Fine-Tuning of GLI Activity through Arginine Methylation: Its Mechanisms and Function


{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
:1. Introduction
2. Regulation of GLI Transcriptional Activity
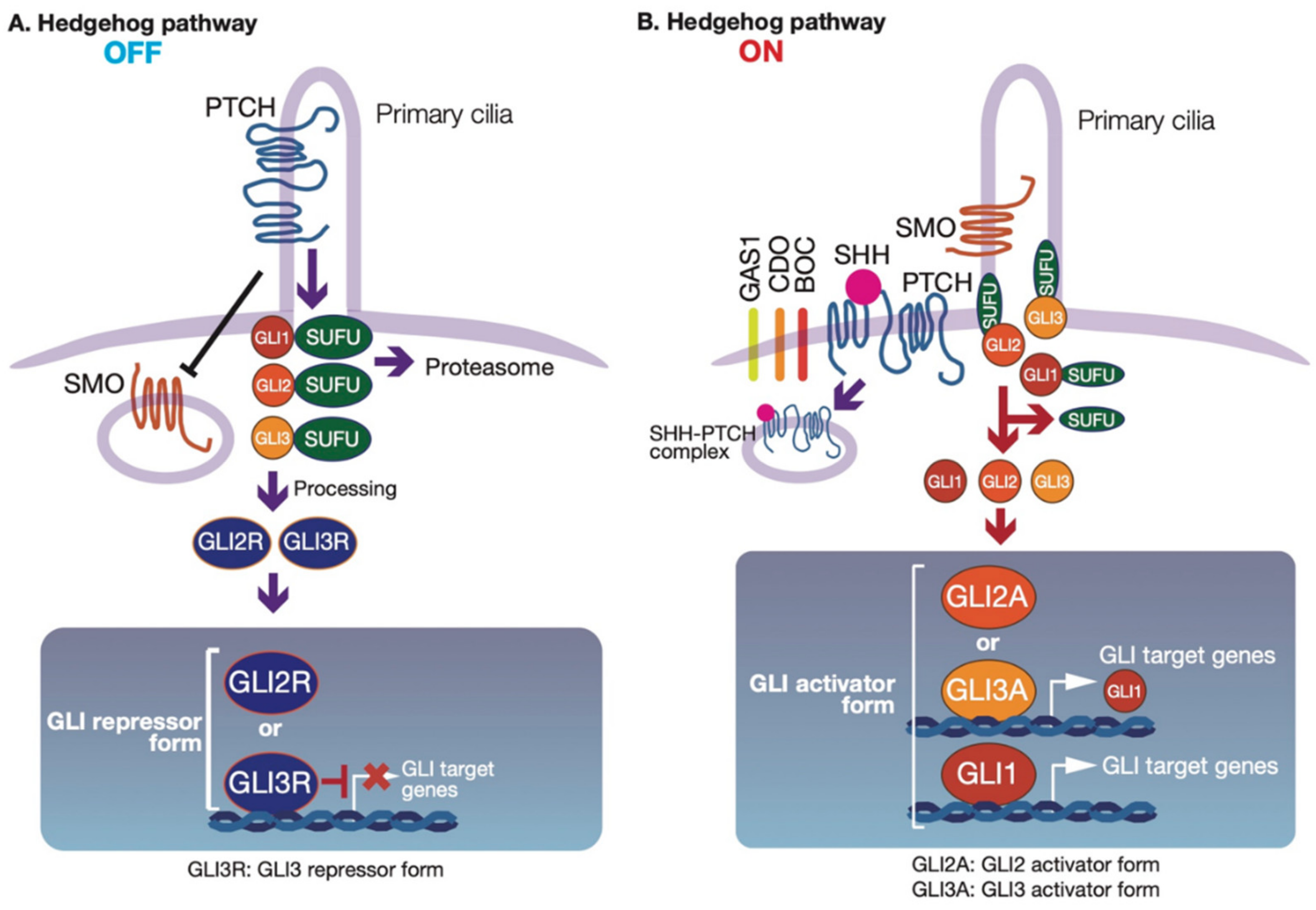
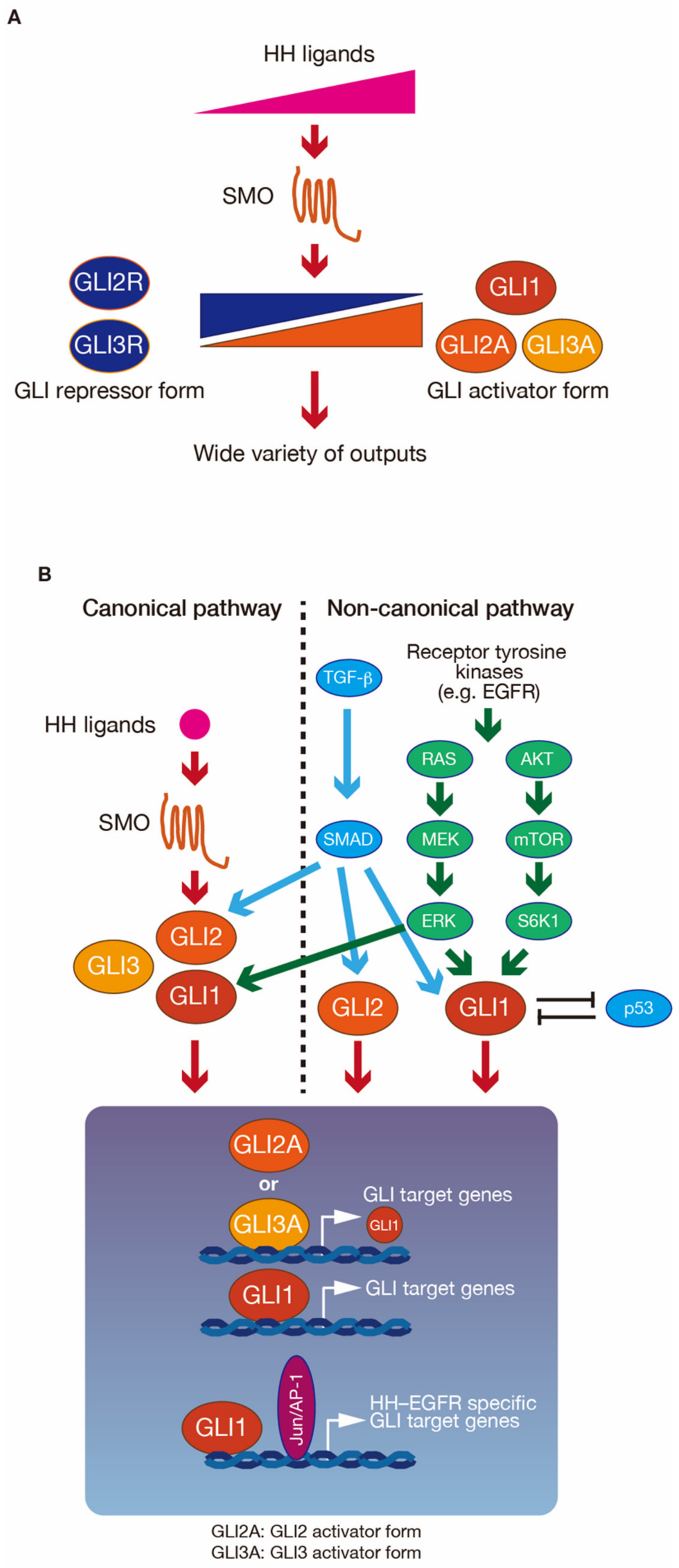
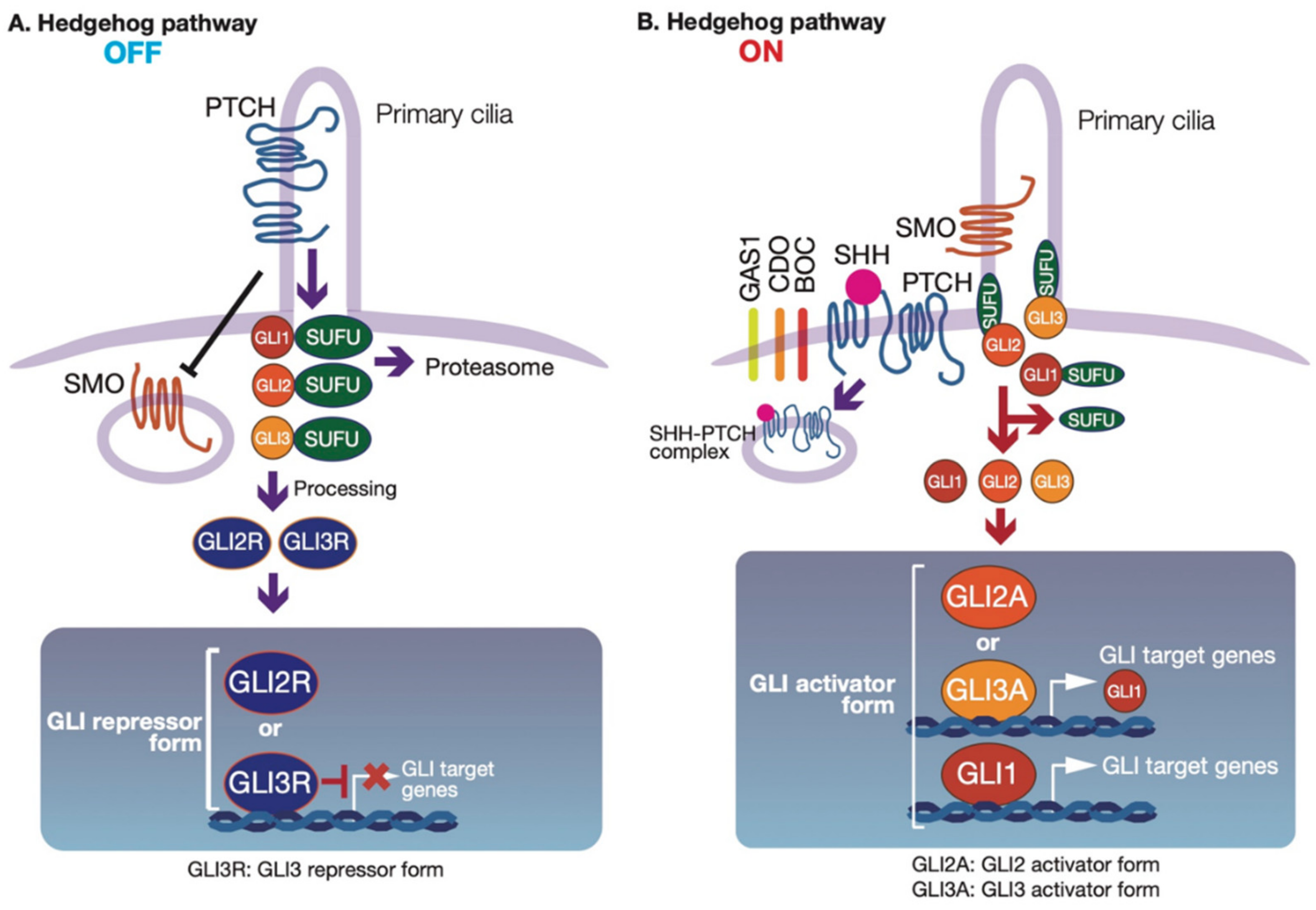
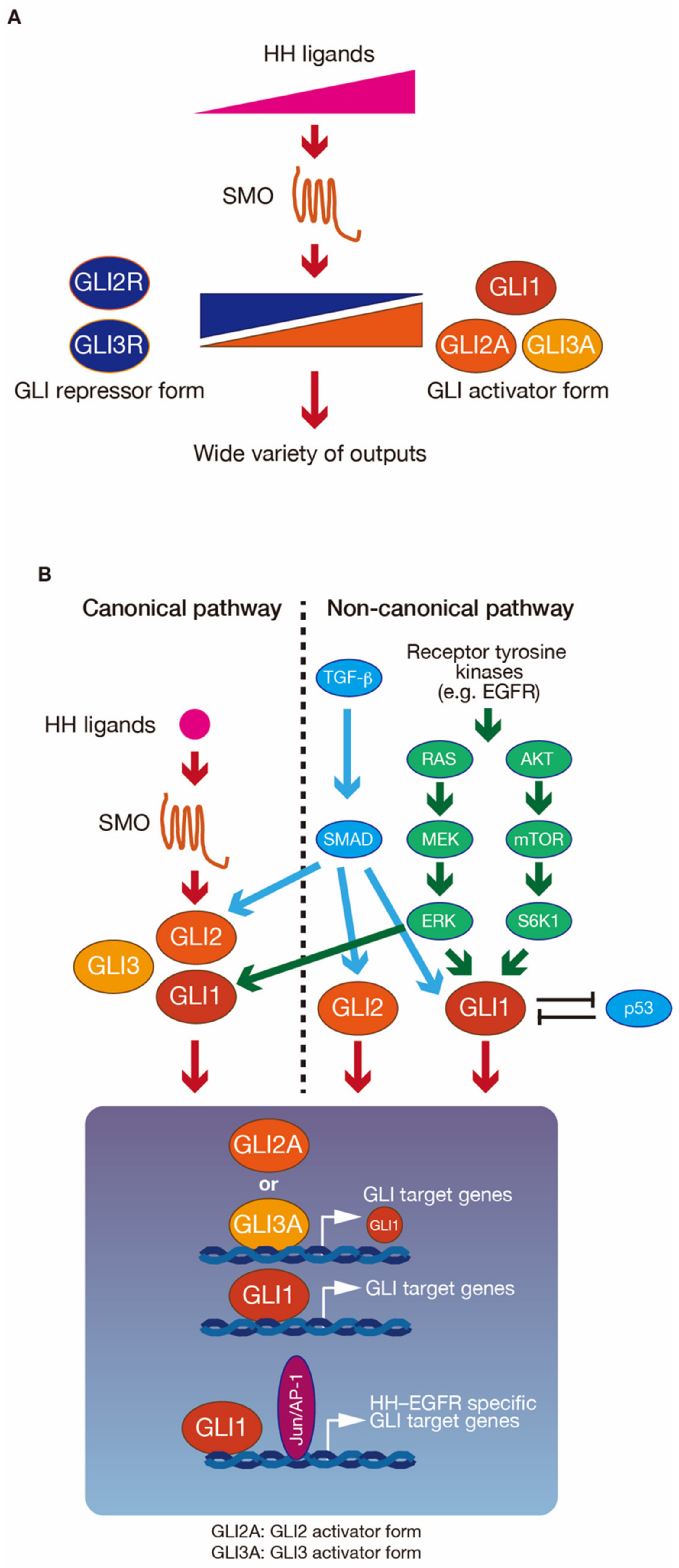
2.1. Regulation of GLI Proteins by HH-Dependent and -Independent Signaling Pathways
2.2. Modulation of GLI Activity
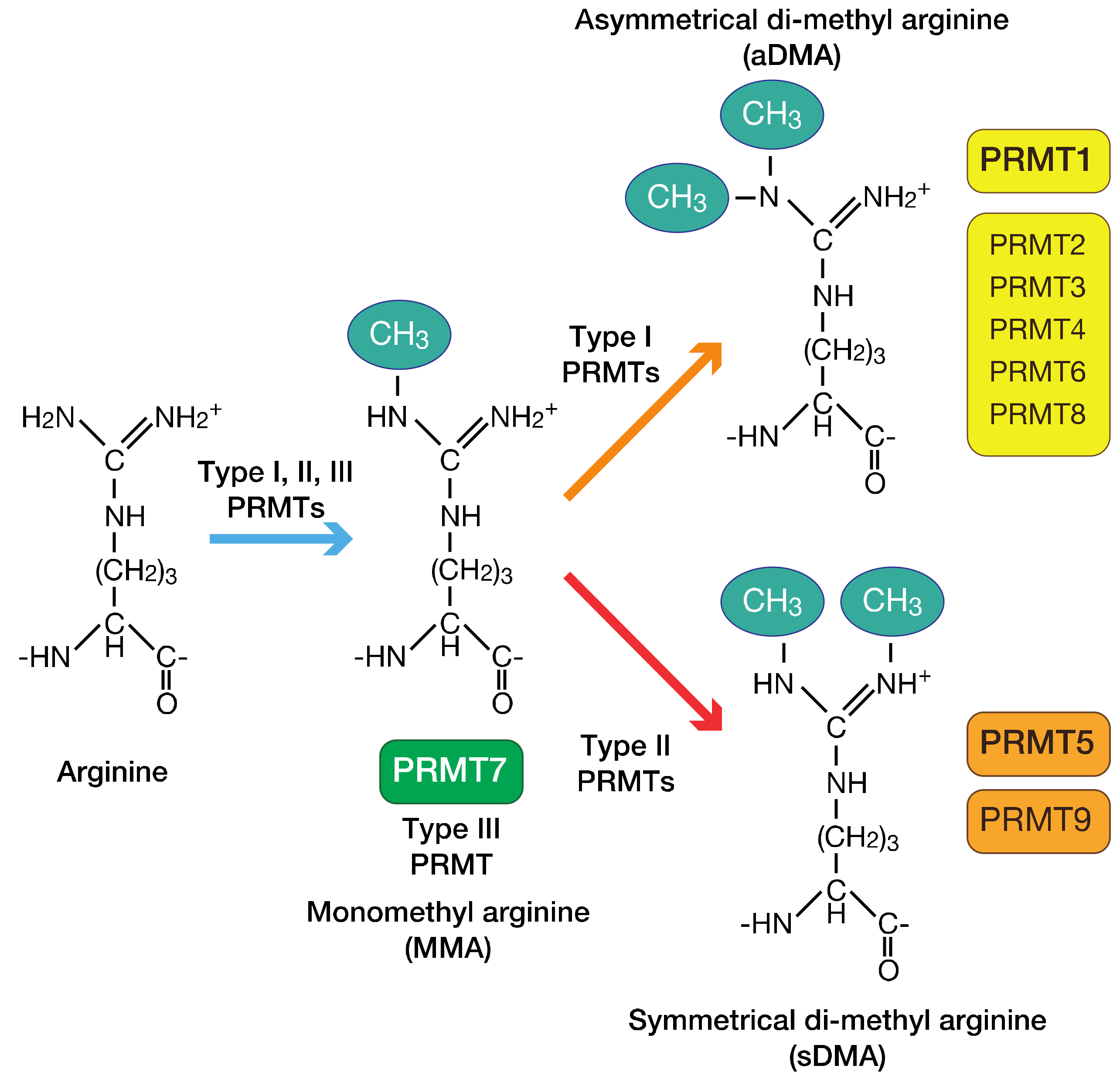
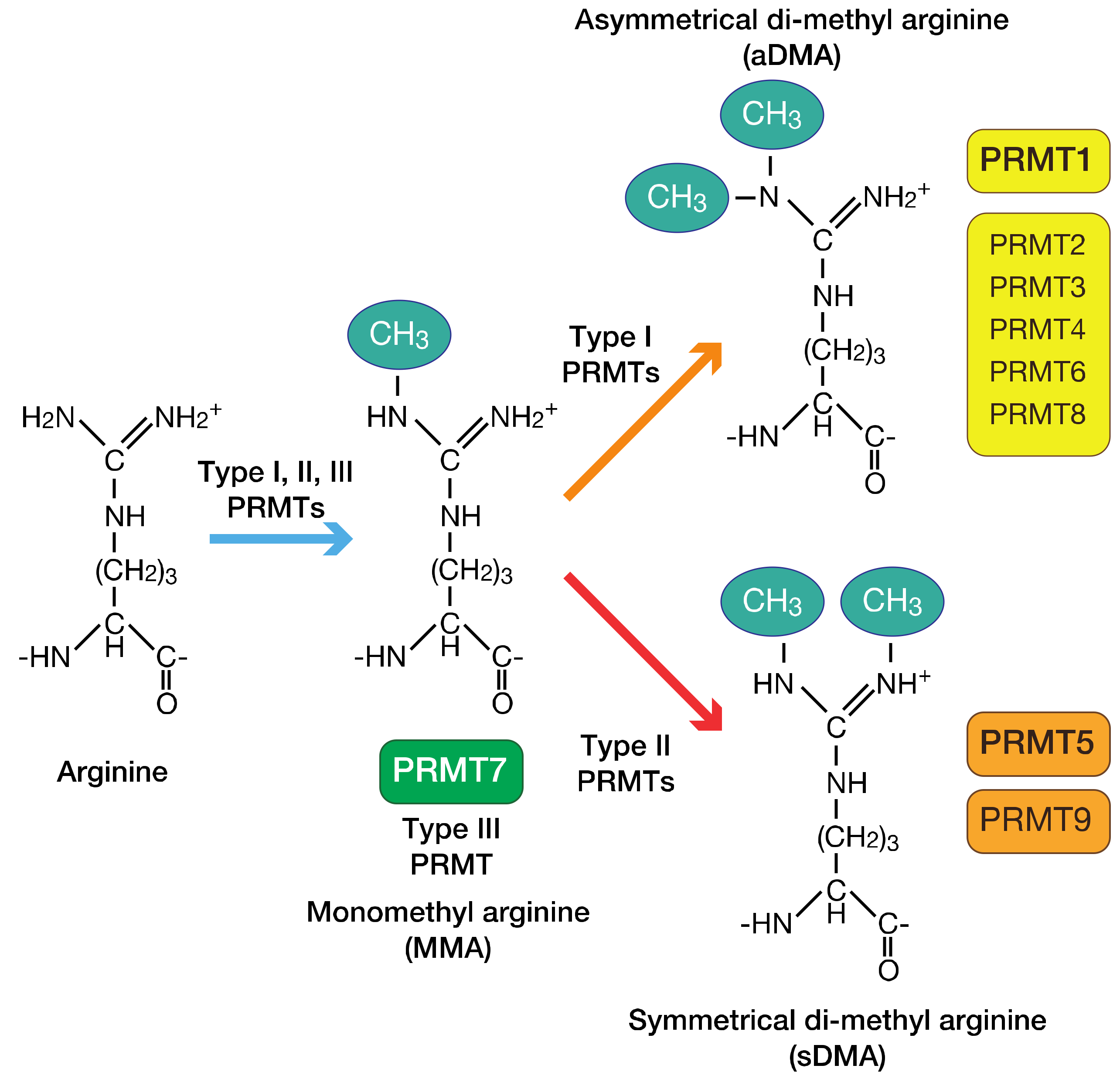
3. PRMTs
3.1. Overview of the PRMT Family of Proteins
3.2. Regulation of PRMT Activity by Post-Translational Modifications
3.3. Roles of PRMTs in Signaling Pathways
3.3.1. Roles of PRMT1 in the TGF-β and Bone Morphogenetic Protein (BMP) Signaling Pathways
3.3.2. Roles of PRMT5 in the EGFR Signaling Pathway
3.4. Roles of PRMTs in Genes Transcription via Histone Arginine Methylation
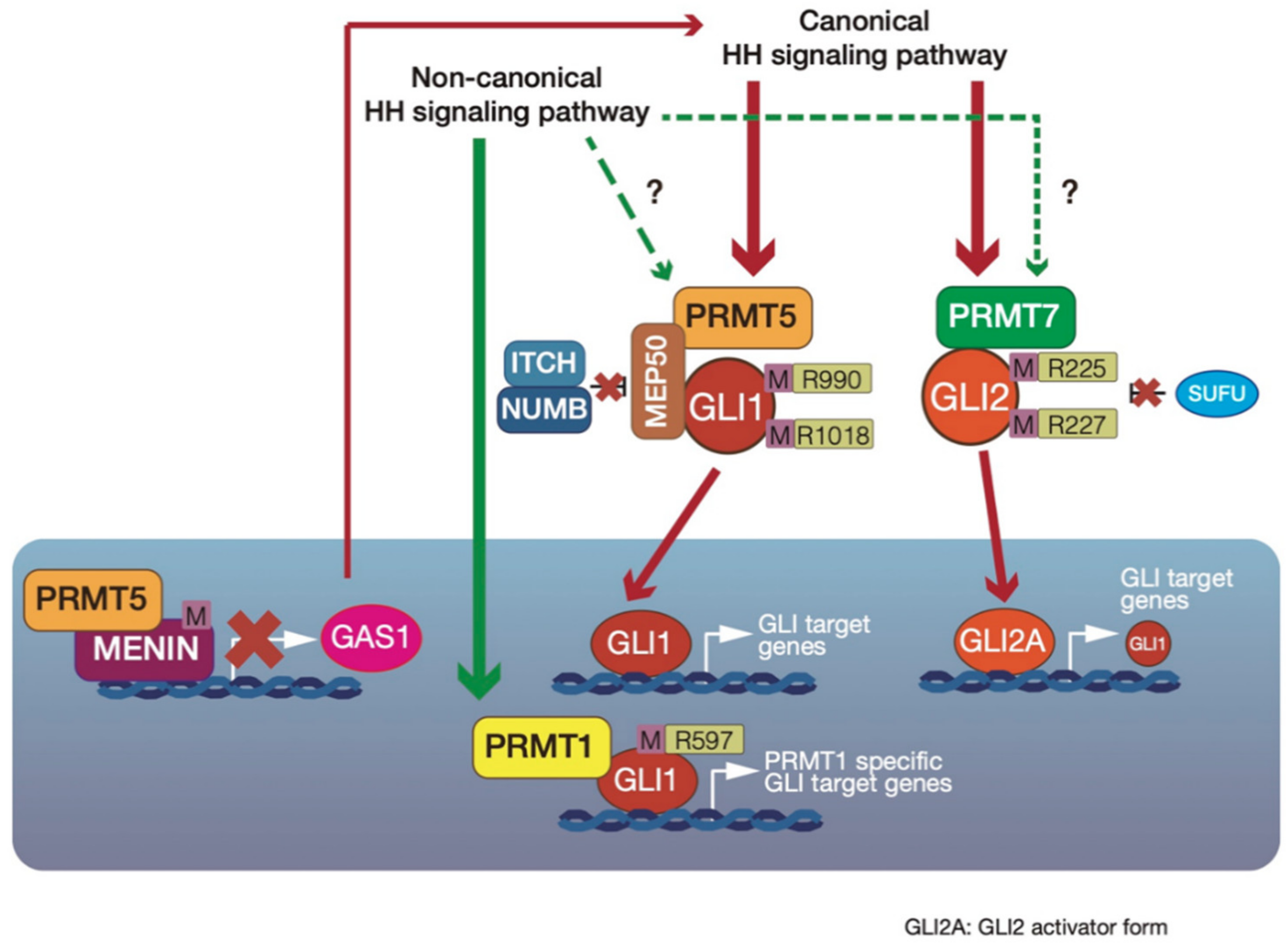
4. PRMT-Mediated Regulation of GLI Activity
4.1. Upregulation of GLI1 Transcriptional Activity by PRMT1-Mediated GLI1 Methylation
4.2. GLI1 Protein Stabilization by Cytosolic PRMT5-Mediated GLI1 Methylation
4.3. Indirect Suppression of GLI1 Activity by PRMT5–Menin Complex-Mediated Epigenetic Modification in Multiple Endocrine Neoplasia Type 1 (MEN1) Tumor Syndrome
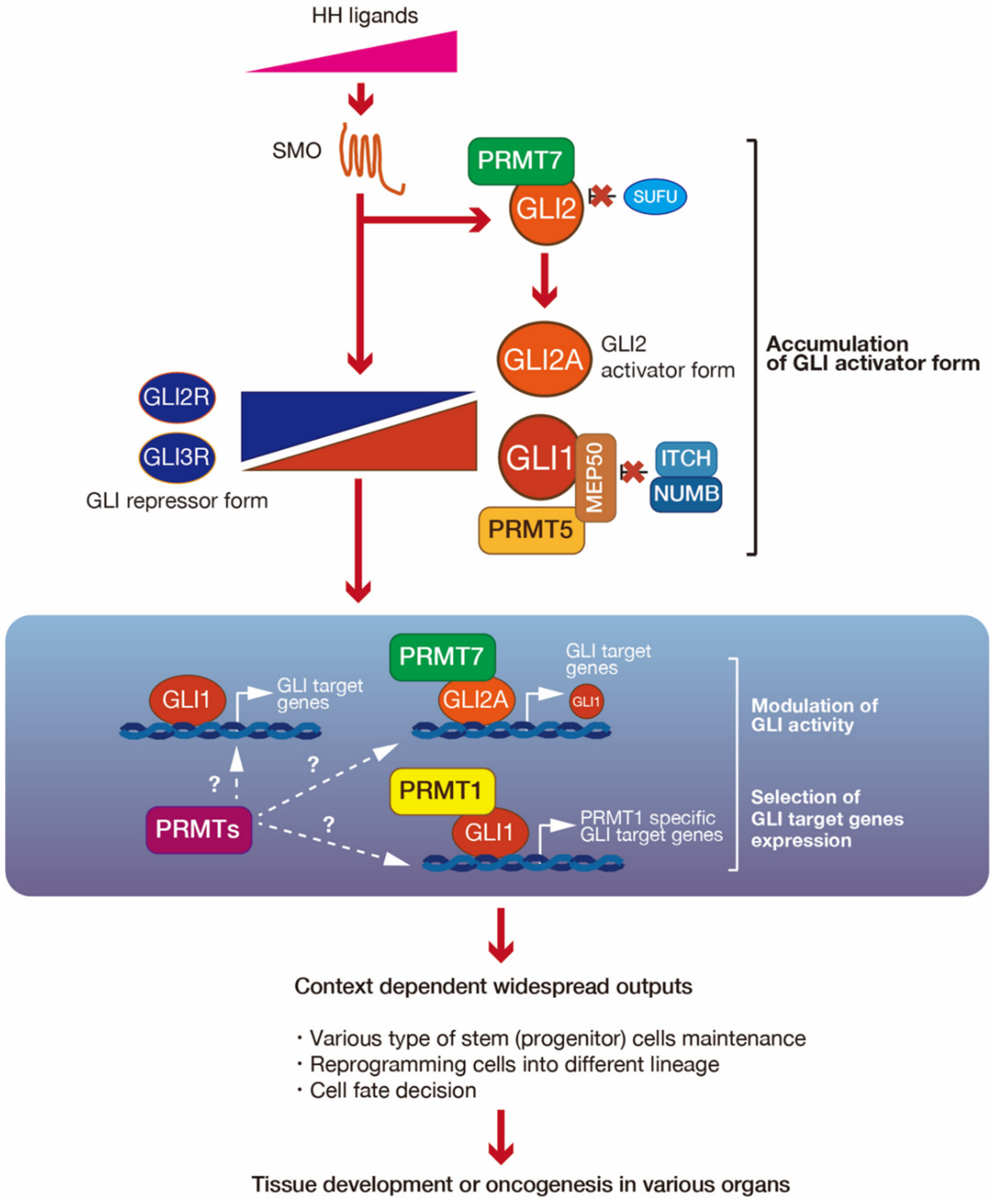
4.4. PRMT7 Upregulates GLI2 Activity via Suppression of SUFU binding
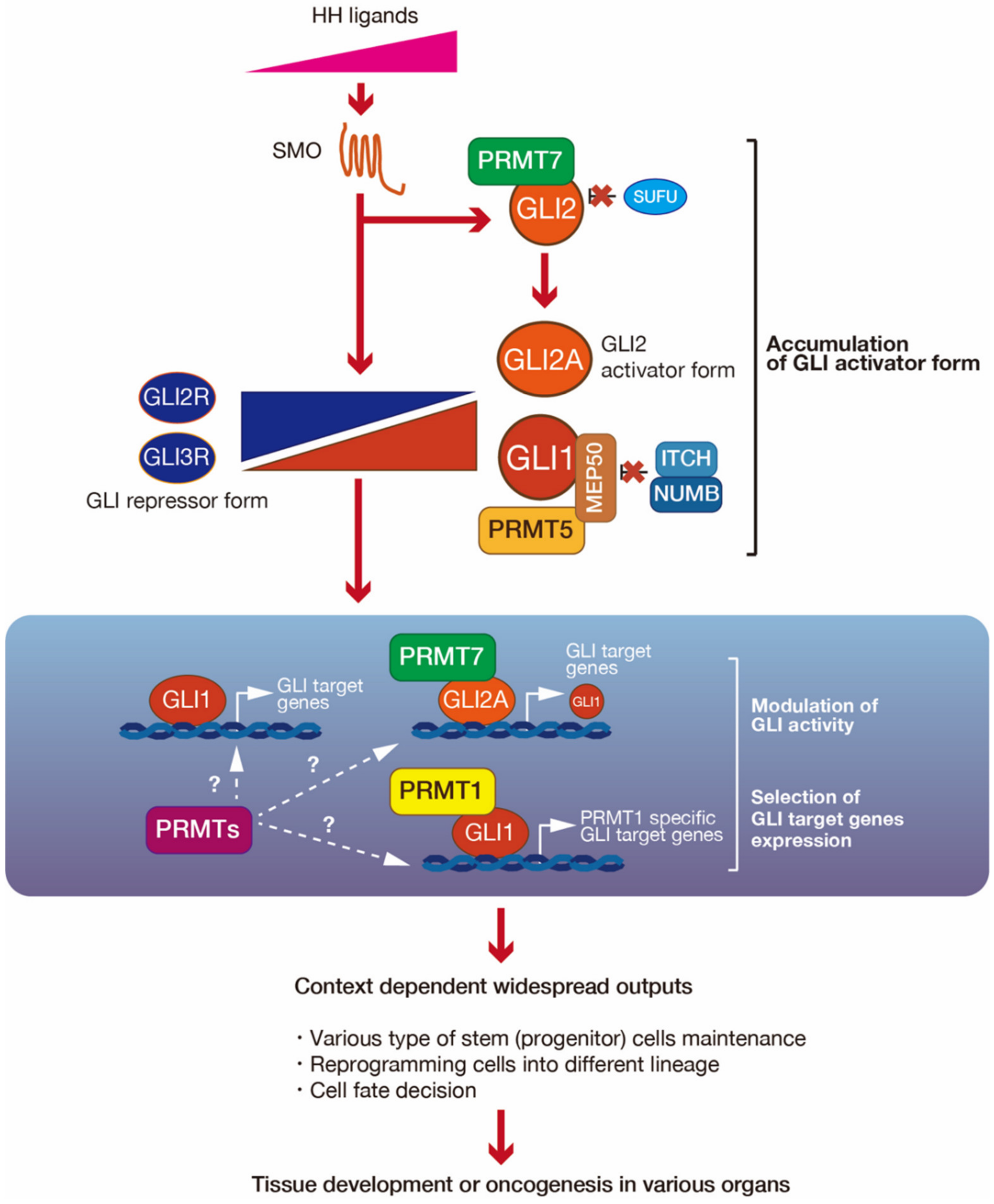
5. Consideration of the Roles of PRMT-Mediated GLI Regulation in the Development and Cancers, with a Focus on Stem Cell Maintenance
6. Outlook for PRMT-Mediated GLI1 Regulatory Mechanisms
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Ingham, P.W.; McMahon, A.P. Hedgehog signaling in animal development: Paradigms and principles. Genes Dev. 2001, 15, 3059–3087. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beachy, P.A.; Karhadkar, S.S.; Berman, D.M. Tissue repair and stem cell renewal in carcinogenesis. Nature 2004, 432, 324–331. [Google Scholar] [CrossRef] [PubMed]
- Ng, J.M.Y.; Curran, T. The Hedgehog’s tale: Developing strategies for targeting cancer. Nat. Rev. Cancer 2011, 11, 493–501. [Google Scholar] [CrossRef] [Green Version]
- Hui, C.; Angers, S. Gli Proteins in Development and Disease. Annu. Rev. Cell Dev. Biol. 2011, 27, 513–537. [Google Scholar] [CrossRef] [Green Version]
- Briscoe1, J.; Thérond, P.P.; Briscoe, J.; Thérond, P.P. The mechanisms of Hedgehog signalling and its roles in development and disease. Nat. Rev. Mol. Cell Biol. 2013, 14, 416–429. [Google Scholar] [CrossRef] [PubMed]
- Aberger, F.; Ruiz i Altaba, A. Context-dependent signal integration by the GLI code: The oncogenic load, pathways, modifiers and implications for cancer therapy. Semin. Cell Dev. Biol. 2014, 33, 93–104. [Google Scholar] [CrossRef]
- Pietrobono, S.; Gagliardi, S.; Stecca, B. Non-canonical hedgehog signaling pathway in cancer: Activation of GLI transcription factors beyond smoothened. Front. Genet. 2019, 10, 556. [Google Scholar] [CrossRef] [Green Version]
- Blanc, R.S.; Richard, S. Arginine Methylation: The Coming of Age. Mol. Cell 2017, 65, 8–24. [Google Scholar] [CrossRef] [Green Version]
- Yang, Y.; Bedford, M.T. Protein arginine methyltransferases and cancer. Nat. Rev. Cancer 2013, 13, 37–50. [Google Scholar] [CrossRef]
- Jarrold, J.; Davies, C.C. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends Mol. Med. 2019, 25, 993–1009. [Google Scholar] [CrossRef]
- Rohatgi, R.; Milenkovic, L.; Scott, M.P. Patched1 regulates hedgehog signaling at the primary cilium. Science 2007, 317, 372–376. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Ding, Q.; Fukami, S.; Meng, X.; Nishizaki, Y.; Zhang, X.; Sasaki, H.; Dlugosz, A.; Nakafuku, M.; Hui, C. Mouse Suppressor of fused is a negative regulator of Sonic hedgehog signaling and alters the subcellular distribution of Gli1. Curr. Biol. 1999, 9, 1119–1122. [Google Scholar] [CrossRef] [Green Version]
