1. Introduction
Incidences of malignant thyroid tumors are increasing worldwide. Follicular thyroid cancer (FTC) is the second most common type, accounting for about 5–10% of all thyroid malignancies. The courses of FTC differ from the course of most common papillary thyroid cancers. FTC affects younger people, has greater metastatic potency, and a higher mortality rate. Besides an aggressive clinical behavior, the management of FTC is challenged by a problematic diagnostic process [
1]. The most important issue is distinguishing between truly malignant FTC and benign follicular adenomas (FA), which are encountered approximately 5 times more frequently. The thyroid node size or growth rate does not differ between FTC and FA [
2]. Ultrasound elastography does not provide an ultimate diagnosis [
3]. Due to the absence of cellular or nuclear atypia, cytological analysis of the material obtained by fine-needle aspiration can only reveal the follicular nature of the thyroid node but cannot rule out malignancy [
4]. Thus, there is currently no method to confidently exclude a malignant diagnosis, which is considered as an indication for surgery. Moreover, a post-surgery histological diagnosis of FA does not guarantee a good prognosis [
5]. This reveals the need for the development of a new, possibly objective, accurate method for the differential diagnosis of the follicular thyroid node.
Potential benefits of “liquid biopsies” in diagnosis, prognosis, and personalization of thyroid cancer therapy have been discussed recently [
6]. Among different types of circulating markers, nano-scaled membrane extracellular vesicles (EVs) attract special interest [
7]. The smallest vesicles of endocytic origin, like exosomes, are secreted by apparently all types of cells and reflect their biochemical properties and condition. Since circulating EVs present a natural, extremely stable, and cell-specific complexes of membrane lipids, proteins, and nucleic acids (mRNA, lncRNA, miRNA), they are considered as a promising marker for cancer diagnosis [
8]. For example, a few years ago, we reported the diagnostic potency of exosomal miRNAs (miR-31, miR-21, miR-181a) for discrimination between benign FA, FTC, and papillary thyroid cancer (PTC) [
9].
However, the development and clinical implementation of EV-based diagnostic tests are still hampered. One issue is an extreme heterogeneity of plasma EV populations with the majority of vesicles derived from the endothelium, platelets, blood, and stromal cells. A certain portion of circulating EVs may account for vesicles secreted by cells of specific tissues, e.g., the thyroid gland. Apparently, the fraction of tissue-derived EVs is defined by the number of specific cells, their secretory activity, and perhaps other factors. In the case of tumor development, the portion of malignant tissue-specific vesicles may increase. For instance, in an in vivo model of glioblastomas, tumor cell-derived exosomes accounted for 35–50% of the total population of circulating vesicles [
10]. Analysis of the isolated fraction of tumor- or even tissue-specific vesicles would have greatly improved the diagnostic potency. Recently, this approach has been explored in the context of different types of cancer. For instance, EVs secreted by head and neck squamous carcinoma cells have been isolated via immune-binding of CD44v3 on their surface [
11], while melanoma-derived exosomes have been captured via binding to CSPG4 [
12].
By planning the presented study, we hypothesize that the isolation of thyroid cell-derived EVs, followed by the analysis of the vesicular miRNAs, may help to distinguish patients with FA and FTC. The hypothesis is based on the reported involvement of thyroid-derived EVs in thyroglobulin processing [
13] and the detection of thyroid peroxidase (TPO) within vesicles [
14,
15]. Consequent steps of the study are schematically presented in
Figure 1. After characterization of EVs isolated from plasma, the reduction of TPO-positive EVs (herein referred to as TPO(+)EVs) in the plasma of FA patients after a thyroidectomy was detected, confirming the thyroid origin of these vesicles. Next, TPO(+)EVs were isolated from the plasma of patients with FA and FTC and the vesicular miRNA profiled. Finally, the diagnostic potency of marker miRNAs (members of Let-7 family) in total and in the TPO(+) population of EVs was compared.
