Role of Inflammasomes in HIV-1 and Drug Abuse Mediated Neuroinflammaging


 , , and
, , and
Abstract

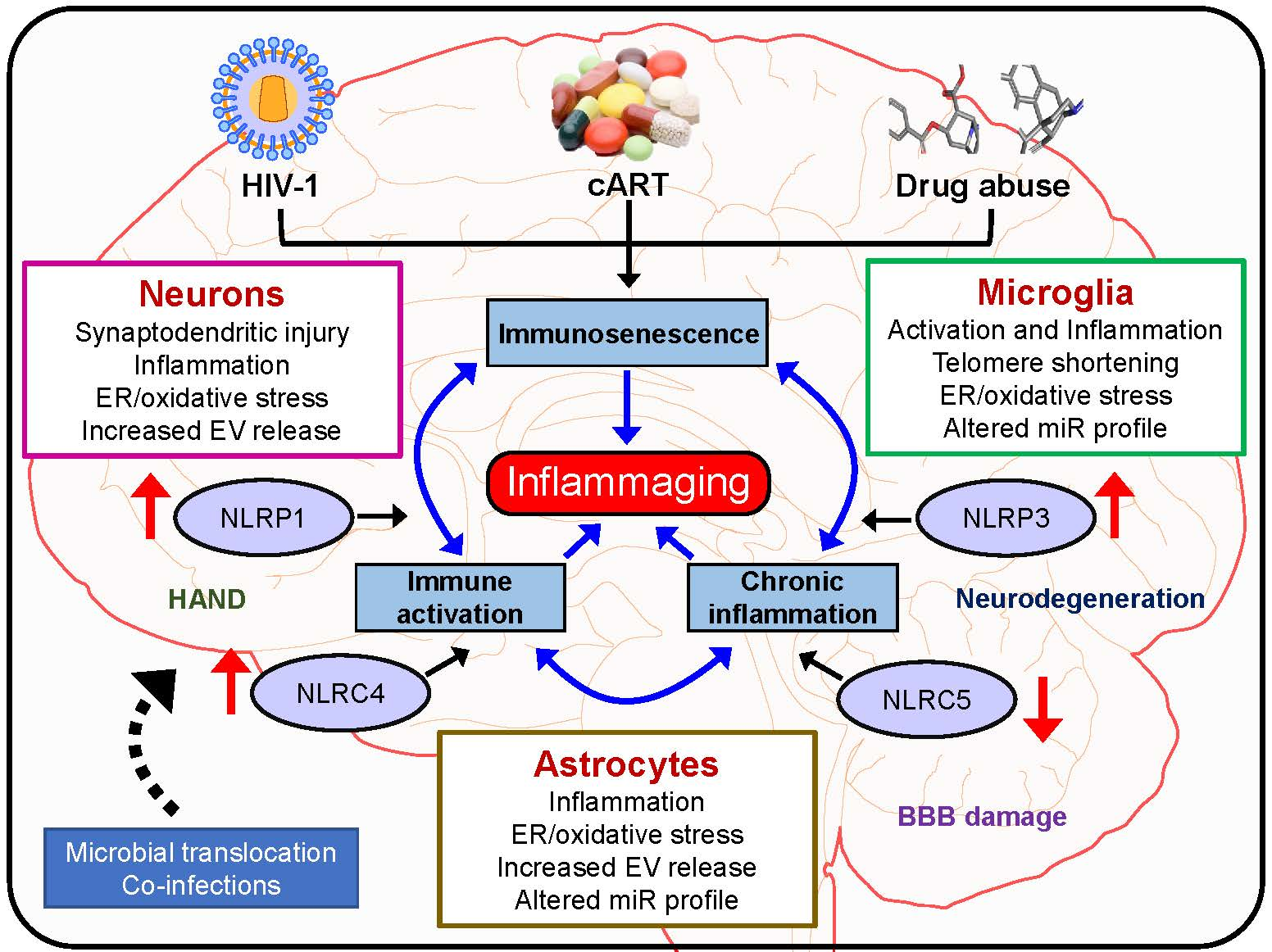{kind=link}
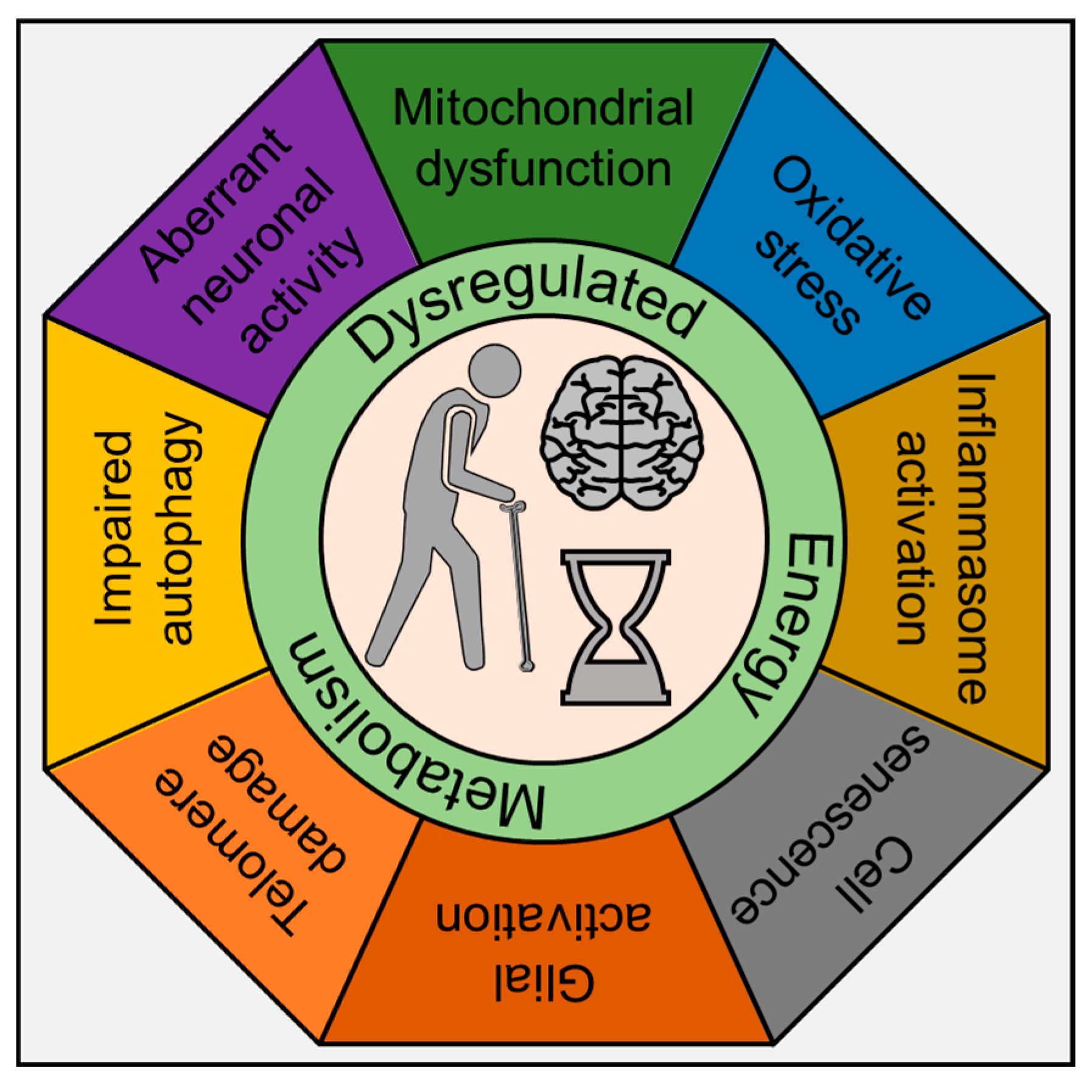{kind=link}
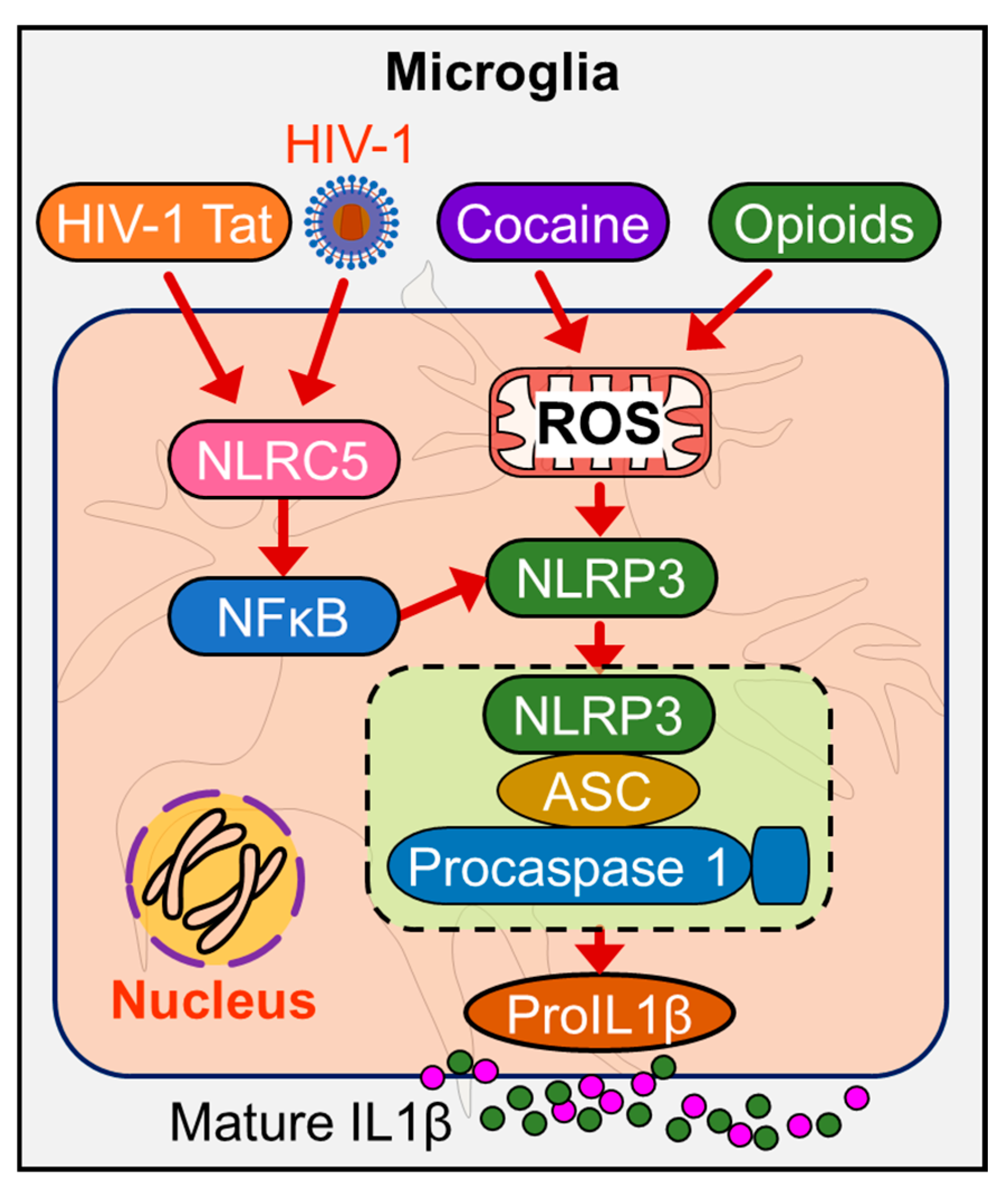{kind=link}
{kind=link}
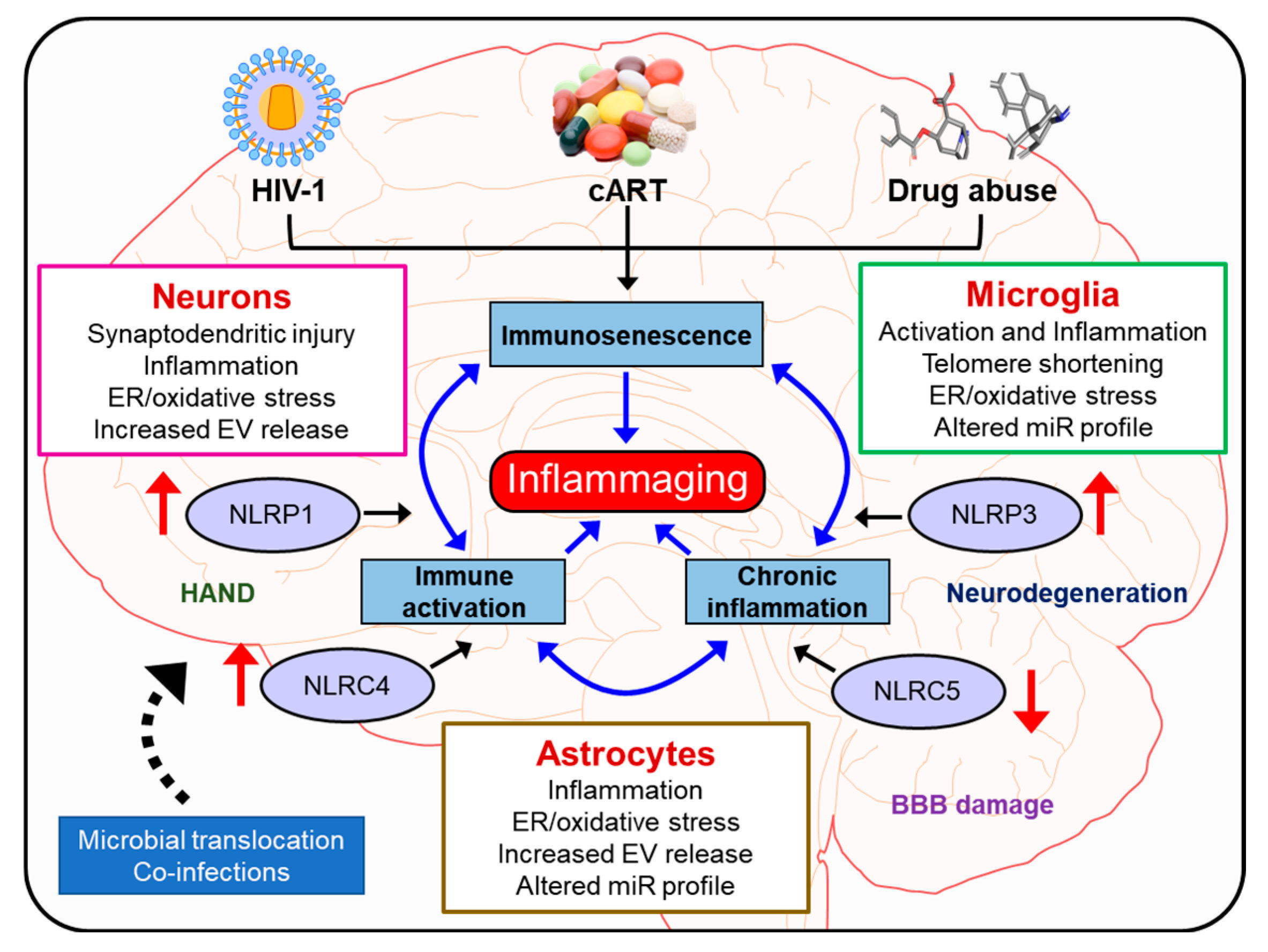{kind=link}
1. Introduction
2. Inflammaging
3. Inflammasomes
3.1. NLRP1 Inflammasome
3.2. NLRP3 Inflammasome
3.3. NLRC4 Inflammasome
3.4. NLRC5 Inflammasome
3.5. AIM2 Inflammasome
4. Neuroinflammation in the Context of HIV-1 and Drug Abuse
5. Role of Microglia—Inflammasomes and Aging
6. Role of Astrocytes—Inflammasomes and Aging
7. Role of Neurons—Inflammasomes and Aging
Role of Extracellular Vesicles (EVs) in Aging
8. Conclusions and Future Perspectives
Funding
Acknowledgments
Conflicts of Interest
References
- Lohse, N.; Obel, N. Update of Survival for Persons with HIV Infection in Denmark. Ann. Intern. Med. 2016, 165, 749–750. [Google Scholar] [CrossRef] [PubMed]
- Legarth, R.A.; Ahlstrom, M.G.; Kronborg, G.; Larsen, C.S.; Pedersen, C.; Pedersen, G.; Mohey, R.; Gerstoft, J.; Obel, N. Long-Term Mortality in HIV-Infected Individuals 50 Years or Older: A Nationwide, Population-Based Cohort Study. J. Acquir. Immune Defic. Syndr. 2016, 71, 213–218. [Google Scholar] [CrossRef] [PubMed]
- Gross, A.M.; Jaeger, P.A.; Kreisberg, J.F.; Licon, K.; Jepsen, K.L.; Khosroheidari, M.; Morsey, B.M.; Swindells, S.; Shen, H.; Ng, C.T.; et al. Methylome-wide Analysis of Chronic HIV Infection Reveals Five-Year Increase in Biological Age and Epigenetic Targeting of HLA. Mol. Cell. 2016, 62, 157–168. [Google Scholar] [CrossRef] [PubMed]
- McArthur, J.C.; Johnson, T.P. Chronic inflammation mediates brain injury in HIV infection: Relevance for cure strategies. Curr. Opin. Neurol. 2020, 33, 397–404. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV infection, inflammation, immunosenescence, and aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G.; Tracy, R.; Douek, D.C. Systemic effects of inflammation on health during chronic HIV infection. Immunity 2013, 39, 633–645. [Google Scholar] [CrossRef] [PubMed]
- Leng, S.X.; Margolick, J.B. Aging, sex, inflammation, frailty, and CMV and HIV infections. Cell. Immunol. 2019, 348, 104024. [Google Scholar] [CrossRef]
- High, K.P.; Brennan-Ing, M.; Clifford, D.B.; Cohen, M.H.; Currier, J.; Deeks, S.G.; Deren, S.; Effros, R.B.; Gebo, K.; Goronzy, J.J.; et al. HIV and aging: State of knowledge and areas of critical need for research. A report to the NIH Office of AIDS Research by the HIV and Aging Working Group. J. Acquir. Immune Defic. Syndr. 2012, 60 (Suppl. 1), S1–S18. [Google Scholar] [CrossRef]
- Guaraldi, G.; Prakash, M.; Moecklinghoff, C.; Stellbrink, H.J. Morbidity in older HIV-infected patients: Impact of long-term antiretroviral use. Aids Rev. 2014, 16, 75–89. [Google Scholar]
- Steele, A.K.; Lee, E.J.; Vestal, B.; Hecht, D.; Dong, Z.; Rapaport, E.; Koeppe, J.; Campbell, T.B.; Wilson, C.C. Contribution of intestinal barrier damage, microbial translocation and HIV-1 infection status to an inflammaging signature. PloS ONE 2014, 9, e97171. [Google Scholar] [CrossRef]
- Sundermann, E.E.; Hussain, M.A.; Moore, D.J.; Horvath, S.; Lin, D.T.S.; Kobor, M.S.; Levine, A.; Group, H. Inflammation-related genes are associated with epigenetic aging in HIV. J. Neurovirol. 2019, 25, 853–865. [Google Scholar] [CrossRef] [PubMed]
- Palmer, C.S.; Palchaudhuri, R.; Albargy, H.; Abdel-Mohsen, M.; Crowe, S.M. Exploiting immune cell metabolic machinery for functional HIV cure and the prevention of inflammaging. F1000Research 2018, 7, 125. [Google Scholar] [CrossRef] [PubMed]
- Kaplan-Lewis, E.; Aberg, J.A.; Lee, M. Aging with HIV in the ART era. Semin. Diagn. Pathol. 2017, 34, 384–397. [Google Scholar] [CrossRef] [PubMed]
- Nasi, M.; De Biasi, S.; Gibellini, L.; Bianchini, E.; Pecorini, S.; Bacca, V.; Guaraldi, G.; Mussini, C.; Pinti, M.; Cossarizza, A. Ageing and inflammation in patients with HIV infection. Clin. Exp. Immunol. 2017, 187, 44–52. [Google Scholar] [CrossRef]
- Xia, C.; Luo, D.; Yu, X.; Jiang, S.; Liu, S. HIV-associated dementia in the era of highly active antiretroviral therapy (HAART). Microbes Infect. 2011, 13, 419–425. [Google Scholar] [CrossRef]
- Singh, D. What’s in a name? AIDS dementia complex, HIV-associated dementia, HIV-associated neurocognitive disorder or HIV encephalopathy. Afr. J. Psychiatry (Johannesbg.) 2012, 15, 172–175. [Google Scholar] [CrossRef][Green Version]
- Schouten, J.; Cinque, P.; Gisslen, M.; Reiss, P.; Portegies, P. HIV-1 infection and cognitive impairment in the cART era: A review. AIDS 2011, 25, 561–575. [Google Scholar] [CrossRef]
- Aquaro, S.; Svicher, V.; Ronga, L.; Perno, C.F.; Pollicita, M. HIV-1-associated dementia during HAART therapy. Recent Pat. Cns Drug Discov. 2008, 3, 23–33. [Google Scholar] [CrossRef]
- Heaton, R.K.; Clifford, D.B.; Franklin, D.R., Jr.; Woods, S.P.; Ake, C.; Vaida, F.; Ellis, R.J.; Letendre, S.L.; Marcotte, T.D.; Atkinson, J.H.; et al. HIV-associated neurocognitive disorders persist in the era of potent antiretroviral therapy: CHARTER Study. Neurology 2010, 75, 2087–2096. [Google Scholar] [CrossRef]
- Bonnet, F.; Amieva, H.; Marquant, F.; Bernard, C.; Bruyand, M.; Dauchy, F.A.; Mercie, P.; Greib, C.; Richert, L.; Neau, D.; et al. Cognitive disorders in HIV-infected patients: Are they HIV-related? AIDS 2013, 27, 391–400. [Google Scholar] [CrossRef]
- Elbirt, D.; Mahlab-Guri, K.; Bezalel-Rosenberg, S.; Gill, H.; Attali, M.; Asher, I. HIV-associated neurocognitive disorders (HAND). Isr. Med. Assoc. J. 2015, 17, 54–59. [Google Scholar] [PubMed]
- Simioni, S.; Cavassini, M.; Annoni, J.M.; Rimbault Abraham, A.; Bourquin, I.; Schiffer, V.; Calmy, A.; Chave, J.P.; Giacobini, E.; Hirschel, B.; et al. Cognitive dysfunction in HIV patients despite long-standing suppression of viremia. AIDS 2010, 24, 1243–1250. [Google Scholar] [CrossRef] [PubMed]
- Goodwin, L.D.; Goodwin, W.L.; Cantrill, J.L. The mental health needs of elementary schoolchildren. J. Sch. Health 1988, 58, 282–287. [Google Scholar] [CrossRef] [PubMed]
- Cotto, B.; Natarajanseenivasan, K.; Langford, D. HIV-1 infection alters energy metabolism in the brain: Contributions to HIV-associated neurocognitive disorders. Prog. Neurobiol. 2019, 181, 101616. [Google Scholar] [CrossRef] [PubMed]
- Bougea, A.; Spantideas, N.; Galanis, P.; Gkekas, G.; Thomaides, T. Optimal treatment of HIV-associated neurocognitive disorders: Myths and reality. A critical review. Adv. Infect. Dis. 2019, 6. [Google Scholar] [CrossRef]