- Svärd, J.; Heby-Henricson, K.; Henricson, K.H.; Persson-Lek, M.; Rozell, B.; Lauth, M.; Bergström, A.; Ericson, J.; Toftgård, R.; Teglund, S. Genetic elimination of Suppressor of fused reveals an essential repressor function in the mammalian Hedgehog signaling pathway. Dev. Cell 2006, 10, 187–197. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Allen, B.L.; Tenzen, T.; McMahon, A.P. The Hedgehog-binding proteins Gas1 and Cdo cooperate to positively regulate Shh signaling during mouse development. Genes Dev. 2007, 21, 1244–1257. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Tanaka, N. Roles of the Hedgehog Signaling Pathway in Epidermal and Hair Follicle Development, Homeostasis, and Cancer. J. Dev. Biol. 2017, 5, 12. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Abe, Y.; Oda-Sato, E.; Tobiume, K.; Kawauchi, K.; Taya, Y.; Okamoto, K.; Oren, M.; Tanaka, N. Hedgehog signaling overrides p53-mediated tumor suppression by activating Mdm2. Proc. Natl. Acad. Sci. USA 2008, 105, 4838–4843. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Ruiz i Altaba, A. A GLI1-p53 inhibitory loop controls neural stem cell and tumour cell numbers. EMBO J. 2009, 28, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Riobó, N.A.; Lu, K.; Ai, X.; Haines, G.M.; Emerson, C.P. Phosphoinositide 3-kinase and Akt are essential for Sonic Hedgehog signaling. Proc. Natl. Acad. Sci. USA 2006, 103, 4505–4510. [Google Scholar] [CrossRef] [Green Version]
- Stecca, B.; Mas, C.; Clement, V.; Zbinden, M.; Correa, R.; Piguet, V.; Beermann, F.; Ruiz, I.; Altaba, A. Melanomas require HEDGEHOG-GLI signaling regulated by interactions between GLI1 and the RAS-MEK/AKT pathways. Proc. Natl. Acad. Sci. USA 2007, 104, 5895–5900. [Google Scholar] [CrossRef] [Green Version]
- Dennler, S.; André, J.; Alexaki, I.; Li, A.; Magnaldo, T.; Ten Dijke, P.; Wang, X.-J.; Verrecchia, F.; Mauviel, A. Induction of Sonic Hedgehog Mediators by Transforming Growth Factor-β: Smad3-Dependent Activation of Gli2 and Gli1 Expression In vitro and In vivo. Cancer Res. 2007, 67, 6981–6986. [Google Scholar] [CrossRef] [Green Version]
- Nye, M.D.; Almada, L.L.; Fernandez-Barrena, M.G.; Marks, D.L.; Elsawa, S.F.; Vrabel, A.; Tolosa, E.J.; Ellenrieder, V.; Fernandez-Zapico, M.E. The Transcription Factor GLI1 Interacts with SMAD Proteins to Modulate Transforming Growth Factor β-Induced Gene Expression in a p300/CREB-binding Protein-associated Factor (PCAF)-dependent Manner. J. Biol. Chem. 2014, 289, 15495–15506. [Google Scholar] [CrossRef] [Green Version]
- Riobo, N.A.; Haines, G.M.; Emerson, C.P. Protein Kinase C-δ and Mitogen-Activated Protein/Extracellular Signal–Regulated Kinase-1 Control GLI Activation in Hedgehog Signaling. Cancer Res. 2006, 66, 839–845. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Whisenant, T.C.; Ho, D.T.; Benz, R.W.; Rogers, J.S.; Kaake, R.M.; Gordon, E.A.; Huang, L.; Baldi, P.; Bardwell, L. Computational Prediction and Experimental Verification of New MAP Kinase Docking Sites and Substrates Including Gli Transcription Factors. PLoS Comput. Biol. 2010, 6, e1000908. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Palma, V.; Lim, D.A.; Dahmane, N.; Sánchez, P.; Brionne, T.C.; Herzberg, C.D.; Gitton, Y.; Carleton, A.; Alvarez-Buylla, A.; Ruiz i Altaba, A. Sonic hedgehog controls stem cell behavior in the postnatal and adult brain. Development 2005, 132, 335–344. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kasper, M.; Schnidar, H.; Neill, G.W.; Hanneder, M.; Klingler, S.; Blaas, L.; Schmid, C.; Hauser-Kronberger, C.; Regl, G.; Philpott, M.P.; et al. Selective modulation of Hedgehog/GLI target gene expression by epidermal growth factor signaling in human keratinocytes. Mol. Cell. Biol. 2006, 26, 6283–6298. [Google Scholar] [CrossRef] [Green Version]
- Eberl, M.; Klingler, S.; Mangelberger, D.; Loipetzberger, A.; Damhofer, H.; Zoidl, K.; Schnidar, H.; Hache, H.; Bauer, H.-C.; Solca, F.; et al. Hedgehog-EGFR cooperation response genes determine the oncogenic phenotype of basal cell carcinoma and tumour-initiating pancreatic cancer cells. EMBO Mol. Med. 2012, 4, 218–233. [Google Scholar] [CrossRef]
- Stamataki, D.; Ulloa, F.; Tsoni, S.V.; Mynett, A.; Briscoe, J. A gradient of Gli activity mediates graded Sonic Hedgehog signaling in the neural tube. Genes Dev. 2005, 19, 626–641. [Google Scholar] [CrossRef] [Green Version]
