Considering Cause and Effect of Immune Cell Aging on Cardiac Repair after Myocardial Infarction


Abstract
1. Introduction
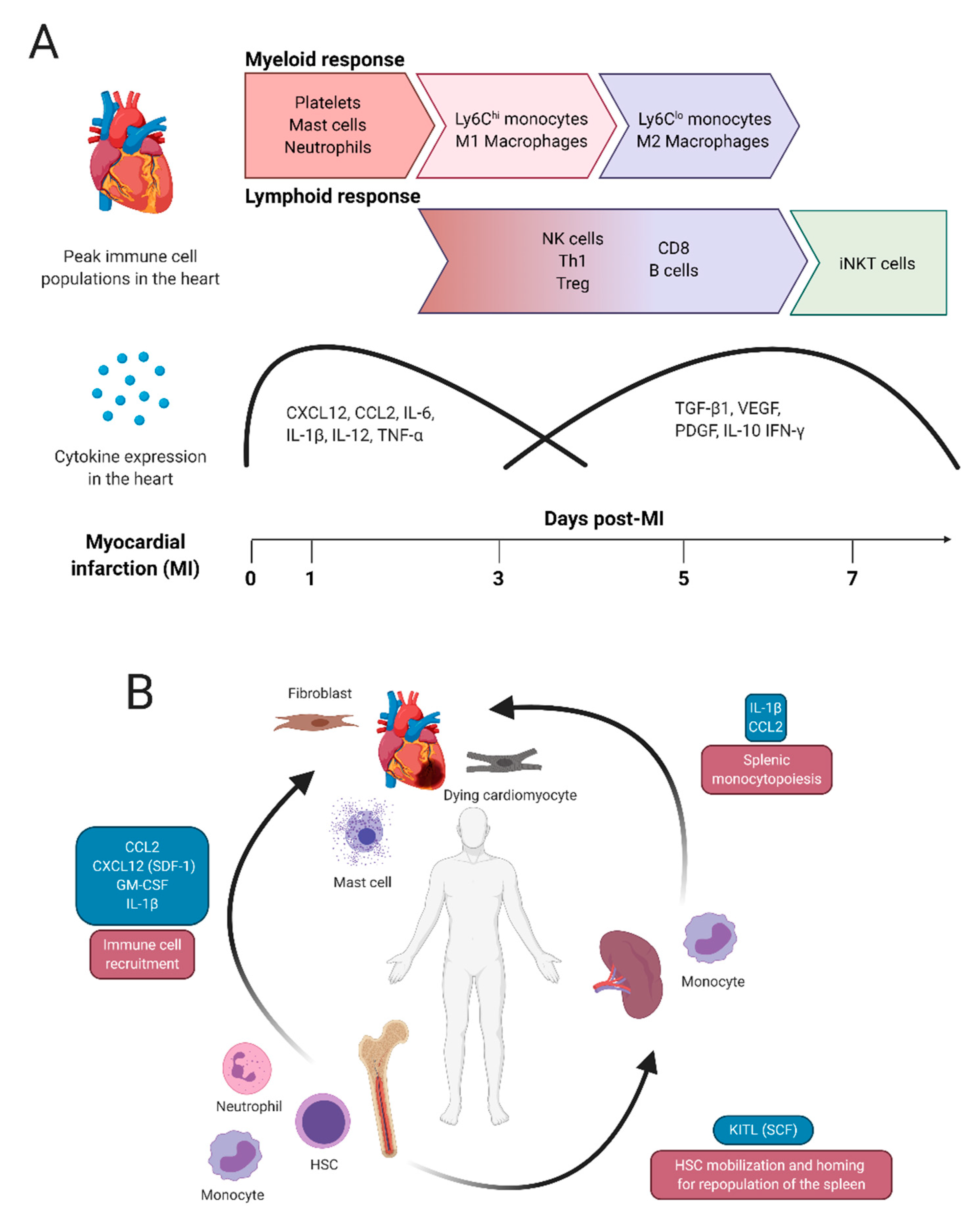
2. Typical Immune Responses after Myocardial Infarction
2.1. Myeloid Cell Activity after Myocardial Infarction
2.2. Lymphoid Cell Activity after Myocardial Infarction
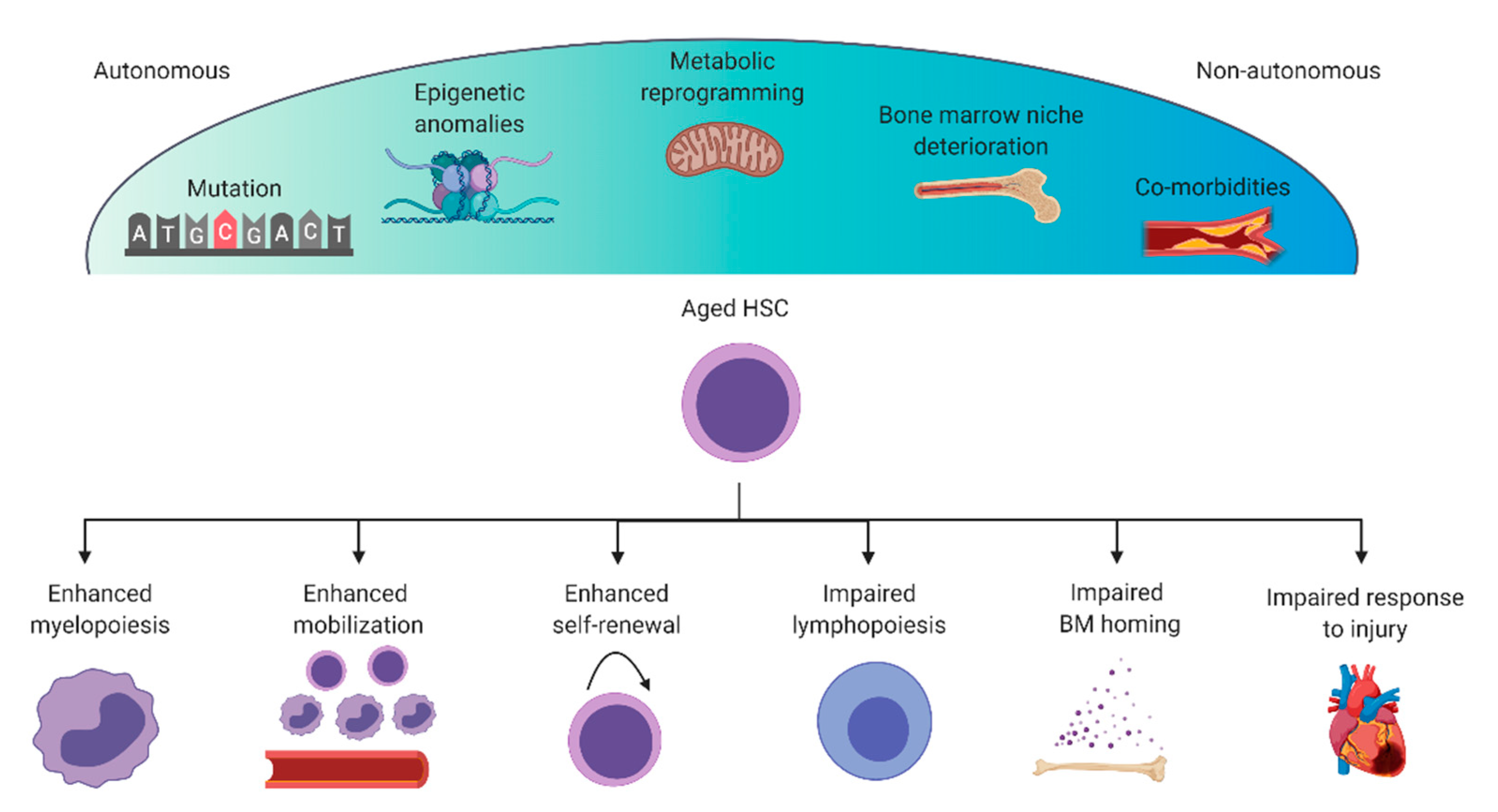
3. Dysregulation of the Immune System during Aging
3.1. Mutations and Epigenetic Anomalies
3.2. Deterioration of the Bone Marrow Niche
3.3. Metabolic Adaptations
3.4. Co-Morbidities and Sex
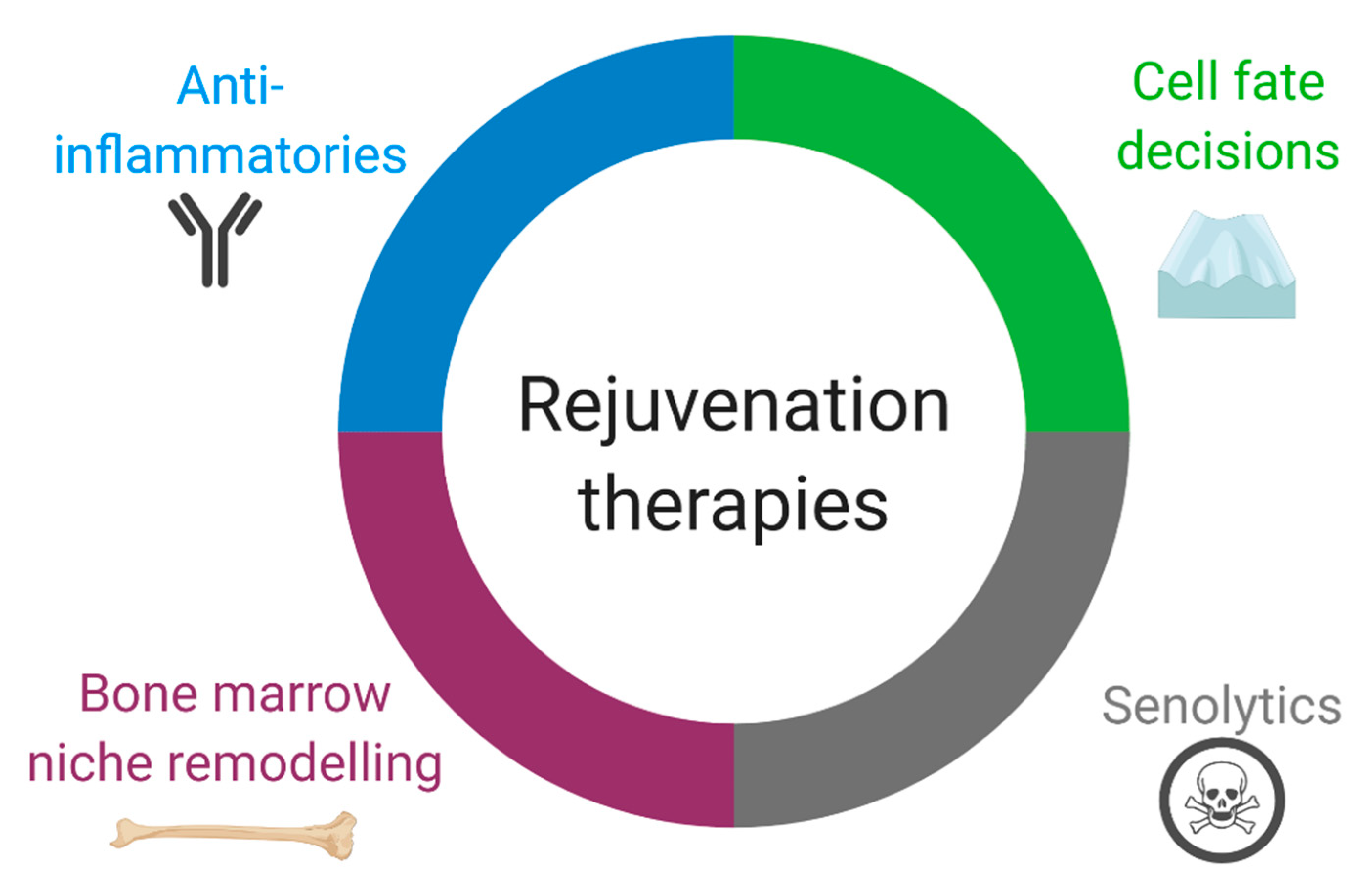
4. Consequences of HSC Aging on Heart Repair and Prospective Therapies
4.1. Anti-Inflammatories
4.2. Regulation of Immune Cell Quantity and Diversity by Directing Cell Fate Decisions
4.3. Bone Marrow Transplant and Niche Remodeling
4.4. Senolytics
5. Conclusions and Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Wei, L.; MacDonald, T.M.; Walker, B.R. Taking Glucocorticoids by Prescription Is Associated with Subsequent Cardiovascular Disease. Ann. Intern. Med. 2004, 141, 764. [Google Scholar] [CrossRef] [PubMed]
- Souverein, P.C.; Berard, A.; Van Staa, T.P.; Cooper, C.; Egberts, A.C.G.; Leufkens, H.G.M.; Walker, B.R. Use of oral glucocorticoids and risk of cardiovascular and cerebrovascular disease in a population based case-control study. Heart 2004, 90, 859–865. [Google Scholar] [CrossRef] [PubMed]
- Bally, M.; Dendukuri, N.; Rich, B.; Nadeau, L.; Helin-Salmivaara, A.; Garbe, E.; Brophy, J.M. Risk of acute myocardial infarction with NSAIDs in real world use: Bayesian meta-analysis of individual patient data. BMJ 2017, 357, j1909. [Google Scholar] [CrossRef] [PubMed]
- Zindel, J.; Kubes, P. DAMPs, PAMPs, and LAMPs in Immunity and Sterile Inflammation. Annu. Rev. Pathol. Mech. Dis. 2020, 15, 493–518. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M. Cardioimmunology: The immune system in cardiac homeostasis and disease. Nat. Rev. Immunol. 2018, 18, 733–744. [Google Scholar] [CrossRef]
- Prabhu, S.D.; Frangogiannis, N.G. The Biological Basis for Cardiac Repair after Myocardial Infarction. Circ. Res. 2016, 119, 91–112. [Google Scholar] [CrossRef]
- Pinto, A.R.; Ilinykh, A.; Ivey, M.J.; Kuwabara, J.T.; D’antoni, M.L.; Debuque, R.; Chandran, A.; Wang, L.; Arora, K.; Rosenthal, N.A.; et al. Revisiting cardiac cellular composition. Circ. Res. 2016, 118, 400–409. [Google Scholar] [CrossRef]
- Hulsmans, M.; Clauss, S.; Xiao, L.; Aguirre, A.D.; King, K.R.; Hanley, A.; Hucker, W.J.; Wülfers, E.M.; Seemann, G.; Courties, G.; et al. Macrophages Facilitate Electrical Conduction in the Heart. Cell 2017, 169, 510–522.e20. [Google Scholar] [CrossRef]
- Mendis, S.; Thygesen, K.; Kuulasmaa, K.; Giampaoli, S.; Mahonen, M.; Blackett, K.N.; Lisheng, L. World Health Organization definition of myocardial infarction: 2008–09 revision. Int. J. Epidemiol. 2011, 40, 139–146. [Google Scholar] [CrossRef]
- Gawaz, M. Role of platelets in coronary thrombosis and reperfusion of ischemic myocardium. Cardiovasc. Res. 2004, 61, 498–511. [Google Scholar] [CrossRef]
- Fishbein, M.C.; Maclean, D.; Maroko, P.R. The histopathologic evolution of myocardial infarction. Chest 1978, 73, 843–849. [Google Scholar] [CrossRef]
- Zuurbier, C.J.; Abbate, A.; Cabrera-Fuentes, H.A.; Cohen, M.V.; Collino, M.; De Kleijn, D.P.V.; Downey, J.M.; Pagliaro, P.; Preissner, K.T.; Takahashi, M.; et al. Innate immunity as a target for acute cardioprotection. Cardiovasc. Res. 2019, 115, 1131–1142. [Google Scholar] [CrossRef]
- Yan, X.; Anzai, A.; Katsumata, Y.; Matsuhashi, T.; Ito, K.; Endo, J.; Yamamoto, T.; Takeshima, A.; Shinmura, K.; Shen, W.; et al. Temporal dynamics of cardiac immune cell accumulation following acute myocardial infarction. J. Mol. Cell. Cardiol. 2013, 62, 24–35. [Google Scholar] [CrossRef]
- Ghigo, A.; Franco, I.; Morello, F.H.E. Myocyte signalling in leucocyte recruitment to the heart. Cardiovasc. Res. 2014, 102, 270–280. [Google Scholar] [CrossRef]
- Anzai, A.; Choi, J.L.; He, S.; Fenn, A.M.; Nairz, M.; Rattik, S.; McAlpine, C.S.; Mindur, J.E.; Chan, C.T.; Iwamoto, Y.; et al. The infarcted myocardium solicits GM-CSF for the detrimental oversupply of inflammatory leukocytes. J. Exp. Med. 2017, 214, 3293–3310. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Courties, G.; Wei, Y.; Leuschner, F.; Gorbatov, R.; Robbins, C.S.; Iwamoto, Y.; Thompson, B.; Carlson, A.L.; Heidt, T.; et al. Myocardial infarction accelerates atherosclerosis. Nature 2012, 487, 325–329. [Google Scholar] [CrossRef] [PubMed]
- Dutta, P.; Sager, H.B.; Stengel, K.R.; Naxerova, K.; Courties, G.; Saez, B.; Silberstein, L.; Heidt, T.; Sebas, M.; Sun, Y.; et al. Myocardial infarction activates CCR2+ hematopoietic stem and progenitor cells. Cell Stem Cell 2015, 16, 477–487. [Google Scholar] [CrossRef] [PubMed]
- Fazel, S.S.; Chen, L.; Angoulvant, D.; Li, S.; Weisel, R.D.; Keating, A.; Li, R. Activation of c-kit is necessary for mobilization of reparative bone marrow progenitor cells in response to cardiac injury. FASEB J. 2008, 22, 930–940. [Google Scholar] [CrossRef] [PubMed]
- Fazel, S.; Cimini, M.; Chen, L.; Li, S.; Angoulvant, D.; Fedak, P.; Verma, S.; Weisel, R.D.; Keating, A.; Li, R.K. Cardioprotective c-kit+ cells are from the bone marrow and regulate the myocardial balance of angiogenic cytokines. J. Clin. Investig. 2006, 116, 1865–1877. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Gao, X.M.; Fang, L.; Jennings, N.L.; Su, Y.; Xu, Q.; Samson, A.L.; Kiriazis, H.; Wang, X.F.; Shan, L.; et al. Novel role of platelets in mediating inflammatory responses and ventricular rupture or remodeling following myocardial infarction. Arterioscler. Thromb. Vasc. Biol. 2011, 31, 834–841. [Google Scholar] [CrossRef]
- Ziegler, M.; Alt, K.; Paterson, B.M.; Kanellakis, P.; Bobik, A.; Donnelly, P.S.; Hagemeyer, C.E.; Peter, K. Highly Sensitive Detection of Minimal Cardiac Ischemia using Positron Emission Tomography Imaging of Activated Platelets. Sci. Rep. 2016, 6, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Santos-Martínez, M.J.; Medina, C.; Jurasz, P.; Radomski, M.W. Role of metalloproteinases in platelet function. Thromb. Res. 2008, 121, 535–542. [Google Scholar] [CrossRef] [PubMed]
- Schiechl, G.; Hermann, F.J.; Rodriguez Gomez, M.; Kutzi, S.; Schmidbauer, K.; Talke, Y.; Neumayer, S.; Goebel, N.; Renner, K.; Brühl, H.; et al. Basophils Trigger Fibroblast Activation in Cardiac Allograft Fibrosis Development. Am. J. Transplant. 2016, 16, 2574–2588. [Google Scholar] [CrossRef] [PubMed]
- Nakashima, C.; Otsuka, A.; Kitoh, A.; Honda, T.; Egawa, G.; Nakajima, S.; Nakamizo, S.; Arita, M.; Kubo, M.; Miyachi, Y.; et al. Basophils regulate the recruitment of eosinophils in a murine model of irritant contact dermatitis. J. Allergy Clin. Immunol. 2014, 134, 100–107. [Google Scholar] [CrossRef]
- Rodriguez Gomez, M.; Talke, Y.; Hofmann, C.; Ketelsen, I.; Hermann, F.; Reich, B.; Goebel, N.; Schmidbauer, K.; Dunger, N.; Brühl, H.; et al. Basophils control T-cell responses and limit disease activity in experimental murine colitis. Mucosal Immunol. 2014, 7, 188–199. [Google Scholar] [CrossRef] [PubMed]
- Toor, I.S.; Rückerl, D.; Mair, I.; Ainsworth, R.; Meloni, M.; Spiroski, A.M.; Benezech, C.; Felton, J.M.; Thomson, A.; Caporali, A.; et al. Eosinophil Deficiency Promotes Aberrant Repair and Adverse Remodeling Following Acute Myocardial Infarction. JACC 2020, 5, 665–681. [Google Scholar] [CrossRef]
- Frangogiannis, N.G.; Lindsey, M.L.; Michael, L.H.; Youker, K.A.; Bressler, R.B.; Mendoza, L.H.; Spengler, R.N.; Smith, C.W.; Entman, M.L. Resident Cardiac Mast Cells Degranulate and Release Preformed TNF-α, Initiating the Cytokine Cascade in Experimental Canine Myocardial Ischemia/Reperfusion. Circulation 1998, 98, 699–710. [Google Scholar] [CrossRef]
- Hara, M.; Matsumori, A.; Ono, K.; Kido, H.; Hwang, M.W.; Miyamoto, T.; Iwasaki, A.; Okada, M.; Nakatani, K.; Sasayama, S. Mast cells cause apoptosis of cardiomyocytes and proliferation of other intramyocardial cells in vitro. Circulation 1999, 100, 1443–1449. [Google Scholar] [CrossRef]
- Tejada, T.; Tan, L.; Torres, R.A.; Calvert, J.W.; Lambert, J.P.; Zaidi, M.; Husain, M.; Berce, M.D.; Naib, H.; Pejler, G.; et al. IGF-1 degradation by mouse mast cell protease 4 promotes cell death and adverse cardiac remodeling days after a myocardial infarction. Proc. Natl. Acad. Sci. USA 2016, 113, 6949–6954. [Google Scholar] [CrossRef]
