Conventional and Non-Conventional Roles of Non-Muscle Myosin II-Actin in Neuronal Development and Degeneration



Abstract
1. Introduction
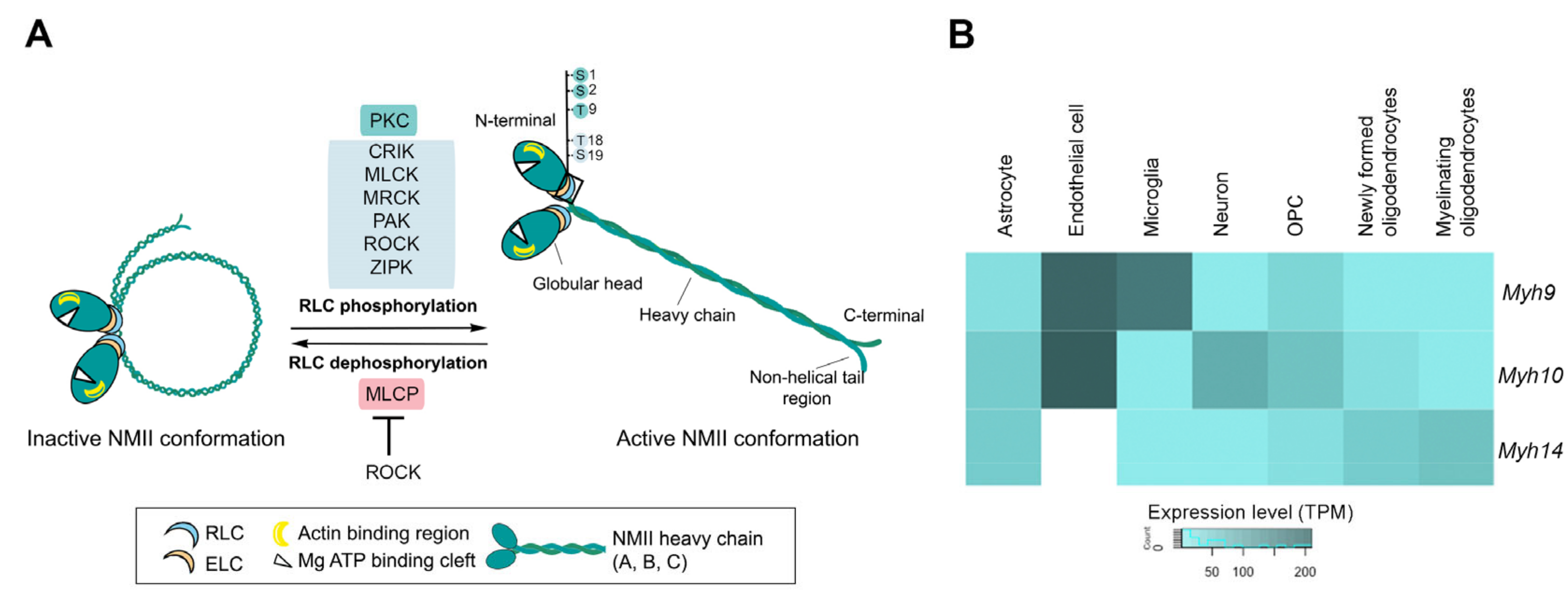
2. Non-Muscle Myosin II in the Nervous System
The Cerebral Expression and Cellular Localization of NMIIs
3. Functions of Non-Muscle Myosin II in the Nervous System
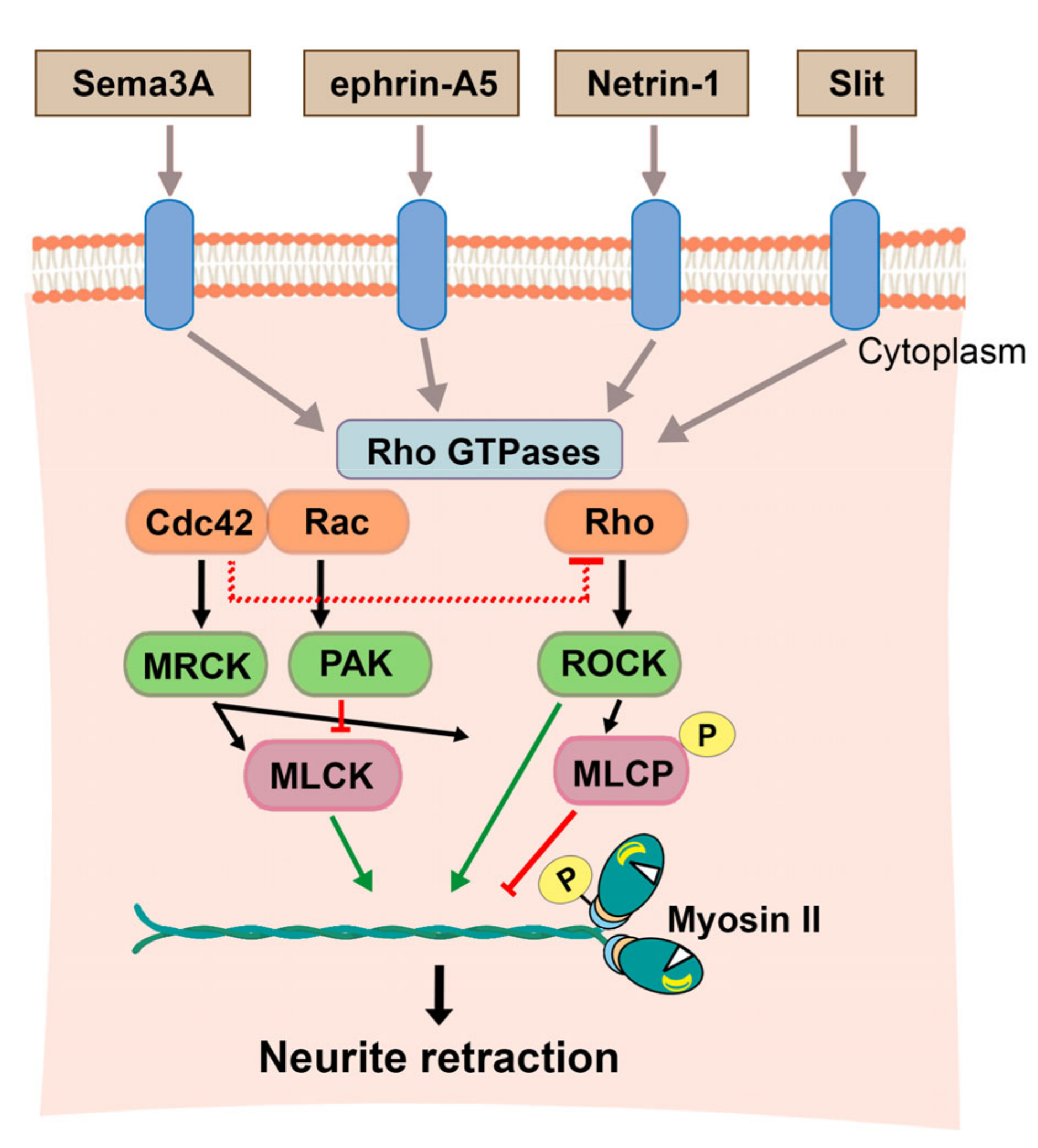
3.1. Non-Muscle Myosin II in Neuronal Polarization
3.1.1. The Role of NMII During Brain Development
3.1.2. The Function of NMII in Neuronal Polarization
3.2. Non-Muscle Myosin II in Neuronal Migration
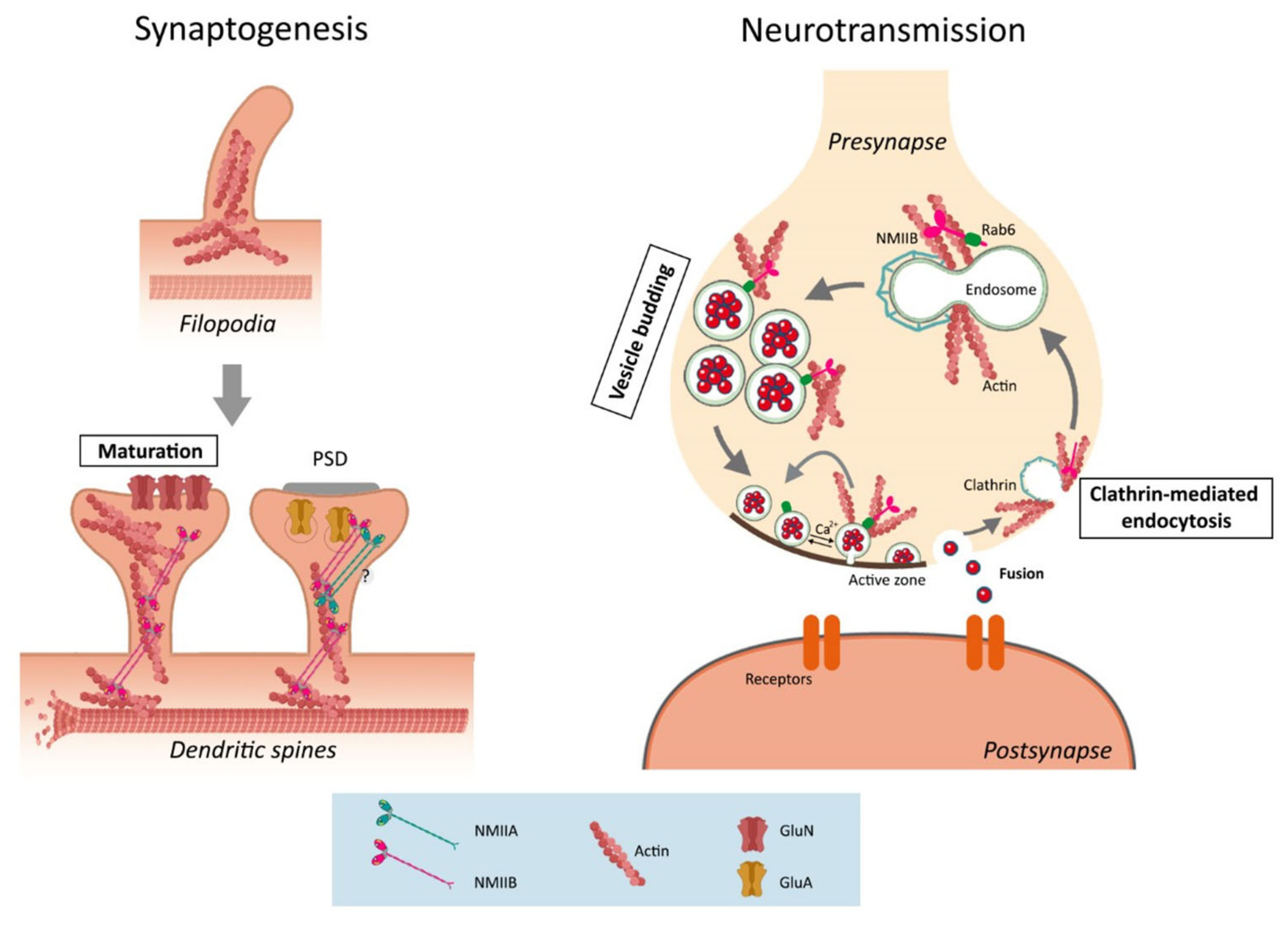
3.3. The Synaptic Function of Non-Muscle Myosin II
3.3.1. The Role of F-Actin in Synapse Morphology
3.3.2. NMII in Synapse Formation and Function
3.3.3. Roles of NMII in Synaptic Plasticity and Memory
4. Non-Muscle Myosin II in Neurological and Neurodegenerative Diseases
4.1. NMIIs in Neurodevelopmental Diseases
4.2. NMIIs in Neurodegenerative Diseases
4.3. NMIIs in Nervous System Injuries
4.4. NMII in Glial Cells and Inflammation
5. Concluding Remarks
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Sebé-Pedrós, A.; Grau-Bove, X.; Richards, T.A.; Ruiz-Trillo, I. Evolution and classification of myosins, a paneukaryotic whole-genome approach. Genome Biol. Evol. 2014, 6, 290–305. [Google Scholar] [CrossRef]
- Trivedi, D.V.; Nag, S.; Spudich, A.; Ruppel, K.M.; Spudich, J.A. The myosin family of mechanoenzymes: From mechanisms to therapeutic approaches. Annu. Rev. Biochem. 2020, 89, 667–693. [Google Scholar] [CrossRef] [PubMed]
- Heissler, S.M.; Manstein, D.J. Nonmuscle myosin-2: Mix and match. Cell. Mol. Life Sci. 2013, 70, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Vicente-Manzanares, M.; Ma, X.; Adelstein, R.S.; Horwitz, A.R. Non-muscle myosin II takes centre stage in cell adhesion and migration. Nat. Rev. Mol. Cell. Biol. 2009, 10, 778–790. [Google Scholar] [CrossRef] [PubMed]
- Newell-Litwa, K.A.; Horwitz, R.; Lamers, M.L. Non-muscle myosin II in disease: Mechanisms and therapeutic opportunities. Dis. Model. Mech. 2015, 8, 1495–1515. [Google Scholar] [CrossRef]
- Heissler, S.M.; Sellers, J.R. Myosin light chains: Teaching old dogs new tricks. Bioarchitecture 2014, 4, 169–188. [Google Scholar] [CrossRef]
- Heissler, S.M.; Sellers, J.R. Various themes of myosin regulation. J. Mol. Biol. 2016, 428, 1927–1946. [Google Scholar] [CrossRef]
- Kneussel, M.; Wagner, W. Myosin motors at neuronal synapses: Drivers of membrane transport and actin dynamics. Nat. Rev. Neurosci. 2013, 14, 233–247. [Google Scholar] [CrossRef]
- Vallee, R.B.; Seale, G.E.; Tsai, J.W. Emerging roles for myosin II and cytoplasmic dynein in migrating neurons and growth cones. Trends Cell Biol. 2009, 19, 347–355. [Google Scholar] [CrossRef]
- Kawamoto, S.; Adelstein, R.S. Chicken nonmuscle myosin heavy chains: Differential expression of two mRNAs and evidence for two different polypeptides. J. Cell Biol. 1991, 112, 915–924. [Google Scholar] [CrossRef]
- Kimura, A.; Nakashima, S.; Uda, T.; Ikeda, H.; Yasuda, S.; Tsuji, T.; Matsumura, S. Heavy-chain isoforms of non-muscle myosin in human tissues. Eur. J. Biochem. 1993, 213, 5–10. [Google Scholar] [CrossRef] [PubMed]
- Takahashi, M.; Kawamoto, S.; Adelstein, R.S. Evidence for inserted sequences in the head region of nonmuscle myosin specific to the nervous system. Cloning of the cDNA encoding the myosin heavy chain-B isoform of vertebrate nonmuscle myosin. J. Biol. Chem. 1992, 267, 17864–17871. [Google Scholar] [PubMed]
- Golomb, E.; Ma, X.; Jana, S.S.; Preston, Y.A.; Kawamoto, S.; Shoham, N.G.; Goldin, E.; Conti, M.A.; Sellers, J.R.; Adelstein, R.S. Identification and characterization of nonmuscle myosin II-C, a new member of the myosin II family. J. Biol. Chem. 2004, 279, 2800–2808. [Google Scholar] [CrossRef] [PubMed]
- Ma, X.; Jana, S.S.; Conti, M.A.; Kawamoto, S.; Claycomb, W.C.; Adelstein, R.S. Ablation of nonmuscle myosin II-B and II-C reveals a role for nonmuscle myosin II in cardiac myocyte karyokinesis. Mol. Biol. Cell 2010, 21, 3952–3962. [Google Scholar] [CrossRef]
- Itoh, K.; Adelstein, R.S. Neuronal cell expression of inserted isoforms of vertebrate nonmuscle myosin heavy chain II-B. J. Biol. Chem. 1995, 270, 14533–14540. [Google Scholar] [CrossRef]
- Zhang, Y.; Chen, K.; Sloan, S.A.; Bennett, M.L.; Scholze, A.R.; O’Keeffe, S.; Phatnani, H.P.; Guarnieri, P.; Caneda, C.; Ruderisch, N.; et al. An RNA-sequencing transcriptome and splicing database of glia, neurons, and vascular cells of the cerebral cortex. J. Neurosci. 2014, 34, 11929–11947. [Google Scholar] [CrossRef]
- Lv, Y.; Liu, W.; Ruan, Z.; Xu, Z.; Fu, L. Myosin IIA regulated tight junction in oxygen glucose-deprived brain endothelial cells via Activation of TLR4/PI3K/Akt/JNK1/2/14-3-3epsilon/NF-kappaB/MMP9 signal transduction pathway. Cell. Mol. Neurobiol. 2019, 39, 301–319. [Google Scholar] [CrossRef]
- Murakami, N.; Elzinga, M. Immunohistochemical studies on the distribution of cellular myosin II isoforms in brain and aorta. Cell Motil. Cytoskelet. 1992, 22, 281–295. [Google Scholar] [CrossRef]
- Janßen, S.; Gudi, V.; Prajeeth, C.K.; Singh, V.; Stahl, K.; Heckers, S.; Skripuletz, T.; Pul, R.; Trebst, C.; Tsiavaliaris, G.; et al. A pivotal role of nonmuscle myosin II during microglial activation. Exp. Neurol. 2014, 261, 666–676. [Google Scholar] [CrossRef]
- Wang, H.; Rusielewicz, T.; Tewari, A.; Leitman, E.M.; Einheber, S.; Melendez-Vasquez, C.V. Myosin II is a negative regulator of oligodendrocyte morphological differentiation. J. Neurosci. Res. 2012, 90, 1547–1556. [Google Scholar] [CrossRef]
- Wang, H.; Tewari, A.; Einheber, S.; Salzer, J.L.; Melendez-Vasquez, C.V. Myosin II has distinct functions in PNS and CNS myelin sheath formation. J. Cell Biol. 2008, 182, 1171–1184. [Google Scholar] [CrossRef] [PubMed]
- Ronen, D.; Ravid, S. Myosin II tailpiece determines its paracrystal structure, filament assembly properties, and cellular localization. J. Biol. Chem. 2009, 284, 24948–24957. [Google Scholar] [CrossRef] [PubMed]