- Wang, B.; Fallon, J. Hedgehog-Regulated Processing of Gli3 Produces an Anterior/Posterior Repressor Gradient in the Developing Vertebrate Limb. Cell 2000, 100, 423–434. [Google Scholar] [CrossRef] [Green Version]
- Götschel, F.; Berg, D.; Gruber, W.; Bender, C.; Eberl, M.; Friedel, M.; Sonntag, J.; Rüngeler, E.; Hache, H.; Wierling, C.; et al. Synergism between Hedgehog-GLI and EGFR Signaling in Hedgehog-Responsive Human Medulloblastoma Cells Induces Downregulation of Canonical Hedgehog-Target Genes and Stabilized Expression of GLI1. PLoS ONE 2013, 8, e65403. [Google Scholar] [CrossRef]
- Wang, Y.; Ding, Q.; Yen, C.-J.; Xia, W.; Izzo, J.G.; Lang, J.-Y.; Li, C.-W.; Hsu, J.L.; Miller, S.A.; Wang, X.; et al. The crosstalk of mTOR/S6K1 and Hedgehog pathways. Cancer Cell 2012, 21, 374–387. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Li, M.; Lee, A.; Tang, J.Y.; Oro, A.E. GLI activation by atypical protein kinase C ι/λ regulates the growth of basal cell carcinomas. Nature 2013, 494, 484–488. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Niewiadomski, P.; Kong, J.; Ahrends, R.; Ma, Y.; Humke, E.; Khan, S.; Teruel, M.; Novitch, B.; Rohatgi, R. Gli protein activity is controlled by multisite phosphorylation in vertebrate hedgehog signaling. Cell Rep. 2014, 6, 168–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Canettieri, G.; Di Marcotullio, L.; Greco, A.; Coni, S.; Antonucci, L.; Infante, P.; Pietrosanti, L.; De Smaele, E.; Ferretti, E.; Miele, E.; et al. Histone deacetylase and Cullin3-REN(KCTD11) ubiquitin ligase interplay regulates Hedgehog signalling through Gli acetylation. Nat. Cell Biol. 2010, 12, 132–142. [Google Scholar] [CrossRef] [PubMed]
- Bedford, M.T.; Richard, S. Arginine methylation an emerging regulator of protein function. Mol. Cell 2005, 18, 263–272. [Google Scholar] [CrossRef]
- Guccione, E.; Richard, S. The regulation, functions and clinical relevance of arginine methylation. Nat. Rev. Mol. Cell Biol. 2019, 20, 642–657. [Google Scholar] [CrossRef]
- Jain, K.; Clarke, S.G. PRMT7 as a unique member of the protein arginine methyltransferase family: A review. Arch. Biochem. Biophys. 2019, 665, 36–45. [Google Scholar] [CrossRef]
- Sipos, A.; Iván, J.; Bécsi, B.; Darula, Z.; Tamás, I.; Horváth, D.; Medzihradszky, K.F.; Erdődi, F.; Lontay, B. Myosin phosphatase and RhoA-activated kinase modulate arginine methylation by the regulation of protein arginine methyltransferase 5 in hepatocellular carcinoma cells. Sci. Rep. 2017, 7, 1–15. [Google Scholar] [CrossRef]
- Lattouf, H.; Kassem, L.; Jacquemetton, J.; Choucair, A.; Poulard, C.; Trédan, O.; Corbo, L.; Diab-Assaf, M.; Hussein, N.; Treilleux, I.; et al. LKB1 regulates PRMT5 activity in breast cancer. Int. J. Cancer 2019, 144, 595–606. [Google Scholar] [CrossRef] [Green Version]
- Hosohata, K.; Li, P.; Hosohata, Y.; Qin, J.; Roeder, R.G.; Wang, Z. Purification and Identification of a Novel Complex Which Is Involved in Androgen Receptor-Dependent Transcription. Mol. Cell. Biol. 2003, 23, 7019–7029. [Google Scholar] [CrossRef] [Green Version]
- Friesen, W.J.; Wyce, A.; Paushkin, S.; Abel, L.; Rappsilber, J.; Mann, M.; Dreyfuss, G. A novel WD repeat protein component of the methylosome binds Sm proteins. J. Biol. Chem. 2002, 277, 8243–8247. [Google Scholar] [CrossRef] [Green Version]
- Aggarwal, P.; Vaites, L.P.; Kim, J.K.; Mellert, H.; Gurung, B.; Nakagawa, H.; Herlyn, M.; Hua, X.; Rustgi, A.K.; McMahon, S.B.; et al. Nuclear cyclin D1/CDK4 kinase regulates CUL4 expression and triggers neoplastic growth via activation of the PRMT5 methyltransferase. Cancer Cell 2010, 18, 329–340. [Google Scholar] [CrossRef] [Green Version]
- Baxter, E.J.; Scott, L.M.; Campbell, P.J.; East, C.; Fourouclas, N.; Swanton, S.; Vassiliou, G.S.; Bench, A.J.; Boyd, E.M.; Curtin, N.; et al. Cancer Genome Project Acquired mutation of the tyrosine kinase JAK2 in human myeloproliferative disorders. Lancet 2005, 365, 1054–1061. [Google Scholar] [CrossRef]
- Liu, F.; Zhao, X.; Perna, F.; Wang, L.; Koppikar, P.; Abdel-Wahab, O.; Harr, M.W.; Levine, R.L.; Xu, H.; Tefferi, A.; et al. JAK2V617F-Mediated Phosphorylation of PRMT5 Downregulates Its Methyltransferase Activity and Promotes Myeloproliferation. Cancer Cell 2011, 19, 283–294. [Google Scholar] [CrossRef] [Green Version]