- Janicki, J.S.; Brower, G.L.; Levick, S.P. The Emerging Prominence of the Cardiac Mast Cell as a Potent Mediator of Adverse Myocardial Remodeling. In Methods in Molecular Biology; Humana Press Inc.: Totowa, NJ, USA, 2015; Volume 1220, pp. 121–139. [Google Scholar]
- Ma, Y.; Yabluchanskiy, A.; Iyer, R.P.; Cannon, P.L.; Flynn, E.R.; Jung, M.; Henry, J.; Cates, C.A.; Deleon-Pennell, K.Y.; Lindsey, M.L. Temporal neutrophil polarization following myocardial infarction. Cardiovasc. Res. 2016, 110, 51–61. [Google Scholar] [CrossRef]
- Soehnlein, O.; Zernecke, A.; Eriksson, E.E.; Rothfuchs, A.G.; Pham, C.T.; Herwald, H.; Bidzhekov, K.; Rottenberg, M.E.; Weber, C.; Lindbom, L. Neutrophil secretion products pave the way for inflammatory monocytes. Blood 2008, 112, 1461–1471. [Google Scholar] [CrossRef] [PubMed]
- Frangogiannis, N.G. Regulation of the Inflammatory Response in Cardiac Repair. Circ. Res. 2012, 110, 159–173. [Google Scholar] [CrossRef]
- Horckmans, M.; Ring, L.; Duchene, J.; Santovito, D.; Schloss, M.J.; Drechsler, M.; Weber, C.; Soehnlein, O.; Steffens, S. Neutrophils Orchestrate Post-Myocardial Infarction Healing by Polarizing Macrophages Towards a Reparative Phenotype. Eur. Heart J. 2017, 38, 187–197. [Google Scholar] [CrossRef] [PubMed]
- Leuschner, F.; Rauch, P.J.; Ueno, T.; Gorbatov, R.; Marinelli, B.; Lee, W.W.; Dutta, P.; Wei, Y.; Robbins, C.; Iwamoto, Y.; et al. Rapid monocyte kinetics in acute myocardial infarction are sustained by extramedullary monocytopoiesis. J. Exp. Med. 2012, 209, 123–137. [Google Scholar] [CrossRef] [PubMed]
- Swirski, F.K.; Nahrendorf, M.; Etzrodt, M.; Wildgruber, M.; Cortez-Retamozo, V.; Panizzi, P.; Figueiredo, J.L.; Kohler, R.H.; Chudnovskiy, A.; Waterman, P.; et al. Identification of splenic reservoir monocytes and their deployment to inflammatory sites. Science 2009, 325, 612–616. [Google Scholar] [CrossRef]
- Saxena, A.; Chen, W.; Su, Y.; Rai, V.; Uche, O.U.; Li, N.; Frangogiannis, N.G. IL-1 Induces Proinflammatory Leukocyte Infiltration and Regulates Fibroblast Phenotype in the Infarcted Myocardium. J. Immunol. 2013, 191, 4838–4848. [Google Scholar] [CrossRef]
- Sager, H.B.; Heidt, T.; Hulsmans, M.; Dutta, P.; Courties, G.; Sebas, M.; Wojtkiewicz, G.R.; Tricot, B.; Iwamoto, Y.; Sun, Y.; et al. Targeting interleukin-1β reduces leukocyte production after acute myocardial infarction. Circulation 2015, 132, 1880–1890. [Google Scholar] [CrossRef]
- Patel, A.A.; Zhang, Y.; Fullerton, J.N.; Boelen, L.; Rongvaux, A.; Maini, A.A.; Bigley, V.; Flavell, R.A.; Gilroy, D.W.; Asquith, B.; et al. The fate and lifespan of human monocyte subsets in steady state and systemic inflammation. J. Exp. Med. 2017, 214, 1913–1923. [Google Scholar] [CrossRef]
- Dewald, O.; Zymek, P.; Winkelmann, K.; Koerting, A.; Ren, G.; Abou-Khamis, T.; Michael, L.H.; Rollins, B.J.; Entman, M.L.; Frangogiannis, N.G. CCL2/monocyte chemoattractant protein-1 regulates inflammatory responses critical to healing myocardial infarcts. Circ. Res. 2005, 96, 881–889. [Google Scholar] [CrossRef]
- Deshmane, S.L.; Kremlev, S.; Amini, S.; Sawaya, B.E. Monocyte Chemoattractant Protein-1 (MCP-1): An Overview. J. Interferon Cytokine Res. 2009, 29, 313–326. [Google Scholar] [CrossRef]
- Nahrendorf, M.; Swirski, F.K.; Aikawa, E.; Stangenberg, L.; Wurdinger, T.; Figueiredo, J.-L.; Libby, P.; Weissleder, R.; Pittet, M.J. The healing myocardium sequentially mobilizes two monocyte subsets with divergent and complementary functions. J. Exp. Med. 2007, 204, 3037–3047. [Google Scholar] [CrossRef] [PubMed]
- Lavine, K.J.; Epelman, S.; Uchida, K.; Weber, K.J.; Nichols, C.G.; Schilling, J.D.; Ornitz, D.M.; Randolph, G.J.; Mann, D.L. Distinct macrophage lineages contribute to disparate patterns of cardiac recovery and remodeling in the neonatal and adult heart. Proc. Natl. Acad. Sci. USA 2014, 111, 16029–16034. [Google Scholar] [CrossRef] [PubMed]
- Ma, Y.; Mouton, A.J.; Lindsey, M.L. Cardiac macrophage biology in the steady-state heart, the aging heart, and following myocardial infarction. Transl. Res. 2018, 191, 15–28. [Google Scholar] [CrossRef] [PubMed]
- Auffray, C.; Fogg, D.; Garfa, M.; Elain, G.; Join-Lambert, O.; Kayal, S.; Sarnacki, S.; Cumano, A.; Lauvau, G.; Geissmann, F. Monitoring of blood vessels and tissues by a population of monocytes with patrolling behavior. Science 2007, 317, 666–670. [Google Scholar] [CrossRef] [PubMed]
- Carlin, L.M.; Stamatiades, E.G.; Auffray, C.; Hanna, R.N.; Glover, L.; Vizcay-Barrena, G.; Hedrick, C.C.; Cook, H.T.; Diebold, S.; Geissmann, F. Nr4a1-dependent Ly6Clow monocytes monitor endothelial cells and orchestrate their disposal. Cell 2013, 153, 362–375. [Google Scholar] [CrossRef]
- Hou, X.; Chen, G.; Bracamonte-Baran, W.; Choi, H.S.; Diny, N.L.; Sung, J.; Hughes, D.; Won, T.; Wood, M.K.; Talor, M.V.; et al. The Cardiac Microenvironment Instructs Divergent Monocyte Fates and Functions in Myocarditis. Cell Rep. 2019, 28, 172–189.e7. [Google Scholar] [CrossRef]
- Hanna, R.N.; Carlin, L.M.; Hubbeling, H.G.; Nackiewicz, D.; Green, A.M.; Punt, J.A.; Geissmann, F.; Hedrick, C.C. The transcription factor NR4A1 (Nur77) controls bone marrow differentiation and the survival of Ly6C- monocytes. Nat. Immunol. 2011, 12, 778–785. [Google Scholar] [CrossRef]
- Hilgendorf, I.; Gerhardt, L.M.S.; Tan, T.C.; Winter, C.; Holderried, T.A.W.; Chousterman, B.G.; Iwamoto, Y.; Liao, R.; Zirlik, A.; Scherer-Crosbie, M.; et al. Ly-6c high monocytes depend on Nr4a1 to balance both inflammatory and reparative phases in the infarcted myocardium. Circ. Res. 2014, 114, 1611–1622. [Google Scholar] [CrossRef]
- Heidt, T.; Courties, G.; Dutta, P.; Sager, H.B.; Sebas, M.; Iwamoto, Y.; Sun, Y.; Da Silva, N.; Panizzi, P.; Van Der Lahn, A.M.; et al. Differential contribution of monocytes to heart macrophages in steady-state and after myocardial infarction. Circ. Res. 2014, 115, 284–295. [Google Scholar] [CrossRef]
- Epelman, S.; Lavine, K.J.; Beaudin, A.E.; Sojka, D.K.; Carrero, J.A.; Calderon, B.; Brija, T.; Gautier, E.L.; Ivanov, S.; Satpathy, A.T.; et al. Embryonic and adult-derived resident cardiac macrophages are maintained through distinct mechanisms at steady state and during inflammation. Immunity 2014, 40, 91–104. [Google Scholar] [CrossRef]
- Dick, S.A.; Macklin, J.A.; Nejat, S.; Momen, A.; Clemente-Casares, X.; Althagafi, M.G.; Chen, J.; Kantores, C.; Hosseinzadeh, S.; Aronoff, L.; et al. Self-renewing resident cardiac macrophages limit adverse remodeling following myocardial infarction. Nat. Immunol. 2019, 20, 29–39. [Google Scholar] [CrossRef] [PubMed]
- King, K.R.; Aguirre, A.D.; Ye, Y.X.; Sun, Y.; Roh, J.D.; Ng, R.P.; Kohler, R.H.; Arlauckas, S.P.; Yoshiko, V.; Savo, A.; et al. IRF3 and type i interferons fuel a fatal response to myocardial infarction. Nat. Med. 2017, 23, 1481–1487. [Google Scholar] [CrossRef] [PubMed]
- Troidl, C.; Möllmann, H.; Nef, H.; Masseli, F.; Voss, S.; Szardien, S.; Willmer, M.; Rolf, A.; Rixe, J.; Troidl, K.; et al. Classically and alternatively activated macrophages contribute to tissue remodelling after myocardial infarction. J. Cell. Mol. Med. 2009, 13, 3485–3496. [Google Scholar] [CrossRef] [PubMed]
- Rőszer, T. Understanding the Mysterious M2 Macrophage through Activation Markers and Effector Mechanisms. Mediat. Inflamm. 2015, 2015, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Ouyang, W.; Rutz, S.; Crellin, N.K.; Valdez, P.A.; Hymowitz, S.G. Regulation and Functions of the IL-10 Family of Cytokines in Inflammation and Disease. Annu. Rev. Immunol. 2011, 29, 71–109. [Google Scholar] [CrossRef]
- Ong, S.; Ligons, D.L.; Barin, J.G.; Wu, L.; Talor, M.V.; Diny, N.; Fontes, J.A.; Gebremariam, E.; Kass, D.A.; Rose, N.R.; et al. Natural killer cells limit cardiac inflammation and fibrosis by halting eosinophil infiltration. Am. J. Pathol. 2015, 185, 847–861. [Google Scholar] [CrossRef]
- Westman, P.C.; Lipinski, M.J.; Luger, D.; Waksman, R.; Bonow, R.O.; Wu, E.; Epstein, S.E. Inflammation as a Driver of Adverse Left Ventricular Remodeling After Acute Myocardial Infarction. J. Am. Coll. Cardiol. 2016, 67, 2050–2060. [Google Scholar] [CrossRef]