- Sandquist, J.C.; Means, A.R. The C-terminal tail region of nonmuscle myosin II directs isoform-specific distribution in migrating cells. Mol. Biol. Cell 2008, 19, 5156–5167. [Google Scholar] [CrossRef] [PubMed]
- Kolega, J. Cytoplasmic dynamics of myosin IIA and IIB: Spatial ’sorting’ of isoforms in locomoting cells. J. Cell Sci. 1998, 111, 2085–2095. [Google Scholar] [PubMed]
- Cheng, T.P.; Murakami, N.; Elzinga, M. Localization of myosin IIB at the leading edge of growth cones from rat dorsal root ganglionic cells. FEBS Lett. 1992, 311, 91–94. [Google Scholar] [CrossRef]
- Miller, M.; Bower, E.; Levitt, P.; Li, D.; Chantler, P.D. Myosin II distribution in neurons is consistent with a role in growth cone motility but not synaptic vesicle mobilization. Neuron 1992, 8, 25–44. [Google Scholar] [CrossRef]
- Rochlin, M.W.; Itoh, K.; Adelstein, R.S.; Bridgman, P.C. Localization of myosin II A and B isoforms in cultured neurons. J. Cell Sci. 1995, 108, 3661–3670. [Google Scholar]
- Hur, E.M.; Yang, I.H.; Kim, D.H.; Byun, J.; Saijilafu Xu, W.L.; Nicovich, P.R.; Cheong, R.; Levchenko, A.; Thakor, N.; Zhou, F.Q. Engineering neuronal growth cones to promote axon regeneration over inhibitory molecules. Proc. Natl. Acad. Sci. USA 2011, 108, 5057–5062. [Google Scholar] [CrossRef]
- Medeiros, N.A.; Burnette, D.T.; Forscher, P. Myosin II functions in actin-bundle turnover in neuronal growth cones. Nat. Cell Biol. 2006, 8, 215–226. [Google Scholar] [CrossRef]
- Burnette, D.T.; Ji, L.; Schaefer, A.W.; Medeiros, N.A.; Danuser, G.; Forscher, P. Myosin II activity facilitates microtubule bundling in the neuronal growth cone neck. Dev. Cell 2008, 15, 163–169. [Google Scholar] [CrossRef]
- Straight, A.F.; Cheung, A.; Limouze, J.; Chen, I.; Westwood, N.J.; Sellers, J.R.; Mitchison, T.J. Dissecting temporal and spatial control of cytokinesis with a myosin II Inhibitor. Science 2003, 299, 1743–1747. [Google Scholar] [CrossRef] [PubMed]
- Kovacs, M.; Toth, J.; Hetenyi, C.; Malnasi-Csizmadia, A.; Sellers, J.R. Mechanism of blebbistatin inhibition of myosin II. J. Biol. Chem. 2004, 279, 35557–35563. [Google Scholar] [CrossRef] [PubMed]
- He, M.; Zhang, Z.H.; Guan, C.B.; Xia, D.; Yuan, X.B. Leading tip drives soma translocation via forward F-actin flow during neuronal migration. J. Neurosci. 2010, 30, 10885–10898. [Google Scholar] [CrossRef] [PubMed]
- Solecki, D.J.; Trivedi, N.; Govek, E.E.; Kerekes, R.A.; Gleason, S.S.; Hatten, M.E. Myosin II motors and F-actin dynamics drive the coordinated movement of the centrosome and soma during CNS glial-guided neuronal migration. Neuron 2009, 63, 63–80. [Google Scholar] [CrossRef]
- Jiang, J.; Zhang, Z.H.; Yuan, X.B.; Poo, M.M. Spatiotemporal dynamics of traction forces show three contraction centers in migratory neurons. J. Cell Biol. 2015, 209, 759–774. [Google Scholar] [CrossRef]
- Ryu, J.; Liu, L.; Wong, T.P.; Wu, D.C.; Burette, A.; Weinberg, R.; Wang, Y.T.; Sheng, M. A critical role for myosin IIb in dendritic spine morphology and synaptic function. Neuron 2006, 49, 175–182. [Google Scholar] [CrossRef]
- Korobova, F.; Svitkina, T. Molecular architecture of synaptic actin cytoskeleton in hippocampal neurons reveals a mechanism of dendritic spine morphogenesis. Mol. Biol. Cell 2010, 21, 165–176. [Google Scholar] [CrossRef]
- Berger, S.L.; Leo-Macias, A.; Yuen, S.; Khatri, L.; Pfennig, S.; Zhang, Y.; Agullo-Pascual, E.; Caillol, G.; Zhu, M.S.; Rothenberg, E.; et al. Localized myosin II activity regulates assembly and plasticity of the axon initial segment. Neuron 2018, 97, 555–570.e6. [Google Scholar] [CrossRef]
- Leterrier, C. The Axon Initial Segment: An Updated Viewpoint. J. Neurosci. 2018, 38, 2135–2145. [Google Scholar] [CrossRef]
- Evans, M.D.; Tufo, C.; Dumitrescu, A.S.; Grubb, M.S. Myosin II activity is required for structural plasticity at the axon initial segment. Eur. J. Neurosci. 2017, 46, 1751–1757. [Google Scholar] [CrossRef]
- Buffington, S.A.; Rasband, M.N. The axon initial segment in nervous system disease and injury. Eur. J. Neurosci. 2011, 34, 1609–1619. [Google Scholar] [CrossRef] [PubMed]
- Conti, M.A.; Even-Ram, S.; Liu, C.; Yamada, K.M.; Adelstein, R.S. Defects in cell adhesion and the visceral endoderm following ablation of nonmuscle myosin heavy chain II-A in mice. J. Biol. Chem. 2004, 279, 41263–41266. [Google Scholar] [CrossRef] [PubMed]
- Wang, A.; Ma, X.; Conti, M.A.; Liu, C.; Kawamoto, S.; Adelstein, R.S. Nonmuscle myosin II isoform and domain specificity during early mouse development. Proc. Natl. Acad. Sci. USA 2010, 107, 14645–14650. [Google Scholar] [CrossRef] [PubMed]
- Tullio, A.N.; Accili, D.; Ferrans, V.J.; Yu, Z.X.; Takeda, K.; Grinberg, A.; Westphal, H.; Preston, Y.A.; Adelstein, R.S. Nonmuscle myosin II-B is required for normal development of the mouse heart. Proc. Natl. Acad. Sci. USA 1997, 94, 12407–12412. [Google Scholar] [CrossRef] [PubMed]
- Tullio, A.N.; Bridgman, P.C.; Tresser, N.J.; Chan, C.C.; Conti, M.A.; Adelstein, R.S.; Hara, Y. Structural abnormalities develop in the brain after ablation of the gene encoding nonmuscle myosin II-B heavy chain. J. Comp. Neurol. 2001, 433, 62–74. [Google Scholar] [CrossRef] [PubMed]
- Uren, D.; Hwang, H.K.; Hara, Y.; Takeda, K.; Kawamoto, S.; Tullio, A.N.; Yu, Z.X.; Ferrans, V.J.; Tresser, N.; Grinberg, A.; et al. Gene dosage affects the cardiac and brain phenotype in nonmuscle myosin II-B-depleted mice. J. Clin. Investig. 2000, 105, 663–671. [Google Scholar] [CrossRef]
- Bao, J.; Ma, X.; Liu, C.; Adelstein, R.S. Replacement of nonmuscle myosin II-B with II-A rescues brain but not cardiac defects in mice. J. Biol. Chem. 2007, 282, 22102–22111. [Google Scholar] [CrossRef]
- Ma, X.; Bao, J.; Adelstein, R.S. Loss of cell adhesion causes hydrocephalus in nonmuscle myosin II-B-ablated and mutated mice. Mol. Biol. Cell 2007, 18, 2305–2312. [Google Scholar] [CrossRef]
- Wang, A.; Ma, X.; Conti, M.A.; Adelstein, R.S. Distinct and redundant roles of the non-muscle myosin II isoforms and functional domains. Biochem. Soc. Trans. 2011, 39, 1131–1135. [Google Scholar] [CrossRef]