- Nie, M.; Wang, Y.; Guo, C.; Li, X.; Wang, Y.; Deng, Y.; Yao, B.; Gui, T.; Ma, C.; Liu, M.; et al. CARM1-mediated methylation of protein arginine methyltransferase 5 represses human γ-globin gene expression in erythroleukemia cells. J. Biol. Chem. 2018, 293, 17454–17463. [Google Scholar] [CrossRef] [Green Version]
- Bao, X.; Siprashvili, Z.; Zarnegar, B.J.; Shenoy, R.M.; Rios, E.J.; Nady, N.; Qu, K.; Mah, A.; Webster, D.E.; Rubin, A.J.; et al. CSNK1a1 Regulates PRMT1 to Maintain the Progenitor State in Self-Renewing Somatic Tissue. Dev. Cell 2017, 43, 227–239.e5. [Google Scholar] [CrossRef] [Green Version]
- Chang, N.C.; Sincennes, M.-C.; Chevalier, F.P.; Brun, C.E.; Lacaria, M.; Segalés, J.; Muñoz-Cánoves, P.; Ming, H.; Rudnicki, M.A. The Dystrophin Glycoprotein Complex Regulates the Epigenetic Activation of Muscle Stem Cell Commitment. Cell Stem Cell 2018, 22, 755–768.e6. [Google Scholar] [CrossRef] [Green Version]
- Tang, J.; Kao, P.N.; Herschman, H.R. Protein-arginine methyltransferase I, the predominant protein-arginine methyltransferase in cells, interacts with and is regulated by interleukin enhancer-binding factor 3. J. Biol. Chem. 2000, 275, 19866–19876. [Google Scholar] [CrossRef] [Green Version]
- Xu, J.; Wang, A.H.; Oses-Prieto, J.; Makhijani, K.; Katsuno, Y.; Pei, M.; Yan, L.; Zheng, Y.G.; Burlingame, A.; Brückner, K.; et al. Arginine methylation initiates BMP-induced Smad signaling. Mol. Cell 2013. [Google Scholar] [CrossRef] [Green Version]
- Katsuno, Y.; Qin, J.; Oses-Prieto, J.; Wang, H.; Jackson-Weaver, O.; Zhang, T.; Lamouille, S.; Wu, J.; Burlingame, A.; Xu, J.; et al. Arginine methylation of SMAD7 by PRMT1 in TGF-β–induced epithelial–mesenchymal transition and epithelial stem-cell generation. J. Biol. Chem. 2018, 293, 13059–13072. [Google Scholar] [CrossRef] [Green Version]
- Zhang, T.; Wu, J.; Ungvijanpunya, N.; Jackson-Weaver, O.; Gou, Y.; Feng, J.; Ho, T.V.; Shen, Y.; Liu, J.; Richard, S.; et al. Smad6 Methylation Represses NFκB Activation and Periodontal Inflammation. J. Dent. Res. 2018. [Google Scholar] [CrossRef]
- Hsu, J.-M.; Chen, C.-T.; Chou, C.-K.; Kuo, H.-P.; Li, L.-Y.; Lin, C.-Y.; Lee, H.-J.; Wang, Y.-N.; Liu, M.; Liao, H.-W.; et al. Crosstalk between Arg 1175 methylation and Tyr 1173 phosphorylation negatively modulates EGFR-mediated ERK activation. Nat. Cell Biol. 2011, 13, 174–181. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Andreu-Perez, P.; Esteve-Puig, R.; de Torre-Minguela, C.; Lopez-Fauqued, M.; Bech-Serra, J.J.; Tenbaum, S.; Garcia-Trevijano, E.R.; Canals, F.; Merlino, G.; Avila, M.A.; et al. Protein Arginine Methyltransferase 5 Regulates ERK1/2 Signal Transduction Amplitude and Cell Fate Through CRAF. Sci. Signal. 2011, 4, ra58. [Google Scholar] [CrossRef] [PubMed]
- Huang, S.; Litt, M.; Felsenfeld, G. Methylation of histone H4 by arginine methyltransferase PRMT1 is essential in vivo for many subsequent histone modifications. Genes Dev. 2005. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Bedford, M.T.; Clarke, S.G. Protein Arginine Methylation in Mammals: Who, What, and Why. Mol. Cell 2009, 33, 1–13. [Google Scholar] [CrossRef] [Green Version]
- Zhao, Q.; Rank, G.; Tan, Y.T.; Li, H.; Moritz, R.L.; Simpson, R.J.; Cerruti, L.; Curtis, D.J.; Patel, D.J.; Allis, C.D.; et al. PRMT5-mediated methylation of histone H4R3 recruits DNMT3A, coupling histone and DNA methylation in gene silencing. Nat. Struct. Mol. Biol. 2009, 16, 304–311. [Google Scholar] [CrossRef] [Green Version]
- Migliori, V.; Müller, J.; Phalke, S.; Low, D.; Bezzi, M.; Mok, W.C.; Sahu, S.K.; Gunaratne, J.; Capasso, P.; Bassi, C.; et al. Symmetric dimethylation of H3R2 is a newly identified histone mark that supports euchromatin maintenance. Nat. Struct. Mol. Biol. 2012, 19, 136–145. [Google Scholar] [CrossRef]
- Morris, J.P.; Wang, S.C.; Hebrok, M. KRAS, Hedgehog, Wnt and the twisted developmental biology of pancreatic ductal adenocarcinoma. Nat. Rev. Cancer 2010, 10, 683–695. [Google Scholar] [CrossRef]