- Zouggari, Y.; Ait-Oufella, H.; Bonnin, P.; Simon, T.; Sage, A.P.; Guérin, C.; Vilar, J.; Caligiuri, G.; Tsiantoulas, D.; Laurans, L.; et al. B lymphocytes trigger monocyte mobilization and impair heart function after acute myocardial infarction. Nat. Med. 2013, 19, 1273–1280. [Google Scholar] [CrossRef]
- Vignali, D.A.A.; Collison, L.W.; Workman, C.J. How regulatory T cells work. Nat. Rev. Immunol. 2008, 8, 523–532. [Google Scholar] [CrossRef]
- Hofmann, U.; Beyersdorf, N.; Weirather, J.; Podolskaya, A.; Bauersachs, J.; Ertl, G.; Kerkau, T.; Frantz, S. Activation of CD4+ T lymphocytes improves wound healing and survival after experimental myocardial infarction in mice. Circulation 2012, 125, 1652–1663. [Google Scholar] [CrossRef]
- Yang, Z.; Day, Y.J.; Toufektsian, M.C.; Xu, Y.; Ramos, S.I.; Marshall, M.A.; French, B.A.; Linden, J. Myocardial infarct-sparing effect of adenosine A2A receptor activation is due to its action on CD4+ T lymphocytes. Circulation 2006, 114, 2056–2064. [Google Scholar] [CrossRef] [PubMed]
- Weirather, J.; Hofmann, U.D.W.; Beyersdorf, N.; Ramos, G.C.; Vogel, B.; Frey, A.; Ertl, G.; Kerkau, T.; Frantz, S. Foxp3+ CD4+ T cells improve healing after myocardial infarction by modulating monocyte/macrophage differentiation. Circ. Res. 2014, 115, 55–67. [Google Scholar] [CrossRef] [PubMed]
- Varda-Bloom, N.; Leor, J.; Ohad, D.G.; Hasin, Y.; Amar, M.; Fixler, R.; Battler, A.; Eldar, M.; Hasin, D. Cytotoxic T lymphocytes are activated following myocardial infarction and can recognize and kill healthy myocytes in vitro. J. Mol. Cell. Cardiol. 2000, 32, 2141–2149. [Google Scholar] [CrossRef] [PubMed]
- Ilatovskaya, D.V.; Pitts, C.; Clayton, J.; Domondon, M.; Troncoso, M.; Pippin, S.; DeLeon-Pennell, K.Y. CD8+ T-cells negatively regulate inflammation post-myocardial infarction. Am. J. Physiol. Hear. Circ. Physiol. 2019, 317, H581–H596. [Google Scholar] [CrossRef]
- Frenkel, D.; Pachori, A.S.; Zhang, L.; Dembinsky-Vaknin, A.; Farfara, D.; Petrovic-Stojkovic, S.; Dzau, V.J.; Weiner, H.L. Nasal vaccination with troponin reduces troponin specific T-cell responses and improves heart function in myocardial ischemia-reperfusion injury. Int. Immunol. 2009, 21, 817–829. [Google Scholar] [CrossRef]
- Rieckmann, M.; Delgobo, M.; Gaal, C.; Büchner, L.; Steinau, P.; Reshef, D.; Gil-Cruz, C.; ter Horst, E.N.; Kircher, M.; Reiter, T.; et al. Myocardial infarction triggers cardioprotective antigen-specific T helper cell responses. J. Clin. Investig. 2019, 129, 4922–4936. [Google Scholar] [CrossRef]
- Zhong, Z.; Wu, H.; Zhang, Q.; Zhong, W.; Zhao, P. Characteristics of T cell receptor repertoires of patients with acute myocardial infarction through high-throughput sequencing. J. Transl. Med. 2019, 17, 21. [Google Scholar] [CrossRef]
- Adamo, L.; Rocha-Resende, C.; Lin, C.-Y.; Evans, S.; Williams, J.; Dun, H.; Li, W.; Mpoy, C.; Andhey, P.S.; Rogers, B.E.; et al. Myocardial B cells are a subset of circulating lymphocytes with delayed transit through the heart. JCI Insight 2020, 5, e134700. [Google Scholar] [CrossRef]
- Wu, L.; Dalal, R.; Cao, C.D.; Postoak, J.L.; Yang, G.; Zhang, Q.; Wang, Z.; Lal, H.; van Kaer, L. IL-10–producing B cells are enriched in murine pericardial adipose tissues and ameliorate the outcome of acute myocardial infarction. Proc. Natl. Acad. Sci. USA 2019, 116, 21673–21684. [Google Scholar] [CrossRef]
- Sobirin, M.A.; Kinugawa, S.; Takahashi, M.; Fukushima, A.; Homma, T.; Ono, T.; Hirabayashi, K.; Suga, T.; Azalia, P.; Takada, S.; et al. Activation of natural killer T cells ameliorates postinfarct cardiac remodeling and failure in mice. Circ. Res. 2012, 111, 1037–1047. [Google Scholar] [CrossRef]
- Eberl, G.; Colonna, M.; Di Santo, J.P.; McKenzie, A.N.J. Innate lymphoid cells: A new paradigm in immunology. Science 2015, 348, 6566. [Google Scholar] [CrossRef] [PubMed]
- Gasteiger, G.; Fan, X.; Dikiy, S.; Lee, S.Y.; Rudensky, A.Y. Tissue residency of innate lymphoid cells in lymphoid and nonlymphoid organs. Science 2015, 350, 981–985. [Google Scholar] [CrossRef] [PubMed]
- Li, J.; Wu, J.; Zhang, M.; Zheng, Y. Dynamic changes of innate lymphoid cells in acute ST-segment elevation myocardial infarction and its association with clinical outcomes. Sci. Rep. 2020, 10, 5099. [Google Scholar] [CrossRef] [PubMed]
- Choi, H.S.; Hou, X.; Bracamonte-Baran, W.; Diny, N.; Talor, M.; Cihakova, D. The role of innate lymphoid cells in the heart and cardiac inflammation. J. Immunol. 2018, 200, 42. [Google Scholar]
- Choi, H.S.; Won, T.; Hou, X.; Chen, G.; Bracamonte-Baran, W.; Talor, M.V.; Jurčová, I.; Szárszoi, O.; Čurnova, L.; Stříž, I.; et al. Innate Lymphoid Cells Play a Pathogenic Role in Pericarditis. Cell Rep. 2020, 30, 2989–3003. [Google Scholar] [CrossRef]
- Zhao, T.X.; Newland, S.A.; Mallat, Z. 2019 ATVB Plenary Lecture. Arterioscler. Thromb. Vasc. Biol. 2020, 40, 853–864. [Google Scholar] [CrossRef]
- Xu, J.-Y.; Xiong, Y.-Y.; Lu, X.-T.; Yang, Y.-J. Regulation of Type 2 Immunity in Myocardial Infarction. Front. Immunol. 2019, 10, 62. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonage, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging: An Evolutionary Perspective on Immunosenescence. Ann. N. Y. Acad. Sci. 2006, 908, 244–254. [Google Scholar] [CrossRef]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. 2014, 69, S4–S9. [Google Scholar] [CrossRef]
- Schaum, N.; Lehallier, B.; Hahn, O.; Hosseinzadeh, S.; Lee, S.E.; Sit, R.; Lee, D.P.; Losada, P.M.; Zardeneta, M.E.; Pálovics, R.; et al. The murine transcriptome reveals global aging nodes with organ-specific phase and amplitude. bioRxiv 2019. [Google Scholar] [CrossRef]
- Consortium, T.T.M.; Pisco, A.O.; McGeever, A.; Schaum, N.; Karkanias, J.; Neff, N.F.; Darmanis, S.; Wyss-Coray, T.; Quake, S.R. A Single Cell Transcriptomic Atlas Characterizes Aging Tissues in the Mouse. bioRxiv 2020. [Google Scholar] [CrossRef]
- Fulop, T.; Larbi, A.; Dupuis, G.; Le Page, A.; Frost, E.H.; Cohen, A.A.; Witkowski, J.M.; Franceschi, C. Immunosenescence and Inflamm-Aging As Two Sides of the Same Coin: Friends or Foes? Front. Immunol. 2018, 8. [Google Scholar] [CrossRef] [PubMed]
- Michaud, M.; Balardy, L.; Moulis, G.; Gaudin, C.; Peyrot, C.; Vellas, B.; Cesari, M.; Nourhashemi, F. Proinflammatory Cytokines, Aging, and Age-Related Diseases. J. Am. Med. Dir. Assoc. 2013, 14, 877–882. [Google Scholar] [CrossRef] [PubMed]
- Cesari, M.; Penninx, B.W.J.H.; Newman, A.B.; Kritchevsky, S.B.; Nicklas, B.J.; Sutton-Tyrrell, K.; Rubin, S.M.; Ding, J.; Simonsick, E.M.; Harris, T.B.; et al. Inflammatory Markers and Onset of Cardiovascular Events: Results from the Health ABC Study. Circulation 2003, 108, 2317–2322. [Google Scholar] [CrossRef] [PubMed]
- Sudo, K.; Ema, H.; Morita, Y.; Nakauchi, H. Age-associated characteristics of murine hematopoietic stem cells. J. Exp. Med. 2000, 192, 1273–1280. [Google Scholar] [CrossRef] [PubMed]
- Pang, W.W.; Price, E.A.; Sahoo, D.; Beerman, I.; Maloney, W.J.; Rossi, D.J.; Schrier, S.L.; Weissman, I.L. Human bone marrow hematopoietic stem cells are increased in frequency and myeloid-biased with age. Proc. Natl. Acad. Sci. USA 2011, 108, 20012–20017. [Google Scholar] [CrossRef]
- Gekas, C.; Graf, T. CD41 expression marks myeloid-biased adult hematopoietic stem cells and increases with age. Blood 2013, 121, 4463–4472. [Google Scholar] [CrossRef]
- Grover, A.; Sanjuan-Pla, A.; Thongjuea, S.; Carrelha, J.; Giustacchini, A.; Gambardella, A.; Macaulay, I.; Mancini, E.; Luis, T.C.; Mead, A.; et al. Single-cell RNA sequencing reveals molecular and functional platelet bias of aged haematopoietic stem cells. Nat. Commun. 2016, 7, 1–12. [Google Scholar] [CrossRef]
- Oliver, G.; Kipnis, J.; Randolph, G.J.; Harvey, N.L. The Lymphatic Vasculature in the 21st Century: Novel Functional Roles in Homeostasis and Disease. Cell 2020, 182, 270–296. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Zahn, J.M.; Ahlenius, H.; Sonu, R.; Wagers, A.J.; Weissman, I.L. Cell intrinsic alterations underlie hematopoietic stem cell aging. Proc. Natl. Acad. Sci. USA 2005, 102, 9194–9199. [Google Scholar] [CrossRef]
- Rossi, D.J.; Bryder, D.; Seita, J.; Nussenzweig, A.; Hoeijmakers, J.; Weissman, I.L. Deficiencies in DNA damage repair limit the function of haematopoietic stem cells with age. Nature 2007, 447, 725–729. [Google Scholar] [CrossRef] [PubMed]