- Alfahad, T.B.; Nath, A. Update on HIV-associated neurocognitive disorders. Curr. Neurol. Neurosci. Rep. 2013, 13, 387. [Google Scholar] [CrossRef][Green Version]
- Gelman, B.B. Neuropathology of HAND With Suppressive Antiretroviral Therapy: Encephalitis and Neurodegeneration Reconsidered. Curr. Hiv/Aids Rep. 2015, 12, 272–279. [Google Scholar] [CrossRef]
- Chan, P.; Hellmuth, J.; Spudich, S.; Valcour, V. Cognitive Impairment and Persistent CNS Injury in Treated HIV. Curr. Hiv/Aids Rep. 2016, 13, 209–217. [Google Scholar] [CrossRef]
- Marra, C.M. HIV-associated neurocognitive disorders and central nervous system drug penetration: What next? Antivir. Ther. 2015, 20, 365–367. [Google Scholar] [CrossRef]
- Letendre, S. Central nervous system complications in HIV disease: HIV-associated neurocognitive disorder. Top. Antivir. Med. 2011, 19, 137–142. [Google Scholar]
- Lanman, T.; Letendre, S.; Ma, Q.; Bang, A.; Ellis, R. CNS Neurotoxicity of Antiretrovirals. J. Neuroimmunepharmacol 2019. [Google Scholar] [CrossRef] [PubMed]
- Dash, S.; Balasubramaniam, M.; Villalta, F.; Dash, C.; Pandhare, J. Impact of cocaine abuse on HIV pathogenesis. Front. Microbiol 2015, 6, 1111. [Google Scholar] [CrossRef] [PubMed]
- Tyagi, M.; Bukrinsky, M.; Simon, G.L. Mechanisms of HIV Transcriptional Regulation by Drugs of Abuse. Curr. Hiv Res. 2016, 14, 442–454. [Google Scholar] [CrossRef] [PubMed]
- Gurwell, J.A.; Nath, A.; Sun, Q.; Zhang, J.; Martin, K.M.; Chen, Y.; Hauser, K.F. Synergistic neurotoxicity of opioids and human immunodeficiency virus-1 Tat protein in striatal neurons in vitro. Neuroscience 2001, 102, 555–563. [Google Scholar] [CrossRef]
- Turchan, J.; Anderson, C.; Hauser, K.F.; Sun, Q.; Zhang, J.; Liu, Y.; Wise, P.M.; Kruman, I.; Maragos, W.; Mattson, M.P.; et al. Estrogen protects against the synergistic toxicity by HIV proteins, methamphetamine and cocaine. BMC Neurosci. 2001, 2, 3. [Google Scholar] [CrossRef]
- Degenhardt, L.; Whiteford, H.; Hall, W.D. The Global Burden of Disease projects: What have we learned about illicit drug use and dependence and their contribution to the global burden of disease? Drug Alcohol. Rev. 2014, 33, 4–12. [Google Scholar] [CrossRef]
- Degenhardt, L.; Whiteford, H.A.; Ferrari, A.J.; Baxter, A.J.; Charlson, F.J.; Hall, W.D.; Freedman, G.; Burstein, R.; Johns, N.; Engell, R.E.; et al. Global burden of disease attributable to illicit drug use and dependence: Findings from the Global Burden of Disease Study 2010. Lancet 2013, 382, 1564–1574. [Google Scholar] [CrossRef]
- Holt, J.L.; Kraft-Terry, S.D.; Chang, L. Neuroimaging studies of the aging HIV-1-infected brain. J. Neurovirol. 2012, 18, 291–302. [Google Scholar] [CrossRef][Green Version]
- Chang, L.; Shukla, D.K. Imaging studies of the HIV-infected brain. Handb. Clin. Neurol. 2018, 152, 229–264. [Google Scholar] [CrossRef]
- Vera, J.H.; Ridha, B.; Gilleece, Y.; Amlani, A.; Thorburn, P.; Dizdarevic, S. PET brain imaging in HIV-associated neurocognitive disorders (HAND) in the era of combination antiretroviral therapy. Eur. J. Nucl. Med. Mol. Imaging 2017, 44, 895–902. [Google Scholar] [CrossRef]
- Campbell, A.N.; Tross, S.; Calsyn, D.A. Substance use disorders and HIV/AIDS prevention and treatment intervention: Research and practice considerations. Soc. Work Public Health 2013, 28, 333–348. [Google Scholar] [CrossRef] [PubMed]
- Dahal, S.; Chitti, S.V.; Nair, M.P.; Saxena, S.K. Interactive effects of cocaine on HIV infection: Implication in HIV-associated neurocognitive disorder and neuroAIDS. Front. Microbiol. 2015, 6, 931. [Google Scholar] [CrossRef] [PubMed]
- Chang, S.L.; Connaghan, K.P.; Wei, Y.; Li, M.D. NeuroHIV and use of addictive substances. Int. Rev. Neurobiol. 2014, 118, 403–440. [Google Scholar] [CrossRef] [PubMed]
- Ene, L. Human Immunodeficiency Virus in the Brain-Culprit or Facilitator? Infect. Dis. (Auckl) 2018, 11. [Google Scholar] [CrossRef]
- Gougeon, M.L. Alarmins and Mol. Cell. Proteom central nervous system inflammation in HIV-associated neurological disorders. J. Intern. Med. 2017, 281, 433–447. [Google Scholar] [CrossRef]
- Hong, S.; Banks, W.A. Role of the immune system in HIV-associated neuroinflammation and neurocognitive implications. Brain Behav. Immun. 2015, 45, 1–12. [Google Scholar] [CrossRef]
- Clifford, D.B.; Ances, B.M. HIV-associated neurocognitive disorder. Lancet Infect. Dis. 2013, 13, 976–986. [Google Scholar] [CrossRef]
- Saylor, D.; Dickens, A.M.; Sacktor, N.; Haughey, N.; Slusher, B.; Pletnikov, M.; Mankowski, J.L.; Brown, A.; Volsky, D.J.; McArthur, J.C. HIV-associated neurocognitive disorder—Pathogenesis and prospects for treatment. Nat. Rev. Neurol. 2016, 12, 234–248. [Google Scholar] [CrossRef]
- Bell, J.E. An update on the neuropathology of HIV in the HAART era. Histopathology 2004, 45, 549–559. [Google Scholar] [CrossRef]
- Gonzalez-Scarano, F.; Martin-Garcia, J. The neuropathogenesis of AIDS. Nat. Rev. Immunol. 2005, 5, 69–81. [Google Scholar] [CrossRef]
- Lawrence, D.M.; Major, E.O. HIV-1 and the brain: Connections between HIV-1-associated dementia, neuropathology and neuroimmunology. Microbes Infect. 2002, 4, 301–308. [Google Scholar] [CrossRef]
- Calder, P.C.; Bosco, N.; Bourdet-Sicard, R.; Capuron, L.; Delzenne, N.; Dore, J.; Franceschi, C.; Lehtinen, M.J.; Recker, T.; Salvioli, S.; et al. Health relevance of the modification of low grade inflammation in ageing (inflammageing) and the role of nutrition. Ageing Res. Rev. 2017, 40, 95–119. [Google Scholar] [CrossRef] [PubMed]
- Gruevska, A.; Moragrega, A.B.; Galindo, M.J.; Esplugues, J.V.; Blas-Garcia, A.; Apostolova, N. p53 and p53-related mediators PAI-1 and IGFBP-3 are downregulated in peripheral blood mononuclear cells of HIV-patients exposed to non-nucleoside reverse transcriptase inhibitors. Antivir. Res. 2020, 178. [Google Scholar] [CrossRef] [PubMed]
- Lopez-Otin, C.; Blasco, M.A.; Partridge, L.; Serrano, M.; Kroemer, G. The hallmarks of aging. Cell 2013, 153, 1194–1217. [Google Scholar] [CrossRef]
- Mackiewicz, M.M.; Overk, C.; Achim, C.L.; Masliah, E. Pathogenesis of age-related HIV neurodegeneration. J. Neurovirol. 2019, 25, 622–633. [Google Scholar] [CrossRef]
- Horvath, S.; Levine, A.J. HIV-1 Infection Accelerates Age According to the Epigenetic Clock. J. Infect. Dis. 2015, 212, 1563–1573. [Google Scholar] [CrossRef]
- Horvath, S.; Stein, D.J.; Phillips, N.; Heany, S.J.; Kobor, M.S.; Lin, D.T.S.; Myer, L.; Zar, H.J.; Levine, A.J.; Hoare, J. Perinatally acquired HIV infection accelerates epigenetic aging in South African adolescents. AIDS 2018, 32, 1465–1474. [Google Scholar] [CrossRef]