2. Materials and Methods
2.1. Patients
Patients with thyroid nodules were treated at the North-West Centre of Endocrinology and Endocrine Surgery and the N.N. Petrov National Medical Research Centre of Oncology, Saint-Petersburg. Evaluation of patients included ultrasound and fine-needle aspiration biopsy followed by a cytological exam. Patients who were classified as Bethesda category III-IV and were planned for thyroidectomy were enrolled in the study. Patients with multiple thyroid nodules and/or any endocrine or metabolic comorbidity were excluded from the study. Patients in whom the post-operative histological diagnosis did not confirm the follicular nature of the node or histological diagnosis was not unambiguous (FTC vs. FA), were excluded from the study as well. Relevant clinical characteristics of patients presented in
Table 1.
The study was approved by the local ethics committee of N.N. Petrov NMRC of Oncology (10.11.2017, protocol AAAA-A18-118012390156-5) and conducted in accordance with the ethical guidelines outlined in the Declaration of Helsinki. All patients signed an informed consent form before being included in the study. Biological material and clinical data have been depersonalized. Plasma samples from patients with histologically confirmed benign follicular adenoma (n = 30) and thyroid follicular cancer (n = 30) were used. Additionally, the plasma of healthy female donors (n = 5) was obtained from the Department of Blood Transfusion of N.N. Petrov National Medical Research Centre of Oncology.
2.2. Plasma Sampling and EV Isolation
Blood samples were collected 3 times, namely, before the operation (a), after 5 days (b), and 4 weeks (c) after surgery. Peripheral blood was collected in vacutainers with EDTA (SARSTEDT AG & Co. KG, Nümbrecht, NO. 32.332). The plasma was separated by centrifugation within 10–15 min and stored at −80 °C. Further analysis proceeded with only plasma samples from patients with a histological diagnosis (FA or FTC) that was independently confirmed by two pathologists.
EVs were isolated from plasma by ultra-centrifugation (UC). Briefly, the samples (2 mL) were diluted by PBS 1:1, centrifuged at 12,000×
g for 30 min at +4 °C. The supernatant was filtered using a 0.22-μm filter, then ultra-centrifuged at 110,000× g (Ultracentrifuge Optima XPN 80, rotor 70.1 Ti/k-factor 36) for 4 h at +4 °C. The pellet was washed once in 3.6 mL of PBS and then ultra-centrifuged again at 110,000 × g for 4 h at 4 °C. The pelleted vesicles were resuspended in 200 uL of PBS and kept at −80 °C. We have submitted all relevant data of our experiments to the EV-TRACK knowledgebase (EV-TRACK ID: EV200077) [
16].
2.3. Analysis of Physical Characteristics of EV
Nanoparticle Tracking Analysis (NTA) was used to determine the size and the concentration of isolated vesicles. The measurements were carried out with a Nanosight NS300 analyzer (Malvern Panalytical Ltd., Malvern, UK) and the data obtained were processed by the Nanosight NTA 3.2 software package. Atomic force microscopy (AFM) was performed with a NT-MDT Solver Bio scanning probe microscope (Molecular Devices and Tools for Nanotechnology / NT-MDT, Moscow, Russia) in the tapping mode, using probe NSG01_DLC. The samples were applied to the mica surface (2SPI, West Chester, PA, USA), then immediately after that the upper layers were removed, incubated for 30 s, washed twice with distilled water, then dried using compressed air. Image processing was performed with the Gwyddion 2.56 (gwyddion.net) and Image Analysis (NT-MDT, Russia) programs.