- Rübe, C.E.; Fricke, A.; Widmann, T.A.; Fürst, T.; Madry, H.; Pfreundschuh, M.; Rübe, C. Accumulation of DNA Damage in Hematopoietic Stem and Progenitor Cells during Human Aging. PLoS ONE 2011, 6, e17487. [Google Scholar] [CrossRef] [PubMed]
- Horvath, S. DNA methylation age of human tissues and cell types. Genome Biol. 2013, 14, R115. [Google Scholar] [CrossRef] [PubMed]
- Ju, Z.; Jiang, H.; Jaworski, M.; Rathinam, C.; Gompf, A.; Klein, C.; Trumpp, A.; Rudolph, K.L. Telomere dysfunction induces environmental alterations limiting hematopoietic stem cell function and engraftment. Nat. Med. 2007, 13, 742–747. [Google Scholar] [CrossRef]
- Chambers, S.M.; Shaw, C.A.; Gatza, C.; Fisk, C.J.; Donehower, L.A.; Goodell, M.A. Aging hematopoietic stem cells decline in function and exhibit epigenetic dysregulation. PLoS Biol. 2007, 5, e201. [Google Scholar] [CrossRef]
- Jaiswal, S.; Fontanillas, P.; Flannick, J.; Manning, A.; Grauman, P.V.; Mar, B.G.; Lindsley, R.C.; Mermel, C.H.; Burtt, N.; Chavez, A.; et al. Age-related clonal hematopoiesis associated with adverse outcomes. N. Engl. J. Med. 2014, 371, 2488–2498. [Google Scholar] [CrossRef]
- Jaiswal, S.; Natarajan, P.; Silver, A.J.; Gibson, C.J.; Bick, A.G.; Shvartz, E.; McConkey, M.; Gupta, N.; Gabriel, S.; Ardissino, D.; et al. Clonal Hematopoiesis and risk of atherosclerotic cardiovascular disease. N. Engl. J. Med. 2017, 377, 111–121. [Google Scholar] [CrossRef]
- Fuster, J.J.; MacLauchlan, S.; Zuriaga, M.A.; Polackal, M.N.; Ostriker, A.C.; Chakraborty, R.; Wu, C.L.; Sano, S.; Muralidharan, S.; Rius, C.; et al. Clonal hematopoiesis associated with TET2 deficiency accelerates atherosclerosis development in mice. Science 2017, 355, 842–847. [Google Scholar] [CrossRef]
- Sano, S.; Oshima, K.; Wang, Y.; MacLauchlan, S.; Katanasaka, Y.; Sano, M.; Zuriaga, M.A.; Yoshiyama, M.; Goukassian, D.; Cooper, M.A.; et al. Tet2-Mediated Clonal Hematopoiesis Accelerates Heart Failure Through a Mechanism Involving the IL-1β/NLRP3 Inflammasome. J. Am. Coll. Cardiol. 2018, 71, 875–886. [Google Scholar] [CrossRef]
- Busch, K.; Klapproth, K.; Barile, M.; Flossdorf, M.; Holland-Letz, T.; Schlenner, S.M.; Reth, M.; Höfer, T.; Rodewald, H.R. Fundamental properties of unperturbed haematopoiesis from stem cells in vivo. Nature 2015, 518, 542–546. [Google Scholar] [CrossRef]
- Min, H.; Montecino-Rodriguez, E.; Dorshkind, K. Effects of Aging on the Common Lymphoid Progenitor to Pro-B Cell Transition. J. Immunol. 2006, 176, 1007–1012. [Google Scholar] [CrossRef] [PubMed]
- Sun, D.; Luo, M.; Jeong, M.; Rodriguez, B.; Xia, Z.; Hannah, R.; Wang, H.; Le, T.; Faull, K.F.; Chen, R.; et al. Epigenomic profiling of young and aged HSCs reveals concerted changes during aging that reinforce self-renewal. Cell Stem Cell 2014, 14, 673–688. [Google Scholar] [CrossRef] [PubMed]
- Kowalczyk, M.S.; Tirosh, I.; Heckl, D.; Rao, T.N.; Dixit, A.; Haas, B.J.; Schneider, R.K.; Wagers, A.J.; Ebert, B.L.; Regev, A. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015, 25, 1860–1872. [Google Scholar] [CrossRef] [PubMed]
- Rodriguez-Fraticelli, A.E.; Wolock, S.L.; Weinreb, C.S.; Panero, R.; Patel, S.H.; Jankovic, M.; Sun, J.; Calogero, R.A.; Klein, A.M.; Camargo, F.D. Clonal analysis of lineage fate in native haematopoiesis. Nature 2018, 553, 212–216. [Google Scholar] [CrossRef] [PubMed]
- Liang, Y.; Van Zant, G.; Szilvassy, S.J. Effects of aging on the homing and engraftment of murine hematopoietic stem and progenitor cells. Blood 2005, 106, 1479–1487. [Google Scholar] [CrossRef]
- Xing, Z.; Ryan, M.A.; Daria, D.; Nattamai, K.J.; Van Zant, G.; Wang, L.; Zheng, Y.; Geiger, H. Increased hematopoietic stem cell mobilization in aged mice. Blood 2006, 108, 2190–2197. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Filippi, M.D.; Williams, D.A.; Zheng, Y. Genetic deletion of Cdc42GAP reveals a role of Cdc42 in erythropoiesis and hematopoietic stem/progenitor cell survival, adhesion, and engraftment. Blood 2006, 107, 98–105. [Google Scholar] [CrossRef]
- Wang, L.; Yang, L.; Debidda, M.; Witte, D.; Zheng, Y. Cdc42 GTPase-activating protein deficiency promotes genomic instability and premature aging-like phenotypes. Proc. Natl. Acad. Sci. USA 2007, 104, 1248–1253. [Google Scholar] [CrossRef]
- Florian, M.C.; Dörr, K.; Niebel, A.; Daria, D.; Schrezenmeier, H.; Rojewski, M.; Filippi, M.D.; Hasenberg, A.; Gunzer, M.; Scharffetter-Kochanek, K.; et al. Cdc42 activity regulates hematopoietic stem cell aging and rejuvenation. Cell Stem Cell 2012, 10, 520–530. [Google Scholar] [CrossRef]
- Florian, M.C.; Nattamai, K.J.; Dörr, K.; Marka, G.; Überle, B.; Vas, V.; Eckl, C.; Andrä, I.; Schiemann, M.; Oostendorp, R.A.J.; et al. A canonical to non-canonical Wnt signalling switch in haematopoietic stem-cell ageing. Nature 2013, 503, 392–396. [Google Scholar] [CrossRef]
- Ho, Y.H.; del Toro, R.; Rivera-Torres, J.; Rak, J.; Korn, C.; García-García, A.; Macías, D.; González-Gómez, C.; del Monte, A.; Wittner, M.; et al. Remodeling of Bone Marrow Hematopoietic Stem Cell Niches Promotes Myeloid Cell Expansion during Premature or Physiological Aging. Cell Stem Cell 2019, 25, 407–418. [Google Scholar] [CrossRef] [PubMed]
- Ergen, A.V.; Boles, N.C.; Goodell, M.A. Rantes/Ccl5 influences hematopoietic stem cell subtypes and causes myeloid skewing. Blood 2012, 119, 2500–2509. [Google Scholar] [CrossRef] [PubMed]
- Sugiyama, T.; Kohara, H.; Noda, M.; Nagasawa, T. Maintenance of the Hematopoietic Stem Cell Pool by CXCL12-CXCR4 Chemokine Signaling in Bone Marrow Stromal Cell Niches. Immunity 2006, 25, 977–988. [Google Scholar] [CrossRef] [PubMed]
- Nakamura-Ishizu, A.; Takubo, K.; Fujioka, M.; Suda, T. Megakaryocytes are essential for HSC quiescence through the production of thrombopoietin. Biochem. Biophys. Res. Commun. 2014, 454, 353–357. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Perry, J.M.; Marshall, H.; Venkatraman, A.; Qian, P.; He, X.C.; Ahamed, J.; Li, L. Megakaryocytes maintain homeostatic quiescence and promote post-injury regeneration of hematopoietic stem cells. Nat. Med. 2014, 20, 1321–1326. [Google Scholar] [CrossRef] [PubMed]
- Bruns, I.; Lucas, D.; Pinho, S.; Ahmed, J.; Lambert, M.P.; Kunisaki, Y.; Scheiermann, C.; Schiff, L.; Poncz, M.; Bergman, A.; et al. Megakaryocytes regulate hematopoietic stem cell quiescence through CXCL4 secretion. Nat. Med. 2014, 20, 1315–1320. [Google Scholar] [CrossRef] [PubMed]
- Katayama, Y.; Battista, M.; Kao, W.M.; Hidalgo, A.; Peired, A.J.; Thomas, S.A.; Frenette, P.S. Signals from the sympathetic nervous system regulate hematopoietic stem cell egress from bone marrow. Cell 2006, 124, 407–421. [Google Scholar] [CrossRef]
- Méndez-Ferrer, S.; Lucas, D.; Battista, M.; Frenette, P.S. Haematopoietic stem cell release is regulated by circadian oscillations. Nature 2008, 452, 442–447. [Google Scholar] [CrossRef]
- Kunisaki, Y.; Bruns, I.; Scheiermann, C.; Ahmed, J.; Pinho, S.; Zhang, D.; Mizoguchi, T.; Wei, Q.; Lucas, D.; Ito, K.; et al. Arteriolar niches maintain haematopoietic stem cell quiescence. Nature 2013, 502, 637–643. [Google Scholar] [CrossRef]
- Pinho, S.; Marchand, T.; Yang, E.; Wei, Q.; Nerlov, C.; Frenette, P.S. Lineage-Biased Hematopoietic Stem Cells Are Regulated by Distinct Niches. Dev. Cell 2018, 44, 634–641. [Google Scholar] [CrossRef]
- Ding, L.; Morrison, S.J. Haematopoietic stem cells and early lymphoid progenitors occupy distinct bone marrow niches. Nature 2013, 495, 231–235. [Google Scholar] [CrossRef] [PubMed]
- Spencer, J.A.; Ferraro, F.; Roussakis, E.; Klein, A.; Wu, J.; Runnels, J.M.; Zaher, W.; Mortensen, L.J.; Alt, C.; Turcotte, R.; et al. Direct measurement of local oxygen concentration in the bone marrow of live animals. Nature 2014, 508, 269–273. [Google Scholar] [CrossRef] [PubMed]
- Frisch, B.J.; Hoffman, C.M.; Latchney, S.E.; LaMere, M.W.; Myers, J.; Ashton, J.; Li, A.J.; Saunders, J.; Palis, J.; Perkins, A.S.; et al. Aged marrow macrophages expand platelet-biased hematopoietic stem cells via interleukin-1B. JCI Insight 2019, 4, e124213. [Google Scholar] [CrossRef] [PubMed]
- Rundberg Nilsson, A.; Soneji, S.; Adolfsson, S.; Bryder, D.; Pronk, C.J. Human and Murine Hematopoietic Stem Cell Aging Is Associated with Functional Impairments and Intrinsic Megakaryocytic/Erythroid Bias. PLoS ONE 2016, 11, e0158369. [Google Scholar] [CrossRef] [PubMed]