- Johnson, T.P.; Patel, K.; Johnson, K.R.; Maric, D.; Calabresi, P.A.; Hasbun, R.; Nath, A. Induction of IL-17 and nonclassical T-cell activation by HIV-Tat protein. Proc. Natl. Acad. Sci. USA 2013, 110, 13588–13593. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Inflammasomes and their roles in health and disease. Annu. Rev. Cell Dev. Biol. 2012, 28, 137–161. [Google Scholar] [CrossRef]
- Davis, B.K.; Wen, H.; Ting, J.P. The inflammasome NLRs in immunity, inflammation, and associated diseases. Annu. Rev. Immunol. 2011, 29, 707–735. [Google Scholar] [CrossRef]
- Kayagaki, N.; Warming, S.; Lamkanfi, M.; Vande Walle, L.; Louie, S.; Dong, J.; Newton, K.; Qu, Y.; Liu, J.; Heldens, S.; et al. Non-canonical inflammasome activation targets caspase-11. Nature 2011, 479, 117–121. [Google Scholar] [CrossRef] [PubMed]
- Martinon, F.; Burns, K.; Tschopp, J. The inflammasome: A molecular platform triggering activation of inflammatory caspases and processing of proIL-beta. Mol. Cell. 2002, 10, 417–426. [Google Scholar] [CrossRef]
- Salvesen, G.S.; Dixit, V.M. Caspase activation: The induced-proximity model. Proc. Natl. Acad. Sci. USA 1999, 96, 10964–10967. [Google Scholar] [CrossRef] [PubMed]
- Shi, Y. Caspase activation: Revisiting the induced proximity model. Cell 2004, 117, 855–858. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Kanneganti, T.D.; Van Damme, P.; Vanden Berghe, T.; Vanoverberghe, I.; Vandekerckhove, J.; Vandenabeele, P.; Gevaert, K.; Nunez, G. Targeted peptidecentric proteomics reveals caspase-7 as a substrate of the caspase-1 inflammasomes. Mol. Cell. Proteom. 2008, 7, 2350–2363. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Declercq, W.; Vanden Berghe, T.; Vandenabeele, P. Caspases leave the beaten track: Caspase-mediated activation of NF-kappaB. J. Cell Biol. 2006, 173, 165–171. [Google Scholar] [CrossRef]
- Poyet, J.L.; Srinivasula, S.M.; Tnani, M.; Razmara, M.; Fernandes-Alnemri, T.; Alnemri, E.S. Identification of Ipaf, a human caspase-1-activating protein related to Apaf-1. J. Biol. Chem. 2001, 276, 28309–28313. [Google Scholar] [CrossRef]
- Faustin, B.; Lartigue, L.; Bruey, J.M.; Luciano, F.; Sergienko, E.; Bailly-Maitre, B.; Volkmann, N.; Hanein, D.; Rouiller, I.; Reed, J.C. Reconstituted NALP1 inflammasome reveals two-step mechanism of caspase-1 activation. Mol. Cell 2007, 25, 713–724. [Google Scholar] [CrossRef]
- Duncan, J.A.; Bergstralh, D.T.; Wang, Y.; Willingham, S.B.; Ye, Z.; Zimmermann, A.G.; Ting, J.P. Cryopyrin/NALP3 binds ATP/dATP, is an ATPase, and requires ATP binding to mediate inflammatory signaling. Proc. Natl. Acad. Sci. USA 2007, 104, 8041–8046. [Google Scholar] [CrossRef]
- Mariathasan, S.; Weiss, D.S.; Newton, K.; McBride, J.; O’Rourke, K.; Roose-Girma, M.; Lee, W.P.; Weinrauch, Y.; Monack, D.M.; Dixit, V.M. Cryopyrin activates the inflammasome in response to toxins and ATP. Nature 2006, 440, 228–232. [Google Scholar] [CrossRef]
- Bouchier-Hayes, L.; Martin, S.J. CARD games in apoptosis and immunity. Embo Rep. 2002, 3, 616–621. [Google Scholar] [CrossRef] [PubMed]
- Strowig, T.; Henao-Mejia, J.; Elinav, E.; Flavell, R. Inflammasomes in health and disease. Nature 2012, 481, 278–286. [Google Scholar] [CrossRef] [PubMed]
- Broz, P.; Dixit, V.M. Inflammasomes: Mechanism of assembly, regulation and signalling. Nat Rev Immunol 2016, 16, 407–420. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Callaway, J.B.; Ting, J.P. Inflammasomes: Mechanism of action, role in disease, and therapeutics. Nat. Med. 2015, 21, 677–687. [Google Scholar] [CrossRef]
- Lamkanfi, M.; Dixit, V.M. Mechanisms and functions of inflammasomes. Cell 2014, 157, 1013–1022. [Google Scholar] [CrossRef]
- Walsh, J.G.; Muruve, D.A.; Power, C. Inflammasomes in the CNS. Nat. Rev. Neurosci. 2014, 15, 84–97. [Google Scholar] [CrossRef]
- Gustin, A.; Kirchmeyer, M.; Koncina, E.; Felten, P.; Losciuto, S.; Heurtaux, T.; Tardivel, A.; Heuschling, P.; Dostert, C. NLRP3 Inflammasome Is Expressed and Functional in Mouse Brain Microglia but Not in Astrocytes. PloS ONE 2015, 10. [Google Scholar] [CrossRef]
- Freeman, L.; Guo, H.; David, C.N.; Brickey, W.J.; Jha, S.; Ting, J.P. NLR members NLRC4 and NLRP3 mediate sterile inflammasome activation in microglia and astrocytes. J. Exp. Med. 2017, 214, 1351–1370. [Google Scholar] [CrossRef]
- Wu, J.; Fernandes-Alnemri, T.; Alnemri, E.S. Involvement of the AIM2, NLRC4, and NLRP3 inflammasomes in caspase-1 activation by Listeria monocytogenes. J. Clin. Immunol. 2010, 30, 693–702. [Google Scholar] [CrossRef]
- Minkiewicz, J.; de Rivero Vaccari, J.P.; Keane, R.W. Human astrocytes express a novel NLRP2 inflammasome. Glia 2013, 61, 1113–1121. [Google Scholar] [CrossRef]
- Kaushal, V.; Dye, R.; Pakavathkumar, P.; Foveau, B.; Flores, J.; Hyman, B.; Ghetti, B.; Koller, B.H.; LeBlanc, A.C. Neuronal NLRP1 inflammasome activation of Caspase-1 coordinately regulates inflammatory interleukin-1-beta production and axonal degeneration-associated Caspase-6 activation. Cell Death Differ. 2015, 22, 1676–1686. [Google Scholar] [CrossRef] [PubMed]
- Saresella, M.; La Rosa, F.; Piancone, F.; Zoppis, M.; Marventano, I.; Calabrese, E.; Rainone, V.; Nemni, R.; Mancuso, R.; Clerici, M. The NLRP3 and NLRP1 inflammasomes are activated in Alzheimer’s disease. Mol. Neurodegener. 2016, 11, 23. [Google Scholar] [CrossRef] [PubMed]
- Kummer, J.A.; Broekhuizen, R.; Everett, H.; Agostini, L.; Kuijk, L.; Martinon, F.; van Bruggen, R.; Tschopp, J. Inflammasome components NALP 1 and 3 show distinct but separate expression profiles in human tissues suggesting a site-specific role in the inflammatory response. J. Histochem. Cytochem. 2007, 55, 443–452. [Google Scholar] [CrossRef] [PubMed]
- Hlaing, T.; Guo, R.F.; Dilley, K.A.; Loussia, J.M.; Morrish, T.A.; Shi, M.M.; Vincenz, C.; Ward, P.A. Molecular cloning and characterization of DEFCAP-L and -S, two isoforms of a novel member of the mammalian Ced-4 family of apoptosis proteins. J. Biol. Chem. 2001, 276, 9230–9238. [Google Scholar] [CrossRef] [PubMed]
- Levinsohn, J.L.; Newman, Z.L.; Hellmich, K.A.; Fattah, R.; Getz, M.A.; Liu, S.; Sastalla, I.; Leppla, S.H.; Moayeri, M. Anthrax lethal factor cleavage of Nlrp1 is required for activation of the inflammasome. PloS Pathog. 2012, 8. [Google Scholar] [CrossRef]
- Chavarria-Smith, J.; Vance, R.E. Direct proteolytic cleavage of NLRP1B is necessary and sufficient for inflammasome activation by anthrax lethal factor. PloS Pathog. 2013, 9. [Google Scholar] [CrossRef]