- Nolan-Stevaux, O.; Lau, J.; Truitt, M.L.; Chu, G.C.; Hebrok, M.; Fernández-Zapico, M.E.; Hanahan, D. GLI1 is regulated through Smoothened-independent mechanisms in neoplastic pancreatic ducts and mediates PDAC cell survival and transformation. Genes Dev. 2009, 23, 24–36. [Google Scholar] [CrossRef] [Green Version]
- Wang, Y.; Hsu, J.M.; Kang, Y.; Wei, Y.; Lee, P.C.; Chang, S.J.; Hsu, Y.H.; Hsu, J.L.; Wang, H.L.; Chang, W.C.; et al. Oncogenic functions of Gli1 in pancreatic adenocarcinoma are supported by its PRMT1-mediated methylation. Cancer Res. 2016, 76, 7049–7058. [Google Scholar] [CrossRef] [Green Version]
- Wang, X.-P.; Xu, X.-F.; Guo, C.-Y.; Liu, J.; Yang, W.-J.; Xia, Y.-J.; Xu, L.; Yu, Y.-C. Gli1 maintains cell survival by up-regulating IGFBP6 and Bcl-2 through promoter regions in parallel manner in pancreatic cancer cells. J. Carcinog. 2009, 8, 13. [Google Scholar] [CrossRef]
- Abe, Y.; Suzuki, Y.; Kawamura, K.; Tanaka, N. MEP50/PRMT5-mediated methylation activates GLI1 in Hedgehog signalling through inhibition of ubiquitination by the ITCH/NUMB complex. Commun. Biol. 2019, 2, 23. [Google Scholar] [CrossRef] [Green Version]
- Di Marcotullio, L.; Ferretti, E.; Greco, A.; De Smaele, E.; Po, A.; Sico, M.A.; Alimandi, M.; Giannini, G.; Maroder, M.; Screpanti, I.; et al. Numb is a suppressor of Hedgehog signalling and targets Gli1 for Itch-dependent ubiquitination. Nat. Cell Biol. 2006, 8, 1415–1423. [Google Scholar] [CrossRef]
- Di Marcotullio, L.; Greco, A.; Mazzà, D.; Canettieri, G.; Pietrosanti, L.; Infante, P.; Coni, S.; Moretti, M.; De Smaele, E.; Ferretti, E.; et al. Numb activates the E3 ligase Itch to control Gli1 function through a novel degradation signal. Oncogene 2011, 30, 65–76. [Google Scholar] [CrossRef] [Green Version]
- Berman, D.M.; Karhadkar, S.S.; Maitra, A.; Montes De Oca, R.; Gerstenblith, M.R.; Briggs, K.; Parker, A.R.; Shimada, Y.; Eshleman, J.R.; Watkins, D.N.; et al. Widespread requirement for Hedgehog ligand stimulation in growth of digestive tract tumours. Nature 2003, 425, 846–851. [Google Scholar] [CrossRef]
- Watkins, D.N.; Berman, D.M.; Burkholder, S.G.; Wang, B.; Beachy, P.A.; Baylin, S.B. Hedgehog signalling within airway epithelial progenitors and in small-cell lung cancer. Nature 2003, 422, 313–317. [Google Scholar] [CrossRef]
- Taipale, J.; Chen, J.K.; Cooper, M.K.; Wang, B.; Mann, R.K.; Milenkovic, L.; Scott, M.P.; Beachy, P.A. Effects of oncogenic mutations in Smoothened and Patched can be reversed by cyclopamine. Nature 2000, 406, 1005–1009. [Google Scholar] [CrossRef]
- Oro, A.E.; Higgins, K.M.; Hu, Z.; Bonifas, J.M.; Epstein, E.H.; Scott, M.P. Basal cell carcinomas in mice overexpressing sonic hedgehog. Science 1997, 276, 817–821. [Google Scholar] [CrossRef] [Green Version]
- Liu, S.; Dontu, G.; Mantle, I.D.; Patel, S.; Ahn, N.S.; Jackson, K.W.; Suri, P.; Wicha, M.S. Hedgehog signaling and Bmi-1 regulate self-renewal of normal and malignant human mammary stem cells. Cancer Res. 2006. [Google Scholar] [CrossRef] [Green Version]
- Gurung, B.; Feng, Z.; Iwamoto, D.V.; Thiel, A.; Jin, G.; Fan, C.M.; Ng, J.M.Y.; Curran, T.; Hua, X. Menin epigenetically represses hedgehog signaling in MEN1 tumor syndrome. Cancer Res. 2013, 73, 2650–2658. [Google Scholar] [CrossRef] [Green Version]
- Chandrasekharappa, S.C.; Guru, S.C.; Manickam, P.; Olufemi, S.E.; Collins, F.S.; Emmert-Buck, M.R.; Debelenko, L.V.; Zhuang, Z.; Lubensky, I.A.; Liotta, L.A.; et al. Positional cloning of the gene for multiple endocrine neoplasia-type 1. Science 1997, 276, 404–407. [Google Scholar] [CrossRef]
- Milne, T.A.; Hughes, C.M.; Lloyd, R.; Yang, Z.; Rozenblatt-Rosen, O.; Dou, Y.; Schnepp, R.W.; Krankel, C.; LiVolsi, V.A.; Gibbs, D.; et al. Menin and MLL cooperatively regulate expression of cyclin-dependent kinase inhibitors. Proc. Natl. Acad. Sci. USA 2005. [Google Scholar] [CrossRef] [Green Version]
- Karnik, S.K.; Hughes, C.M.; Gu, X.; Rozenblatt-Rosen, O.; McLean, G.W.; Xiong, Y.; Meyerson, M.; Kim, S.K. Menin regulates pancreatic islet growth by promoting histone methylation and expression of genes encoding p27Kip1 and p18INK4c. Proc. Natl. Acad. Sci. USA 2005. [Google Scholar] [CrossRef] [Green Version]
- Marx, S.J. Molecular genetics of multiple endocrine neoplasia types 1 and 2. Nat. Rev. Cancer 2005, 5, 367–375. [Google Scholar] [CrossRef]