- Ma, X.; Kawamoto, S.; Hara, Y.; Adelstein, R.S. A point mutation in the motor domain of nonmuscle myosin II-B impairs migration of distinct groups of neurons. Mol. Biol. Cell 2004, 15, 2568–2579. [Google Scholar] [CrossRef]
- Ma, X.; Takeda, K.; Singh, A.; Yu, Z.X.; Zerfas, P.; Blount, A.; Liu, C.; Towbin, J.A.; Schneider, M.D.; Adelstein, R.S.; et al. Conditional ablation of nonmuscle myosin II-B delineates heart defects in adult mice. Circ. Res. 2009, 105, 1102–1109. [Google Scholar] [CrossRef] [PubMed]
- Turney, S.G.; Bridgman, P.C. Laminin stimulates and guides axonal outgrowth via growth cone myosin II activity. Nat. Neurosci. 2005, 8, 717–719. [Google Scholar] [CrossRef] [PubMed]
- Kollins, K.M.; Hu, J.; Bridgman, P.C.; Huang, Y.Q.; Gallo, G. Myosin-II negatively regulates minor process extension and the temporal development of neuronal polarity. Dev. Neurobiol. 2009, 69, 279–298. [Google Scholar] [CrossRef] [PubMed]
- Tamariz, E.; Varela-Echavarria, A. The discovery of the growth cone and its influence on the study of axon guidance. Front. Neuroanat. 2015, 9, 51. [Google Scholar] [CrossRef] [PubMed]
- Brown, J.; Bridgman, P.C. Role of myosin II in axon outgrowth. J. Histochem. Cytochem. 2003, 51, 421–428. [Google Scholar] [CrossRef] [PubMed]
- Lin, C.H.; Espreafico, E.M.; Mooseker, M.S.; Forscher, P. Myosin drives retrograde F-actin flow in neuronal growth cones. Neuron 1996, 16, 769–782. [Google Scholar] [CrossRef]
- Stankiewicz, T.R.; Linseman, D.A. Rho family GTPases: Key players in neuronal development, neuronal survival, and neurodegeneration. Front. Cell. Neurosci. 2014, 8, 314. [Google Scholar] [CrossRef]
- Roland, A.B.; Ricobaraza, A.; Carrel, D.; Jordan, B.M.; Rico, F.; Simon, A.; Humbert-Claude, M.; Ferrier, J.; McFadden, M.H.; Scheuring, S.; et al. Cannabinoid-induced actomyosin contractility shapes neuronal morphology and growth. Elife 2014, 3, e03159. [Google Scholar] [CrossRef]
- Javier-Torrent, M.; Marco, S.; Rocandio, D.; Pons-Vizcarra, M.; Janes, P.W.; Lackmann, M.; Egea, J.; Saura, C.A. Presenilin/γ-secretase-dependent EphA3 processing mediates axon elongation through non-muscle myosin IIA. Elife 2019, 8. [Google Scholar] [CrossRef]
- Costa, A.R.; Pinto-Costa, R.; Sousa, S.C.; Sousa, M.M. The regulation of axon diameter: From axonal circumferential contractility to activity-dependent axon swelling. Front. Mol. Neurosci. 2018, 11, 319. [Google Scholar] [CrossRef]
- Wang, T.; Li, W.; Martin, S.; Papadopulos, A.; Joensuu, M.; Liu, C.; Jiang, A.; Shamsollahi, G.; Amor, R.; Lanoue, V.; et al. Radial contractility of actomyosin rings facilitates axonal trafficking and structural stability. J. Cell Biol. 2020, 219, e201902001. [Google Scholar] [CrossRef] [PubMed]
- Wylie, S.R.; Chantler, P.D. Myosin IIC: A third molecular motor driving neuronal dynamics. Mol. Biol. Cell 2008, 19, 3956–3968. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Wylie, S.R.; Chantler, P.D. Separate but linked functions of conventional myosins modulate adhesion and neurite outgrowth. Nat. Cell Biol. 2001, 3, 88–92. [Google Scholar] [CrossRef] [PubMed]
- Yuan, X.B.; Jin, M.; Xu, X.; Song, Y.Q.; Wu, C.P.; Poo, M.M.; Duan, S. Signalling and crosstalk of Rho GTPases in mediating axon guidance. Nat. Cell Biol. 2003, 5, 38–45. [Google Scholar] [CrossRef] [PubMed]
- Wylie, S.R.; Chantler, P.D. Myosin IIA drives neurite retraction. Mol. Biol. Cell 2003, 14, 4654–4666. [Google Scholar] [CrossRef] [PubMed]
- Kubo, T.; Endo, M.; Hata, K.; Taniguchi, J.; Kitajo, K.; Tomura, S.; Yamaguchi, A.; Mueller, B.K.; Yamashita, T. Myosin IIA is required for neurite outgrowth inhibition produced by repulsive guidance molecule. J. Neurochem. 2008, 105, 113–126. [Google Scholar] [CrossRef]
- Gallo, G. RhoA-kinase coordinates F-actin organization and myosin II activity during semaphorin-3A-induced axon retraction. J. Cell Sci. 2006, 119, 3413–3423. [Google Scholar] [CrossRef]
- Brown, J.A.; Wysolmerski, R.B.; Bridgman, P.C. Dorsal root ganglion neurons react to semaphorin 3A application through a biphasic response that requires multiple myosin II isoforms. Mol. Biol. Cell 2009, 20, 1167–1179. [Google Scholar] [CrossRef][Green Version]
- Roy, A.; Lordier, L.; Mazzi, S.; Chang, Y.; Lapierre, V.; Larghero, J.; Debili, N.; Raslova, H.; Vainchenker, W. Activity of nonmuscle myosin II isoforms determines localization at the cleavage furrow of megakaryocytes. Blood 2016, 128, 3137–3145. [Google Scholar] [CrossRef]
- Murray, A.; Naeem, A.; Barnes, S.H.; Drescher, U.; Guthrie, S. Slit and Netrin-1 guide cranial motor axon pathfinding via Rho-kinase, myosin light chain kinase and myosin II. Neural Dev. 2010, 5, 16. [Google Scholar] [CrossRef]
- Yue, X.; Dreyfus, C.; Kong, T.A.; Zhou, R. A subset of signal transduction pathways is required for hippocampal growth cone collapse induced by ephrin-A5. Dev. Neurobiol. 2008, 68, 1269–1286. [Google Scholar] [CrossRef] [PubMed]
- Wahl, S.; Barth, H.; Ciossek, T.; Aktories, K.; Mueller, B.K. Ephrin-A5 induces collapse of growth cones by activating Rho and Rho kinase. J. Cell Biol. 2000, 149, 263–270. [Google Scholar] [CrossRef] [PubMed]
- Kostic, A.; Sap, J.; Sheetz, M.P. RPTPalpha is required for rigidity-dependent inhibition of extension and differentiation of hippocampal neurons. J. Cell Sci. 2007, 120, 3895–3904. [Google Scholar] [CrossRef] [PubMed]
- Norman, L.L.; Aranda-Espinoza, H. Cortical neuron outgrowth is insensitive to substrate stiffness. Cell. Mol. Bioeng. 2010, 3, 398–414. [Google Scholar] [CrossRef]
- Nichol, R.H.T.; Catlett, T.S.; Onesto, M.M.; Hollender, D.; Gomez, T.M. Environmental elasticity regulates cell-type specific RhoA signaling and neuritogenesis of human neurons. Stem Cell Rep. 2019, 13, 1006–1021. [Google Scholar] [CrossRef] [PubMed]
- Engler, A.J.; Sen, S.; Sweeney, H.L.; Discher, D.E. Matrix elasticity directs stem cell lineage specification. Cell 2006, 126, 677–689. [Google Scholar] [CrossRef] [PubMed]
- Li, Z.; Aizenman, C.D.; Cline, H.T. Regulation of Rho GTPases by crosstalk and neuronal activity in vivo. Neuron 2002, 33, 741–750. [Google Scholar] [CrossRef]