- Feria, M.G.; Taborda, N.A.; Hernandez, J.C.; Rugeles, M.T. HIV replication is associated to inflammasomes activation, IL-1beta, IL-18 and caspase-1 expression in GALT and peripheral blood. PloS ONE 2018, 13. [Google Scholar] [CrossRef]
- Lippai, D.; Bala, S.; Petrasek, J.; Csak, T.; Levin, I.; Kurt-Jones, E.A.; Szabo, G. Alcohol-induced IL-1beta in the brain is mediated by NLRP3/ASC inflammasome activation that amplifies neuroinflammation. J. Leukoc. Biol. 2013, 94, 171–182. [Google Scholar] [CrossRef]
- Lowe, P.P.; Cho, Y.; Tornai, D.; Coban, S.; Catalano, D.; Szabo, G. Inhibition of the Inflammasome Signaling Cascade Reduces Alcohol Consumption in Female But Not Male Mice. Alcohol. Clin. Exp. Res. 2020, 44, 567–578. [Google Scholar] [CrossRef]
- Zou, J.; Crews, F.T. Inflammasome-IL-1beta Signaling Mediates Ethanol Inhibition of Hippocampal Neurogenesis. Front Neurosci. 2012, 6, 77. [Google Scholar] [CrossRef]
- Hoffman, H.M.; Mueller, J.L.; Broide, D.H.; Wanderer, A.A.; Kolodner, R.D. Mutation of a new gene encoding a putative pyrin-like protein causes familial cold autoinflammatory syndrome and Muckle-Wells syndrome. Nat. Genet. 2001, 29, 301–305. [Google Scholar] [CrossRef] [PubMed]
- Vajjhala, P.R.; Mirams, R.E.; Hill, J.M. Multiple binding sites on the pyrin domain of ASC protein allow self-association and interaction with NLRP3 protein. J. Biol. Chem. 2012, 287, 41732–41743. [Google Scholar] [CrossRef] [PubMed]
- Walsh, J.G.; Reinke, S.N.; Mamik, M.K.; McKenzie, B.A.; Maingat, F.; Branton, W.G.; Broadhurst, D.I.; Power, C. Rapid inflammasome activation in microglia contributes to brain disease in HIV/AIDS. Retrovirology 2014, 11, 35. [Google Scholar] [CrossRef]
- Mamik, M.K.; Hui, E.; Branton, W.G.; McKenzie, B.A.; Chisholm, J.; Cohen, E.A.; Power, C. HIV-1 Viral Protein R Activates NLRP3 Inflammasome in Microglia: Implications for HIV-1 Associated Neuroinflammation. J. Neuroimmune Pharm. 2017, 12, 233–248. [Google Scholar] [CrossRef] [PubMed]
- Guo, H.; Gao, J.; Taxman, D.J.; Ting, J.P.; Su, L. HIV-1 infection induces interleukin-1beta production via TLR8 protein-dependent and NLRP3 inflammasome mechanisms in human monocytes. J. Biol. Chem. 2014, 289, 21716–21726. [Google Scholar] [CrossRef]
- Chivero, E.T.; Guo, M.L.; Periyasamy, P.; Liao, K.; Callen, S.E.; Buch, S. HIV-1 Tat Primes and Activates Microglial NLRP3 Inflammasome-Mediated Neuroinflammation. J. Neurosci. 2017, 37, 3599–3609. [Google Scholar] [CrossRef]
- Pontillo, A.; Oshiro, T.M.; Girardelli, M.; Kamada, A.J.; Crovella, S.; Duarte, A.J. Polymorphisms in inflammasome’ genes and susceptibility to HIV-1 infection. J. Acquir. Immune Defic. Syndr. 2012, 59, 121–125. [Google Scholar] [CrossRef]
- Atluri, V.S.; Pilakka-Kanthikeel, S.; Garcia, G.; Jayant, R.D.; Sagar, V.; Samikkannu, T.; Yndart, A.; Nair, M. Effect of Cocaine on HIV Infection and Inflammasome Gene Expression Profile in HIV Infected Macrophages. Sci. Rep. 2016, 6, 27864. [Google Scholar] [CrossRef]
- Franchi, L.; Amer, A.; Body-Malapel, M.; Kanneganti, T.D.; Ozoren, N.; Jagirdar, R.; Inohara, N.; Vandenabeele, P.; Bertin, J.; Coyle, A.; et al. Cytosolic flagellin requires Ipaf for activation of caspase-1 and interleukin 1beta in salmonella-infected macrophages. Nat. Immunol. 2006, 7, 576–582. [Google Scholar] [CrossRef]
- Miao, E.A.; Alpuche-Aranda, C.M.; Dors, M.; Clark, A.E.; Bader, M.W.; Miller, S.I.; Aderem, A. Cytoplasmic flagellin activates caspase-1 and secretion of interleukin 1beta via Ipaf. Nat. Immunol. 2006, 7, 569–575. [Google Scholar] [CrossRef]
- Ren, T.; Zamboni, D.S.; Roy, C.R.; Dietrich, W.F.; Vance, R.E. Flagellin-deficient Legionella mutants evade caspase-1- and Naip5-mediated macrophage immunity. PloS Pathog. 2006, 2. [Google Scholar] [CrossRef] [PubMed]
- Dos Reis, E.C.; Leal, V.N.C.; Soares, J.; Fernandes, F.P.; Souza de Lima, D.; de Alencar, B.C.; Pontillo, A. Flagellin/NLRC4 Pathway Rescues NLRP3-Inflammasome Defect in Dendritic Cells From HIV-Infected Patients: Perspective for New Adjuvant in Immunocompromised Individuals. Front Immunol. 2019, 10, 1291. [Google Scholar] [CrossRef] [PubMed]
- Kuenzel, S.; Till, A.; Winkler, M.; Hasler, R.; Lipinski, S.; Jung, S.; Grotzinger, J.; Fickenscher, H.; Schreiber, S.; Rosenstiel, P. The nucleotide-binding oligomerization domain-like receptor NLRC5 is involved in IFN-dependent antiviral immune responses. J. Immunol. 2010, 184, 1990–2000. [Google Scholar] [CrossRef] [PubMed]
- Lamkanfi, M.; Kanneganti, T.D. Regulation of immune pathways by the NOD-like receptor NLRC5. Immunobiology 2012, 217, 13–16. [Google Scholar] [CrossRef]
- Benko, S.; Magalhaes, J.G.; Philpott, D.J.; Girardin, S.E. NLRC5 limits the activation of inflammatory pathways. J. Immunol. 2010, 185, 1681–1691. [Google Scholar] [CrossRef]
- Cui, J.; Zhu, L.; Xia, X.; Wang, H.Y.; Legras, X.; Hong, J.; Ji, J.; Shen, P.; Zheng, S.; Chen, Z.J.; et al. NLRC5 negatively regulates the NF-kappaB and type I interferon signaling pathways. Cell 2010, 141, 483–496. [Google Scholar] [CrossRef]
- Hacker, H.; Karin, M. Regulation and function of IKK and IKK-related kinases. Sci. Stke 2006. [Google Scholar] [CrossRef]
- Periyasamy, P.; Thangaraj, A.; Bendi, V.S.; Buch, S. HIV-1 Tat-mediated microglial inflammation involves a novel miRNA-34a-NLRC5-NFkappaB signaling axis. Brain Behav. Immun. 2019, 80, 227–237. [Google Scholar] [CrossRef]
- DiSabato, D.J.; Quan, N.; Godbout, J.P. Neuroinflammation: The devil is in the details. J. Neurochem. 2016, 139 (Suppl. 2), 136–153. [Google Scholar] [CrossRef]
- Garvey, L.J.; Pavese, N.; Politis, M.; Ramlackhansingh, A.; Brooks, D.J.; Taylor-Robinson, S.D.; Winston, A. Increased microglia activation in neurologically asymptomatic HIV-infected patients receiving effective ART. AIDS 2014, 28, 67–72. [Google Scholar] [CrossRef]
- Rubin, L.H.; Sacktor, N.; Creighton, J.; Du, Y.; Endres, C.J.; Pomper, M.G.; Coughlin, J.M. Microglial activation is inversely associated with cognition in individuals living with HIV on effective antiretroviral therapy. AIDS 2018, 32, 1661–1667. [Google Scholar] [CrossRef] [PubMed]
- Vera, J.H.; Guo, Q.; Cole, J.H.; Boasso, A.; Greathead, L.; Kelleher, P.; Rabiner, E.A.; Kalk, N.; Bishop, C.; Gunn, R.N.; et al. Neuroinflammation in treated HIV-positive individuals: A TSPO PET study. Neurology 2016, 86, 1425–1432. [Google Scholar] [CrossRef] [PubMed]