- Chen, C.; Liu, Y.; Liu, Y.; Zheng, P. mTOR Regulation and Therapeutic Rejuvenation of Aging Hematopoietic Stem Cells. Sci. Signal. 2009, 2, ra75. [Google Scholar] [CrossRef] [PubMed]
- Cheng, C.W.; Adams, G.B.; Perin, L.; Wei, M.; Zhou, X.; Lam, B.S.; Da Sacco, S.; Mirisola, M.; Quinn, D.I.; Dorff, T.B.; et al. Prolonged fasting reduces IGF-1/PKA to promote hematopoietic-stem-cell- based regeneration and reverse immunosuppression. Cell Stem Cell 2014, 14, 810–823. [Google Scholar] [CrossRef]
- Ho, T.T.; Warr, M.R.; Adelman, E.R.; Lansinger, O.M.; Flach, J.; Verovskaya, E.V.; Figueroa, M.E.; Passegué, E. Autophagy maintains the metabolism and function of young and old stem cells. Nature 2017, 543, 205–210. [Google Scholar] [CrossRef]
- Warr, M.R.; Binnewies, M.; Flach, J.; Reynaud, D.; Garg, T.; Malhotra, R.; Debnath, J.; Passegué, E. FOXO3A directs a protective autophagy program in haematopoietic stem cells. Nature 2013, 494, 323–327. [Google Scholar] [CrossRef]
- Vannini, N.; Girotra, M.; Naveiras, O.; Nikitin, G.; Campos, V.; Giger, S.; Roch, A.; Auwerx, J.; Lutolf, M.P. Specification of haematopoietic stem cell fate via modulation of mitochondrial activity. Nat. Commun. 2016, 7, 1312. [Google Scholar] [CrossRef]
- Simsek, T.; Kocabas, F.; Zheng, J.; Deberardinis, R.J.; Mahmoud, A.I.; Olson, E.N.; Schneider, J.W.; Zhang, C.C.; Sadek, H.A. The distinct metabolic profile of hematopoietic stem cells reflects their location in a hypoxic niche. Cell Stem Cell 2010, 7, 380–390. [Google Scholar] [CrossRef]
- Vannini, N.; Campos, V.; Girotra, M.; Trachsel, V.; Rojas-Sutterlin, S.; Tratwal, J.; Ragusa, S.; Stefanidis, E.; Ryu, D.; Rainer, P.Y.; et al. The NAD-Booster Nicotinamide Riboside Potently Stimulates Hematopoiesis through Increased Mitochondrial Clearance. Cell Stem Cell 2019, 24, 405–418. [Google Scholar] [CrossRef] [PubMed]
- Zhao, M.; Wang, D.D.-H.; Liu, X.; Tian, R. Metabolic Modulation of Macrophage Function Post Myocardial Infarction. Front. Physiol. 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- van Beek, A.A.; Van den Bossche, J.; Mastroberardino, P.G.; de Winther, M.P.J.; Leenen, P.J.M. Metabolic Alterations in Aging Macrophages: Ingredients for Inflammaging? Trends Immunol. 2019, 40, 113–127. [Google Scholar] [CrossRef] [PubMed]
- Bharath, L.P.; Agrawal, M.; McCambridge, G.; Nicholas, D.A.; Hasturk, H.; Liu, J.; Jiang, K.; Liu, R.; Guo, Z.; Deeney, J.; et al. Metformin Enhances Autophagy and Normalizes Mitochondrial Function to Alleviate Aging-Associated Inflammation. Cell Metab. 2020, 32, 44–55. [Google Scholar] [CrossRef]
- Schloss, M.J.; Swirski, F.K.; Nahrendorf, M. Modifiable Cardiovascular Risk, Hematopoiesis, and Innate Immunity. Circ. Res. 2020, 126, 1242–1259. [Google Scholar] [CrossRef]
- Netea, M.G.; Quintin, J.; van der Meer, J.W.M. Trained Immunity: A Memory for Innate Host Defense. Cell Host Microbe 2011, 9, 355–361. [Google Scholar] [CrossRef]
- Mulder, W.J.M.; Ochando, J.; Joosten, L.A.B.; Fayad, Z.A.; Netea, M.G. Therapeutic targeting of trained immunity. Nat. Rev. Drug Discov. 2019, 18, 553–566. [Google Scholar] [CrossRef]
- Fan, H.; Cook, J.A. Molecular mechanisms of endotoxin tolerance. J. Endotoxin Res. 2004, 10, 71–84. [Google Scholar] [CrossRef]
- Ifrim, D.C.; Quintin, J.; Joosten, L.A.B.; Jacobs, C.; Jansen, T.; Jacobs, L.; Gow, N.A.R.; Williams, D.L.; Van Der Meer, J.W.M.; Netea, M.G. Trained immunity or tolerance: Opposing functional programs induced in human monocytes after engagement of various pattern recognition receptors. Clin. Vaccine Immunol. 2014, 21, 534–545. [Google Scholar] [CrossRef]
- Saeed, S.; Quintin, J.; Kerstens, H.H.D.; Rao, N.A.; Aghajanirefah, A.; Matarese, F.; Cheng, S.C.; Ratter, J.; Berentsem, K.; Van Der Ent, M.A.; et al. Epigenetic programming of monocyte-to-macrophage differentiation and trained innate immunity. Science 2014, 345, 1251086. [Google Scholar] [CrossRef]
- Cremer, S.; Schloss, M.J.; Vinegoni, C.; Foy, B.H.; Zhang, S.; Rohde, D.; Hulsmans, M.; Fumene Feruglio, P.; Schmidt, S.; Wojtkiewicz, G.; et al. Diminished Reactive Hematopoiesis and Cardiac Inflammation in a Mouse Model of Recurrent Myocardial Infarction. J. Am. Coll. Cardiol. 2020, 75, 901–915. [Google Scholar] [CrossRef] [PubMed]
- Halter, J.B.; Musi, N.; McFarland Horne, F.; Crandall, J.P.; Goldberg, A.; Harkless, L.; Hazzard, W.R.; Huang, E.S.; Kirkman, M.S.; Plutzky, J.; et al. Diabetes and Cardiovascular Disease in Older Adults: Current Status and Future Directions. Diabetes 2014, 63, 2578–2589. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Murphy, A.J.; Stirzaker, R.A.; Hu, Y.; Yu, S.; Miller, R.G.; Ramkhelawon, B.; Distel, E.; Westerterp, M.; Huang, L.S.; et al. Hyperglycemia promotes myelopoiesis and impairs the resolution of atherosclerosis. Cell Metab. 2013, 17, 695–708. [Google Scholar] [CrossRef] [PubMed]
- Nagareddy, P.R.; Kraakman, M.; Masters, S.L.; Stirzaker, R.A.; Gorman, D.J.; Grant, R.W.; Dragoljevic, D.; Hong, E.S.; Abdel-Latif, A.; Smyth, S.S.; et al. Adipose tissue macrophages promote myelopoiesis and monocytosis in obesity. Cell Metab. 2014, 19, 821–835. [Google Scholar] [CrossRef] [PubMed]
- Albiero, M.; Ciciliot, S.; Tedesco, S.; Menegazzo, L.; D’Anna, M.; Scattolini, V.; Cappellari, R.; Zuccolotto, G.; Rosato, A.; Cignarella, A.; et al. Diabetes-associated myelopoiesis drives stem cell mobilopathy through an OSM-p66Shc signaling pathway. Diabetes 2019, 68, 1303–1314. [Google Scholar] [CrossRef] [PubMed]
- Hoyer, F.F.; Zhang, X.; Coppin, E.; Vasamsetti, S.B.; Modugu, G.; Schloss, M.J.; Rohde, D.; McAlpine, C.S.; Iwamoto, Y.; Libby, P.; et al. Bone Marrow Endothelial Cells Regulate Myelopoiesis in Diabetes Mellitus. Circulation 2020, 142, 244–258. [Google Scholar] [CrossRef]
- Spinetti, G.; Cordella, D.; Fortunato, O.; Sangalli, E.; Losa, S.; Gotti, A.; Carnelli, F.; Rosa, F.; Riboldi, S.; Sessa, F.; et al. Global remodeling of the vascular stem cell niche in bone marrow of diabetic patients: Implication of the microRNA-155/FOXO3a signaling pathway. Circ. Res. 2013, 112, 510–522. [Google Scholar] [CrossRef]
- Ferraro, F.; Lymperi, S.; Mendez-Ferrer, S.; Saez, B.; Spencer, J.A.; Yeap, B.Y.; Masselli, E.; Graiani, G.; Prezioso, L.; Rizzini, E.L.; et al. Diabetes Impairs Hematopoietic Stem Cell Mobilization by Altering Niche Function. Sci. Transl. Med. 2011, 3, 104ra101. [Google Scholar] [CrossRef]
- Edner, N.M.; Heuts, F.; Thomas, N.; Wang, C.J.; Petersone, L.; Kenefeck, R.; Kogimtzis, A.; Ovcinnikovs, V.; Ross, E.M.; Ntavli, E.; et al. Follicular helper T cell profiles predict response to costimulation blockade in type 1 diabetes. Nat. Immunol. 2020, 1–12. [Google Scholar] [CrossRef]
- Santopaolo, M.; Sullivan, N.; Thomas, A.C.; Alvino, V.; Nicholson, L.; Gu, Y.; Spinetti, G.; Marinos Kallikourdis, M.; Blom, A.; Madeddu, P. Activation of bone marrow adaptive immunity in type 2 diabetes: Rescue by co-stimulation modulator Abatacept. bioRxiv 2020. [Google Scholar] [CrossRef]
- Keller, K.M.; Howlett, S.E. Sex Differences in the Biology and Pathology of the Aging Heart. Can. J. Cardiol. 2016, 32, 1065–1073. [Google Scholar] [CrossRef]
- Kovats, S. Estrogen receptors regulate innate immune cells and signaling pathways. Cell. Immunol. 2015, 294, 63–69. [Google Scholar] [CrossRef] [PubMed]
- Iorga, A.; Cunningham, C.M.; Moazeni, S.; Ruffenach, G.; Umar, S.; Eghbali, M. The protective role of estrogen and estrogen receptors in cardiovascular disease and the controversial use of estrogen therapy. Biol. Sex. Differ. 2017, 8, 33. [Google Scholar] [CrossRef] [PubMed]
- Nakada, D.; Oguro, H.; Levi, B.P.; Ryan, N.; Kitano, A.; Saitoh, Y.; Takeichi, M.; Wendt, G.R.; Morrison, S.J. Oestrogen increases haematopoietic stem-cell self-renewal in females and during pregnancy. Nature 2014, 505, 555–558. [Google Scholar] [CrossRef] [PubMed]
- Fish, E.N. The X-files in immunity: Sex-based differences predispose immune responses. Nat. Rev. Immunol. 2008, 8, 737–744. [Google Scholar] [CrossRef]
- Dalgård, C.; Benetos, A.; Verhulst, S.; Labat, C.; Kark, J.D.; Christensen, K.; Kimura, M.; Kyvik, K.O.; Aviv, A. Leukocyte telomere length dynamics in women and men: Menopause vs age effects. Int. J. Epidemiol. 2015, 44, 1688–1695. [Google Scholar] [CrossRef]