- Ming, G.; Henley, J.; Tessier-Lavigne, M.; Song, H.; Poo, M. Electrical activity modulates growth cone guidance by diffusible factors. Neuron 2001, 29, 441–452. [Google Scholar] [CrossRef]
- Giannone, G.; Dubin-Thaler, B.J.; Dobereiner, H.G.; Kieffer, N.; Bresnick, A.R.; Sheetz, M.P. Periodic lamellipodial contractions correlate with rearward actin waves. Cell 2004, 116, 431–443. [Google Scholar] [CrossRef]
- Peng, H.; Ong, Y.M.; Shah, W.A.; Holland, P.C.; Carbonetto, S. Integrins regulate centrosome integrity and astrocyte polarization following a wound. Dev. Neurobiol. 2013, 73, 333–353. [Google Scholar] [CrossRef]
- Bellion, A.; Baudoin, J.P.; Alvarez, C.; Bornens, M.; Metin, C. Nucleokinesis in tangentially migrating neurons comprises two alternating phases: Forward migration of the Golgi/centrosome associated with centrosome splitting and myosin contraction at the rear. J. Neurosci. 2005, 25, 5691–5699. [Google Scholar] [CrossRef] [PubMed]
- Silva, C.G.; Peyre, E.; Adhikari, M.H.; Tielens, S.; Tanco, S.; Van Damme, P.; Magno, L.; Krusy, N.; Agirman, G.; Magiera, M.M.; et al. Cell-intrinsic control of interneuron migration drives cortical morphogenesis. Cell 2018, 172, 1063–1078.e19. [Google Scholar] [CrossRef] [PubMed]
- Tsai, J.W.; Bremner, K.H.; Vallee, R.B. Dual subcellular roles for LIS1 and dynein in radial neuronal migration in live brain tissue. Nat. Neurosci. 2007, 10, 970–979. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.K.; Umeshima, H.; Kurisu, J.; Kengaku, M. Nesprins and opposing microtubule motors generate a point force that drives directional nuclear motion in migrating neurons. Development 2018, 145, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Kumar, A.; Maitra, A.; Sumit, M.; Ramaswamy, S.; Shivashankar, G.V. Actomyosin contractility rotates the cell nucleus. Sci. Rep. 2014, 4, 3781. [Google Scholar] [CrossRef] [PubMed]
- Petrie, R.J.; Koo, H.; Yamada, K.M. Generation of compartmentalized pressure by a nuclear piston governs cell motility in a 3D matrix. Science 2014, 345, 1062–1065. [Google Scholar] [CrossRef]
- Maniotis, A.J.; Chen, C.S.; Ingber, D.E. Demonstration of mechanical connections between integrins, cytoskeletal filaments, and nucleoplasm that stabilize nuclear structure. Proc. Natl. Acad. Sci. USA 1997, 94, 849–854. [Google Scholar] [CrossRef]
- Kanellos, G.; Zhou, J.; Patel, H.; Ridgway, R.A.; Huels, D.; Gurniak, C.B.; Sandilands, E.; Carragher, N.O.; Sansom, O.J.; Witke, W.; et al. ADF and Cofilin1 control actin stress fibers, nuclear integrity, and cell survival. Cell Rep. 2015, 13, 1949–1964. [Google Scholar] [CrossRef]
- Wiggan, O.; Schroder, B.; Krapf, D.; Bamburg, J.R.; DeLuca, J.G. Cofilin regulates nuclear rchitecture through a Myosin-II dependent mechanotransduction module. Sci. Rep. 2017, 7, 40953. [Google Scholar] [CrossRef]
- Keeling, M.C.; Flores, L.R.; Dodhy, A.H.; Murray, E.R.; Gavara, N. Actomyosin and vimentin cytoskeletal networks regulate nuclear shape, mechanics and chromatin organization. Sci. Rep. 2017, 7, 5219. [Google Scholar] [CrossRef]
- Li, Q.; Sarna, S.K. Nuclear myosin II regulates the assembly of preinitiation complex for ICAM-1 gene transcription. Gastroenterology 2009, 137, 1051–1060.e3. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; He, H.; Li, W.; Phay, J.; Shen, R.; Yu, L.; Hancioglu, B.; de la Chapelle, A. MYH9 binds to lncRNA gene PTCSC2 and regulates FOXE1 in the 9q22 thyroid cancer risk locus. Proc. Natl. Acad. Sci. USA 2017, 114, 474–479. [Google Scholar] [CrossRef] [PubMed]
- Coaxum, S.D.; Tiedeken, J.; Garrett-Mayer, E.; Myers, J.; Rosenzweig, S.A.; Neskey, D.M. The tumor suppressor capability of p53 is dependent on non-muscle myosin IIA function in head and neck cancer. Oncotarget 2017, 8, 22991–23007. [Google Scholar] [CrossRef] [PubMed]
- Bucher, M.; Fanutza, T.; Mikhaylova, M. Cytoskeletal makeup of the synapse: Shaft versus spine. Cytoskeleton 2019. [Google Scholar] [CrossRef]
- Bourne, J.; Harris, K.M. Do thin spines learn to be mushroom spines that remember? Curr. Opin. Neurobiol. 2007, 17, 381–386. [Google Scholar] [CrossRef]
- Caroni, P.; Donato, F.; Muller, D. Structural plasticity upon learning: Regulation and functions. Nat. Rev. Neurosci. 2012, 13, 478–490. [Google Scholar] [CrossRef]
- Cingolani, L.A.; Goda, Y. Actin in action: The interplay between the actin cytoskeleton and synaptic efficacy. Nat. Rev. Neurosci. 2008, 9, 344–356. [Google Scholar] [CrossRef]
- Fu, A.K.; Ip, N.Y. Regulation of postsynaptic signaling in structural synaptic plasticity. Curr. Opin. Neurobiol. 2017, 45, 148–155. [Google Scholar] [CrossRef]
- Krucker, T.; Siggins, G.R.; Halpain, S. Dynamic actin filaments are required for stable long-term potentiation (LTP) in area CA1 of the hippocampus. Proc. Natl. Acad. Sci. USA 2000, 97, 6856–6861. [Google Scholar] [CrossRef]
- Kim, C.H.; Lisman, J.E. A role of actin filament in synaptic transmission and long-term potentiation. J. Neurosci. 1999, 19, 4314–4324. [Google Scholar] [CrossRef]
- Borovac, J.; Bosch, M.; Okamoto, K. Regulation of actin dynamics during structural plasticity of dendritic spines: Signaling messengers and actin-binding proteins. Mol. Cell. Neurosci. 2018, 91, 122–130. [Google Scholar] [CrossRef] [PubMed]
- Bosch, M.; Castro, J.; Saneyoshi, T.; Matsuno, H.; Sur, M.; Hayashi, Y. Structural and molecular remodeling of dendritic spine substructures during long-term potentiation. Neuron 2014, 82, 444–459. [Google Scholar] [CrossRef] [PubMed]
- Morales, M.; Fifkova, E. In situ localization of myosin and actin in dendritic spines with the immunogold technique. J. Comp. Neurol. 1989, 279, 666–674. [Google Scholar] [CrossRef] [PubMed]
- Jordan, B.A.; Fernholz, B.D.; Boussac, M.; Xu, C.; Grigorean, G.; Ziff, E.B.; Neubert, T.A. Identification and verification of novel rodent postsynaptic density proteins. Mol. Cell. Proteom. 2004, 3, 857–871. [Google Scholar] [CrossRef] [PubMed]
- Blitz, A.L.; Fine, R.E. Muscle-like contractile proteins and tubulin in synaptosomes. Proc. Natl. Acad. Sci. USA 1974, 71, 4472–4476. [Google Scholar] [CrossRef] [PubMed]
