Discovery of Targetable Genetic Alterations in NSCLC Patients with Different Metastatic Patterns Using a MassARRAY-Based Circulating Tumor DNA Assay

 , , , and
, , , and
Abstract
1. Introduction
2. Materials and Methods
2.1. Patients and Blood Samples
2.2. Plasma Isolation and cfDNA Extraction
3. Results
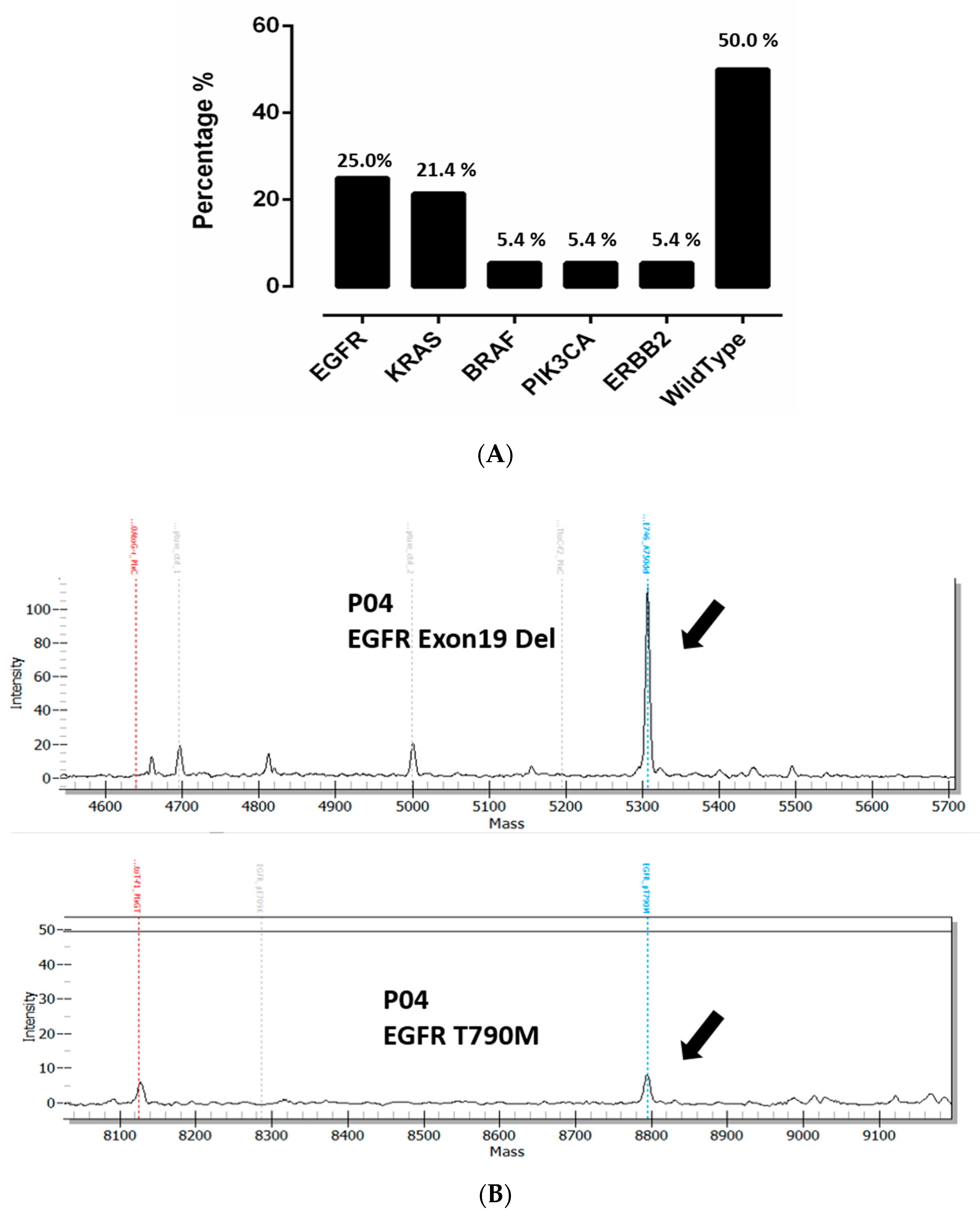
3.1. Overall Detection of Mutations in Cell Free DNA Using UltraSEEK™ Lung Panel in Advanced NSCLC Patients
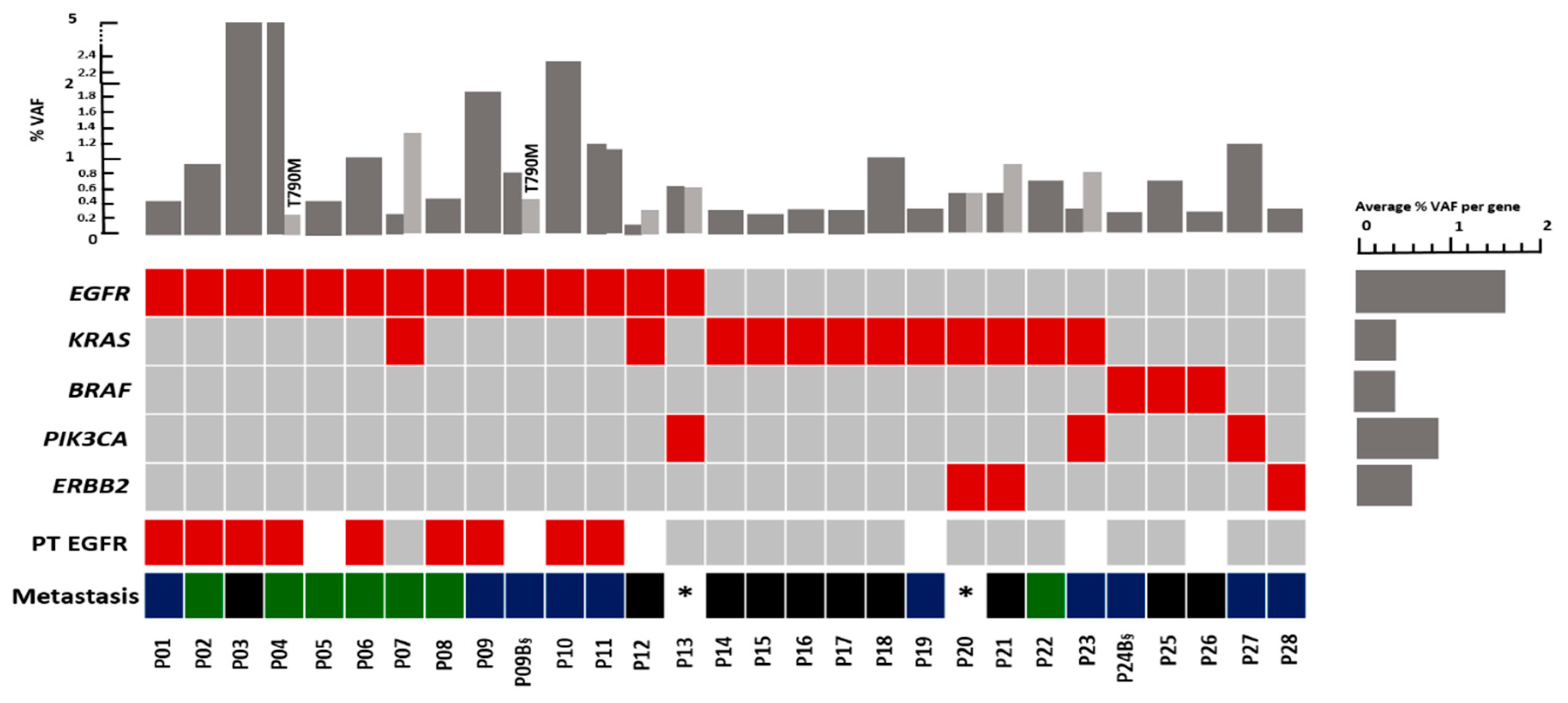
3.2. Distribution of Mutations in the Plasma of NSCLC Patients with Different Metastatic Patterns
3.3. Comparison of EGFR Mutation Status in Tumor Tissue and Plasma
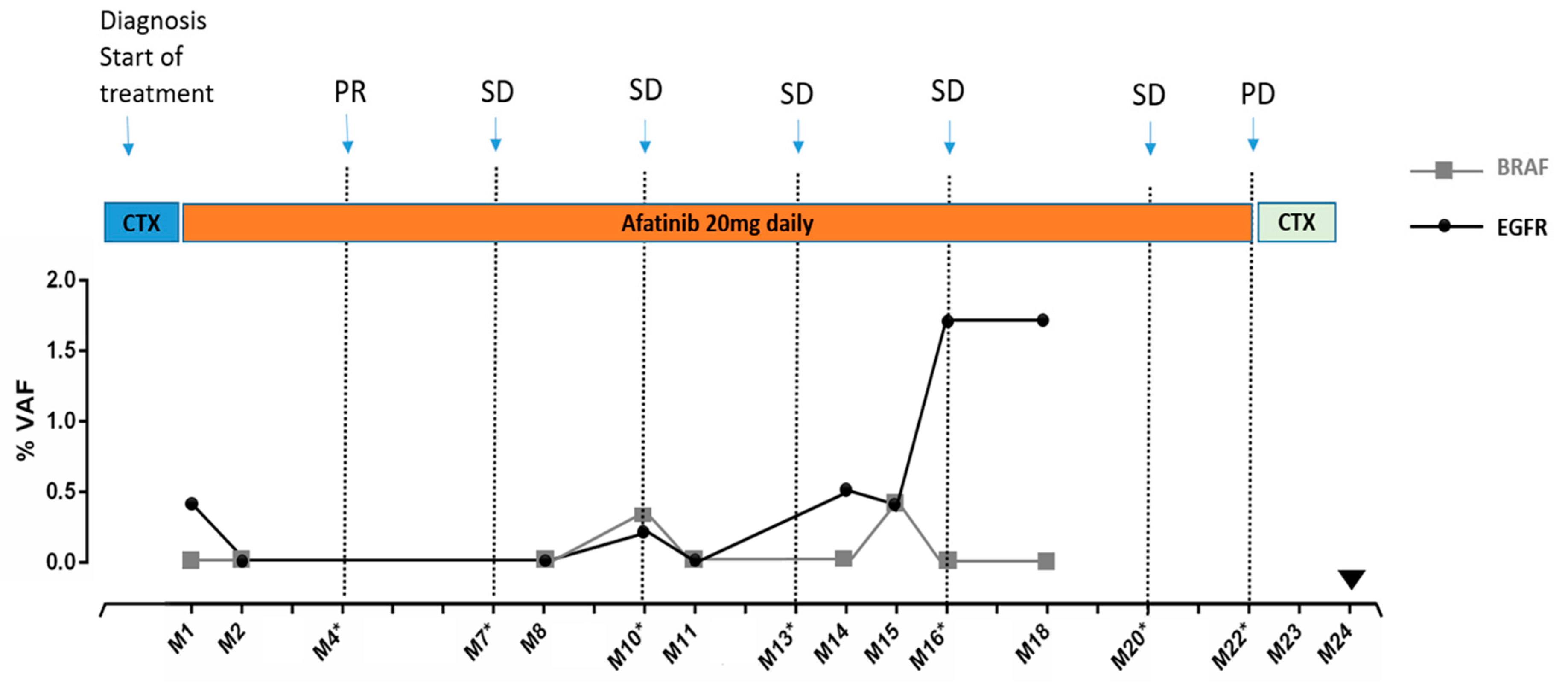
3.4. Monitoring Patient’s Disease by Tracking Mutations in Plasma ctDNA: A Case Report
4. Discussion
5. Conclusions
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Bray, F.; Me, J.F.; Soerjomataram, I.; Siegel, R.L.; Torre, L.A.; Jemal, A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J. Clin. 2018, 68, 394–424. [Google Scholar] [CrossRef] [PubMed]
- Heigener, D.F.; Kerr, K.M.; Laing, G.M.; Mok, T.; Moiseyenko, F.V.; Reck, M. Redefining Treatment Paradigms in First-line Advanced Non-Small-Cell Lung Cancer. Clin. Cancer Res. 2019, 25, 4881–4887. [Google Scholar] [CrossRef] [PubMed]
- Boire, A.; Brastianos, P.K.; Garzia, L.; Valiente, M. Brain metastasis. Nat. Rev. Cancer 2020, 20, 4–11. [Google Scholar] [CrossRef] [PubMed]
- Hohensee, I.; Lamszus, K.; Riethdorf, S.; Meyer-Staeckling, S.; Glatzel, M.; Matschke, J.; Witzel, I.; Westphal, M.; Brandt, B.; Müller, V.; et al. Frequent Genetic Alterations in EGFR- and HER2-Driven Pathways in Breast Cancer Brain Metastases. Am. J. Pathol. 2013, 183, 83–95. [Google Scholar] [CrossRef] [PubMed]
- Brastianos, P.K.; Carter, S.L.; Santagata, S.; Cahill, D.P.; Taylor-Weiner, A.; Jones, R.T.; van Allen, E.M.; Lawrence, M.S.; Horowitz, P.M.; Cibulskis, K.; et al. Genomic Characterization of Brain Metastases Reveals Branched Evolution and Potential Therapeutic Targets. Cancer Discov. 2015, 5, 1164–1177. [Google Scholar] [CrossRef] [PubMed]