- Coughlin, J.M.; Wang, Y.; Ma, S.; Yue, C.; Kim, P.K.; Adams, A.V.; Roosa, H.V.; Gage, K.L.; Stathis, M.; Rais, R.; et al. Regional brain distribution of translocator protein using [(11)C]DPA-713 PET in individuals infected with HIV. J. Neurovirol. 2014, 20, 219–232. [Google Scholar] [CrossRef] [PubMed]
- Hammoud, D.A.; Endres, C.J.; Chander, A.R.; Guilarte, T.R.; Wong, D.F.; Sacktor, N.C.; McArthur, J.C.; Pomper, M.G. Imaging glial cell activation with [11C]-R-PK11195 in patients with AIDS. J. Neurovirol. 2005, 11, 346–355. [Google Scholar] [CrossRef] [PubMed]
- Alakkas, A.; Ellis, R.J.; Watson, C.W.; Umlauf, A.; Heaton, R.K.; Letendre, S.; Collier, A.; Marra, C.; Clifford, D.B.; Gelman, B.; et al. White matter damage, neuroinflammation, and neuronal integrity in HAND. J. Neurovirol. 2019, 25, 32–41. [Google Scholar] [CrossRef] [PubMed]
- Tavazzi, E.; Morrison, D.; Sullivan, P.; Morgello, S.; Fischer, T. Brain inflammation is a common feature of HIV-infected patients without HIV encephalitis or productive brain infection. Curr. Hiv Res. 2014, 12, 97–110. [Google Scholar] [CrossRef]
- Chao, J.; Zhang, Y.; Du, L.; Zhou, R.; Wu, X.; Shen, K.; Yao, H. Molecular mechanisms underlying the involvement of the sigma-1 receptor in methamphetamine-mediated microglial polarization. Sci. Rep. 2017, 7, 11540. [Google Scholar] [CrossRef]
- Najera, J.A.; Bustamante, E.A.; Bortell, N.; Morsey, B.; Fox, H.S.; Ravasi, T.; Marcondes, M.C. Methamphetamine abuse affects gene expression in brain-derived microglia of SIV-infected macaques to enhance inflammation and promote virus targets. BMC Immunol. 2016, 17, 7. [Google Scholar] [CrossRef]
- Liao, K.; Guo, M.; Niu, F.; Yang, L.; Callen, S.E.; Buch, S. Cocaine-mediated induction of microglial activation involves the ER stress-TLR2 axis. J. Neuroinflamm. 2016, 13, 33. [Google Scholar] [CrossRef]
- Guo, M.L.; Liao, K.; Periyasamy, P.; Yang, L.; Cai, Y.; Callen, S.E.; Buch, S. Cocaine-mediated microglial activation involves the ER stress-autophagy axis. Autophagy 2015, 11, 995–1009. [Google Scholar] [CrossRef]
- Kettenmann, H.; Hanisch, U.K.; Noda, M.; Verkhratsky, A. Physiology of microglia. Physiol. Rev. 2011, 91, 461–553. [Google Scholar] [CrossRef] [PubMed]
- Harry, G.J. Microglia during development and aging. Pharm. Ther. 2013, 139, 313–326. [Google Scholar] [CrossRef] [PubMed]
- Chen, N.C.; Partridge, A.T.; Sell, C.; Torres, C.; Martin-Garcia, J. Fate of microglia during HIV-1 infection: From activation to senescence? Glia 2017, 65, 431–446. [Google Scholar] [CrossRef] [PubMed]
- Spudich, S.; Gonzalez-Scarano, F. HIV-1-related central nervous system disease: Current issues in pathogenesis, diagnosis, and treatment. Cold Spring Harb. Perspect. Med. 2012, 2, a007120. [Google Scholar] [CrossRef] [PubMed]
- Lacagnina, M.J.; Rivera, P.D.; Bilbo, S.D. Glial and Neuroimmune Mechanisms as Critical Modulators of Drug Use and Abuse. Neuropsychopharmacology 2017, 42, 156–177. [Google Scholar] [CrossRef]
- Gampierakis, I.A.; Koutmani, Y.; Semitekolou, M.; Morianos, I.; Charalampopoulos, I.; Xanthou, G.; Gravanis, A.; Karalis, K.P. Hippocampal neural stem cells and microglia response to experimental inflammatory bowel disease (IBD). Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Yap, J.K.Y.; Pickard, B.S.; Chan, E.W.L.; Gan, S.Y. The Role of Neuronal NLRP1 Inflammasome in Alzheimer’s Disease: Bringing Neurons into the Neuroinflammation Game. Mol. Neurobiol. 2019, 56, 7741–7753. [Google Scholar] [CrossRef]
- Poh, L.; Kang, S.W.; Baik, S.H.; Ng, G.Y.Q.; She, D.T.; Balaganapathy, P.; Dheen, S.T.; Magnus, T.; Gelderblom, M.; Sobey, C.G.; et al. Evidence that NLRC4 inflammasome mediates apoptotic and pyroptotic microglial death following ischemic stroke. Brain Behav. Immun. 2019, 75, 34–47. [Google Scholar] [CrossRef]
- Heneka, M.T.; McManus, R.M.; Latz, E. Inflammasome signalling in brain function and neurodegenerative disease. Nat. Rev. Neurosci. 2018, 19, 610–621. [Google Scholar] [CrossRef]
- Furman, D.; Chang, J.; Lartigue, L.; Bolen, C.R.; Haddad, F.; Gaudilliere, B.; Ganio, E.A.; Fragiadakis, G.K.; Spitzer, M.H.; Douchet, I.; et al. Expression of specific inflammasome gene modules stratifies older individuals into two extreme clinical and immunological states. Nat. Med. 2017, 23, 174–184. [Google Scholar] [CrossRef]
- Hu, M.Y.; Lin, Y.Y.; Zhang, B.J.; Lu, D.L.; Lu, Z.Q.; Cai, W. Update of inflammasome activation in microglia/macrophage in aging and aging-related disease. Cns Neurosci. 2019, 25, 1299–1307. [Google Scholar] [CrossRef] [PubMed]
- Scheiblich, H.; Trombly, M.; Ramirez, A.; Heneka, M.T. Neuroimmune Connections in Aging and Neurodegenerative Diseases. Trends Immunol. 2020, 41, 300–312. [Google Scholar] [CrossRef] [PubMed]
- Mecca, C.; Giambanco, I.; Donato, R.; Arcuri, C. Microglia and Aging: The Role of the TREM2-DAP12 and CX3CL1-CX3CR1 Axes. Int. J. Mol. Sci. 2018, 19, 318. [Google Scholar] [CrossRef] [PubMed]
- Cunningham, C. Microglia and neurodegeneration: The role of systemic inflammation. Glia 2013, 61, 71–90. [Google Scholar] [CrossRef]
- Hickman, S.; Izzy, S.; Sen, P.; Morsett, L.; El Khoury, J. Microglia in neurodegeneration. Nat. Neurosci. 2018, 21, 1359–1369. [Google Scholar] [CrossRef]
- Lebeaupin, C.; Proics, E.; de Bieville, C.H.; Rousseau, D.; Bonnafous, S.; Patouraux, S.; Adam, G.; Lavallard, V.J.; Rovere, C.; Le Thuc, O.; et al. ER stress induces NLRP3 inflammasome activation and hepatocyte death. Cell Death Dis. 2015, 6, e1879. [Google Scholar] [CrossRef]
- Zhou, R.; Yazdi, A.S.; Menu, P.; Tschopp, J. A role for mitochondria in NLRP3 inflammasome activation. Nature 2011, 469, 221–225. [Google Scholar] [CrossRef]
- Kelley, N.; Jeltema, D.; Duan, Y.; He, Y. The NLRP3 Inflammasome: An Overview of Mechanisms of Activation and Regulation. Int. J. Mol. Sci. 2019, 20, 3328. [Google Scholar] [CrossRef]
- Youm, Y.H.; Kanneganti, T.D.; Vandanmagsar, B.; Zhu, X.; Ravussin, A.; Adijiang, A.; Owen, J.S.; Thomas, M.J.; Francis, J.; Parks, J.S.; et al. The Nlrp3 inflammasome promotes age-related thymic demise and immunosenescence. Cell Rep. 2012, 1, 56–68. [Google Scholar] [CrossRef]
- Munoz-Planillo, R.; Kuffa, P.; Martinez-Colon, G.; Smith, B.L.; Rajendiran, T.M.; Nunez, G. K(+) efflux is the common trigger of NLRP3 inflammasome activation by bacterial toxins and particulate matter. Immunity 2013, 38, 1142–1153. [Google Scholar] [CrossRef]