- Cheng, D.; Hoogenraad, C.C.; Rush, J.; Ramm, E.; Schlager, M.A.; Duong, D.M.; Xu, P.; Wijayawardana, S.R.; Hanfelt, J.; Nakagawa, T.; et al. Relative and absolute quantification of postsynaptic density proteome isolated from rat forebrain and cerebellum. Mol. Cell. Proteom. 2006, 5, 1158–1170. [Google Scholar] [CrossRef]
- Peng, J.; Kim, M.J.; Cheng, D.; Duong, D.M.; Gygi, S.P.; Sheng, M. Semiquantitative proteomic analysis of rat forebrain postsynaptic density fractions by mass spectrometry. J. Biol. Chem. 2004, 279, 21003–21011. [Google Scholar] [CrossRef]
- Zhang, H.; Webb, D.J.; Asmussen, H.; Niu, S.; Horwitz, A.F. A GIT1/PIX/Rac/PAK signaling module regulates spine morphogenesis and synapse formation through MLC. J. Neurosci. 2005, 25, 3379–3388. [Google Scholar] [CrossRef]
- Chandrasekar, I.; Huettner, J.E.; Turney, S.G.; Bridgman, P.C. Myosin II regulates activity dependent compensatory endocytosis at central synapses. J. Neurosci. 2013, 33, 16131–16145. [Google Scholar] [CrossRef]
- Marrs, G.S.; Green, S.H.; Dailey, M.E. Rapid formation and remodeling of postsynaptic densities in developing dendrites. Nat. Neurosci. 2001, 4, 1006–1013. [Google Scholar] [CrossRef]
- Honkura, N.; Matsuzaki, M.; Noguchi, J.; Ellis-Davies, G.C.; Kasai, H. The subspine organization of actin fibers regulates the structure and plasticity of dendritic spines. Neuron 2008, 57, 719–729. [Google Scholar] [CrossRef] [PubMed]
- Koskinen, M.; Bertling, E.; Hotulainen, R.; Tanhuanpaa, K.; Hotulainen, P. Myosin IIb controls actin dynamics underlying the dendritic spine maturation. Mol. Cell. Neurosci. 2014, 61, 56–64. [Google Scholar] [CrossRef] [PubMed]
- Polo-Parada, L.; Plattner, F.; Bose, C.; Landmesser, L.T. NCAM 180 acting via a conserved C-terminal domain and MLCK is essential for effective transmission with repetitive stimulation. Neuron 2005, 46, 917–931. [Google Scholar] [CrossRef] [PubMed]
- Seabrooke, S.; Stewart, B.A. Synaptic transmission and plasticity are modulated by nonmuscle myosin II at the neuromuscular junction of Drosophila. J. Neurophysiol. 2011, 105, 1966–1976. [Google Scholar] [CrossRef][Green Version]
- Mochida, S.; Kobayashi, H.; Matsuda, Y.; Yuda, Y.; Muramoto, K.; Nonomura, Y. Myosin II is involved in transmitter release at synapses formed between rat sympathetic neurons in culture. Neuron 1994, 13, 1131–1142. [Google Scholar] [CrossRef]
- Peng, A.; Rotman, Z.; Deng, P.Y.; Klyachko, V.A. Differential motion dynamics of synaptic vesicles undergoing spontaneous and activity-evoked endocytosis. Neuron 2012, 73, 1108–1115. [Google Scholar] [CrossRef]
- Miserey-Lenkei, S.; Chalancon, G.; Bardin, S.; Formstecher, E.; Goud, B.; Echard, A. Rab and actomyosin-dependent fission of transport vesicles at the Golgi complex. Nat. Cell Biol. 2010, 12, 645–654. [Google Scholar] [CrossRef]
- Hodges, J.L.; Newell-Litwa, K.; Asmussen, H.; Vicente-Manzanares, M.; Horwitz, A.R. Myosin IIb activity and phosphorylation status determines dendritic spine and post-synaptic density morphology. PLoS ONE 2011, 6, e24149. [Google Scholar] [CrossRef]
- Rubio, M.D.; Johnson, R.; Miller, C.A.; Huganir, R.L.; Rumbaugh, G. Regulation of synapse structure and function by distinct myosin II motors. J. Neurosci. 2011, 31, 1448–1460. [Google Scholar] [CrossRef]
- Kim, K.Y.; Kawamoto, S.; Bao, J.; Sellers, J.R.; Adelstein, R.S. The B2 alternatively spliced isoform of nonmuscle myosin II-B lacks actin-activated MgATPase activity and in vitro motility. Biochem. Biophys. Res. Commun. 2008, 369, 124–134. [Google Scholar] [CrossRef]
- Ma, X.; Kawamoto, S.; Uribe, J.; Adelstein, R.S. Function of the neuron-specific alternatively spliced isoforms of nonmuscle myosin II-B during mouse brain development. Mol. Biol. Cell 2006, 17, 2138–2149. [Google Scholar] [CrossRef] [PubMed]
- Rex, C.S.; Gavin, C.F.; Rubio, M.D.; Kramar, E.A.; Chen, L.Y.; Jia, Y.; Huganir, R.L.; Muzyczka, N.; Gall, C.M.; Miller, C.A.; et al. Myosin IIb regulates actin dynamics during synaptic plasticity and memory formation. Neuron 2010, 67, 603–617. [Google Scholar] [CrossRef] [PubMed]
- Mizui, T.; Sekino, Y.; Yamazaki, H.; Ishizuka, Y.; Takahashi, H.; Kojima, N.; Kojima, M.; Shirao, T. Myosin II ATPase activity mediates the long-term potentiation-induced exodus of stable F-actin bound by drebrin A from dendritic spines. PLoS ONE 2014, 9, e85367. [Google Scholar] [CrossRef] [PubMed]
- Ozkan, E.D.; Aceti, M.; Creson, T.K.; Rojas, C.S.; Hubbs, C.R.; McGuire, M.N.; Kakad, P.P.; Miller, C.A.; Rumbaugh, G. Input-specific regulation of hippocampal circuit maturation by non-muscle myosin IIB. J. Neurochem. 2015, 134, 429–444. [Google Scholar] [CrossRef] [PubMed]
- Amparan, D.; Avram, D.; Thomas, C.G.; Lindahl, M.G.; Yang, J.; Bajaj, G.; Ishmael, J.E. Direct interaction of myosin regulatory light chain with the NMDA receptor. J. Neurochem. 2005, 92, 349–361. [Google Scholar] [CrossRef]
- Bajaj, G.; Zhang, Y.; Schimerlik, M.I.; Hau, A.M.; Yang, J.; Filtz, T.M.; Kioussi, C.; Ishmael, J.E. N-methyl-D-aspartate receptor subunits are non-myosin targets of myosin regulatory light chain. J. Biol. Chem. 2009, 284, 1252–1266. [Google Scholar] [CrossRef]
- Bu, Y.; Wang, N.; Wang, S.; Sheng, T.; Tian, T.; Chen, L.; Pan, W.; Zhu, M.; Luo, J.; Lu, W. Myosin IIb-dependent regulation of actin dynamics Is required for N-Methyl-D-aspartate Receptor trafficking during synaptic plasticity. J. Biol. Chem. 2015, 290, 25395–25410. [Google Scholar] [CrossRef]
- Yagi, H.; Nagano, T.; Xie, M.J.; Ikeda, H.; Kuroda, K.; Komada, M.; Iguchi, T.; Tariqur, R.M.; Morikubo, S.; Noguchi, K.; et al. Filamin A-interacting protein (FILIP) is a region-specific modulator of myosin 2b and controls spine morphology and NMDA receptor accumulation. Sci. Rep. 2014, 4, 6353. [Google Scholar] [CrossRef]
- Gavin, C.F.; Rubio, M.D.; Young, E.; Miller, C.; Rumbaugh, G. Myosin II motor activity in the lateral amygdala is required for fear memory consolidation. Learn. Mem. 2012, 19, 9–14. [Google Scholar] [CrossRef]
- Lamprecht, R.; Margulies, D.S.; Farb, C.R.; Hou, M.; Johnson, L.R.; LeDoux, J.E. Myosin light chain kinase regulates synaptic plasticity and fear learning in the lateral amygdala. Neuroscience 2006, 139, 821–829. [Google Scholar] [CrossRef]