- Shih, D.J.H.; Nayyar, N.; Bihun, I.; Dagogo-Jack, I.; Gill, C.M.; Aquilanti, E.; Bertalan, M.; Kaplan, A.; D’Andrea, M.R.; Chukwueke, U.; et al. Genomic characterization of human brain metastases identifies drivers of metastatic lung adenocarcinoma. Nat. Genet. 2020, 52, 371–377. [Google Scholar] [CrossRef] [PubMed]
- Heitzer, E.; Haque, I.S.; Roberts, C.E.S.; Speicher, M.R. Current and future perspectives of liquid biopsies in genomics-driven oncology. Nat. Rev. Genet. 2018, 20, 71–88. [Google Scholar] [CrossRef] [PubMed]
- Marques, J.F.; Reis, J.P.; Fernandes, G.; Hespanhol, V.; Machado, J.C.; Costa, J.L. Circulating Tumor DNA: A Step into the Future of Cancer Management. Acta Cytol. 2019, 63, 456–465. [Google Scholar]
- Wan, J.C.M.; Massie, C.; Garcia-Corbacho, J.; Mouliere, F.; Brenton, J.D.; Caldas, C.M.; Pacey, S.; Baird, R.; Rosenfeld, N. Liquid biopsies come of age: Towards implementation of circulating tumour DNA. Nat. Rev. Cancer 2017, 17, 223–238. [Google Scholar] [CrossRef]
- Keller, L.; Belloum, Y.; Wikman, H.; Pantel, K. Clinical relevance of blood-based ctDNA analysis: Mutation detection and beyond. Br. J. Cancer 2020, 1–14. [Google Scholar] [CrossRef]
- Keller, L.; Pantel, K. Unravelling tumour heterogeneity by single-cell profiling of circulating tumour cells. Nat. Rev. Cancer 2019, 19, 553–567. [Google Scholar] [CrossRef] [PubMed]
- Pantel, K.; Alix-Panabières, C. Liquid biopsy and minimal residual disease—latest advances and implications for cure. Nat. Rev. Clin. Oncol. 2019, 16, 409–424. [Google Scholar] [CrossRef] [PubMed]
- Gerlinger, M.; Rowan, A.J.; Horswell, S.; Larkin, J.; Endesfelder, D.; Gronroos, E.; Martinez, P.; Matthews, N.; Stewart, A.; Tarpey, P.; et al. Intratumor Heterogeneity and Branched Evolution Revealed by Multiregion Sequencing. N. Engl. J. Med. 2012, 366, 883–892. [Google Scholar] [CrossRef] [PubMed]
- Murtaza, M.; Dawson, S.-J.; Pogrebniak, K.; Rueda, O.M.; Provenzano, E.; Grant, J.; Chin, S.-F.; Tsui, D.W.Y.; Marass, F.; Gale, D.; et al. Multifocal clonal evolution characterized using circulating tumour DNA in a case of metastatic breast cancer. Nat. Commun. 2015, 6, 8760. [Google Scholar] [CrossRef]
- Marusyk, A.; Janiszewska, M.; Polyak, K. Intratumor Heterogeneity: The Rosetta Stone of Therapy Resistance. Cancer Cell 2020, 37, 471–484. [Google Scholar] [CrossRef]