2.4. Analysis of Biochemical Characteristics of EV (FACS, Dot-Blot)
In order to assay the presence of exosomal markers on the surface of vesicles, we used an Exo-FACS Kit (HansaBioMed, Tallinn, Estonia, NO. HBM-FACS-PEP). The EVs isolated by UC were incubated with 4 µm aldehyde-sulfate latex beads inPA accordance with the producer’s protocol. The tetraspanins CD63 and CD9 were detected with a monoclonal FITC-labelled antibody to CD63 (AbCam, Cambridge, MA, USA, ab8319) and a monoclonal PE-labelled antibody to CD9 (AbCam, Cambridge, MA, USA, ab82394), correspondently. Incubation was performed at a working concentration of antibodies 1 of mg/mL for 2 h at +4 °C in the dark. The complexes of “latex bead-EV-antibodies” were washed twice and diluted in PBS (200 uL). The samples were analyzed in a CytoFLEX (Beckman Coulter, Miami, FL, USA) flow cytometer equipped for multi-parametric and multi-color analysis, including a 488 nm argon laser for measurement of forward light scattering (FSC) and orthogonal scattering (SSC). The complexes assembled without EVs were used as a negative control. The samples were acquired and gated by FSC and SSC and then analyzed. FSC and SSC, as well as the fluorescence FITC and PE signals, were collected and stored as list mode files. The data were analyzed with the CytExpert (Beckman Coulter, Miami, FL, USA) software package.
In order to assay the presence of markers of interest in the whole vesicles, they were incubated with a 1x RIPA buffer solution (Thermo Scientific, Wilmington, DE, USA, NO.89900) for 10 min at +4 °C. The total amount of proteins was normalized by a Bradford assay, and 1 uL of the equilibrated lysate was spotted onto the nitrocellulose membrane. The membranes with spotted samples were then blocked with 5% BSA in TBS-T for 1 h at room temperature, then washed twice and incubated with the primary antibodies against TPO (AbCam, Cambridge, MA, USA, ab109383) or TSG-Rec (Santa Cruz, California, CA, USA, sc7964) diluted at 1:1000 in 0.1% BSA in TBS-T. Then, the membranes were washed with TBS-T and incubated with the HRP-labelled secondary antibodies (AbCam, USA-ab6789, ab6721), diluted at 1:10000 in 0.1% BSA in TBS-T for 30 min, then washed with TBS-T and evaluated with a Pierce ECL Western blotting substrate (AbCam, USA-ab133406) using the iBright FL1500 Imaging System (Invitrogen, Carlsbad, CA, USA). The received signal data were processed in the ImageJ program.
2.5. Isolation of TPO(+)EVs with Immune Beads
Superparamagnetic beads (SPMB) coupled with streptavidin (“Sileks Ltd.”, Moscow, Russia) and a biotinylated antibody to TPO (clon TPO28 “HyTest Ltd.”, Turku, Finland) were used. For this, 1 uL of the SPMB suspension (1 mg/mL) and 10 uL of the antibody (0.6 mg/mL) were mixed in 50 uL of PBS and incubated for 1 h at +4 °C with slow rotation. Then, the supernatants were removed using a magnetic separator, and the SPMB-antibody complexes were washed twice with PBS, then mixed with a suspension of EVs (50 uL) isolated from 0.5 mL of plasma. Incubation was carried out overnight at +4 °C with slow rotation. After incubation, the supernatants were placed into clean tubes and used for a second round of isolation. Complexes of SPMB-antibodies-TPO(+)EVs were washed by PBS and used for further experiments (flow cytometry, dot-blot, or RNA isolation).
In order to estimate the efficacy of TPO(+)EV attachment onto the SPMBs, complexes of SPMB-antibodies-TPO(+)EV were blocked with 0.2% I-Block Protein-Based Blocking Reagent, washed twice by PBS, and then incubated with the monoclonal FITC-labelled antibody to CD63 (AbCam, Cambridge, MA, USA, ab8319) for 2 h at +4 °C in the dark. Complexes were washed twice, diluted in PBS (200 uL), and then analyzed with a flow cytometer, as described above. Once the efficacy of the TPO(+)EV isolation was confirmed, the SPMB-antibodies-TPO(+)EV complexes were directly used for total protein or RNA isolation with a RIPA buffer or the Lyra reagent, respectively.