- Olivieri, F.; Rippo, M.R.; Monsurro, V.; Salvioli, S.; Capri, M.; Procopio, A.D.; Franceschi, C. MicroRNAs linking inflamm-aging, cellular senescence and cancer. Ageing Res. Rev. 2013, 12, 1056–1068. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.H.; Wang, T.L. MicroRNAs of microglia: Wrestling with central nervous system disease. Neural Regen. Res. 2018, 13, 2067–2072. [Google Scholar] [CrossRef] [PubMed]
- Soreq, H.; Wolf, Y. NeurimmiRs: microRNAs in the neuroimmune interface. Trends Mol. Med. 2011, 17, 548–555. [Google Scholar] [CrossRef] [PubMed]
- Periyasamy, P.; Liao, K.; Kook, Y.H.; Niu, F.; Callen, S.E.; Guo, M.L.; Buch, S. Cocaine-Mediated Downregulation of miR-124 Activates Microglia by Targeting KLF4 and TLR4 Signaling. Mol. Neurobiol. 2018, 55, 3196–3210. [Google Scholar] [CrossRef]
- Liu, J.; Xu, C.; Chen, L.; Xu, P.; Xiong, H. Involvement of Kv1.3 and p38 MAPK signaling in HIV-1 glycoprotein 120-induced microglia neurotoxicity. Cell Death Dis. 2012, 3, e254. [Google Scholar] [CrossRef]
- Costa, B.M.; Yao, H.; Yang, L.; Buch, S. Role of endoplasmic reticulum (ER) stress in cocaine-induced microglial cell death. J. Neuroimmune Pharm. 2013, 8, 705–714. [Google Scholar] [CrossRef][Green Version]
- Rawat, P.; Teodorof-Diedrich, C.; Spector, S.A. Human immunodeficiency virus Type-1 single-stranded RNA activates the NLRP3 inflammasome and impairs autophagic clearance of damaged mitochondria in human microglia. Glia 2019, 67, 802–824. [Google Scholar] [CrossRef]
- Du, L.; Shen, K.; Bai, Y.; Chao, J.; Hu, G.; Zhang, Y.; Yao, H. Involvement of NLRP3 inflammasome in methamphetamine-induced microglial activation through miR-143/PUMA axis. Toxicol. Lett. 2019, 301, 53–63. [Google Scholar] [CrossRef]
- Lippai, D.; Bala, S.; Csak, T.; Kurt-Jones, E.A.; Szabo, G. Chronic alcohol-induced microRNA-155 contributes to neuroinflammation in a TLR4-dependent manner in mice. PloS ONE 2013, 8, e70945. [Google Scholar] [CrossRef]
- Wyczechowska, D.; Lin, H.Y.; LaPlante, A.; Jeansonne, D.; Lassak, A.; Parsons, C.H.; Molina, P.E.; Peruzzi, F. A miRNA Signature for Cognitive Deficits and Alcohol Use Disorder in Persons Living with HIV/AIDS. Front. Mol. Neurosci. 2017, 10, 385. [Google Scholar] [CrossRef]
- Chen, Y.; Swanson, R.A. Astrocytes and brain injury. J. Cereb. Blood Flow Metab. 2003, 23, 137–149. [Google Scholar] [CrossRef] [PubMed]
- Farina, C.; Aloisi, F.; Meinl, E. Astrocytes are active players in cerebral innate immunity. Trends Immunol. 2007, 28, 138–145. [Google Scholar] [CrossRef] [PubMed]
- Kielian, T. Toll-like receptors in central nervous system glial inflammation and homeostasis. J. Neurosci. Res. 2006, 83, 711–730. [Google Scholar] [CrossRef] [PubMed]
- Xiao, C.; Yang, B.F.; Asadi, N.; Beguinot, F.; Hao, C. Tumor necrosis factor-related apoptosis-inducing ligand-induced death-inducing signaling complex and its modulation by c-FLIP and PED/PEA-15 in glioma cells. J. Biol. Chem. 2002, 277, 25020–25025. [Google Scholar] [CrossRef] [PubMed]
- Song, J.H.; Bellail, A.; Tse, M.C.; Yong, V.W.; Hao, C. Human astrocytes are resistant to Fas ligand and tumor necrosis factor-related apoptosis-inducing ligand-induced apoptosis. J. Neurosci. 2006, 26, 3299–3308. [Google Scholar] [CrossRef]
- Cohen, J.; Torres, C. Astrocyte senescence: Evidence and significance. Aging Cell 2019, 18, e12937. [Google Scholar] [CrossRef]
- Matias, I.; Morgado, J.; Gomes, F.C.A. Astrocyte Heterogeneity: Impact to Brain Aging and Disease. Front. Aging Neurosci. 2019, 11, 59. [Google Scholar] [CrossRef]
- Zhang, J.; Jiang, N.; Zhang, L.; Meng, C.; Zhao, J.; Wu, J. NLRP6 expressed in astrocytes aggravates neurons injury after OGD/R through activating the inflammasome and inducing pyroptosis. Int. Immunopharmacol. 2020, 80, 106183. [Google Scholar] [CrossRef]
- Voet, S.; Srinivasan, S.; Lamkanfi, M.; van Loo, G. Inflammasomes in neuroinflammatory and neurodegenerative diseases. Embo Mol. Med. 2019, 11, e10248. [Google Scholar] [CrossRef]
- Park, J.H.; Choi, J.Y.; Jo, C.; Koh, Y.H. Involvement of ADAM10 in acrolein-induced astrocytic inflammation. Toxicol. Lett. 2020, 318, 44–49. [Google Scholar] [CrossRef]
- Hong, Y.; Liu, Y.; Yu, D.; Wang, M.; Hou, Y. The neuroprotection of progesterone against Abeta-induced NLRP3-Caspase-1 inflammasome activation via enhancing autophagy in astrocytes. Int. Immunopharmacol. 2019, 74, 105669. [Google Scholar] [CrossRef] [PubMed]
- Pacheco, A.L.; Vicentini, G.; Matteucci, K.C.; Ribeiro, R.R.; Weinlich, R.; Bortoluci, K.R. The impairment in the NLRP3-induced NO secretion renders astrocytes highly permissive to T. cruzi replication. J. Leukoc. Biol. 2019, 106, 201–207. [Google Scholar] [CrossRef] [PubMed]
- Sun, Y.B.; Zhao, H.; Mu, D.L.; Zhang, W.; Cui, J.; Wu, L.; Alam, A.; Wang, D.X.; Ma, D. Dexmedetomidine inhibits astrocyte pyroptosis and subsequently protects the brain in in vitro and in vivo models of sepsis. Cell Death Dis. 2019, 10, 167. [Google Scholar] [CrossRef] [PubMed]
- Ojeda, D.S.; Grasso, D.; Urquiza, J.; Till, A.; Vaccaro, M.I.; Quarleri, J. Cell Death Is Counteracted by Mitophagy in HIV-Productively Infected Astrocytes but Is Promoted by Inflammasome Activation among Non-productively Infected Cells. Front. Immunol. 2018, 9, 2633. [Google Scholar] [CrossRef] [PubMed]
- Ebrahimi, T.; Rust, M.; Kaiser, S.N.; Slowik, A.; Beyer, C.; Koczulla, A.R.; Schulz, J.B.; Habib, P.; Bach, J.P. Alpha1-Antitrypsin mitigates NLRP3-inflammasome activation in amyloid beta1-42-stimulated murine astrocytes. J. Neuroinflamm. 2018, 15, 282. [Google Scholar] [CrossRef] [PubMed]
- Cheon, S.Y.; Kim, E.J.; Kim, S.Y.; Kim, J.M.; Kam, E.H.; Park, J.K.; Koo, B.N. Apoptosis Signal-regulating Kinase 1 Silencing on Astroglial Inflammasomes in an Experimental Model of Ischemic Stroke. Neuroscience 2018, 390, 218–230. [Google Scholar] [CrossRef]
- Slowik, A.; Lammerding, L.; Zendedel, A.; Habib, P.; Beyer, C. Impact of steroid hormones E2 and P on the NLRP3/ASC/Casp1 axis in primary mouse astroglia and BV-2 cells after in vitro hypoxia. J. Steroid. Biochem. Mol. Biol. 2018, 183, 18–26. [Google Scholar] [CrossRef]
- de Paula Martins, R.; Ghisoni, K.; Lim, C.K.; Aguiar, A.S., Jr.; Guillemin, G.J.; Latini, A. Neopterin preconditioning prevents inflammasome activation in mammalian astrocytes. Free Radic. Biol. Med. 2018, 115, 371–382. [Google Scholar] [CrossRef]
- Du, R.H.; Wu, F.F.; Lu, M.; Shu, X.D.; Ding, J.H.; Wu, G.; Hu, G. Uncoupling protein 2 modulation of the NLRP3 inflammasome in astrocytes and its implications in depression. Redox Biol. 2016, 9, 178–187. [Google Scholar] [CrossRef]