- Parks, R.J.; Fares, E.; MacDonald, J.K.; Ernst, M.C.; Sinal, C.J.; Rockwood, K.; Howlett, S.E. A procedure for creating a frailty index based on deficit accumulation in aging mice. J. Gerontol. 2012, 67, 217–227. [Google Scholar] [CrossRef]
- Kane, A.E.; Keller, K.M.; Heinze-Milne, S.; Grandy, S.A.; Howlett, S.E. A murine frailty index based on clinical and laboratory measurements: Links between frailty and pro-inflammatory cytokines differ in a sex-specific manner. J. Gerontol. 2019, 74, 275–282. [Google Scholar] [CrossRef]
- Singer, K.; Maley, N.; Mergian, T.; DelProposto, J.; Cho, K.W.; Zamarron, B.F.; Martinez-Santibanez, G.; Geletka, L.; Muir, L.; Wachowiak, P.; et al. Differences in hematopoietic stem cells contribute to sexually dimorphic inflammatory responses to high fat diet-induced obesity. J. Biol. Chem. 2015, 290, 13250–13262. [Google Scholar] [CrossRef]
- Madjid, M.; Awan, I.; Willerson, J.T.; Casscells, S.W. Leukocyte count and coronary heart disease: Implications for risk assessment. J. Am. Coll. Cardiol. 2004, 44, 1945–1956. [Google Scholar] [CrossRef]
- Olson, N.C.; Sitlani, C.M.; Doyle, M.F.; Huber, S.A.; Landay, A.L.; Tracy, R.P.; Psaty, B.M.; Delaney, J.A. Innate and adaptive immune cell subsets as risk factors for coronary heart disease in two population-based cohorts. Atherosclerosis 2020, 300, 47–53. [Google Scholar] [CrossRef] [PubMed]
- Haycock, P.C.; Heydon, E.E.; Kaptoge, S.; Butterworth, A.S.; Thompson, A.; Willeit, P. Leucocyte telomere length and risk of cardiovascular disease: Systematic review and meta-analysis. BMJ 2014, 349, g4227. [Google Scholar] [CrossRef] [PubMed]
- Mahara, K.; Anzai, T.; Yoshikawa, T.; Maekawa, Y.; Okabe, T.; Asakura, Y.; Satoh, T.; Mitamura, H.; Suzuki, M.; Murayama, A.; et al. Aging adversely affects postinfarction inflammatory response and early left ventricular remodeling after reperfused acute anterior myocardial infarction. Cardiology 2006, 105, 67–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, M.; Zhang, P.; Chen, M.; Zhang, W.; Yu, L.; Yang, X.C.; Fan, Q. Aging might increase myocardial ischemia / reperfusion-induced apoptosis in humans and rats. Age 2012, 34, 621–632. [Google Scholar] [CrossRef]
- Bujak, M.; Kweon, H.J.; Chatila, K.; Li, N.; Taffet, G.; Frangogiannis, N.G. Aging-Related Defects Are Associated With Adverse Cardiac Remodeling in a Mouse Model of Reperfused Myocardial Infarction. J. Am. Coll. Cardiol. 2008, 51, 1384–1392. [Google Scholar] [CrossRef]
- Hulsmans, M.; Sager, H.B.; Roh, J.D.; Valero-Muñoz, M.; Houstis, N.E.; Iwamoto, Y.; Sun, Y.; Wilson, R.M.; Wojtkiewicz, G.; Tricot, B.; et al. Cardiac macrophages promote diastolic dysfunction. J. Exp. Med. 2018, 215, 423–440. [Google Scholar] [CrossRef]
- Ramosa, G.C.; Berg, A.V.D.; Nunes-Silva, V.; Weirather, J.; Peters, L.; Burkard, M.; Friedrich, M.; Pinnecker, J.; Abeer, M.; Heinzed, K.G.; et al. Myocardial aging as a T-cell-mediated phenomenon. Proc. Natl. Acad. Sci. USA 2017, 114, E2420–E2429. [Google Scholar] [CrossRef]
- Pinto, A.R.; Godwin, J.W.; Chandran, A.; Hersey, L.; Ilinykh, A.; Debuque, R.; Wang, L.; Rosenthal, N.A. Age-related changes in tissue macrophages precede cardiac functional impairment. Aging 2014, 6, 399–413. [Google Scholar] [CrossRef]
- Loffredo, F.S.; Steinhauser, M.L.; Jay, S.M.; Gannon, J.; Pancoast, J.R.; Yalamanchi, P.; Sinha, M.; Dall’Osso, C.; Khong, D.; Shadrach, J.L.; et al. Growth differentiation factor 11 is a circulating factor that reverses age-related cardiac hypertrophy. Cell 2013, 153, 828–839. [Google Scholar] [CrossRef]
- Walaszczyk, A.; Dookun, E.; Redgrave, R.; Tual-Chalot, S.; Victorelli, S.; Spyridopoulos, I.; Owens, A.; Arthur, H.M.; Passos, J.F.; Richardson, G.D. Pharmacological clearance of senescent cells improves survival and recovery in aged mice following acute myocardial infarction. Aging Cell 2019, 18. [Google Scholar] [CrossRef]
- Li, S.-H.; Sun, Z.; Brunt, K.R.; Shi, X.; Chen, M.-S.; Weisel, R.D.; Li, R.-K. Reconstitution of aged bone marrow with young cells repopulates cardiac-resident bone marrow-derived progenitor cells and prevents cardiac dysfunction after a myocardial infarction. Eur. Heart J. 2013, 34, 1157–1167. [Google Scholar] [CrossRef] [PubMed][Green Version]
- Li, S.-H.; Sun, L.; Yang, L.; Li, J.; Shao, Z.; Du, G.-Q.; Wu, J.; Weisel, R.D.; Li, R.-K. Young Bone-Marrow Sca-1+ Stem Cells Rejuvenate the Aged Heart and Improve Function after Injury through PDGFRβ-Akt pathway. Sci. Rep. 2017, 7, 41756. [Google Scholar] [CrossRef] [PubMed]
- Tobin, S.W.; Li, S.; Li, J.; Wu, J.; Yeganeh, A.; Yu, P.; Weisel, R.D.; Li, R.-K. Dual roles for bone marrow-derived Sca-1 cells in cardiac function. FASEB J. 2017, 31, 2905–2915. [Google Scholar] [CrossRef] [PubMed]
- Yabluchanskiy, A.; Ma, Y.; Deleon-Pennell, K.Y.; Altara, R.; Halade, G.V.; Voorhees, A.P.; Nguyen, N.T.; Jin, Y.F.; Winniford, M.D.; Hall, M.E.; et al. Myocardial infarction superimposed on aging: MMP-9 deletion promotes M2 macrophage polarization. J. Gerontol. 2016, 71, 475–483. [Google Scholar] [CrossRef]
- Mannisi, J.A.; Weisman, H.F.; Bush, D.E.; Dudeck, P.; Healy, B. Steroid administration after myocardial infarction promotes early infarct expansion. A study in the rat. J. Clin. Investig. 1987, 79, 1431–1439. [Google Scholar] [CrossRef]
- Hammerman, H.; Kloner, R.A.; Hale, S.; Schoen, F.J.; Braunwald, E. Dose-dependent effects of short-term methylprednisolone on myocardial infarct extent, scar formation, and ventricular function. Circulation 1983, 68, 446–452. [Google Scholar] [CrossRef]
- Bulkley, B.H.; Roberts, W.C. Steroid therapy during acute myocardial infarction. A cause of delayed healing and of ventricular aneurysm. Am. J. Med. 1974, 56, 244–250. [Google Scholar] [CrossRef]
- Padfield, G.J.; Din, J.N.; Koushiappi, E.; Mills, N.L.; Robinson, S.D.; Le May Cruden, N.; Lucking, A.J.; Chia, S.; Harding, S.A.; Newby, D.E. Cardiovascular effects of tumour necrosis factor α antagonism in patients with acute myocardial infarction: A first in human study. Heart 2013, 99, 1330–1335. [Google Scholar] [CrossRef]
- Kleveland, O.; Kunszt, G.; Bratlie, M.; Ueland, T.; Broch, K.; Holte, E.; Michelsen, A.E.; Bendz, B.; Amundsen, B.H.; Espevik, T.; et al. Effect of a single dose of the interleukin-6 receptor antagonist tocilizumab on inflammation and troponin T release in patients with non-ST-elevation myocardial infarction: A double-blind, randomized, placebo-controlled phase 2 trial † Tocilizumab attenuated the inflammatory response and primarily PCI-related TnT release in NSTEMI patients. Eur. Heart J. 2016, 37, 2406–2413. [Google Scholar] [CrossRef]
- Carroll, M.B.; Haller, C.; Smith, C. Short-term application of tocilizumab during myocardial infarction (STAT-MI). Rheumatol. Int. 2018, 38, 59–66. [Google Scholar] [CrossRef]
- Newby, L.K.; Marber, M.S.; Melloni, C.; Sarov-Blat, L.; Aberle, L.H.; Aylward, P.E.; Cai, G.; De Winter, R.J.; Hamm, C.W.; Heitner, J.F.; et al. Losmapimod, a novel p38 mitogen-activated protein kinase inhibitor, in non-ST-segment elevation myocardial infarction: A randomised phase 2 trial. Lancet 2014, 384, 1187–1195. [Google Scholar] [CrossRef]
- Ridker, P.M.; Everett, B.M.; Pradhan, A.; MacFadyen, J.G.; Solomon, D.H.; Zaharris, E.; Mam, V.; Hasan, A.; Rosenberg, Y.; Iturriaga, E.; et al. Low-Dose Methotrexate for the Prevention of Atherosclerotic Events. N. Engl. J. Med. 2019, 380, 752–762. [Google Scholar] [CrossRef] [PubMed]
- O’Donoghue, M.L.; Braunwald, E.; White, H.D.; Steen, D.P.; Lukas, M.A.; Tarka, E.; Steg, P.G.; Hochman, J.S.; Bode, C.; Maggioni, A.P.; et al. Effect of darapladib on major coronary events after an acute coronary syndrome: The SOLID-TIMI 52 randomized clinical trial. JAMA 2014, 312, 1006–1015. [Google Scholar] [CrossRef] [PubMed]
- Ridker, P.M.; Everett, B.M.; Thuren, T.; MacFadyen, J.G.; Chang, W.H.; Ballantyne, C.; Fonseca, F.; Nicolau, J.; Koenig, W.; Anker, S.D.; et al. Antiinflammatory Therapy with Canakinumab for Atherosclerotic Disease. N. Engl. J. Med. 2017, 377, 1119–1131. [Google Scholar] [CrossRef]
- O’Riordan, M. Hopes Fade for a CV Indication for Canakinumab: What’s Next for the Inflammatory Hypothesis? Available online: https://www.tctmd.com/news/hopes-fade-cv-indication-canakinumab-whats-next-inflammatory-hypothesis (accessed on 7 July 2020).
