Underlie Gene Expression Differences between Microsatellite Stable and Unstable Colorectal Cancers. Clin. Cancer Res. 2008, 14, 8061–8069. [Google Scholar] [CrossRef] [Green Version]
- Gröne, J.; Lenze, D.; Jurinovic, V.; Hummel, M.; Seidel, H.; Leder, G.; Beckmann, G.; Sommer, A.; Grützmann, R.; Pilarsky, C.; et al. Molecular profiles and clinical outcome of stage UICC II colon cancer patients. Int. J. Color. Dis. 2011, 26, 847–858. [Google Scholar] [CrossRef] [PubMed]
- Vilar, E.; Bartnik, C.M.; Stenzel, S.L.; Raskin, L.; Ahn, J.; Moreno, V.; Mukherjee, B.; Iniesta, M.D.; Morgan, M.A.; Rennert, G.; et al. MRE11 deficiency increases sensitivity to poly(ADP-ribose) polymerase inhibition in microsatellite unstable colorectal cancers. Cancer Res. 2011, 71, 2632–2642. [Google Scholar] [CrossRef] [Green Version]
- Sanz-Pamplona, R.; Cordero, D.; Berenguer, A.; Lejbkowicz, F.; Rennert, H.; Salazar, R.; Biondo, S.; Sanjuan, X.; Pujana, M.A.; Rozek, L.; et al. Gene expression differences between colon and rectum tumors. Clin. Cancer Res. 2011, 17, 7303–7312. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Schmit, S.L.; Gollub, J.; Shapero, M.H.; Huang, S.-C.; Rennert, H.S.; Finn, A.; Rennert, G.; Gruber, S.B. MicroRNA polymorphisms and risk of colorectal cancer. Cancer Epidemiol. Biomark. Prev. 2014, 24, 65–72. [Google Scholar] [CrossRef] [Green Version]
- Banerjee, D.; Mandal, S.M.; Das, A.; Hegde, M.L.; Das, S.; Bhakat, K.K.; Boldogh, I.; Sarkar, P.S.; Mitra, S.; Hazra, T.K. Preferential Repair of Oxidized Base Damage in the Transcribed Genes of Mammalian Cells. J. Biol. Chem. 2011, 286, 6006–6016. [Google Scholar] [CrossRef] [Green Version]
- Limpose, K.L.; Trego, K.S.; Li, Z.; Leung, S.W.; Sarker, A.H.; Shah, J.A.; Ramalingam, S.S.; Werner, E.; Dynan, W.S.; Cooper, P.K.; et al. Overexpression of the base excision repair NTHL1 glycosylase causes genomic instability and early cellular hallmarks of cancer. Nucleic Acids Res. 2018, 46, 4515–4532. [Google Scholar] [CrossRef] [Green Version]
- Takao, M.; Kanno, S.; Shiromoto, T.; Hasegawa, R.; Ide, H.; Ikeda, S.; Sarker, A.H.; Seki, S.; Xing, J.Z.; Le, X.C.; et al. Novel nuclear and mitochondrial glycosylases revealed by disruption of the mouse Nth1 gene encoding an endonuclease III homolog for repair of thymine glycols. EMBO J. 2002, 21, 3486–3493. [Google Scholar] [CrossRef] [Green Version]
- Hanada, K.; Uchida, T.; Tsukamoto, Y.; Watada, M.; Yamaguchi, N.; Yamamoto, K.; Shiota, S.; Moriyama, M.; Graham, D.Y.; Yamaoka, Y. Helicobacter pylori Infection Introduces DNA Double-Strand Breaks in Host Cells. Infect. Immun. 2014, 82, 4182–4189. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Nalbantoglu, I.; Blanc, V.; Davidson, N.O. Characterization of Colorectal Cancer Development in Apc (min/+) Mice. Methods Mol. Biol. 2016, 1422, 309–327. [Google Scholar] [CrossRef] [Green Version]
- Wu, S.; Rhee, K.-J.; Albesiano, E.; Rabizadeh, S.; Wu, X.; Yen, H.-R.; Huso, D.L.; Brancati, F.L.; Wick, E.; McAllister, F.; et al. A human colonic commensal promotes colon tumorigenesis via activation of T helper type 17 T cell responses. Nat. Med. 2009, 15, 1016–1022. [Google Scholar] [CrossRef]
- Arthur, J.C.; Perez-Chanona, E.; Mühlbauer, M.; Tomkovich, S.; Uronis, J.M.; Fan, T.-J.; Campbell, B.J.; Abujamel, T.; Dogan, B.; Rogers, A.B.; et al. Intestinal Inflammation Targets Cancer-Inducing Activity of the Microbiota. Sciences 2012, 338, 120–123. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Poulsen, M.L.M.; Bisgaard, M.L. MUTYH Associated Polyposis (MAP). Curr. Genom. 2008, 9, 420–435. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Al-Tassan, N.; Chmiel, N.H.; Maynard, J.; Fleming, N.; Livingston, A.L.; Williams, G.T.; Hodges, A.K.; Davies, D.R.; David, S.S.; Sampson, J.R.; et al. Inherited variants of MYH associated with somatic G:C→T:A mutations in colorectal tumors. Nat. Genet. 2002, 30, 227–232. [Google Scholar] [CrossRef] [PubMed]