- Vuong, T.A.; Jeong, H.-J.; Lee, H.-J.; Kim, B.-G.; Leem, Y.-E.; Cho, H.; Kang, J.-S. PRMT7 methylates and suppresses GLI2 binding to SUFU thereby promoting its activation. Cell Death Differ. 2020, 27, 15–28. [Google Scholar] [CrossRef]
- Blanc, R.S.; Vogel, G.; Chen, T.; Crist, C.; Richard, S. PRMT7 Preserves Satellite Cell Regenerative Capacity. Cell Rep. 2016, 14, 1528–1539. [Google Scholar] [CrossRef] [Green Version]
- Bishop, C.L.; Bergin, A.M.H.; Fessart, D.; Borgdorff, V.; Hatzimasoura, E.; Garbe, J.C.; Stampfer, M.R.; Koh, J.; Beach, D.H. Primary cilium-dependent and -independent hedgehog signaling inhibits p16INK4A. Mol. Cell 2010, 40, 533–547. [Google Scholar] [CrossRef] [Green Version]
- Dunaeva, M.; Michelson, P.; Kogerman, P.; Toftgard, R. Characterization of the physical interaction of Gli proteins with SUFU proteins. J. Biol. Chem. 2003. [Google Scholar] [CrossRef] [Green Version]
- Wu, Q.; Bruce, A.W.; Jedrusik, A.; Ellis, P.D.; Andrews, R.M.; Langford, C.F.; Glover, D.M.; Zernicka-Goetz, M. CARM1 is required in embryonic stem cells to maintain pluripotency and resist differentiation. Stem Cells 2009. [Google Scholar] [CrossRef]
- Tee, W.-W.; Pardo, M.; Theunissen, T.W.; Yu, L.; Choudhary, J.S.; Hajkova, P.; Surani, M.A. Prmt5 is essential for early mouse development and acts in the cytoplasm to maintain ES cell pluripotency. Genes Dev. 2010, 24, 2772–2777. [Google Scholar] [CrossRef] [Green Version]
- Chiang, K.; Zielinska, A.E.; Shaaban, A.M.; Sanchez-Bailon, M.P.; Jarrold, J.; Clarke, T.L.; Zhang, J.; Francis, A.; Jones, L.J.; Smith, S.; et al. PRMT5 Is a Critical Regulator of Breast Cancer Stem Cell Function via Histone Methylation and FOXP1 Expression. Cell Rep. 2017, 21, 3498–3513. [Google Scholar] [CrossRef] [Green Version]
- Lee, S.-H.; Chen, T.-Y.; Dhar, S.S.; Gu, B.; Chen, K.; Kim, Y.Z.; Li, W.; Lee, M.G. A feedback loop comprising PRMT7 and miR-24-2 interplays with Oct4, Nanog, Klf4 and c-Myc to regulate stemness. Nucleic Acids Res. 2016, 44, 10603–10618. [Google Scholar] [CrossRef]
- Chen, T.Y.; Lee, S.H.; Dhar, S.S.; Lee, M.G. Protein arginine methyltransferase 7-mediated microRNA-221 repression maintains Oct4, Nanog, and Sox2 levels in mouse embryonic stem cells. J. Biol. Chem. 2018, 293, 3925–3936. [Google Scholar] [CrossRef] [Green Version]
- Takahashi, K.; Yamanaka, S. Induction of Pluripotent Stem Cells from Mouse Embryonic and Adult Fibroblast Cultures by Defined Factors. Cell 2006, 126, 663–676. [Google Scholar] [CrossRef] [Green Version]
- Nagamatsu, G.; Kosaka, T.; Kawasumi, M.; Kinoshita, T.; Takubo, K.; Akiyama, H.; Sudo, T.; Kobayashi, T.; Oya, M.; Suda, T. A germ cell-specific gene, Prmt5, works in somatic cell reprogramming. J. Biol. Chem. 2011, 286, 10641–10648. [Google Scholar] [CrossRef] [Green Version]
- Chu, Z.; Niu, B.; Zhu, H.; He, X.; Bai, C.; Li, G.; Hua, J. PRMT5 enhances generation of induced pluripotent stem cells from dairy goat embryonic fibroblasts via down-regulation of p53. Cell Prolif. 2015, 48, 29–38. [Google Scholar] [CrossRef]
- Wang, B.; Pfeiffer, M.J.; Drexler, H.C.A.; Fuellen, G.; Boiani, M. Proteomic analysis of mouse oocytes identifies PRMT7 as a reprogramming factor that replaces SOX2 in the induction of pluripotent stem cells. J. Proteome Res. 2016, 15, 2407–2421. [Google Scholar] [CrossRef] [Green Version]
- Moon, J.H.; Heo, J.S.; Kim, J.S.; Jun, E.K.; Lee, J.H.; Kim, A.; Kim, J.; Whang, K.Y.; Kang, Y.K.; Yeo, S.; et al. Reprogramming fibroblasts into induced pluripotent stem cells with Bmi1. Cell Res. 2011, 21, 1305–1315. [Google Scholar] [CrossRef] [Green Version]
- Shia, W.-J.; Okumura, A.J.; Yan, M.; Sarkeshik, A.; Lo, M.-C.; Matsuura, S.; Komeno, Y.; Zhao, X.; Nimer, S.D.; Yates, J.R.; et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood 2012, 119, 4953–4962. [Google Scholar] [CrossRef] [Green Version]
- Jin, Y.; Zhou, J.; Xu, F.; Jin, B.; Cui, L.; Wang, Y.; Du, X.; Li, J.; Li, P.; Ren, R.; et al. Targeting methyltransferase PRMT5 eliminates leukemia stem cells in chronic myelogenous leukemia. J. Clin. Investig. 2016, 126, 3961–3980. [Google Scholar] [CrossRef] [Green Version]