- Oxnard, G.R.; Thress, K.S.; Alden, R.S.; Lawrance, R.; Paweletz, C.P.; Cantarini, M.; Yang, J.C.-H.; Barrett, J.C.; Jänne, P.A. Association Between Plasma Genotyping and Outcomes of Treatment with Osimertinib (AZD9291) in Advanced Non–Small-Cell Lung Cancer. J. Clin. Oncol. 2016, 34, 3375–3382. [Google Scholar] [CrossRef]
- Planchard, D.; Popat, S.; Kerr, K.; Novello, S.; Smit, E.; Faivre-Finn, C.; Mok, T.; Reck, M.; van Schil, P.; Hellmann, M.; et al. Metastatic non-small cell lung cancer: ESMO Clinical Practice Guidelines for diagnosis, treatment and follow-up. Ann. Oncol. 2018, 29, iv192–iv237. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Alix-Panabières, C.; Müller, I.; Letang, N.; Vendrell, J.P.; Rebillard, X.; Pantel, K. Cell-free Tumor DNA in Blood Plasma as a Marker for Circulating Tumor Cells in Prostate Cancer. Clin. Cancer Res. 2009, 15, 1032–1038. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Hoon, D.S.B.; Pantel, K. Cell-free nucleic acids as biomarkers in cancer patients. Nat. Rev. Cancer 2011, 11, 426–437. [Google Scholar] [CrossRef]
- Schwarzenbach, H.; Stoehlmacher, J.; Pantel, K.; Goekkurt, E. Detection and Monitoring of Cell-Free DNA in Blood of Patients with Colorectal Cancer. Ann. N. Y. Acad. Sci. 2008, 1137, 190–196. [Google Scholar] [CrossRef]
- Salvianti, F.; Pinzani, P.; Verderio, P.; Ciniselli, C.M.; Massi, D.; de Giorgi, V.; Grazzini, M.; Pazzagli, M.; Orlando, C. Multiparametric Analysis of Cell-Free DNA in Melanoma Patients. PLoS ONE 2012, 7, e49843. [Google Scholar] [CrossRef] [PubMed]
- Qin, Z.; Ljubimov, V.A.; Zhou, C.; Tong, Y.; Liang, J. Cell-free circulating tumor DNA in cancer. Chin. J. Cancer 2016, 35, 1–9. [Google Scholar] [CrossRef]
- Smith, C.G.; Moser, T.; Mouliere, F.; Field-Rayner, J.; Eldridge, M.; Riediger, A.L.; Chandrananda, D.; Heider, K.; Wan, J.C.M.; Warren, A.Y.; et al. Comprehensive characterization of cell-free tumor DNA in plasma and urine of patients with renal tumors. Genome Med. 2020, 12, 17–23. [Google Scholar] [CrossRef] [PubMed]
- Elazezy, M.; Joosse, S.A. Techniques of using circulating tumor DNA as a liquid biopsy component in cancer management. Comput. Struct. Biotechnol. J. 2018, 16, 370–378. [Google Scholar] [CrossRef] [PubMed]