2.6. RNA Isolation and miRNA Profiling
RNA from the total population of EVs or TPO(+)EVs was isolated by the Lyra reagent (Biolabmix Ltd., Novosibirsk, Russia, NO. LRU-100-50) in accordance with the producer’s protocol, which was assumed to preserve small RNA molecules and microRNA. The concentrations and purities of the extracted RNAs were analyzed with an Implen NanoPhotometr N60 spectrophotometer (Implen Gmb, Munchen, Germany). The RNA concentration ranged from 20 to 130 ng/μL. The preparations were considered pure, with absorption rates of A260/A280 above 1.8. For the purpose of preliminary miRNA screening, 10 samples were chosen from patients with FA and 10 samples from patients with FTC. Aliquots of individual RNA samples were combined in equivalent quantities into 2 pools, representing FA and FTC. In the first step, the RNA was polyadenylated and reverse transcribed using a miRCURY LNA Universal RT microRNA polyadenylation and cDNA synthesis kit (Qiagen, Germantown, MD, USA, cat. number 339340). Then, quantitative PCR analysis was performed using miRCURY LNA miRNA Focus PCR Panels (Qiagen, USA, cat. number 339325) and an ExiLENT SYBR Green Master Mix (Qiagen, USA, cat. number 339345) with a CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, Hercules, CA, USA). Inter-plate amplification rate discrepancies were corrected with interpolated calibrators. Values of Ct higher than 38 were considered as background values and were excluded from the analysis.
2.7. Quantitative Real-Time RT-PCR (qPCR)
For the evaluation of selected miRNAs (Let-7 family) and miR-191 expression, miRNA-specific reverse transcription with stem-looped primers was used, followed by quantitative PCR. The IDs of the miRNAs, sequences of primers, and FAM-labeled probes were presented as
Table S1. Reverse transcription (RT) assays were performed using 1 uL RNA, a RT primers (50 pM) using a M-MuLV-RH RT kit (Biolabmix Ltd., Novosibirsk) with a 20 uL reaction volume. The reaction was carried out at 25 °C for 45 min, followed by incubation at 85 °C for 5 min to inactivate the reverse transcriptase. Quantitative PCR analysis was performed using a 3 uL RT reaction mix, PCR primers (0.3 uM for both), FAM-labeled probe (0.2 uM) with a BioMaster HS-qPCR (2×) kit (Biolabmix Ltd., Novosibirsk). A CFX96 Touch™ Real-Time PCR Detection System (Bio-Rad, USA) was used for all reactions. The conditions for qPCR were as follows: Temperature of 95 °C for 10 min and 45 cycles of 95 °C for 5 s, followed by 65 °C for 15 s. RT-qPCR reactions for each miRNA molecule were repeated independently twice and then averaged. The results of the RT-qPCR were normalized to the total mean of cycle threshold (Ct) and to the reference Ct of miR-191 independently. The obtained data were analyzed with a CFX Manager, SigmaPlot 11.0, and GraphPad Prism 8.0.2.
2.8. Statistics
Statistical evaluation of the differences between comparable groups of samples was done with Mann-Whitney or Kruskal Wallis tests assuming a non-parametric distribution of measured parameters.
4. Discussion
Thyroid cancer’s mortality has increased over the last years that indicates the need to develop new diagnostic approaches. The advantages of new immuno-histochemical (Hector Battifora mesothelial cell 1 (HBME-1), high molecular weight cytokeratin 19 (CK19), galectin-3, c-met) and genetic (TERT, BRAF, PAX8/PPARγ, RAS, and RET/PTC) markers to be tested in the material of cytological or histological biopsy are discussed explicitly [
21,
22,
23]. The development of the noninvasive diagnostic tools for analysis of the circulating plasma components, also known as “liquid biopsy”, brings a new insight for thyroid cancer diagnosis. Thus, traditional markers of thyroid cancer, like BRAF mutation or miRNA expression profile, can be evaluated in plasma [
24]. The circulating DNA methylation pattern of some iodine transporters and DNA methyltransferase were tested as a promising diagnostic biomarker of thyroid cancer as well [
25]. The list of candidates for “liquid biopsy” markers of thyroid cancer includes circulating tumor cell (CTC), exosomes, and extrachromosomal circular DNA (ecDNA) [
6], that indicates active research in this area. Exosomes, important mediators of intercellular communications involved in the development of various thyroid disorders, including auto-immune pathologies [
14] and cancer [
26], are considered to present a new marker for liquid biopsy application.