- Mani, S.A.; Guo, W.; Liao, M.J.; Eaton, E.N.; Ayyanan, A.; Zhou, A.Y.; Brooks, M.; Reinhard, F.; Zhang, C.C.; Shipitsin, M.; et al. The Epithelial-Mesenchymal Transition Generates Cells with Properties of Stem Cells. Cell 2008. [Google Scholar] [CrossRef] [Green Version]
- Chen, H.; Lorton, B.; Gupta, V.; Shechter, D. A TGFβ-PRMT5-MEP50 axis regulates cancer cell invasion through histone H3 and H4 arginine methylation coupled transcriptional activation and repression. Oncogene 2017. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Chen, X.; Chen, R.X.; Wei, W.S.; Li, Y.H.; Feng, Z.H.; Tan, L.; Chen, J.W.; Yuan, G.J.; Chen, S.L.; Guo, S.J.; et al. PRMT5 circular RNA promotes metastasis of urothelial carcinoma of the bladder through sponging miR-30c to induce epithelial–mesenchymal transition. Clin. Cancer Res. 2018. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Zbinden, M.; Duquet, A.; Lorente-Trigos, A.; Ngwabyt, S.-N.; Borges, I.; Ruiz i Altaba, A. NANOG regulates glioma stem cells and is essential in vivo acting in a cross-functional network with GLI1 and p53. EMBO J. 2010, 29, 2659–2674. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Leung, C.; Lingbeek, M.; Shakhova, O.; Liu, J.; Tanger, E.; Saremaslani, P.; Van Lohuizen, M.; Marino, S. Bmi1 is essential for cerebellar development and is overexpressed in human medulloblastomas. Transl. Res. 2004, 70, 337–341. [Google Scholar] [CrossRef]
- Bora-Singhal, N.; Perumal, D.; Nguyen, J.; Chellappan, S. Gli1-Mediated Regulation of Sox2 Facilitates Self-Renewal of Stem-Like Cells and Confers Resistance to EGFR Inhibitors in Non–Small Cell Lung Cancer. Neoplasia 2015, 17, 538–551. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Li, X.; Deng, W.; Nail, C.D.; Bailey, S.K.; Kraus, M.H.; Ruppert, J.M.; Lobo-Ruppert, S.M. Snail induction is an early response to Gli1 that determines the efficiency of epithelial transformation. Oncogene 2006, 25, 609–621. [Google Scholar] [CrossRef] [Green Version]
- Alexaki, V.I.; Javelaud, D.; Van Kempen, L.C.L.; Mohammad, K.S.; Dennler, S.; Luciani, F.; Hoek, K.S.; Jurez, P.; Goydos, J.S.; Fournier, P.J.; et al. GLI2-mediated melanoma invasion and metastasis. J. Natl. Cancer Inst. 2010, 102, 1148–1159. [Google Scholar] [CrossRef] [Green Version]
- Johnson, R.W.; Nguyen, M.P.; Padalecki, S.S.; Grubbs, B.G.; Merkel, A.R.; Oyajobi, B.O.; Matrisian, L.M.; Mundy, G.R.; Sterling, J.A. TGF-β promotion of Gli2-induced expression of parathyroid hormone-related protein, an important osteolytic factor in bone metastasis, is independent of canonical Hedgehog signaling. Cancer Res. 2011. [Google Scholar] [CrossRef] [Green Version]
- Suvà, M.L.; Rheinbay, E.; Gillespie, S.M.; Patel, A.P.; Wakimoto, H.; Rabkin, S.D.; Riggi, N.; Chi, A.S.; Cahill, D.P.; Nahed, B.V.; et al. Reconstructing and reprogramming the tumor-propagating potential of glioblastoma stem-like cells. Cell 2014. [Google Scholar] [CrossRef] [Green Version]
- Pan, X.Y.; Tsai, M.H.; Wuputra, K.; Ku, C.C.; Lin, W.H.; Lin, Y.C.; Kishikawa, S.; Noguchi, M.; Saito, S.; Lin, C.S.; et al. Application of Cancer Cell Reprogramming Technology to Human Cancer Research. Anticancer Res. 2017, 37, 3367–3377. [Google Scholar] [CrossRef] [Green Version]
- Robarge, K.D.; Brunton, S.A.; Castanedo, G.M.; Cui, Y.; Dina, M.S.; Goldsmith, R.; Gould, S.E.; Guichert, O.; Gunzner, J.L.; Halladay, J.; et al. GDC-0449-A potent inhibitor of the hedgehog pathway. Bioorg. Med. Chem. Lett. 2009, 19, 5576–5581. [Google Scholar] [CrossRef]
- Pan, S.; Wu, X.; Jiang, J.; Gao, W.; Wan, Y.; Cheng, D.; Han, D.; Liu, J.; Englund, N.P.; Wang, Y.; et al. Discovery of NVP-LDE225, a potent and selective smoothened antagonist. ACS Med. Chem. Lett. 2010, 1, 130–134. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Yauch, R.L.; Dijkgraaf, G.J.P.; Alicke, B.; Januario, T.; Ahn, C.P.; Holcomb, T.; Pujara, K.; Stinson, J.; Callahan, C.A.; Tang, T.; et al. Smoothened Mutation Confers Resistance to a Hedgehog Pathway Inhibitor in Medulloblastoma. Science 2009, 326, 572–574. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Dijkgraaf, G.J.P.; Alicke, B.; Weinmann, L.; Januario, T.; West, K.; Modrusan, Z.; Burdick, D.; Goldsmith, R.; Robarge, K.; Sutherlin, D.; et al. Small molecule inhibition of GDC-0449 refractory smoothened mutants and downstream mechanisms of drug resistance. Cancer Res. 2011, 71, 435–444. [Google Scholar] [CrossRef] [Green Version]