- Steendam, C.M.; Atmodimedjo, P.; de Jonge, E.; Paats, M.S.; van der Leest, C.; Hoop, E.O.D.; Jansen, M.P.; del Re, M.; von der Thüsen, J.H.; Dinjens, W.N.; et al. Plasma Cell-Free DNA Testing of Patients with EGFR Mutant Non–Small-Cell Lung Cancer: Droplet Digital PCR Versus Next-Generation Sequencing Compared with Tissue-Based Results. JCO Precis. Oncol. 2019, 2019, 1–9. [Google Scholar] [CrossRef]
- Jiang, X.; Liu, W.; Zhu, X.; Xu, X. Evaluation of EGFR mutations in NSCLC with highly sensitive droplet digital PCR assays. Mol. Med. Rep. 2019, 20, 593–603. [Google Scholar] [CrossRef]
- Zhu, G.; Ye, X.; Dong, Z.; Lu, Y.C.; Sun, Y.; Liu, Y.; McCormack, R.; Gu, Y.; Liu, X. Highly Sensitive Droplet Digital PCR Method for Detection of EGFR-Activating Mutations in Plasma Cell–Free DNA from Patients with Advanced Non–Small Cell Lung Cancer. J. Mol. Diagn. 2015, 17, 265–272. [Google Scholar] [CrossRef]
- Hellman, S.; Weichselbaum, R.R. Oligometastases. J. Clin. Oncol. 1995, 13, 8–10. [Google Scholar] [CrossRef] [PubMed]
- Weber, S.; Spiegl, B.; Perakis, S.; Ulz, C.M.; Abuja, P.M.; Kashofer, K.; van der Leest, P.; Azpurua, M.A.; Tamminga, M.; Brudzewsky, D.; et al. Technical Evaluation of Commercial Mutation Analysis Platforms and Reference Materials for Liquid Biopsy Profiling. Cancers 2020, 12, 1588. [Google Scholar] [CrossRef] [PubMed]
- Mosko, M.J.; Nakorchevsky, A.A.; Flores, E.; Metzler, H.; Ehrich, M.; Boom, D.J.V.D.; Sherwood, J.L.; Nygren, A.O. Ultrasensitive Detection of Multiplexed Somatic Mutations Using MALDI-TOF Mass Spectrometry. J. Mol. Diagn. 2016, 18, 23–31. [Google Scholar] [CrossRef]
- Collison, A.E.; Campbell, J.D.; Brooks, A.N.; Berger, A.H.; Lee, W.; Chmielecki, J.; Beer, D.G.; Cope, L.; Creighton, C.J.; Danilova, L.; et al. Comprehensive molecular profiling of lung adenocarcinoma. Nat. Cell Biol. 2014, 511, 543–550. [Google Scholar]
- Sacher, A.G.; Paweletz, C.; Dahlberg, S.E.; Alden, R.S.; O’Connell, A.; Feeney, N.; Mach, S.L.; Jänne, P.A.; Oxnard, G.R. Prospective Validation of Rapid Plasma Genotyping for the Detection ofEGFRandKRASMutations in Advanced Lung Cancer. JAMA Oncol. 2016, 2, 1014–1022. [Google Scholar] [CrossRef] [PubMed]