- Tardif, J.-C.; Kouz, S.; Waters, D.D.; Bertrand, O.F.; Diaz, R.; Maggioni, A.P.; Pinto, F.J.; Ibrahim, R.; Gamra, H.; Kiwan, G.S.; et al. Efficacy and Safety of Low-Dose Colchicine after Myocardial Infarction. N. Engl. J. Med. 2019, 381, 2497–2505. [Google Scholar] [CrossRef]
- Alibhai, F.J.; Tobin, S.W.; Yeganeh, A.; Weisel, R.D.; Li, R.-K. Emerging roles of extracellular vesicles in cardiac repair and rejuvenation. Am. J. Physiol. Heart Circ. Physiol. 2018, 315, H733–H744. [Google Scholar] [CrossRef]
- Biemmi, V.; Milano, G.; Ciullo, A.; Cervio, E.; Burrello, J.; Cas, M.D.; Paroni, R.; Tallone, T.; Moccetti, T.; Pedrazzini, G.; et al. Inflammatory extracellular vesicles prompt heart dysfunction via TRL4-dependent NF-κB activation. Theranostics 2020, 10, 2773–2790. [Google Scholar] [CrossRef]
- Alibhai, F.J.; Lim, F.; Yeganeh, A.; DiStefano, P.V.; Binesh-Marvasti, T.; Belfiore, A.; Wlodarek, L.; Gustafson, D.; Millar, S.; Li, S.H.; et al. Cellular senescence contributes to age-dependent changes in circulating extracellular vesicle cargo and function. Aging Cell 2020, 19, e13103. [Google Scholar] [CrossRef]
- Molawi, K.; Wolf, Y.; Kandalla, P.K.; Favret, J.; Hagemeyer, N.; Frenzel, K.; Pinto, A.R.; Klapproth, K.; Henri, S.; Malissen, B.; et al. Progressive replacement of embryo-derived cardiac macrophages with age. J. Exp. Med. 2014, 211, 2151–2158. [Google Scholar] [CrossRef]
- Ma, Y.; Chiao, Y.A.; Clark, R.; Flynn, E.R.; Yabluchanskiy, A.; Ghasemi, O.; Zouein, F.; Lindsey, M.L.; Jin, Y.F. Deriving a cardiac ageing signature to reveal MMP-9-dependent inflammatory signalling in senescence. Cardiovasc. Res. 2015, 106, 421–431. [Google Scholar] [CrossRef]
- Heidt, T.; Sager, H.B.; Courties, G.; Dutta, P.; Iwamoto, Y.; Zaltsman, A.; Von Zur Muhlen, C.; Bode, C.; Fricchione, G.L.; Denninger, J.; et al. Chronic variable stress activates hematopoietic stem cells. Nat. Med. 2014, 20, 754–758. [Google Scholar] [CrossRef] [PubMed]
- Seidler, S.; Zimmermann, H.W.; Bartneck, M.; Trautwein, C.; Tacke, F. Age-dependent alterations of monocyte subsets and monocyte-related chemokine pathways in healthy adults. BMC Immunol. 2010, 11, 30. [Google Scholar] [CrossRef] [PubMed]
- Courties, G.; Heidt, T.; Sebas, M.; Iwamoto, Y.; Jeon, D.; Truelove, J.; Tricot, B.; Wojtkiewicz, G.; Dutta, P.; Sager, H.B.; et al. In vivo silencing of the transcription factor IRF5 reprograms the macrophage phenotype and improves infarct healing. J. Am. Coll. Cardiol. 2014, 63, 1556–1566. [Google Scholar] [CrossRef] [PubMed]
- Fujiwara, M.; Matoba, T.; Koga, J.I.; Okahara, A.; Funamoto, D.; Nakano, K.; Tsutsui, H.; Egashira, K. Nanoparticle incorporating Toll-like receptor 4 inhibitor attenuates myocardial ischaemia-reperfusion injury by inhibiting monocyte-mediated inflammation in mice. Cardiovasc. Res. 2019, 115, 1244–1255. [Google Scholar] [CrossRef] [PubMed]
- Gast, M.; Rauch, B.H.; Haghikia, A.; Nakagawa, S.; Haas, J.; Stroux, A.; Schmidt, D.; Schumann, P.; Weiss, S.; Jensen, L.; et al. Long noncoding RNA NEAT1 modulates immune cell functions and is suppressed in early onset myocardial infarction patients. Cardiovasc. Res. 2019, 115, 1886–1906. [Google Scholar] [CrossRef]
- Kruidenier, L.; Chung, C.; Cheng, Z.; Liddle, J.; Che, K.; Joberty, G.; Bantscheff, M.; Bountra, C.; Bridges, A.; Diallo, H.; et al. A selective jumonji H3K27 demethylase inhibitor modulates the proinflammatory macrophage response. Nature 2012, 488, 404–408. [Google Scholar] [CrossRef]
- Kittan, N.A.; Allen, R.M.; Dhaliwal, A.; Cavassani, K.A.; Schaller, M.; Gallagher, K.A.; Carson, W.F.; Mukherjee, S.; Grembecka, J.; Cierpicki, T.; et al. Cytokine Induced Phenotypic and Epigenetic Signatures Are Key to Establishing Specific Macrophage Phenotypes. PLoS ONE 2013, 8, e78045. [Google Scholar] [CrossRef]
- Menasché, P. Cardiac cell therapy: Current status, challenges and perspectives. Arch. Cardiovasc. Dis. 2020, 113, 285–292. [Google Scholar] [CrossRef]
- Vagnozzi, R.J.; Maillet, M.; Sargent, M.A.; Khalil, H.; Johansen, A.K.Z.; Schwanekamp, J.A.; York, A.J.; Huang, V.; Nahrendorf, M.; Sadayappan, S.; et al. An acute immune response underlies the benefit of cardiac stem cell therapy. Nature 2020, 577, 405–409. [Google Scholar] [CrossRef]
- Lin, Y.; Damjanovic, A.; Jeffrey Metter, E.; Nguyen, H.; Truong, T.; Najarro, K.; Morris, C.; Longo, D.L.; Zhan, M.; Ferrucci, L.; et al. Age-associated telomere attrition of lymphocytes in vivo is co-ordinated with changes in telomerase activity, composition of lymphocyte subsets and health conditions. Clin. Sci. 2015, 128, 367–377. [Google Scholar] [CrossRef]
- Spyridopoulos, I.; Hoffmann, J.; Aicher, A.; Brümmendorf, T.H.; Doerr, H.W.; Zeiher, A.M.; Dimmeler, S. Accelerated telomere shortening in leukocyte subpopulations of patients with coronary heart disease: Role of cytomegalovirus seropositivity. Circulation 2009, 120, 1364–1372. [Google Scholar] [CrossRef] [PubMed]
- Ucar, D.; Márquez, E.J.; Chung, C.H.; Marches, R.; Rossi, R.J.; Uyar, A.; Wu, T.C.; George, J.; Stitzel, M.L.; Karolina Palucka, A.; et al. The chromatin accessibility signature of human immune aging stems from CD8+ T cells. J. Exp. Med. 2017, 214, 3123–3144. [Google Scholar] [CrossRef] [PubMed]
- Gibbons, C.; Sykes, M. Manipulating the immune system for anti-tumor responses and transplant tolerance via mixed hematopoietic chimerism. Immunol. Rev. 2008, 223, 334–360. [Google Scholar] [CrossRef] [PubMed]
- Sykes, M.; Nikolic, B. Treatment of severe autoimmune disease by stem-cell transplantation. Nature 2005, 435, 620–627. [Google Scholar] [CrossRef]
- Nikolic, B.; Takeuchi, Y.; Leykin, I.; Fudaba, Y.; Smith, R.N.; Sykes, M. Mixed Hematopoietic Chimerism Allows Cure of Autoimmune Diabetes Through Allogeneic Tolerance and Reversal of Autoimmunity. Diabetes 2004, 53, 376–383. [Google Scholar] [CrossRef]
- Li, J.; Li, S.; Dong, J.; Alibhai, F.J.; Zhang, C.; Shao, Z.; Song, H.; He, S.; Yin, W.; Wu, J.; et al. Long-term repopulation of aged bone marrow stem cells using young Sca-1 cells promotes aged heart rejuvenation. Aging Cell 2019, 18, e13026. [Google Scholar] [CrossRef]
- Wlodarek, L.; Cao, F.; Alibhai, F.J.; Fekete, A.; Noyan, N.; Tobin, S.W.; Marvasti, T.B.; Wu, J.; Li, S.-H.; Weisel, R.D.; et al. Rectification of radiotherapy-induced cognitive impairments in aged mice by reconstituted Sca-1+ stem cells from young donors. J. Neuroinflamm. 2020, 17, 51. [Google Scholar] [CrossRef]
- Kang, S.; Moser, V.A.; Svendsen, C.N.; Goodridge, H.S. Rejuvenating the blood and bone marrow to slow aging-associated cognitive decline and Alzheimer’s disease. Commun. Biol. 2020, 3, 69. [Google Scholar] [CrossRef]
- Tobin, S.W.; Alibhai, F.J.; Wlodarek, L.; Yeganeh, A.; Millar, S.; Wu, J.; Li, S.; Weisel, R.D.; Li, R.-K. Delineating the relationship between immune system aging and myogenesis in muscle repair. bioRxiv 2020. [Google Scholar] [CrossRef]
- Das, M.M.; Godoy, M.; Chen, S.; Moser, V.A.; Avalos, P.; Roxas, K.M.; Dang, I.; Yáñez, A.; Zhang, W.; Bresee, C.; et al. Young bone marrow transplantation preserves learning and memory in old mice. Commun. Biol. 2019, 2, 73. [Google Scholar] [CrossRef]
- Villeda, S.A.; Luo, J.; Mosher, K.I.; Zou, B.; Britschgi, M.; Bieri, G.; Stan, T.M.; Fainberg, N.; Ding, Z.; Eggel, A.; et al. The ageing systemic milieu negatively regulates neurogenesis and cognitive function. Nature 2011, 477, 90–96. [Google Scholar] [CrossRef] [PubMed]
- Smith, L.K.; He, Y.; Park, J.S.; Bieri, G.; Snethlage, C.E.; Lin, K.; Gontier, G.; Wabl, R.; Plambeck, K.E.; Udeochu, J.; et al. β2-microglobulin is a systemic pro-aging factor that impairs cognitive function and neurogenesis. Nat. Med. 2015, 21, 932–937. [Google Scholar] [CrossRef] [PubMed]
- Kovina, M.V.; Karnaukhov, A.V.; Krasheninnikov, M.E.; Kovin, A.L.; Gazheev, S.T.; Sergievich, L.A.; Karnaukhova, E.V.; Bogdanenko, E.V.; Balyasin, M.V.; Khodarovich, Y.M.; et al. Extension of Maximal Lifespan and High Bone Marrow Chimerism After Nonmyeloablative Syngeneic Transplantation of Bone Marrow From Young to Old Mice. Front. Genet. 2019, 10, 310. [Google Scholar] [CrossRef] [PubMed]
- Chhabra, A.; Ring, A.M.; Weiskopf, K.; Schnorr, P.J.; Gordon, S.; Le, A.C.; Kwon, H.-S.; Ring, N.G.; Volkmer, J.; Ho, P.Y.; et al. Hematopoietic stem cell transplantation in immunocompetent hosts without radiation or chemotherapy. Sci. Transl. Med. 2016, 8, 351ra105. [Google Scholar] [CrossRef]