- Liu, S.; Li, Q.; Zhang, M.T.; Mao-Ying, Q.L.; Hu, L.Y.; Wu, G.C.; Mi, W.L.; Wang, Y.Q. Curcumin ameliorates neuropathic pain by down-regulating spinal IL-1beta via suppressing astroglial NALP1 inflammasome and JAK2-STAT3 signalling. Sci. Rep. 2016, 6, 28956. [Google Scholar] [CrossRef]
- Jian, Z.; Ding, S.; Deng, H.; Wang, J.; Yi, W.; Wang, L.; Zhu, S.; Gu, L.; Xiong, X. Probenecid protects against oxygen-glucose deprivation injury in primary astrocytes by regulating inflammasome activity. Brain Res. 2016, 1643, 123–129. [Google Scholar] [CrossRef] [PubMed]
- Alfonso-Loeches, S.; Urena-Peralta, J.R.; Morillo-Bargues, M.J.; Oliver-De La Cruz, J.; Guerri, C. Role of mitochondria ROS generation in ethanol-induced NLRP3 inflammasome activation and cell death in astroglial cells. Front. Cell Neurosci. 2014, 8, 216. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chan, C. IPAF inflammasome is involved in interleukin-1beta production from astrocytes, induced by palmitate; implications for Alzheimer’s Disease. Neurobiol. Aging 2014, 35, 309–321. [Google Scholar] [CrossRef] [PubMed]
- Silverman, W.R.; de Rivero Vaccari, J.P.; Locovei, S.; Qiu, F.; Carlsson, S.K.; Scemes, E.; Keane, R.W.; Dahl, G. The pannexin 1 channel activates the inflammasome in neurons and astrocytes. J. Biol. Chem. 2009, 284, 18143–18151. [Google Scholar] [CrossRef]
- Bhat, R.; Crowe, E.P.; Bitto, A.; Moh, M.; Katsetos, C.D.; Garcia, F.U.; Johnson, F.B.; Trojanowski, J.Q.; Sell, C.; Torres, C. Astrocyte senescence as a component of Alzheimer’s disease. PloS ONE 2012, 7, e45069. [Google Scholar] [CrossRef] [PubMed]
- Chinta, S.J.; Lieu, C.A.; Demaria, M.; Laberge, R.M.; Campisi, J.; Andersen, J.K. Environmental stress, ageing and glial cell senescence: A novel mechanistic link to Parkinson’s disease? J. Intern. Med. 2013, 273, 429–436. [Google Scholar] [CrossRef]
- Kang, C.; Xu, Q.; Martin, T.D.; Li, M.Z.; Demaria, M.; Aron, L.; Lu, T.; Yankner, B.A.; Campisi, J.; Elledge, S.J. The DNA damage response induces inflammation and senescence by inhibiting autophagy of GATA4. Science 2015, 349, aaa5612. [Google Scholar] [CrossRef]
- Yu, C.; Narasipura, S.D.; Richards, M.H.; Hu, X.T.; Yamamoto, B.; Al-Harthi, L. HIV and drug abuse mediate astrocyte senescence in a beta-catenin-dependent manner leading to neuronal toxicity. Aging Cell 2017, 16, 956–965. [Google Scholar] [CrossRef]
- Churchill, M.J.; Wesselingh, S.L.; Cowley, D.; Pardo, C.A.; McArthur, J.C.; Brew, B.J.; Gorry, P.R. Extensive astrocyte infection is prominent in human immunodeficiency virus-associated dementia. Ann. Neurol. 2009, 66, 253–258. [Google Scholar] [CrossRef]
- Barat, C.; Proust, A.; Deshiere, A.; Leboeuf, M.; Drouin, J.; Tremblay, M.J. Astrocytes sustain long-term productive HIV-1 infection without establishment of reactivable viral latency. Glia 2018, 66, 1363–1381. [Google Scholar] [CrossRef]
- Li, G.H.; Henderson, L.; Nath, A. Astrocytes as an HIV Reservoir: Mechanism of HIV Infection. Curr. Hiv Res. 2016, 14, 373–381. [Google Scholar] [CrossRef] [PubMed]
- Li, G.H.; Maric, D.; Major, E.O.; Nath, A. Productive HIV infection in astrocytes can be established via a nonclassical mechanism. AIDS 2020, 34, 963–978. [Google Scholar] [CrossRef] [PubMed]
- Al-Harti, L.; Joseph, J.; Nath, A. Astrocytes as an HIV CNS reservoir: Highlights and reflections of an NIMH-sponsored symposium. J. Neurovirol. 2018, 24, 665–669. [Google Scholar] [CrossRef] [PubMed]
- Zhao, X.; Fan, Y.; Vann, P.H.; Wong, J.M.; Sumien, N.; He, J.J. Long-term HIV-1 Tat Expression in the Brain Led to Neurobehavioral, Pathological, and Epigenetic Changes Reminiscent of Accelerated Aging. Aging Dis. 2020, 11, 93–107. [Google Scholar] [CrossRef] [PubMed]
- Cohen, J.; D’Agostino, L.; Wilson, J.; Tuzer, F.; Torres, C. Astrocyte Senescence and Metabolic Changes in Response to HIV Antiretroviral Therapy Drugs. Front. Aging Neurosci. 2017, 9, 281. [Google Scholar] [CrossRef] [PubMed]
- Gonzalez, H.; Podany, A.; Al-Harthi, L.; Wallace, J. The far-reaching HAND of cART: cART effects on astrocytes. J. Neuroimmune Pharm. 2020. [Google Scholar] [CrossRef] [PubMed]
- Dang, J.; Tiwari, S.K.; Agrawal, K.; Hui, H.; Qin, Y.; Rana, T.M. Glial cell diversity and methamphetamine-induced neuroinflammation in human cerebral organoids. Mol. Psychiatry 2020. [Google Scholar] [CrossRef]
- Shi, Y.; Yuan, S.; Tang, S.J. Morphine and HIV-1 gp120 cooperatively promote pathogenesis in the spinal pain neural circuit. Mol. Pain 2019, 15. [Google Scholar] [CrossRef]
- Chang, L.; Andres, M.; Sadino, J.; Jiang, C.S.; Nakama, H.; Miller, E.; Ernst, T. Impact of apolipoprotein E epsilon4 and HIV on cognition and brain atrophy: Antagonistic pleiotropy and premature brain aging. Neuroimage 2011, 58, 1017–1027. [Google Scholar] [CrossRef]
- Chang, L.; Jiang, C.; Cunningham, E.; Buchthal, S.; Douet, V.; Andres, M.; Ernst, T. Effects of APOE epsilon4, age, and HIV on glial metabolites and cognitive deficits. Neurology 2014, 82, 2213–2222. [Google Scholar] [CrossRef]
- Evans, S.; Dowell, N.G.; Tabet, N.; Tofts, P.S.; King, S.L.; Rusted, J.M. Cognitive and neural signatures of the APOE E4 allele in mid-aged adults. Neurobiol. Aging 2014, 35, 1615–1623. [Google Scholar] [CrossRef] [PubMed]
- Crawford, F.; Wood, M.; Ferguson, S.; Mathura, V.; Gupta, P.; Humphrey, J.; Mouzon, B.; Laporte, V.; Margenthaler, E.; O’Steen, B.; et al. Apolipoprotein E-genotype dependent hippocampal and cortical responses to traumatic brain injury. Neuroscience 2009, 159, 1349–1362. [Google Scholar] [CrossRef] [PubMed]
- Theendakara, V.; Peters-Libeu, C.A.; Spilman, P.; Poksay, K.S.; Bredesen, D.E.; Rao, R.V. Direct Transcriptional Effects of Apolipoprotein E. J. Neurosci. 2016, 36, 685–700. [Google Scholar] [CrossRef] [PubMed]
- Babu, H.; Ambikan, A.T.; Gabriel, E.E.; Svensson Akusjarvi, S.; Palaniappan, A.N.; Sundaraj, V.; Mupanni, N.R.; Sperk, M.; Cheedarla, N.; Sridhar, R.; et al. Systemic Inflammation and the Increased Risk of Inflamm-Aging and Age-Associated Diseases in People Living With HIV on Long Term Suppressive Antiretroviral Therapy. Front. Immunol. 2019, 10, 1965. [Google Scholar] [CrossRef] [PubMed]
- Zhang, Y.; Kim, M.S.; Jia, B.; Yan, J.; Zuniga-Hertz, J.P.; Han, C.; Cai, D. Hypothalamic stem cells control ageing speed partly through exosomal miRNAs. Nature 2017, 548, 52–57. [Google Scholar] [CrossRef] [PubMed]
- Olivieri, F.; Albertini, M.C.; Orciani, M.; Ceka, A.; Cricca, M.; Procopio, A.D.; Bonafe, M. DNA damage response (DDR) and senescence: Shuttled inflamma-miRNAs on the stage of inflamm-aging. Oncotarget 2015, 6, 35509–35521. [Google Scholar] [CrossRef]