- Oxnard, G.R.; Paweletz, C.P.; Kuang, Y.; Mach, S.L.; O’Connell, A.; Messineo, M.M.; Luke, J.J.; Butaney, M.; Kirschmeier, P.; Jackman, D.M.; et al. Noninvasive Detection of Response and Resistance in EGFR-Mutant Lung Cancer using Quantitative Next-Generation Genotyping of Cell-Free Plasma DNA. Clin. Cancer Res. 2014, 20, 1698–1705. [Google Scholar] [CrossRef] [PubMed]
- Hanssen, A.; Riebensahm, C.; Mohme, M.; Joosse, S.A.; Velthaus, J.-L.; Berger, L.A.; Bernreuther, C.; Glatzel, M.; Loges, S.; Lamszus, K.; et al. Frequency of Circulating Tumor Cells (CTC) in Patients with Brain Metastases: Implications as a Risk Assessment Marker in Oligo-Metastatic Disease. Cancers 2018, 10, 527. [Google Scholar] [CrossRef]
- Riebensahm, C.; Joosse, S.A.; Mohme, M.; Hanssen, A.; Matschke, J.; Goy, Y.; Witzel, I.; Lamszus, K.; Kropidlowski, J.; Petersen, C.; et al. Clonality of circulating tumor cells in breast cancer brain metastasis patients. Breast Cancer Res. 2019, 21, 1–11. [Google Scholar] [CrossRef]
- Piccioni, D.E.; Achrol, A.S.; Kiedrowski, L.A.; Banks, K.C.; Boucher, N.; Barkhoudarian, G.; Kelly, D.F.; Juarez, T.; Lanman, R.B.; Raymond, V.M.; et al. Analysis of cell-free circulating tumor DNA in 419 patients with glioblastoma and other primary brain tumors. CNS Oncol. 2019, 8, CNS34. [Google Scholar] [CrossRef]
- Aldea, M.; Hendriks, L.; Mezquita, L.; Jovelet, C.; Planchard, D.; Auclin, E.; Remon, J.; Howarth, K.; Benitez, J.C.; Gazzah, A.; et al. Circulating Tumor DNA Analysis for Patients with Oncogene-Addicted NSCLC with Isolated Central Nervous System Progression. J. Thorac. Oncol. 2020, 15, 383–391. [Google Scholar] [CrossRef]
- Miller, A.M.; Shah, R.H.; Pentsova, E.I.; Pourmaleki, M.; Briggs, S.; Distefano, N.; Zheng, Y.; Skakodub, A.; Mehta, S.A.; Campos, C.; et al. Tracking tumour evolution in glioma through liquid biopsies of cerebrospinal fluid. Nat. Cell Biol. 2019, 565, 654–658. [Google Scholar] [CrossRef]
- Pan, C.; Diplas, B.H.; Chen, X.; Wu, Y.; Xiao, X.; Jiang, L.; Geng, Y.; Xu, C.; Sun, Y.; Zhang, P.; et al. Molecular profiling of tumors of the brainstem by sequencing of CSF-derived circulating tumor DNA. Acta Neuropathol. 2018, 137, 297–306. [Google Scholar] [CrossRef]