- Guderyon, M.J.; Chen, C.; Bhattacharjee, A.; Ge, G.; Fernandez, R.A.; Gelfond, J.A.L.; Gorena, K.M.; Cheng, C.J.; Li, Y.; Nelson, J.F.; et al. Mobilization-based transplantation of young-donor hematopoietic stem cells extends lifespan in mice. Aging Cell 2020, 19. [Google Scholar] [CrossRef]
- Kamminga, L.M.; van Os, R.; Ausema, A.; Noach, E.J.K.; Weersing, E.; Dontje, B.; Vellenga, E.; de Haan, G. Impaired Hematopoietic Stem Cell Functioning After Serial Transplantation and During Normal Aging. Stem Cells 2005, 23, 82–92. [Google Scholar] [CrossRef]
- Brown, K.; Xie, S.; Qiu, X.; Mohrin, M.; Shin, J.; Liu, Y.; Zhang, D.; Scadden, D.T.; Chen, D. SIRT3 Reverses Aging-Associated Degeneration. Cell Rep. 2013, 3, 319–327. [Google Scholar] [CrossRef]
- Maryanovich, M.; Zahalka, A.H.; Pierce, H.; Pinho, S.; Nakahara, F.; Asada, N.; Wei, Q.; Wang, X.; Ciero, P.; Xu, J.; et al. Adrenergic nerve degeneration in bone marrow drives aging of the hematopoietic stem cell niche. Nat. Med. 2018, 24, 782–791. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef]
- Tchkonia, T.; Zhu, Y.; van Deursen, J.; Campisi, J.; Kirkland, J.L. Cellular senescence and the senescent secretory phenotype: Therapeutic opportunities. J. Clin. Investig. 2013, 123, 966–972. [Google Scholar] [CrossRef]
- Ruhland, M.K.; Loza, A.J.; Capietto, A.-H.; Luo, X.; Knolhoff, B.L.; Flanagan, K.C.; Belt, B.A.; Alspach, E.; Leahy, K.; Luo, J.; et al. Stromal senescence establishes an immunosuppressive microenvironment that drives tumorigenesis. Nat. Commun. 2016, 7, 11762. [Google Scholar] [CrossRef] [PubMed]
- Vicente, R.; Mausset-Bonnefont, A.-L.; Jorgensen, C.; Louis-Plence, P.; Brondello, J.-M. Cellular senescence impact on immune cell fate and function. Aging Cell 2016, 15, 400–406. [Google Scholar] [CrossRef] [PubMed]
- Signer, R.A.J.; Montecino-Rodriguez, E.; Witte, O.N.; Dorshkind, K. Aging and cancer resistance in lymphoid progenitors are linked processes conferred by p16Ink4a and Arf. Genes Dev. 2008, 22, 3115–3120. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Childs, B.G.; Durik, M.; Wijers, M.E.; Sieben, C.J.; Zhong, J.; Saltness, R.A.; Jeganathan, K.B.; Verzosa, G.C.; Pezeshki, A.; et al. Naturally occurring p16 Ink4a-positive cells shorten healthy lifespan. Nature 2016, 530, 184–189. [Google Scholar] [CrossRef]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; Lebrasseur, N.K.; Childs, B.G.; Van De Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16 Ink4a-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef]
- Chang, J.; Wang, Y.; Shao, L.; Laberge, R.M.; Demaria, M.; Campisi, J.; Janakiraman, K.; Sharpless, N.E.; Ding, S.; Feng, W.; et al. Clearance of senescent cells by ABT263 rejuvenates aged hematopoietic stem cells in mice. Nat. Med. 2016, 22, 78–83. [Google Scholar] [CrossRef]
- Yosef, R.; Pilpel, N.; Tokarsky-Amiel, R.; Biran, A.; Ovadya, Y.; Cohen, S.; Vadai, E.; Dassa, L.; Shahar, E.; Condiotti, R.; et al. Directed elimination of senescent cells by inhibition of BCL-W and BCL-XL. Nat. Commun. 2016, 7, 11190. [Google Scholar] [CrossRef]
- Zhu, Y.; Tchkonia, T.; Pirtskhalava, T.; Gower, A.C.; Ding, H.; Giorgadze, N.; Palmer, A.K.; Ikeno, Y.; Hubbard, G.B.; Lenburg, M.; et al. The Achilles’ heel of senescent cells: From transcriptome to senolytic drugs. Aging Cell 2015, 14, 644–658. [Google Scholar] [CrossRef]
- Xu, M.; Pirtskhalava, T.; Farr, J.N.; Weigand, B.M.; Palmer, A.K.; Weivoda, M.M.; Inman, C.L.; Ogrodnik, M.B.; Hachfeld, C.M.; Fraser, D.G.; et al. Senolytics improve physical function and increase lifespan in old age. Nat. Med. 2018, 24, 1246–1256. [Google Scholar] [CrossRef]
- Roos, C.M.; Zhang, B.; Palmer, A.K.; Ogrodnik, M.B.; Pirtskhalava, T.; Thalji, N.M.; Hagler, M.; Jurk, D.; Smith, L.A.; Casaclang-Verzosa, G.; et al. Chronic senolytic treatment alleviates established vasomotor dysfunction in aged or atherosclerotic mice. Aging Cell 2016, 15, 973–977. [Google Scholar] [CrossRef]
- Amor, C.; Feucht, J.; Leibold, J.; Ho, Y.-J.; Zhu, C.; Alonso-Curbelo, D.; Mansilla-Soto, J.; Boyer, J.A.; Li, X.; Giavridis, T.; et al. Senolytic CAR T cells reverse senescence-associated pathologies. Nature 2020, 583, 127–132. [Google Scholar] [CrossRef] [PubMed]
- Fuhrmann-Stroissnigg, H.; Niedernhofer, L.J.; Robbins, P.D. Hsp90 inhibitors as senolytic drugs to extend healthy aging. Cell Cycle 2018, 17, 1048–1055. [Google Scholar] [CrossRef] [PubMed]
- Guerrero, A.; Herranz, N.; Sun, B.; Wagner, V.; Gallage, S.; Guiho, R.; Wolter, K.; Pombo, J.; Irvine, E.E.; Innes, A.J.; et al. Cardiac glycosides are broad-spectrum senolytics. Nat. Metab. 2019, 1, 1074–1088. [Google Scholar] [CrossRef] [PubMed]
- Yousefzadeh, M.J.; Zhu, Y.; McGowan, S.J.; Angelini, L.; Fuhrmann-Stroissnigg, H.; Xu, M.; Ling, Y.Y.; Melos, K.I.; Pirtskhalava, T.; Inman, C.L.; et al. Fisetin is a senotherapeutic that extends health and lifespan. EBioMedicine 2018, 36, 18–28. [Google Scholar] [CrossRef]
- Anderson, R.; Lagnado, A.; Maggiorani, D.; Walaszczyk, A.; Dookun, E.; Chapman, J.; Birch, J.; Salmonowicz, H.; Ogrodnik, M.; Jurk, D.; et al. Length-independent telomere damage drives post-mitotic cardiomyocyte senescence. EMBO J. 2019, 38, e100492. [Google Scholar] [CrossRef]
- Lewis-McDougall, F.C.; Ruchaya, P.J.; Domenjo-Vila, E.; Shin Teoh, T.; Prata, L.; Cottle, B.J.; Clark, J.E.; Punjabi, P.P.; Awad, W.; Torella, D.; et al. Aged-senescent cells contribute to impaired heart regeneration. Aging Cell 2019, 18, e12931. [Google Scholar] [CrossRef]
- Hickson, L.J.; Langhi Prata, L.G.P.; Bobart, S.A.; Evans, T.K.; Giorgadze, N.; Hashmi, S.K.; Herrmann, S.M.; Jensen, M.D.; Jia, Q.; Jordan, K.L.; et al. Senolytics decrease senescent cells in humans: Preliminary report from a clinical trial of Dasatinib plus Quercetin in individuals with diabetic kidney disease. EBioMedicine 2019, 47, 446–456. [Google Scholar] [CrossRef]
- Justice, J.N.; Nambiar, A.M.; Tchkonia, T.; LeBrasseur, N.K.; Pascual, R.; Hashmi, S.K.; Prata, L.; Masternak, M.M.; Kritchevsky, S.B.; Musi, N.; et al. Senolytics in idiopathic pulmonary fibrosis: Results from a first-in-human, open-label, pilot study. EBioMedicine 2019, 40, 554–563. [Google Scholar] [CrossRef]
- Zhu, F.; Li, Y.; Zhang, J.; Piao, C.; Liu, T.; Li, H.-H.; Du, J. Senescent Cardiac Fibroblast Is Critical for Cardiac Fibrosis after Myocardial Infarction. PLoS ONE 2013, 8, e74535. [Google Scholar] [CrossRef]




{kind=link}
{kind=link}
{kind=link}
| Method | Injury | Outcome | Reference |
|---|---|---|---|
| 8 wk., 18- and 30-mo. (WT) | N/A | Cardiac macrophages and neutrophils increase with age | [167] |
| 2–3 vs. 12–15 mo. (WT) | N/A | Higher T cell activity in the heart draining lymph nodes | [168] |
| Cardiac macrophages from 4-, 8-, or 30-wk. (WT) | N/A | Functional and transcriptional profiling indicate a senescent, fibrotic phenotype forms | [169] |
| Heterochronic parabiosis | N/A | Reduced age-related cardiac hypertrophy | [170] |
| 2–3 and >24 mo. (WT) | I/R | Impaired inflammation and healing; decreased cardiac function | [166] |
| Senolytic (ABT-263) administration; 23 mo. (WT) | P | Improved survival and cardiac function | [171] |
| Heterochronic BMT (2–3 vs. 20–22 mo.) | P | Enhanced angiogenesis, scar thickness and overall cardiac function | [172,173,174] |
| Competitive BMT using Tet2−/− cells | P | Upregulated IL-1β expression; Increased fibrosis; Decreased heart function | [100] |
| MMP9 KO mice; 11–36 mo. | P | Enhanced M2 macrophage activity; improved survival; reduced left ventricular dilatation. | [175] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Tobin, S.W.; Alibhai, F.J.; Weisel, R.D.; Li, R.-K. Considering Cause and Effect of Immune Cell Aging on Cardiac Repair after Myocardial Infarction. Cells 2020, 9, 1894. https://doi.org/10.3390/cells9081894
Tobin SW, Alibhai FJ, Weisel RD, Li R-K. Considering Cause and Effect of Immune Cell Aging on Cardiac Repair after Myocardial Infarction. Cells. 2020; 9(8):1894. https://doi.org/10.3390/cells9081894
Chicago/Turabian StyleTobin, Stephanie W., Faisal J. Alibhai, Richard D. Weisel, and Ren-Ke Li. 2020. "Considering Cause and Effect of Immune Cell Aging on Cardiac Repair after Myocardial Infarction" Cells 9, no. 8: 1894. https://doi.org/10.3390/cells9081894
APA StyleTobin, S. W., Alibhai, F. J., Weisel, R. D., & Li, R.-K. (2020). Considering Cause and Effect of Immune Cell Aging on Cardiac Repair after Myocardial Infarction. Cells, 9(8), 1894. https://doi.org/10.3390/cells9081894