- Farrington, S.M.; Tenesa, A.; Barnetson, R.; Wiltshire, A.; Prendergast, J.G.; Porteous, M.; Campbell, H.; Dunlop, M.G. Germline susceptibility to colorectal cancer due to base-excision repair gene defects. Am. J. Hum. Genet. 2005, 77, 112–119. [Google Scholar] [CrossRef] [Green Version]
- Broderick, P.; Bagratuni, T.; Vijayakrishnan, J.; Lubbe, S.; Chandler, I.; Houlston, R.S. Evaluation of NTHL1, NEIL1, NEIL2, MPG, TDG, UNG and SMUG1genes in familial colorectal cancer predisposition. BMC Cancer 2006, 6, 243. [Google Scholar] [CrossRef] [Green Version]
- Vodenkova, S.; Jiraskova, K.; Urbanová, M.; Kroupa, M.; Slyskova, J.; Schneiderova, M.; Levy, M.; Buchler, T.; Liska, V.; Vodickova, L.; et al. Base excision repair capacity as a determinant of prognosis and therapy response in colon cancer patients. DNA Repair 2018, 72, 77–85. [Google Scholar] [CrossRef]








{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Species | Forward Primer (5’ → 3’) | Reverse Primer (5′ → 3’) |
|---|---|---|---|
| 18srRNA | GTAACCCGTTGAACCCCATT | CCATCCAATCGGTAGTAGCG | |
| NEIL1 | mouse | TCGTAGACATCCGTCGCTTT | TGTCTGATAGGTTCCGAAGTACG |
| NEIL2 | mouse | CTGCCGCCTTTCAGTCTCT | TCTGGATCAAACCGAAGGAA |
| NTH1 | mouse | GCATGAACTCAGGGAAGGAAGA | CCTCACCATTAGCCGCTTCA |
| OGG1 | mouse | TTATCATGGCTTCCCAAACC | GTACCCCAGGCCCAACTT |
| NEIL1 | human | CCAGGCAGTGGGAAGTCA | AGGGAGGGTGGCAGAGTC |
| NEIL2 | human | GGGGCAGCAGTAAGAAGCTA | GGAATAATTTCTTTCCATGGACCT |
| NTH1 | human | GACAGCATCCTGCAGACAGA | TTGATGTATTTCACCTTGCTCCT |
| OGG1 | human | CCAGACCAACAAGGAACTGG | CAAATGCATTGCCAAGGA |
| IL-8/KC | mouse | CGCTTCTCTGTGCAGCGCTGCTGCT | AAGCCTCGCGACCATTCTTGAGTC |
| IL-8 | Human | GAGCACTCCATAAGGCACAAA | ATGGTTCCTTCCGGTGGT |
| MLH1 | Human | GATTACCCCTTCTGATTGACA | ACTGAGGCTTTCAAAACA |
| MLH3 | Human | CGCACGAGCCTCAAGATCC | TCTGACTGGAAATAATTGCCTGGA |
| MSH2 | Human | CAGTATATTGGAGAATCGCA | AGGGCATTTGTTTCACC |
| MSH6 | Human | GCTTCTTCCCCAAGTCTCCG | AGAAGTCACAACTGGTGGGG |
| PMS2 | Human | ACTGAGTCTAAGCACTGCGG | TGACATCGCTCAGTGCACAA |
| Ku70 | Human | CCACAGGAAGAAGAGTTGGA | CTGCTCTGGAGTTGCCATGA |
| Primer Name | Gene Name | Sequences | PCR |
|---|---|---|---|
| m-Polβ-LA-F | Polβ (mouse) | TAT CTC TCT TCC TCT TCA CTT CTC CCC TGG | LA-qPCR |
| m-Polβ-LA-R | Polβ (mouse) | CGT GAT GCC GCC GTT GAG GGT CTC CTG | LA-qPCR |
| m-β Globin-LA-F | β globin (mouse) | TTG AGA CTG TGA TTG GCA ATG CCT | LA-qPCR |
| m-β Globin-LA-R | β globin (mouse) | CCT TTA ATG CCC ATC CCG GAC T | LA-qPCR |
| m-Polβ-SA-F | Polβ (mouse) | TATGGACCCCCATGAGGAACA | SA-PCR |
| m-Polβ-SA-R | Polβ (mouse) | AACCGTCGGCTAAAGACGTG | SA-PCR |
| m-β Globin-SA-F | β globin (mouse) | ACACTACTCAGAGTGAGACCCA | SA-PCR |
| m-β Globin-SA-R | β Globin (mouse) | ATACCCAATGCTGGCTCCTG | SA-PCR |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sayed, I.M.; Chakraborty, A.; Abd El-Hafeez, A.A.; Sharma, A.; Sahan, A.Z.; Huang, W.J.M.; Sahoo, D.; Ghosh, P.; Hazra, T.K.; Das, S. The DNA Glycosylase NEIL2 Suppresses Fusobacterium-Infection-Induced Inflammation and DNA Damage in Colonic Epithelial Cells. Cells 2020, 9, 1980. https://doi.org/10.3390/cells9091980
Sayed IM, Chakraborty A, Abd El-Hafeez AA, Sharma A, Sahan AZ, Huang WJM, Sahoo D, Ghosh P, Hazra TK, Das S. The DNA Glycosylase NEIL2 Suppresses Fusobacterium-Infection-Induced Inflammation and DNA Damage in Colonic Epithelial Cells. Cells. 2020; 9(9):1980. https://doi.org/10.3390/cells9091980
Chicago/Turabian StyleSayed, Ibrahim M., Anirban Chakraborty, Amer Ali Abd El-Hafeez, Aditi Sharma, Ayse Z. Sahan, Wendy Jia Men Huang, Debashis Sahoo, Pradipta Ghosh, Tapas K. Hazra, and Soumita Das. 2020. "The DNA Glycosylase NEIL2 Suppresses Fusobacterium-Infection-Induced Inflammation and DNA Damage in Colonic Epithelial Cells" Cells 9, no. 9: 1980. https://doi.org/10.3390/cells9091980