- Zhao, Z.; Zhang, C.; Li, M.; Shen, Y.; Feng, S.; Liu, J.; Li, F.; Hou, L.; Chen, Z.; Jiang, J.; et al. Applications of cerebrospinal fluid circulating tumor DNA in the diagnosis of gliomas. Jpn. J. Clin. Oncol. 2020, 50, 325–332. [Google Scholar] [CrossRef]
- Ma, C.; Yang, X.; Xing, W.; Yu, H.; Si, T.; Guo, Z. Detection of circulating tumor DNA from non-small cell lung cancer brain metastasis in cerebrospinal fluid samples. Thorac. Cancer 2020, 11, 588–593. [Google Scholar] [CrossRef] [PubMed]
- Huang, R.; Xu, X.; Li, D.; Chen, K.; Zhan, Q.; Ge, M.; Zhou, X.; Liang, X.; Guan, M. Digital PCR-Based Detection of EGFR Mutations in Paired Plasma and CSF Samples of Lung Adenocarcinoma Patients with Central Nervous System Metastases. Target. Oncol. 2019, 14, 343–350. [Google Scholar] [CrossRef] [PubMed]
- Kuang, Y.; Rogers, A.; Yeap, B.Y.; Wang, L.; Makrigiorgos, M.; Vetrand, K.; Thiede, S.; Distel, R.J.; Janne, P.A. Noninvasive Detection of EGFR T790M in Gefitinib or Erlotinib Resistant Non–Small Cell Lung Cancer. Clin. Cancer Res. 2009, 15, 2630–2636. [Google Scholar] [CrossRef]
- Berger, L.A.; Janning, M.; Velthaus, J.L.; Ben-Batalla, I.; Schatz, S.; Falk, M.; Iglauer, P.; Simon, R.; Cao, R.; Forcato, C.; et al. Identification of a High-Level MET Amplification in CTCs and cfTNA of an ALK-Positive NSCLC Patient Developing Evasive Resistance to Crizotinib. J. Thorac. Oncol. 2018, 13, e243–e246. [Google Scholar] [CrossRef] [PubMed]
- Karachaliou, N.; Casas, C.M.D.L.; Queralt, C.; de Aguirre, I.; Melloni, B.; Cardenal, F.; Garcia-Gomez, R.; Massuti, B.; Sánchez, J.M.; Porta, R.; et al. Association ofEGFRL858R Mutation in Circulating Free DNA with Survival in the EURTAC Trial. JAMA Oncol. 2015, 1, 149–157. [Google Scholar] [CrossRef] [PubMed]
- Vendrell, J.A.; Mau-Them, F.T.; Béganton, B.; Godreuil, S.; Coopman, P.; Solassol, J. Circulating Cell Free Tumor DNA Detection as a Routine Tool forLung Cancer Patient Management. Int. J. Mol. Sci. 2017, 18, 264. [Google Scholar] [CrossRef]
