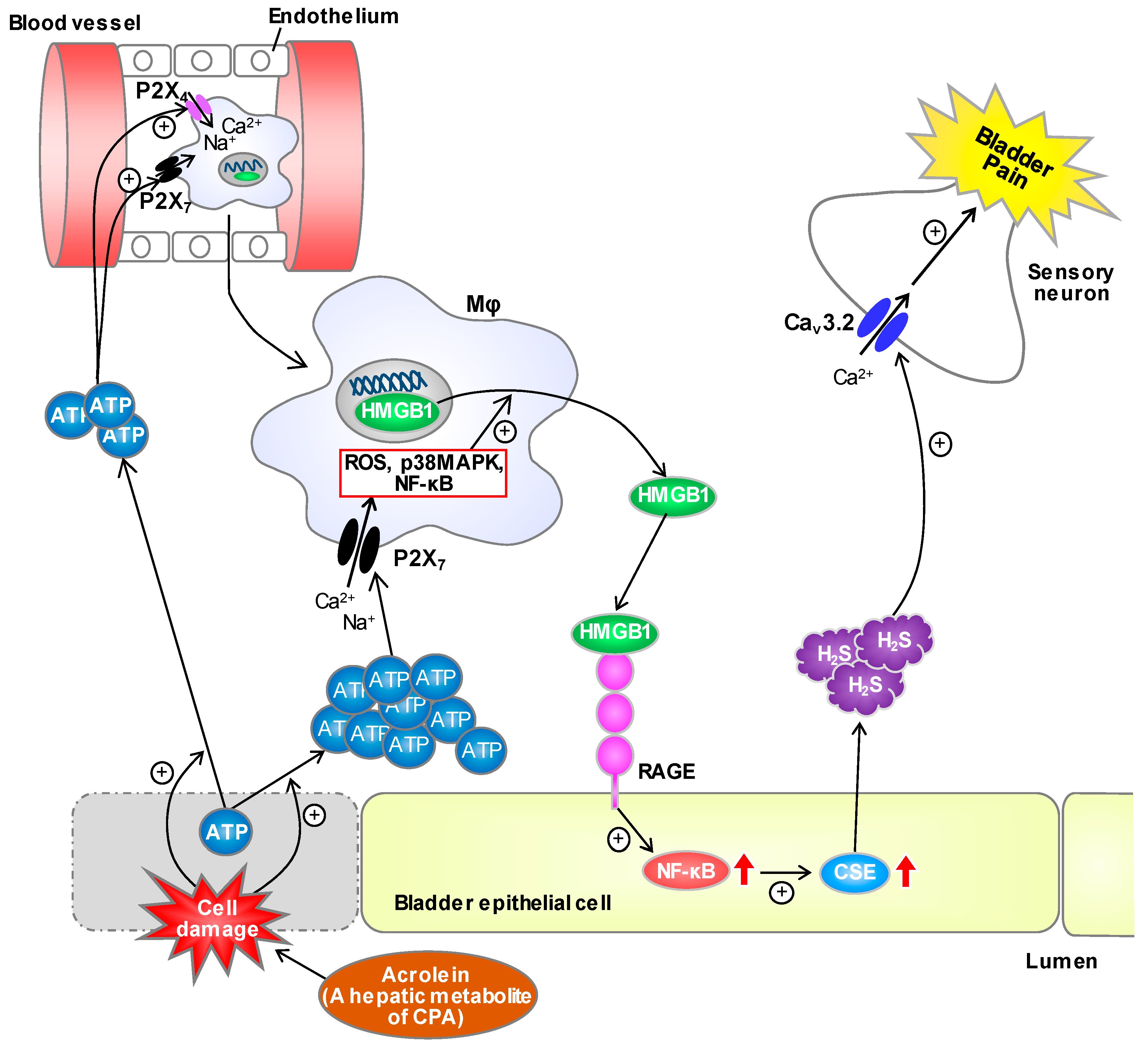
Cystitis-Related Bladder Pain Involves ATP-Dependent HMGB1 Release from Macrophages and Its Downstream H2S/Cav3.2 Signaling in Mice

 , and
, and 
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Materials and Methods
2.1. Animals
2.2. Major Chemicals
2.3. Induction of Cystitis and Related Bladder Pain by CPA, and Assessment of Bladder Pain-Like Nociceptive Behavior, Referred Hyperalgesia and Bladder Swelling in the Mice