The first investigations proposing the diagnostic value of circulating extracellular nano-vesicles or exosomes were published many years ago [
27]. Since then, hundreds or even thousands of studies relevant to vesicular markers of different types of cancer have come out. Diagnostic potency of plasma exosomes in thyroid cancer has been explored in several studies [
9,
28]. However, EV-based methods for cancer diagnosis, prognosis, or management, are still far from clinical implementation. This is caused by obvious technological issues related to EV isolation and analysis. Confident results are still hardly achievable, even with ISEV’s regular efforts to standardize research in this area [
29,
30]. Besides methodological aspects, important biological issues need to be addressed before circulating EVs can be used as feasible cancer diagnostic markers. First of all, the population of circulating nano-vesicles is too complex and heterogenous in terms of the cellular origin to reflect the condition of certain tissues, for example, when focusing on malignant transformation. The development of an approach to isolate fractions of EVs released by cells of certain tissues presents an extremely important issue.
In the presented study, we have attempted to isolate thyroid-derived EVs using a single surface marker, thyroid peroxidase, which is expressed by thyroid follicular cells. Our result reveals the feasibility of isolating TPO(+)EVs from only 2 mL of plasma in an amount sufficient for further analysis. The proposed strategy is very promising because it can be applied for the diagnosis of other types of thyroid cancer or even other thyroid pathologies. However, several methodological issues should still be considered. First, the expression of TPO is not absolutely restricted by the thyroid gland [