- Kriegsmann, M.; Wandernoth, P.; Lisenko, K.; Casadonte, R.; Longuespée, R.; Arens, N.; Kriegsmann, J. Detection of HPV subtypes by mass spectrometry in FFPE tissue specimens: A reliable tool for routine diagnostics. J. Clin. Pathol. 2016, 70, 417–423. [Google Scholar] [CrossRef]
- Toomey, S.; Carr, A.; Mezynski, M.J.; Elamin, Y.; Rafee, S.; Cremona, M.; Morgan, C.; Madden, S.; Abdul-Jalil, K.I.; Gately, K.; et al. Identification and clinical impact of potentially actionable somatic oncogenic mutations in solid tumor samples. J. Transl. Med. 2020, 18, 1–14. [Google Scholar] [CrossRef]
- Scott, S.A.; Scott, E.R.; Seki, Y.; Chen, A.J.; Wallsten, R.; Obeng, A.O.; Botton, M.R.; Cody, N.; Shi, H.; Zhao, G.; et al. Development and Analytical Validation of a 29 Gene Clinical Pharmacogenetic Genotyping Panel: Multi-Ethnic Allele and Copy Number Variant Detection. Clin. Transl. Sci. 2020. [Google Scholar] [CrossRef]
- Miller, J.K.; Buchner, N.; Timms, L.; Tam, S.; Luo, X.; Brown, A.M.; Pasternack, D.; Bristow, R.G.; Fraser, M.; Boutros, P.C.; et al. Use of Sequenom sample ID Plus(R) SNP genotyping in identification of FFPE tumor samples. PLoS ONE 2014, 9, e88163. [Google Scholar] [CrossRef]
- Wandernoth, P.; Kriegsmann, K.; Groh-Mohanu, C.; Daeumer, M.; Gohl, P.; Harzer, O.; Kriegsmann, M.; Kriegsmann, J. Detection of Severe Acute Respiratory Syndrome Coronavirus 2 (SARS-CoV-2) by Mass Spectrometry. Viruses 2020, 12, 849. [Google Scholar] [CrossRef] [PubMed]

 Mutation detected;
Mutation detected;  No mutation detected;
No mutation detected;  Not determined;
Not determined;  Oligo-brain metastases;
Oligo-brain metastases;  Multi-brain metastases;
Multi-brain metastases;  Other metastases; VAF: Variant allele frequency, * Brain metastases in these patients was not documented. §: Follow-up sample, only the first mutation was considered.
Mutation detected; No mutation detected; Not determined; Oligo-brain metastases; Multi-brain metastases; Other metastases; VAF: Variant allele frequency, * Brain metastases in these patients was not documented. §: Follow-up sample, only the first mutation was considered.
Other metastases; VAF: Variant allele frequency, * Brain metastases in these patients was not documented. §: Follow-up sample, only the first mutation was considered.