2.4. Drug Administration Schedule
2.5. Determination of Expression of Cystathionine-γ-Lyase (CSE) Protein in the Bladder of Mice
2.6. Immunohistochemical Analysis of Localization of CSE in the Bladder of Mice
2.7. Macrophage Depletion
2.8. Accumulation of Macrophages in the Bladder of Mice Treated with CPA
2.9. Cell Culture
2.10. Determination of ATP Release from T24 Cells
2.11. Determination of HMGB1 Release from RAW264.7 Cells and T24 Cells
2.12. Knockdown of P2X4 or P2X7 by siRNA
2.13. Determination of Phosphorylation of p38 MAP Kinase and NF-κB p65 in RAW264.7 Cells Stimulated with ATP
2.14. Determination of ATP-Induced ROS Accumulation in RAW264.7 Cells
2.15. Migration Assay
2.16. Statistics Analysis
3. Results
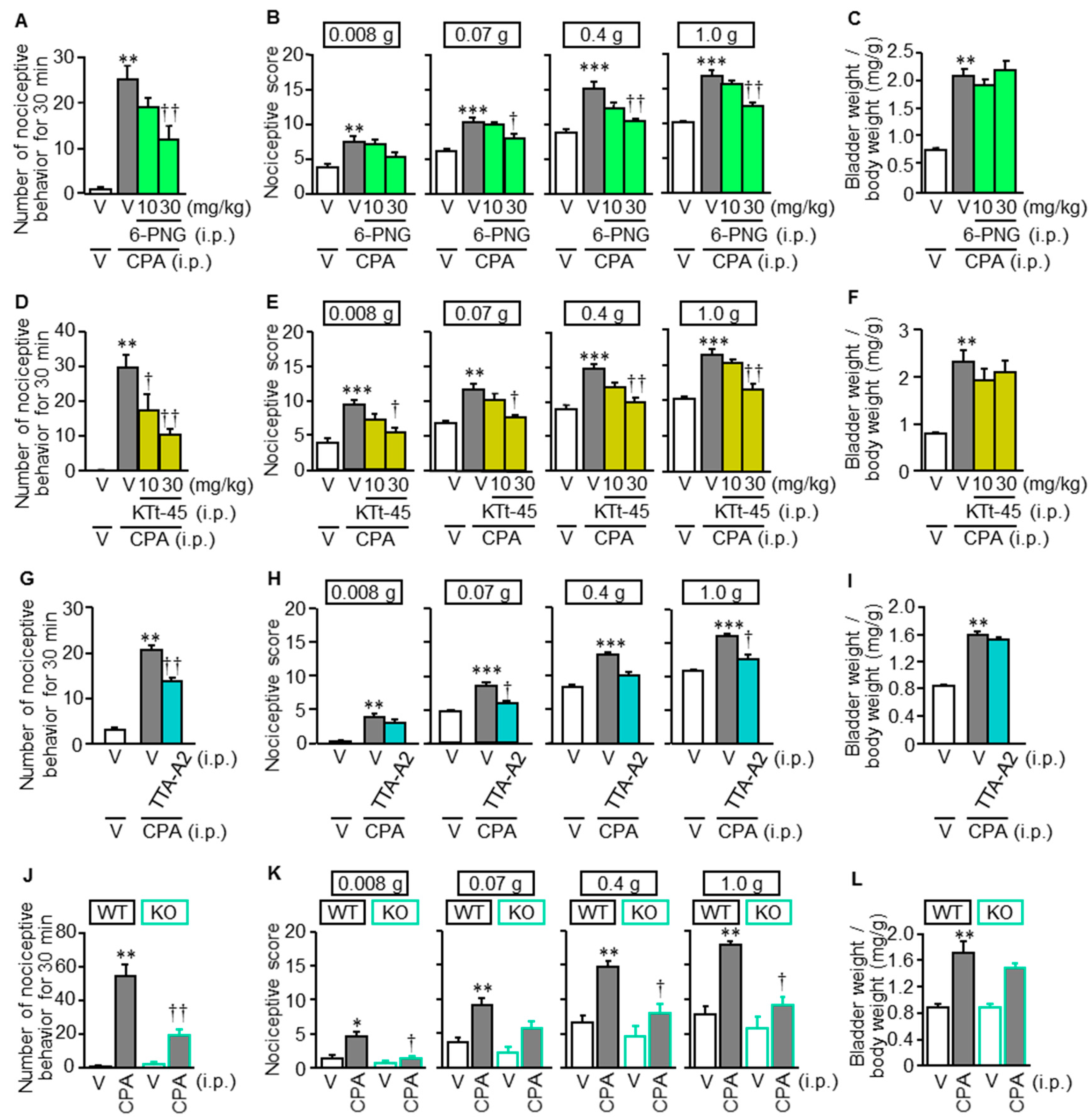
3.1. Novel T-Type Ca2+ Channel Blockers and Genetic Deletion of Cav3.2 Suppress CPA-Induced Cystitis-Related Bladder Pain in Mice