31], and a fraction of the EVs isolated by the proposed technology is enriched by (rather than consists of) thyroid-derived vesicles. The efficacy of the thyroid-derived EV isolation can be optimized by using the ligands to other thyroid-specific molecules. For instance, The protein atlas provides 13 thyroid-enriched genes, including TPO and TSH-Rec [
17]. Moreover, it can be assumed that thyroid-derived vesicles present a miserable fraction of the total population of plasma EV. Thus, the optimal ratio between the specificity and the efficacy of the thyroid-derived EV isolation will result in the necessary purity and quantity of the isolated vesicles. This issue needs to be addressed experimentally.
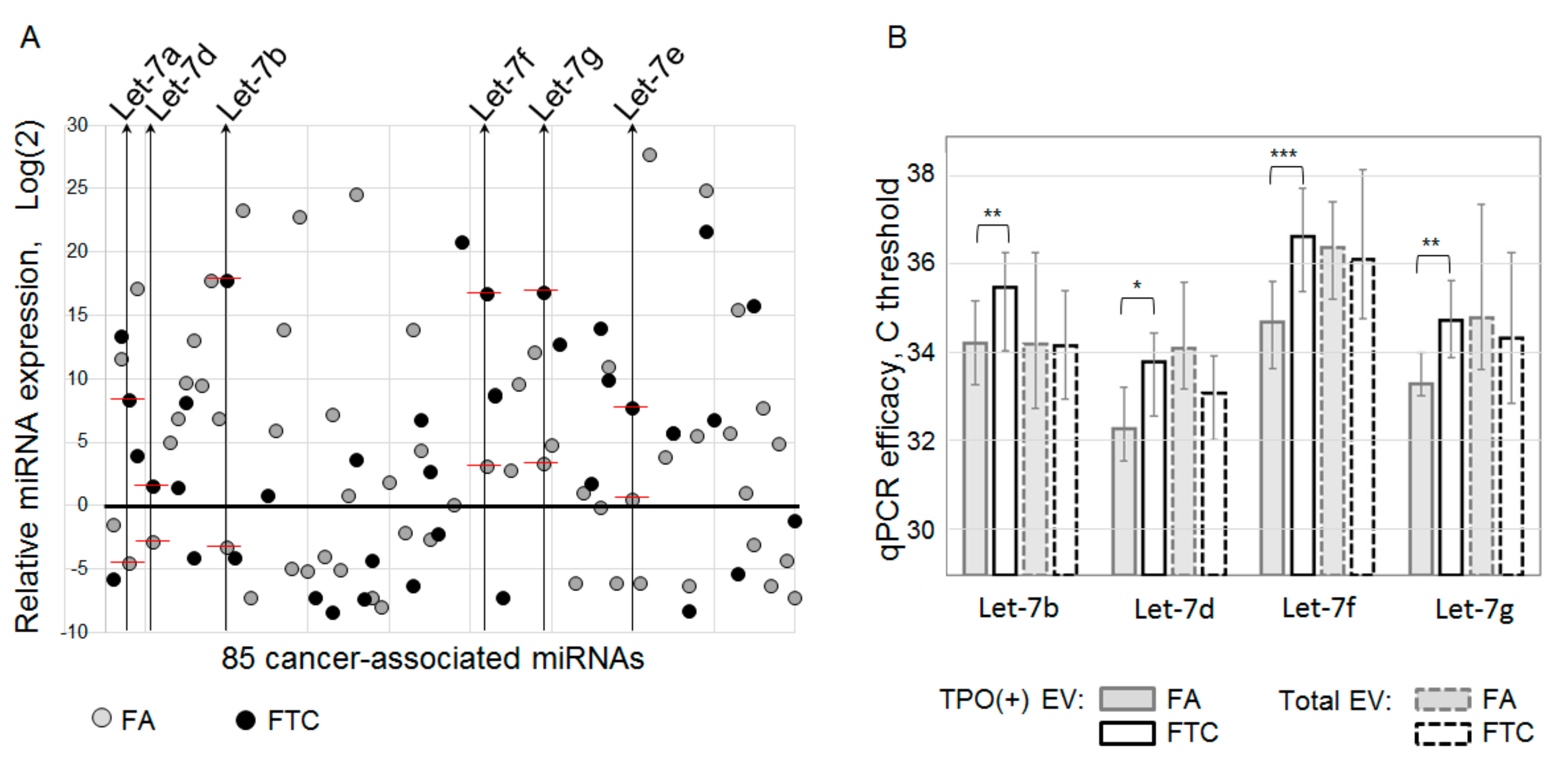
The list of analyzed components of thyroid-derived EVs present one more disputable aspect. In the presented study, we have analyzed vesicular miRNAs only, and our profiling assay was limited by only 85 cancer-associated miRNAs. This definitely restricts the diagnostic potency of the proposed method. In addition to the Let-7 miRNA family, FTC-relevant miRNAs, other types of vesicular RNA, or other vesicular components can serve as a marker of follicular cell malignancy. Moreover, an increase in the plasma level of Let-7 miRNAs was also associated with papillary thyroid cancer [
19]. These data and the design of our study do not allow us to argue about the diagnostic specificity of Let-7, even isolated from TPO(+)EV. Thus, the diagnostic potency of the proposed method can be augmented by the analysis of larger lists of molecules associated with particular thyroid pathologies.