Mutation detected; No mutation detected; Not determined; Oligo-brain metastases; Multi-brain metastases; Other metastases; VAF: Variant allele frequency, * Brain metastases in these patients was not documented. §: Follow-up sample, only the first mutation was considered.
 : Death of the patient. CTX: chemotherapy. Afatinib: 2nd generation EGFR inhibitor.
: Death of the patient. CTX: chemotherapy. Afatinib: 2nd generation EGFR inhibitor.
: Death of the patient. CTX: chemotherapy. Afatinib: 2nd generation EGFR inhibitor.
: Death of the patient. CTX: chemotherapy. Afatinib: 2nd generation EGFR inhibitor.

{kind=link}
{kind=link}
{kind=link}
| Characteristics | Number | Percentage | |
|---|---|---|---|
| Gender | Male | 26 | 46.4% |
| Female | 30 | 53.6% | |
| Histology | Adeno ca. | 49 | 87.5% |
| Squamous cell ca. | 5 | 8.9% | |
| other | 2 | 3.6% | |
| EGFR in tissue * | Mutant | 9 | 20.5% |
| Wild type | 35 | 79.5% | |
| Disease stage | First diagnosis | 39 | 69.6% |
| Progressive disease | 16 | 28.6% | |
| Complete response | 1 | 1.8% | |
| Metastases ** | Brain metastases | 37 | 66.1% |
| Other metastases than brain | 16 | 28.6% | |
| Unknown | 3 | 5.5% | |
| Brain- Metastases *** | Oligo-brain metastases | 20 | 54.0% |
| Multi-brain metastases | 16 | 43.2% | |
| Unknown | 1 | 2.7% |
| BRAF | EGFR | KRAS | ERBB2 | PIK3CA | Number of Pts | Median VAF | |
|---|---|---|---|---|---|---|---|
| n (%) | n (%) | n (%) | n (%) | n (%) | with Mutation (%) | of All Mutations | |
| Oligo-brain metastases (n = 20) * | 2 (10.0%) | 2 (10.0%) | 7 (35.0%) | 1 (5.0%) | 0 | 10 (50.0%) | 0.4 |
| Multi-brain metastases (n = 16) ** | 0 | 6 (37.5%) | 2 (12.5%) | 0 | 0 | 7 (43.8%) | 0.9 |
| Other metastases (n = 16) *** | 0 | 4 (25.0%) | 2 (12.5%) | 1 (6.3%) | 2 (12.5%) | 8 (50.0%) | 1.2 |
Publisher’s Note: MDPI stays neutral with regard to jurisdictional claims in published maps and institutional affiliations. |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Belloum, Y.; Janning, M.; Mohme, M.; Simon, R.; Kropidlowski, J.; Sartori, A.; Irwin, D.; Westphal, M.; Lamszus, K.; Loges, S.; et al. Discovery of Targetable Genetic Alterations in NSCLC Patients with Different Metastatic Patterns Using a MassARRAY-Based Circulating Tumor DNA Assay. Cells 2020, 9, 2337. https://doi.org/10.3390/cells9112337
Belloum Y, Janning M, Mohme M, Simon R, Kropidlowski J, Sartori A, Irwin D, Westphal M, Lamszus K, Loges S, et al. Discovery of Targetable Genetic Alterations in NSCLC Patients with Different Metastatic Patterns Using a MassARRAY-Based Circulating Tumor DNA Assay. Cells. 2020; 9(11):2337. https://doi.org/10.3390/cells9112337
Chicago/Turabian StyleBelloum, Yassine, Melanie Janning, Malte Mohme, Ronald Simon, Jolanthe Kropidlowski, Alexander Sartori, Darryl Irwin, Manfred Westphal, Katrin Lamszus, Sonja Loges, and et al. 2020. "Discovery of Targetable Genetic Alterations in NSCLC Patients with Different Metastatic Patterns Using a MassARRAY-Based Circulating Tumor DNA Assay" Cells 9, no. 11: 2337. https://doi.org/10.3390/cells9112337
APA StyleBelloum, Y., Janning, M., Mohme, M., Simon, R., Kropidlowski, J., Sartori, A., Irwin, D., Westphal, M., Lamszus, K., Loges, S., Riethdorf, S., Pantel, K., & Wikman, H. (2020). Discovery of Targetable Genetic Alterations in NSCLC Patients with Different Metastatic Patterns Using a MassARRAY-Based Circulating Tumor DNA Assay. Cells, 9(11), 2337. https://doi.org/10.3390/cells9112337