- Young, E.J.; Aceti, M.; Griggs, E.M.; Fuchs, R.A.; Zigmond, Z.; Rumbaugh, G.; Miller, C.A. Selective, retrieval-independent disruption of methamphetamine-associated memory by actin depolymerization. Biol. Psychiatry 2014, 75, 96–104. [Google Scholar] [CrossRef] [PubMed]
- Young, E.J.; Briggs, S.B.; Rumbaugh, G.; Miller, C.A. Nonmuscle myosin II inhibition disrupts methamphetamine-associated memory in females and adolescents. Neurobiol. Learn. Mem. 2017, 139, 109–116. [Google Scholar] [CrossRef] [PubMed]
- Young, E.J.; Blouin, A.M.; Briggs, S.B.; Sillivan, S.E.; Lin, L.; Cameron, M.D.; Rumbaugh, G.; Miller, C.A. Nonmuscle myosin IIB as a therapeutic target for the prevention of relapse to methamphetamine use. Mol. Psychiatry 2016, 21, 615–623. [Google Scholar] [CrossRef] [PubMed]
- Briggs, S.B.; Blouin, A.M.; Young, E.J.; Rumbaugh, G.; Miller, C.A. Memory disrupting effects of nonmuscle myosin II inhibition depend on the class of abused drug and brain region. Learn. Mem. 2017, 24, 70–75. [Google Scholar] [CrossRef]
- Heath, K.E.; Campos-Barros, A.; Toren, A.; Rozenfeld-Granot, G.; Carlsson, L.E.; Savige, J.; Denison, J.C.; Gregory, M.C.; White, J.G.; Barker, D.F.; et al. Nonmuscle myosin heavy chain IIA mutations define a spectrum of autosomal dominant macrothrombocytopenias: May-Hegglin anomaly and Fechtner, Sebastian, Epstein, and Alport-like syndromes. Am. J. Hum. Genet. 2001, 69, 1033–1045. [Google Scholar] [CrossRef] [PubMed]
- Donaudy, F.; Snoeckx, R.; Pfister, M.; Zenner, H.P.; Blin, N.; Di Stazio, M.; Ferrara, A.; Lanzara, C.; Ficarella, R.; Declau, F.; et al. Nonmuscle myosin heavy-chain gene MYH14 is expressed in cochlea and mutated in patients affected by autosomal dominant hearing impairment (DFNA4). Am. J. Hum. Genet. 2004, 74, 770–776. [Google Scholar] [CrossRef]
- Krey, J.F.; Pasca, S.P.; Shcheglovitov, A.; Yazawa, M.; Schwemberger, R.; Rasmusson, R.; Dolmetsch, R.E. Timothy syndrome is associated with activity-dependent dendritic retraction in rodent and human neurons. Nat. Neurosci. 2013, 16, 201–209. [Google Scholar] [CrossRef]
- Liu, Y.L.; Fann, C.S.; Liu, C.M.; Chen, W.J.; Wu, J.Y.; Hung, S.I.; Chen, C.H.; Jou, Y.S.; Liu, S.K.; Hwang, T.J.; et al. RASD2, MYH9, and CACNG2 genes at chromosome 22q12 associated with the subgroup of schizophrenia with non-deficit in sustained attention and executive function. Biol. Psychiatry 2008, 64, 789–796. [Google Scholar] [CrossRef]
- Li, J.; Cai, T.; Jiang, Y.; Chen, H.; He, X.; Chen, C.; Li, X.; Shao, Q.; Ran, X.; Li, Z.; et al. Genes with de novo mutations are shared by four neuropsychiatric disorders discovered from NPdenovo database. Mol. Psychiatry 2016, 21, 290–297. [Google Scholar] [CrossRef]
- Wang, G.; Wang, T.; Hu, Y.; Wang, J.; Wang, Y.; Zhang, Y.; Li, F.; Liu, W.; Sun, Y.; Yu, B.; et al. NMMHC IIA triggers neuronal autophagic cell death by promoting F-actin-dependent ATG9A trafficking in cerebral ischemia/reperfusion. Cell Death Dis. 2020, 11, 428. [Google Scholar] [CrossRef]
- Wang, G.Y.; Wang, T.Z.; Zhang, Y.Y.; Li, F.; Yu, B.Y.; Kou, J.P. NMMHC IIA inhibition ameliorates cerebral ischemic/reperfusion-induced neuronal apoptosis through caspase-3/ROCK1/MLC pathway. Drug Des. Devel. 2020, 14, 13–25. [Google Scholar] [CrossRef] [PubMed]
- Tuzovic, L.; Yu, L.; Zeng, W.; Li, X.; Lu, H.; Lu, H.M.; Gonzalez, K.D.; Chung, W.K. A human de novo mutation in MYH10 phenocopies the loss of function mutation in mice. Rare Dis. 2013, 1, e26144. [Google Scholar] [CrossRef] [PubMed]
- Rusielewicz, T.; Nam, J.; Damanakis, E.; John, G.R.; Raine, C.S.; Melendez-Vasquez, C.V. Accelerated repair of demyelinated CNS lesions in the absence of non-muscle myosin IIB. Glia 2014, 62, 580–591. [Google Scholar] [CrossRef] [PubMed]
- Choi, B.O.; Kang, S.H.; Hyun, Y.S.; Kanwal, S.; Park, S.W.; Koo, H.; Kim, S.B.; Choi, Y.C.; Yoo, J.H.; Kim, J.W.; et al. A complex phenotype of peripheral neuropathy, myopathy, hoarseness, and hearing loss is linked to an autosomal dominant mutation in MYH14. Hum. Mutat. 2011, 32, 669–677. [Google Scholar] [CrossRef]
- Iyadurai, S.; Arnold, W.D.; Kissel, J.T.; Ruhno, C.; McGovern, V.L.; Snyder, P.J.; Prior, T.W.; Roggenbuck, J.; Burghes, A.H.; Kolb, S.J. Variable phenotypic expression and onset in MYH14 distal hereditary motor neuropathy phenotype in a large, multigenerational North American family. Muscle Nerve 2017, 56, 341–345. [Google Scholar] [CrossRef]
- Almutawa, W.; Smith, C.; Sabouny, R.; Smit, R.B.; Zhao, T.; Wong, R.; Lee-Glover, L.; Desrochers-Goyette, J.; Ilamathi, H.S.; Care4Rare Canada, C.; et al. The R941L mutation in MYH14 disrupts mitochondrial fission and associates with peripheral neuropathy. EBioMedicine 2019, 45, 379–392. [Google Scholar] [CrossRef]
- Rubio, M.D.; Haroutunian, V.; Meador-Woodruff, J.H. Abnormalities of the Duo/Ras-related C3 botulinum toxin substrate 1/p21-activated kinase 1 pathway drive myosin light chain phosphorylation in frontal cortex in schizophrenia. Biol. Psychiatry 2012, 71, 906–914. [Google Scholar] [CrossRef]
- Kutsche, K.; Yntema, H.; Brandt, A.; Jantke, I.; Nothwang, H.G.; Orth, U.; Boavida, M.G.; David, D.; Chelly, J.; Fryns, J.P.; et al. Mutations in ARHGEF6, encoding a guanine nucleotide exchange factor for Rho GTPases, in patients with X-linked mental retardation. Nat. Genet. 2000, 26, 247–250. [Google Scholar] [CrossRef]
- Vandervore, L.V.; Schot, R.; Kasteleijn, E.; Oegema, R.; Stouffs, K.; Gheldof, A.; Grochowska, M.M.; van der Sterre, M.L.T.; van Unen, L.M.A.; Wilke, M.; et al. Heterogeneous clinical phenotypes and cerebral malformations reflected by rotatin cellular dynamics. Brain 2019, 142, 867–884. [Google Scholar] [CrossRef]
- Wang, X.; Williams, D.; Muller, I.; Lemieux, M.; Dukart, R.; Maia, I.B.L.; Wang, H.; Woerman, A.L.; Schmitt-Ulms, G. Tau interactome analyses in CRISPR-Cas9 engineered neuronal cells reveal ATPase-dependent binding of wild-type but not P301L Tau to non-muscle myosins. Sci. Rep. 2019, 9, 16238. [Google Scholar] [CrossRef]