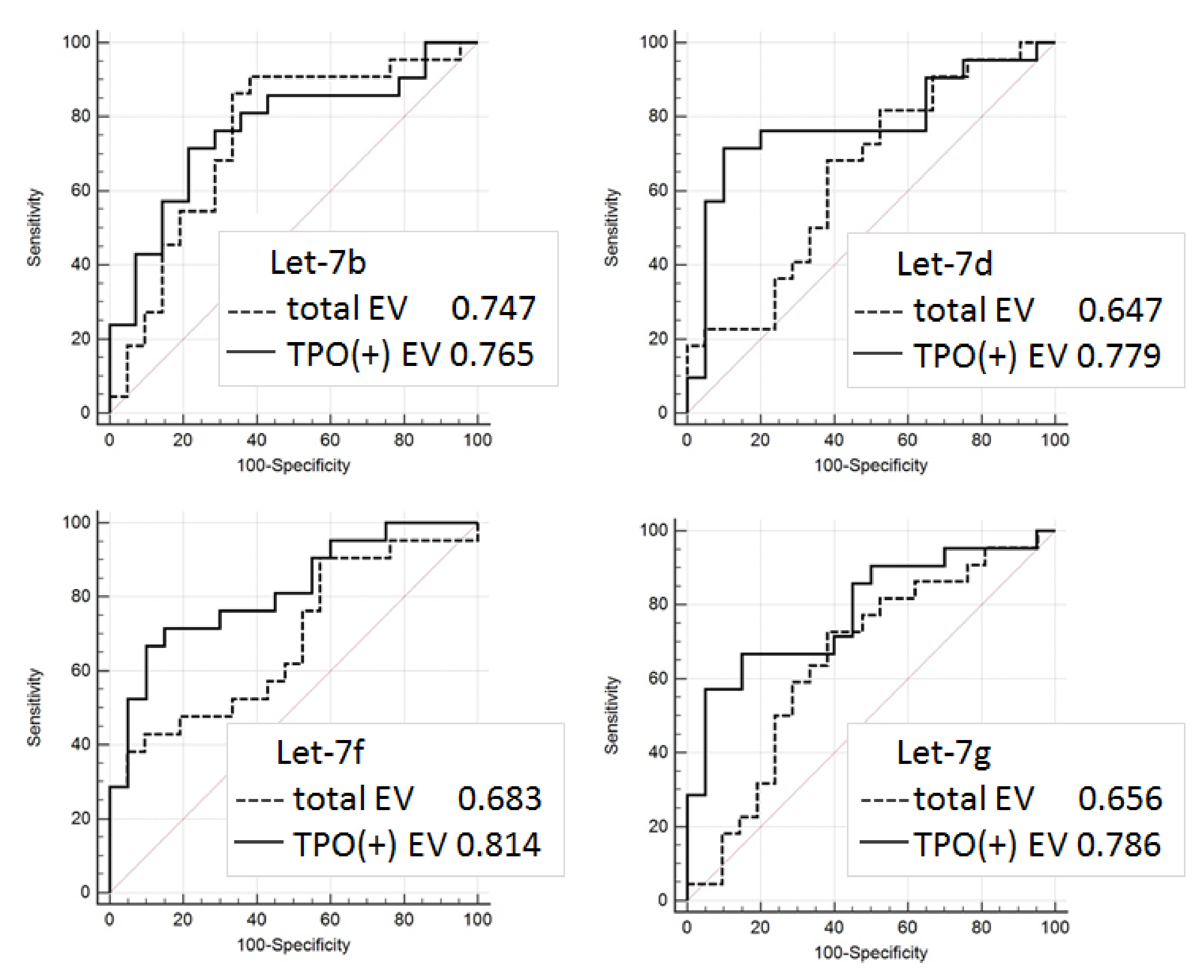
Even considering the limitations mentioned above, the proposed approach allowed us to discriminate FTC from FA with quite a high accuracy (AUC = 0.81). The results obtained are still not reliable enough to be recommended for practical implementations, however, they confirm the validity of the proposed analytical approach and provide consolidation for further research. Diagnostic potency of the described method can be obviously improved by the use of multiple thyroid-specific ligands for EVs isolation and by the analysis of various EV components associated with specific thyroid diseases.


 ,
, 












{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}