- Urbanelli, L.; Buratta, S.; Sagini, K.; Tancini, B.; Emiliani, C. Extracellular Vesicles as New Players in Cellular Senescence. Int. J. Mol. Sci. 2016, 17, 1408. [Google Scholar] [CrossRef]
- Xu, D.; Tahara, H. The role of exosomes and microRNAs in senescence and aging. Adv. Drug Deliv. Rev. 2013, 65, 368–375. [Google Scholar] [CrossRef]
- Bertoldi, K.; Cechinel, L.R.; Schallenberger, B.; Corssac, G.B.; Davies, S.; Guerreiro, I.C.K.; Bello-Klein, A.; Araujo, A.S.R.; Siqueira, I.R. Circulating extracellular vesicles in the aging process: Impact of aerobic exercise. Mol. Cell Biochem. 2018, 440, 115–125. [Google Scholar] [CrossRef]
- Eitan, E.; Green, J.; Bodogai, M.; Mode, N.A.; Baek, R.; Jorgensen, M.M.; Freeman, D.W.; Witwer, K.W.; Zonderman, A.B.; Biragyn, A.; et al. Age-Related Changes in Plasma Extracellular Vesicle Characteristics and Internalization by Leukocytes. Sci. Rep. 2017, 7, 1342. [Google Scholar] [CrossRef]
- Guha, D.; Lorenz, D.R.; Misra, V.; Chettimada, S.; Morgello, S.; Gabuzda, D. Proteomic analysis of cerebrospinal fluid extracellular vesicles reveals synaptic injury, inflammation, and stress response markers in HIV patients with cognitive impairment. J. Neuroinflamm. 2019, 16, 254. [Google Scholar] [CrossRef] [PubMed]
- Dagur, R.S.; Liao, K.; Sil, S.; Niu, F.; Sun, Z.; Lyubchenko, Y.L.; Peeples, E.S.; Hu, G.; Buch, S. Neuronal-derived extracellular vesicles are enriched in the brain and serum of HIV-1 transgenic rats. J. Extracell. Vesicles 2020, 9, 1703249. [Google Scholar] [CrossRef] [PubMed]
- Pulliam, L.; Sun, B.; Mustapic, M.; Chawla, S.; Kapogiannis, D. Plasma neuronal exosomes serve as biomarkers of cognitive impairment in HIV infection and Alzheimer’s disease. J. Neurovirol. 2019, 25, 702–709. [Google Scholar] [CrossRef]
- Sun, B.; Dalvi, P.; Abadjian, L.; Tang, N.; Pulliam, L. Blood neuron-derived exosomes as biomarkers of cognitive impairment in HIV. AIDS 2017, 31, F9–F17. [Google Scholar] [CrossRef]
- Jarvis, R.; Tamashiro-Orrego, A.; Promes, V.; Tu, L.; Shi, J.; Yang, Y. Cocaine Self-administration and Extinction Inversely Alter Neuron to Glia Exosomal Dynamics in the Nucleus Accumbens. Front. Cell Neurosci. 2019, 13, 581. [Google Scholar] [CrossRef] [PubMed]
- de Guglielmo, G.; Fu, Y.; Chen, J.; Larrosa, E.; Hoang, I.; Kawamura, T.; Lorrai, I.; Zorman, B.; Bryant, J.; George, O.; et al. Increases in compulsivity, inflammation, and neural injury in HIV transgenic rats with escalated methamphetamine self-administration under extended-access conditions. Brain Res. 2020, 1726, 146502. [Google Scholar] [CrossRef]
- Sil, S.; Periyasamy, P.; Guo, M.L.; Callen, S.; Buch, S. Morphine-Mediated Brain Region-Specific Astrocytosis Involves the ER Stress-Autophagy Axis. Mol. Neurobiol. 2018, 55, 6713–6733. [Google Scholar] [CrossRef]
- Sil, S.; Niu, F.; Tom, E.; Liao, K.; Periyasamy, P.; Buch, S. Cocaine Mediated Neuroinflammation: Role of Dysregulated Autophagy in Pericytes. Mol. Neurobiol. 2019, 56, 3576–3590. [Google Scholar] [CrossRef]
- Volpe, G.E.; Ward, H.; Mwamburi, M.; Dinh, D.; Bhalchandra, S.; Wanke, C.; Kane, A.V. Associations of cocaine use and HIV infection with the intestinal microbiota, microbial translocation, and inflammation. J. Stud. Alcohol Drugs 2014, 75, 347–357. [Google Scholar] [CrossRef]
- Reiner, B.C.; Keblesh, J.P.; Xiong, H. Methamphetamine abuse, HIV infection, and neurotoxicity. Int. J. Physiol. Pathophysiol. Pharm. 2009, 1, 162–179. [Google Scholar]
- Dave, R.S. Morphine affects HIV-induced inflammatory response without influencing viral replication in human monocyte-derived macrophages. Fems Immunol. Med. Microbiol. 2012, 64, 228–236. [Google Scholar] [CrossRef] [PubMed]
- Strittmatter, W.J.; Weisgraber, K.H.; Huang, D.Y.; Dong, L.M.; Salvesen, G.S.; Pericak-Vance, M.; Schmechel, D.; Saunders, A.M.; Goldgaber, D.; Roses, A.D. Binding of human apolipoprotein E to synthetic amyloid beta peptide: Isoform-specific effects and implications for late-onset Alzheimer disease. Proc. Natl. Acad. Sci. USA 1993, 90, 8098–8102. [Google Scholar] [CrossRef] [PubMed]
- Xu, J.; Ikezu, T. The comorbidity of HIV-associated neurocognitive disorders and Alzheimer’s disease: A foreseeable medical challenge in post-HAART era. J. Neuroimmune Pharm. 2009, 4, 200–212. [Google Scholar] [CrossRef] [PubMed]
- Achim, C.L.; Adame, A.; Dumaop, W.; Everall, I.P.; Masliah, E.; Neurobehavioral Research, C. Increased accumulation of intraneuronal amyloid beta in HIV-infected patients. J. Neuroimmune Pharm. 2009, 4, 190–199. [Google Scholar] [CrossRef] [PubMed]
- Sil, S.; Hu, G.; Liao, K.; Niu, F.; Callen, S.; Periyasamy, P.; Fox, H.S.; Buch, S. HIV-1 Tat-mediated astrocytic amyloidosis involves the HIF-1alpha/lncRNA BACE1-AS axis. Plos Biol. 2020, 18. [Google Scholar] [CrossRef] [PubMed]
- Liu, L.; Chan, C. The role of inflammasome in Alzheimer’s disease. Ageing Res. Rev. 2014, 15, 6–15. [Google Scholar] [CrossRef] [PubMed]
- Sweeney, M.D.; Ayyadurai, S.; Zlokovic, B.V. Pericytes of the neurovascular unit: Key functions and signaling pathways. Nat. Neurosci. 2016, 19, 771–783. [Google Scholar] [CrossRef]
- Bell, R.D.; Winkler, E.A.; Sagare, A.P.; Singh, I.; LaRue, B.; Deane, R.; Zlokovic, B.V. Pericytes control key neurovascular functions and neuronal phenotype in the adult brain and during brain aging. Neuron 2010, 68, 409–427. [Google Scholar] [CrossRef]
- Nelson, A.R.; Sagare, M.A.; Wang, Y.; Kisler, K.; Zhao, Z.; Zlokovic, B.V. Channelrhodopsin Excitation Contracts Brain Pericytes and Reduces Blood Flow in the Aging Mouse Brain in vivo. Front. Aging Neurosci. 2020, 12, 108. [Google Scholar] [CrossRef]




© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Sil, S.; Niu, F.; Chivero, E.T.; Singh, S.; Periyasamy, P.; Buch, S. Role of Inflammasomes in HIV-1 and Drug Abuse Mediated Neuroinflammaging. Cells 2020, 9, 1857. https://doi.org/10.3390/cells9081857
Sil S, Niu F, Chivero ET, Singh S, Periyasamy P, Buch S. Role of Inflammasomes in HIV-1 and Drug Abuse Mediated Neuroinflammaging. Cells. 2020; 9(8):1857. https://doi.org/10.3390/cells9081857
Chicago/Turabian StyleSil, Susmita, Fang Niu, Ernest T. Chivero, Seema Singh, Palsamy Periyasamy, and Shilpa Buch. 2020. "Role of Inflammasomes in HIV-1 and Drug Abuse Mediated Neuroinflammaging" Cells 9, no. 8: 1857. https://doi.org/10.3390/cells9081857
APA StyleSil, S., Niu, F., Chivero, E. T., Singh, S., Periyasamy, P., & Buch, S. (2020). Role of Inflammasomes in HIV-1 and Drug Abuse Mediated Neuroinflammaging. Cells, 9(8), 1857. https://doi.org/10.3390/cells9081857