- Penzes, P.; Cahill, M.E.; Jones, K.A.; VanLeeuwen, J.E.; Woolfrey, K.M. Dendritic spine pathology in neuropsychiatric disorders. Nat. Neurosci. 2011, 14, 285–293. [Google Scholar] [CrossRef] [PubMed]
- Allen, K.M.; Gleeson, J.G.; Bagrodia, S.; Partington, M.W.; MacMillan, J.C.; Cerione, R.A.; Mulley, J.C.; Walsh, C.A. PAK3 mutation in nonsyndromic X-linked mental retardation. Nat. Genet. 1998, 20, 25–30. [Google Scholar] [CrossRef] [PubMed]
- Bienvenu, T.; des Portes, V.; McDonell, N.; Carrie, A.; Zemni, R.; Couvert, P.; Ropers, H.H.; Moraine, C.; van Bokhoven, H.; Fryns, J.P.; et al. Missense mutation in PAK3, R67C, causes X-linked nonspecific mental retardation. Am. J. Med. Genet. 2000, 93, 294–298. [Google Scholar] [CrossRef]
- Qian, Y.; Wu, B.; Lu, Y.; Zhou, W.; Wang, S.; Wang, H. Novel PAK3 gene missense variant associated with two Chinese siblings with intellectual disability: A case report. BMC Med. Genet. 2020, 21, 31. [Google Scholar] [CrossRef]
- Boda, B.; Alberi, S.; Nikonenko, I.; Node-Langlois, R.; Jourdain, P.; Moosmayer, M.; Parisi-Jourdain, L.; Muller, D. The mental retardation protein PAK3 contributes to synapse formation and plasticity in hippocampus. J. Neurosci. 2004, 24, 10816–10825. [Google Scholar] [CrossRef]
- Zhang, H.; Webb, D.J.; Asmussen, H.; Horwitz, A.F. Synapse formation is regulated by the signaling adaptor GIT1. J. Cell Biol. 2003, 161, 131–142. [Google Scholar] [CrossRef]
- Ramakers, G.J.; Wolfer, D.; Rosenberger, G.; Kuchenbecker, K.; Kreienkamp, H.J.; Prange-Kiel, J.; Rune, G.; Richter, K.; Langnaese, K.; Masneuf, S.; et al. Dysregulation of Rho GTPases in the αPix/Arhgef6 mouse model of X-linked intellectual disability is paralleled by impaired structural and synaptic plasticity and cognitive deficits. Hum. Mol. Genet. 2012, 21, 268–286. [Google Scholar] [CrossRef]
- Node-Langlois, R.; Muller, D.; Boda, B. Sequential implication of the mental retardation proteins ARHGEF6 and PAK3 in spine morphogenesis. J. Cell Sci. 2006, 119, 4986–4993. [Google Scholar] [CrossRef]
- Wong, R.O.; Ghosh, A. Activity-dependent regulation of dendritic growth and patterning. Nat. Rev. Neurosci. 2002, 3, 803–812. [Google Scholar] [CrossRef]
- Ba, W.; van der Raadt, J.; Nadif Kasri, N. Rho GTPase signaling at the synapse: Implications for intellectual disability. Exp. Cell Res. 2013, 319, 2368–2374. [Google Scholar] [CrossRef]
- Pinto, D.; Pagnamenta, A.T.; Klei, L.; Anney, R.; Merico, D.; Regan, R.; Conroy, J.; Magalhaes, T.R.; Correia, C.; Abrahams, B.S.; et al. Functional impact of global rare copy number variation in autism spectrum disorders. Nature 2010, 466, 368–372. [Google Scholar] [CrossRef] [PubMed]
- Kim, M.J.; Biag, J.; Fass, D.M.; Lewis, M.C.; Zhang, Q.; Fleishman, M.; Gangwar, S.P.; Machius, M.; Fromer, M.; Purcell, S.M.; et al. Functional analysis of rare variants found in schizophrenia implicates a critical role for GIT1-PAK3 signaling in neuroplasticity. Mol. Psychiatry 2017, 22, 417–429. [Google Scholar] [CrossRef] [PubMed]
- Hayashi-Takagi, A.; Araki, Y.; Nakamura, M.; Vollrath, B.; Duron, S.G.; Yan, Z.; Kasai, H.; Huganir, R.L.; Campbell, D.A.; Sawa, A. PAKs inhibitors ameliorate schizophrenia-associated dendritic spine deterioration in vitro and in vivo during late adolescence. Proc. Natl. Acad. Sci. USA 2014, 111, 6461–6466. [Google Scholar] [CrossRef] [PubMed]
- Dolan, B.M.; Duron, S.G.; Campbell, D.A.; Vollrath, B.; Shankaranarayana Rao, B.S.; Ko, H.Y.; Lin, G.G.; Govindarajan, A.; Choi, S.Y.; Tonegawa, S. Rescue of fragile X syndrome phenotypes in Fmr1 KO mice by the small-molecule PAK inhibitor FRAX486. Proc. Natl. Acad. Sci. USA 2013, 110, 5671–5676. [Google Scholar] [CrossRef]
- Govek, E.E.; Newey, S.E.; Akerman, C.J.; Cross, J.R.; Van der Veken, L.; Van Aelst, L. The X-linked mental retardation protein oligophrenin-1 is required for dendritic spine morphogenesis. Nat. Neurosci. 2004, 7, 364–372. [Google Scholar] [CrossRef]
- Spires-Jones, T.L.; Hyman, B.T. The intersection of amyloid beta and tau at synapses in Alzheimer’s disease. Neuron 2014, 82, 756–771. [Google Scholar] [CrossRef]
- Jämsa, A.; Agerman, K.; Radesäter, A.C.; Ottervald, J.; Malmström, J.; Hiller, G.; Liu, G.; Vasänge, M. Identification of non-muscle myosin heavy chain as a substrate for Cdk5 and tool for drug screening. J. Biomed. Sci. 2009, 16, 55. [Google Scholar] [CrossRef]
- Feuillette, S.; Deramecourt, V.; Laquerriere, A.; Duyckaerts, C.; Delisle, M.B.; Maurage, C.A.; Blum, D.; Buee, L.; Frebourg, T.; Campion, D.; et al. Filamin-A and myosin VI colocalize with fibrillary Tau protein in Alzheimer’s disease and FTDP-17 brains. Brain Res. 2010, 1345, 182–189. [Google Scholar] [CrossRef]
- Peethumnongsin, E.; Yang, L.; Kallhoff-Munoz, V.; Hu, L.; Takashima, A.; Pautler, R.G.; Zheng, H. Convergence of presenilin- and tau-mediated pathways on axonal trafficking and neuronal function. J. Neurosci. 2010, 30, 13409–13418. [Google Scholar] [CrossRef][Green Version]
- Stokin, G.B.; Lillo, C.; Falzone, T.L.; Brusch, R.G.; Rockenstein, E.; Mount, S.L.; Raman, R.; Davies, P.; Masliah, E.; Williams, D.S.; et al. Axonopathy and transport deficits early in the pathogenesis of Alzheimer’s disease. Science 2005, 307, 1282–1288. [Google Scholar] [CrossRef]
- Barthet, G.; Mulle, C. Presynaptic failure in Alzheimer’s disease. Prog. Neurobiol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Lauterborn, J.C.; Cox, C.D.; Chan, S.W.; Vanderklish, P.W.; Lynch, G.; Gall, C.M. Synaptic actin stabilization protein loss in Down syndrome and Alzheimer disease. Brain Pathol. 2020, 30, 319–331. [Google Scholar] [CrossRef] [PubMed]
- Wang, Y.; Xu, Y.; Liu, Q.; Zhang, Y.; Gao, Z.; Yin, M.; Jiang, N.; Cao, G.; Yu, B.; Cao, Z.; et al. Myosin IIA-related actomyosin contractility mediates oxidative stress-induced neuronal apoptosis. Front. Mol. Neurosci. 2017, 10, 75. [Google Scholar] [CrossRef] [PubMed]
- Srivastava, K.; Shao, B.; Bayraktutan, U. PKC-β exacerbates in vitro brain barrier damage in hyperglycemic settings via regulation of RhoA/Rho-kinase/MLC2 pathway. J. Cereb. Blood Flow Metab. 2013, 33, 1928–1936. [Google Scholar] [CrossRef]