- Atwood, S.X.; Sarin, K.Y.; Whitson, R.J.; Li, J.R.; Kim, G.; Rezaee, M.; Ally, M.S.; Kim, J.; Yao, C.; Chang, A.L.S.; et al. Smoothened Variants Explain the Majority of Drug Resistance in Basal Cell Carcinoma. Cancer Cell 2015, 27, 342–353. [Google Scholar] [CrossRef] [Green Version]
- Abe, Y.; Tanaka, N. The Hedgehog Signaling Networks in Lung Cancer: The Mechanisms and Roles in Tumor Progression and Implications for Cancer Therapy. Biomed Res. Int. 2016, 2016, 7969286. [Google Scholar] [CrossRef]
- Lauth, M.; Bergström, Å. Inhibition of GLI-mediated transcription and tumor cell growth by small-molecule antagonists. Proc. Natl. Acad. Sci. USA 2007, 104, 8455–8460. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kim, J.; Lee, J.J.; Kim, J.; Gardner, D.; Beachy, P.A. Arsenic antagonizes the Hedgehog pathway by preventing ciliary accumulation and reducing stability of the Gli2 transcriptional effector. Proc. Natl. Acad. Sci. USA 2010, 107, 13432–13437. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Beauchamp, E.M.; Ringer, L.; Bulut, G.; Sajwan, K.P.; Hall, M.D.; Lee, Y.-C.; Peaceman, D.; Özdemirli, M.; Rodriguez, O.; Macdonald, T.J.; et al. Arsenic trioxide inhibits human cancer cell growth and tumor development in mice by blocking Hedgehog/GLI pathway. J. Clin. Investig. 2011, 121, 148–160. [Google Scholar] [CrossRef] [Green Version]
- Hu, H.; Qian, K.; Ho, M.-C.; Zheng, Y.G. Small Molecule Inhibitors of Protein Arginine Methyltransferases. Expert Opin. Investig. Drugs 2016, 25, 335–358. [Google Scholar] [CrossRef] [Green Version]
- Cheung, N.; Fung, T.K.; Zeisig, B.B.; Holmes, K.; Rane, J.K.; Mowen, K.A.; Finn, M.G.; Lenhard, B.; Chan, L.C.; So, C.W.E. Targeting Aberrant Epigenetic Networks Mediated by PRMT1 and KDM4C in Acute Myeloid Leukemia. Cancer Cell 2016. [Google Scholar] [CrossRef] [PubMed] [Green Version]
- Kaushik, S.; Liu, F.; Veazey, K.J.; Gao, G.; Das, P.; Neves, L.F.; Lin, K.; Zhong, Y.; Lu, Y.; Giuliani, V.; et al. Genetic deletion or small-molecule inhibition of the arginine methyltransferase PRMT5 exhibit anti-tumoral activity in mouse models of MLL-rearranged AML. Leukemia 2018, 32, 499–509. [Google Scholar] [CrossRef] [PubMed]
- Koh, C.M.; Bezzi, M.; Low, D.H.P.; Ang, W.X.; Teo, S.X.; Gay, F.P.H.; Al-Haddawi, M.; Tan, S.Y.; Osato, M.; Sabò, A.; et al. MYC regulates the core pre-mRNA splicing machinery as an essential step in lymphomagenesis. Nature 2015, 523, 96–100. [Google Scholar] [CrossRef] [PubMed]
- Ibrahim, R.; Matsubara, D.; Osman, W.; Morikawa, T.; Goto, A.; Morita, S.; Ishikawa, S.; Aburatani, H.; Takai, D.; Nakajima, J.; et al. Expression of PRMT5 in lung adenocarcinoma and its significance in epithelial-mesenchymal transition. Hum. Pathol. 2014, 45, 1397–1405. [Google Scholar] [CrossRef] [PubMed]
- Sheng, X.; Wang, Z. Protein arginine methyltransferase 5 regulates multiple signaling pathways to promote lung cancer cell proliferation. BMC Cancer 2016, 16, 567. [Google Scholar] [CrossRef] [Green Version]
- Li, X.; Wang, C.; Jiang, H.; Luo, C. A patent review of arginine methyltransferase inhibitors (2010–2018). Expert Opin. Ther. Pat. 2019, 29, 97–114. [Google Scholar] [CrossRef]

© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Abe, Y.; Tanaka, N. Fine-Tuning of GLI Activity through Arginine Methylation: Its Mechanisms and Function. Cells 2020, 9, 1973. https://doi.org/10.3390/cells9091973
Abe Y, Tanaka N. Fine-Tuning of GLI Activity through Arginine Methylation: Its Mechanisms and Function. Cells. 2020; 9(9):1973. https://doi.org/10.3390/cells9091973
Chicago/Turabian StyleAbe, Yoshinori, and Nobuyuki Tanaka. 2020. "Fine-Tuning of GLI Activity through Arginine Methylation: Its Mechanisms and Function" Cells 9, no. 9: 1973. https://doi.org/10.3390/cells9091973
APA StyleAbe, Y., & Tanaka, N. (2020). Fine-Tuning of GLI Activity through Arginine Methylation: Its Mechanisms and Function. Cells, 9(9), 1973. https://doi.org/10.3390/cells9091973