- Ning, G.; Liu, Y.; Xu, H.; Li, Y.; Wu, H.; Wang, X.; Feng, S. Gene silencing NMII promotes axonal regeneration against contusive spinal cord injury in rats. Int. J. Clin. Exp. Pathol. 2017, 10, 11345–11352. [Google Scholar]
- Flippo, K.H.; Strack, S. Mitochondrial dynamics in neuronal injury, development and plasticity. J. Cell Sci. 2017, 130, 671–681. [Google Scholar] [CrossRef]
- Kippert, A.; Fitzner, D.; Helenius, J.; Simons, M. Actomyosin contractility controls cell surface area of oligodendrocytes. BMC Cell Biol. 2009, 10, 71. [Google Scholar] [CrossRef]
- Koch, J.C.; Tatenhorst, L.; Roser, A.E.; Saal, K.A.; Tonges, L.; Lingor, P. ROCK inhibition in models of neurodegeneration and its potential for clinical translation. Pharmacy 2018, 189, 1–21. [Google Scholar] [CrossRef]
- Ginhoux, F.; Garel, S. The mysterious origins of microglia. Nat. Neurosci. 2018, 21, 897–899. [Google Scholar] [CrossRef]
- Gu, B.J.; Saunders, B.M.; Jursik, C.; Wiley, J.S. The P2X7-nonmuscle myosin membrane complex regulates phagocytosis of nonopsonized particles and bacteria by a pathway attenuated by extracellular ATP. Blood 2010, 115, 1621–1631. [Google Scholar] [CrossRef]
- Gu, B.J.; Huang, X.; Ou, A.; Rembach, A.; Fowler, C.; Avula, P.K.; Horton, A.; Doecke, J.D.; Villemagne, V.L.; Macaulay, S.L.; et al. Innate phagocytosis by peripheral blood monocytes is altered in Alzheimer’s disease. Acta Neuropathol. 2016, 132, 377–389. [Google Scholar] [CrossRef] [PubMed]
- Gu, B.J.; Wiley, J.S. P2X7 as a scavenger receptor for innate phagocytosis in the brain. Br. J. Pharm. 2018, 175, 4195–4208. [Google Scholar] [CrossRef] [PubMed]
- Francistiová, L.; Bianchi, C.; Di Lauro, C.; Sebastián-Serrano, A.; de Diego-García, L.; Kobolák, J.; Dinnyés, A.; Díaz-Hernández, M. The role of P2X7 receptor in Alzheimer’s disease. Front. Mol. Neurosci. 2020, 13, 94. [Google Scholar] [CrossRef] [PubMed]
- Barcia, C.; Ros, C.M.; Annese, V.; Carrillo-de Sauvage, M.A.; Ros-Bernal, F.; Gomez, A.; Yuste, J.E.; Campuzano, C.M.; de Pablos, V.; Fernandez-Villalba, E.; et al. ROCK/Cdc42-mediated microglial motility and gliapse formation lead to phagocytosis of degenerating dopaminergic neurons in vivo. Sci. Rep. 2012, 2, 809. [Google Scholar] [CrossRef] [PubMed]
- Socodato, R.; Portugal, C.C.; Canedo, T.; Rodrigues, A.; Almeida, T.O.; Henriques, J.F.; Vaz, S.H.; Magalhaes, J.; Silva, C.M.; Baptista, F.I.; et al. Microglia dysfunction caused by the loss of Rhoa disrupts neuronal physiology and leads to neurodegeneration. Cell Rep. 2020, 31, 107796. [Google Scholar] [CrossRef]
- Beard, R.S., Jr.; Haines, R.J.; Wu, K.Y.; Reynolds, J.J.; Davis, S.M.; Elliott, J.E.; Malinin, N.L.; Chatterjee, V.; Cha, B.J.; Wu, M.H.; et al. Non-muscle Mlck is required for beta-catenin- and FoxO1-dependent downregulation of Cldn5 in IL-1beta-mediated barrier dysfunction in brain endothelial cells. J. Cell Sci. 2014, 127, 1840–1853. [Google Scholar] [CrossRef]
- Burgos, M.; Calvo, S.; Molina, F.; Vaquero, C.F.; Samarel, A.; Llopis, J.; Tranque, P. PKCε induces astrocyte stellation by modulating multiple cytoskeletal proteins and interacting with Rho A signalling pathways: Implications for neuroinflammation. Eur. J. Neurosci. 2007, 25, 1069–1078. [Google Scholar] [CrossRef]
- Betapudi, V.; Licate, L.S.; Egelhoff, T.T. Distinct roles of nonmuscle myosin II isoforms in the regulation of MDA-MB-231 breast cancer cell spreading and migration. Cancer Res. 2006, 66, 4725–4733. [Google Scholar] [CrossRef]





{kind=link}
{kind=link}
{kind=link}
{kind=link}
| Gene | Protein | Central Nervous System | Other Organs |
|---|---|---|---|
| MYH9 | NMIIA | Cerebellum | Colon, kidney |
| Mesencephalon | lung, platelets | ||
| brain stem | spleen, thymus, uterus | ||
| MYH10 | NMIIB | Cerebral cortex | Aorta |
| hippocampus | Kidney | ||
| thalamus, spinal cord | urinary bladder | ||
| MYH14 | NMIIC | Cerebellum | Colon, heart |
| corpus callosum | kidney, liver | ||
| pons, thalamus | skeletal muscle |
| Gene | NMII | Pathological Features | Neurological Diseases | References |
|---|---|---|---|---|
| Cav1.2 | NMII | Dendrite retraction | Timothy syndrome | [137] |
| MYH9 | NMIIA | Neurological impairments | Autism, intellectual disability, schizophrenia | [138,139] |
| MYH9 | NMIIA | Autophagic neuron death, infarct volume, neurological defects | Brain ischemia | [17,140,141] |
| MYH10 | NMIIB | Dysplasia, microcephaly, cerebral atrophy, and hydrocephalus | Mental retardation | [142] |
| MYH10 | NMIIB | Axon demyelination | Multiple sclerosis * | [21,143] |
| MYH14 | NMIIC | Neuropathy, axonal transport deficits, muscle weakness/atrophy | Peripheral neuropathy | [144,145,146] |
| PAK1 | NMII | Synapse pathology | Schizophrenia | [147] |
| PAK3/GIT1/Rac | NMIIB | Synapse pathology | X-linked mental retardation, schizophrenia | [108] |
| αPIX/ARHGEF6 | NMIIB | Synapse pathology | X-linked mental retardation | [108,148] |
| PSEN1 | NMIIA | Axon retraction | Alzheimer’s disease * | [59] |
| RTTN | NMIIA,B,C | Cortical malformation, microcephaly and polymicrogyria | Intellectual disability | [149] |
| Tau | NMIIA,B | Dystrophic neurites and axons | Alzheimer’s disease, FTD * | [150] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Javier-Torrent, M.; Saura, C.A. Conventional and Non-Conventional Roles of Non-Muscle Myosin II-Actin in Neuronal Development and Degeneration. Cells 2020, 9, 1926. https://doi.org/10.3390/cells9091926
Javier-Torrent M, Saura CA. Conventional and Non-Conventional Roles of Non-Muscle Myosin II-Actin in Neuronal Development and Degeneration. Cells. 2020; 9(9):1926. https://doi.org/10.3390/cells9091926
Chicago/Turabian StyleJavier-Torrent, Míriam, and Carlos A. Saura. 2020. "Conventional and Non-Conventional Roles of Non-Muscle Myosin II-Actin in Neuronal Development and Degeneration" Cells 9, no. 9: 1926. https://doi.org/10.3390/cells9091926
APA StyleJavier-Torrent, M., & Saura, C. A. (2020). Conventional and Non-Conventional Roles of Non-Muscle Myosin II-Actin in Neuronal Development and Degeneration. Cells, 9(9), 1926. https://doi.org/10.3390/cells9091926