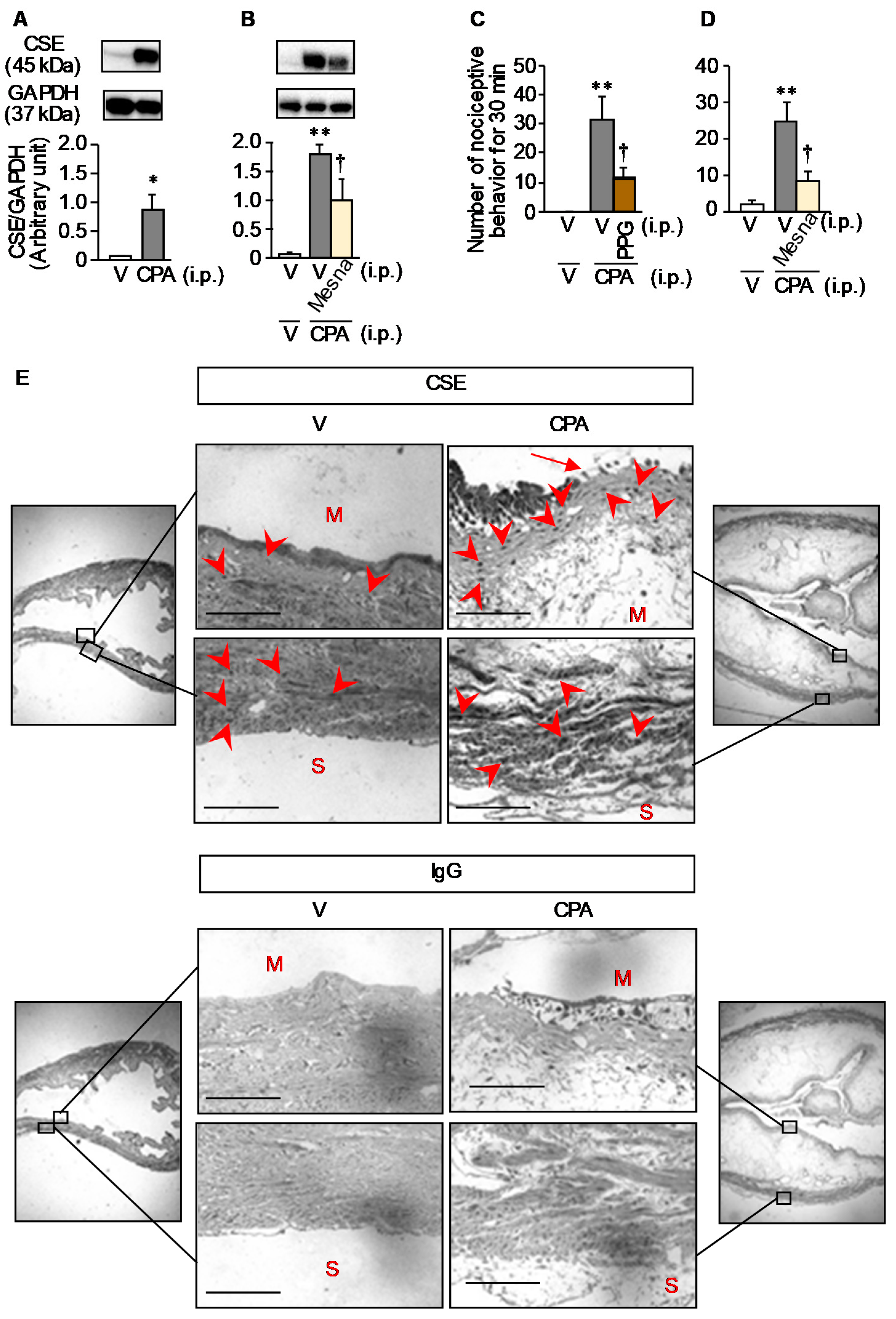
3.2. CPA-Induced Over-Expression of CSE, an Enzyme Responsible for Generation of H2S Capable of Enhancing Cav3.2 Activity, Occurs in the Mucosal Layer of the Bladder in Mice
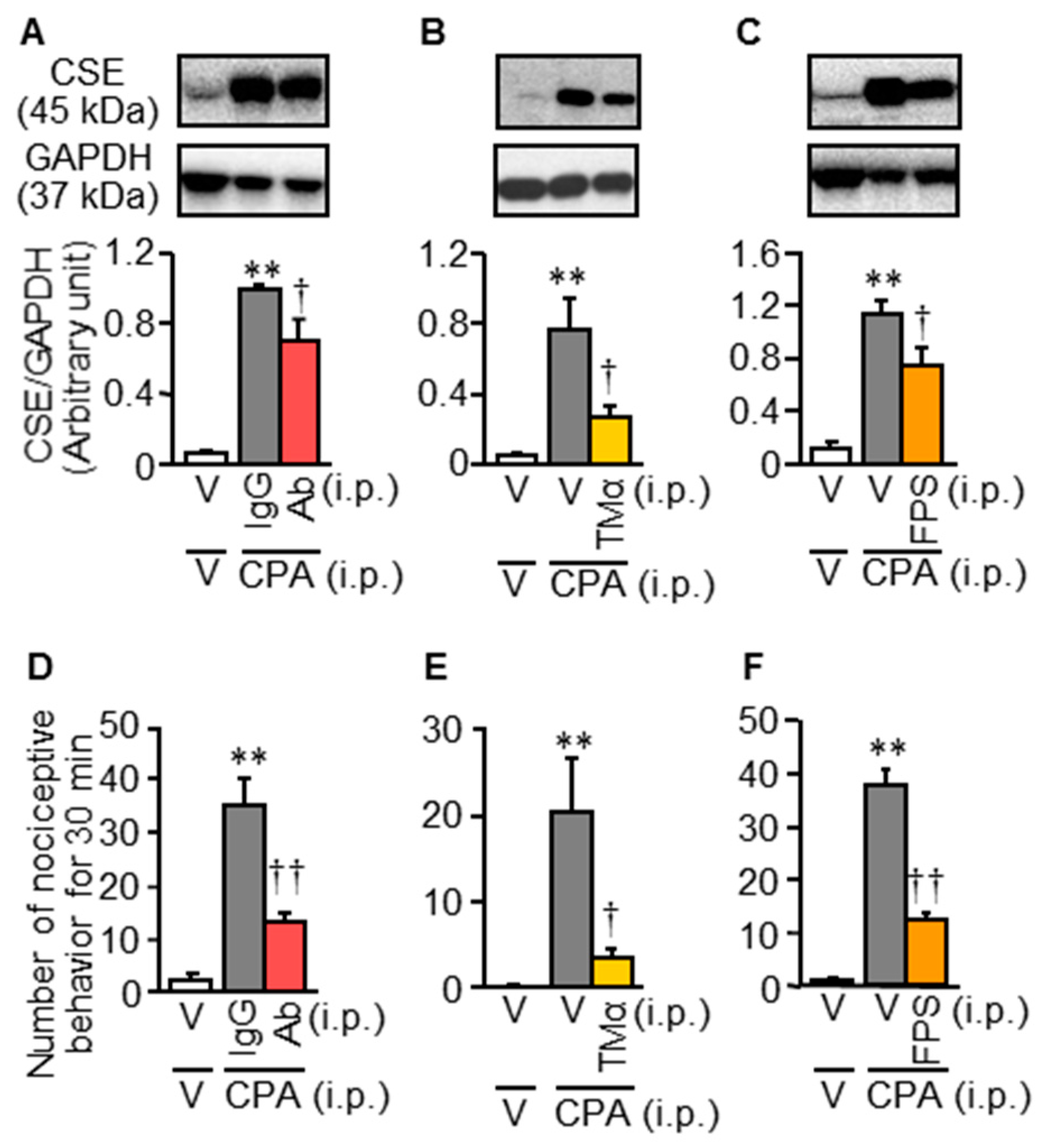
3.3. Inhibition of the HMGB1/RAGE Pathway Reduces the CPA-Induced CSE Over-Expression and Bladder Pain Symptoms in Mice
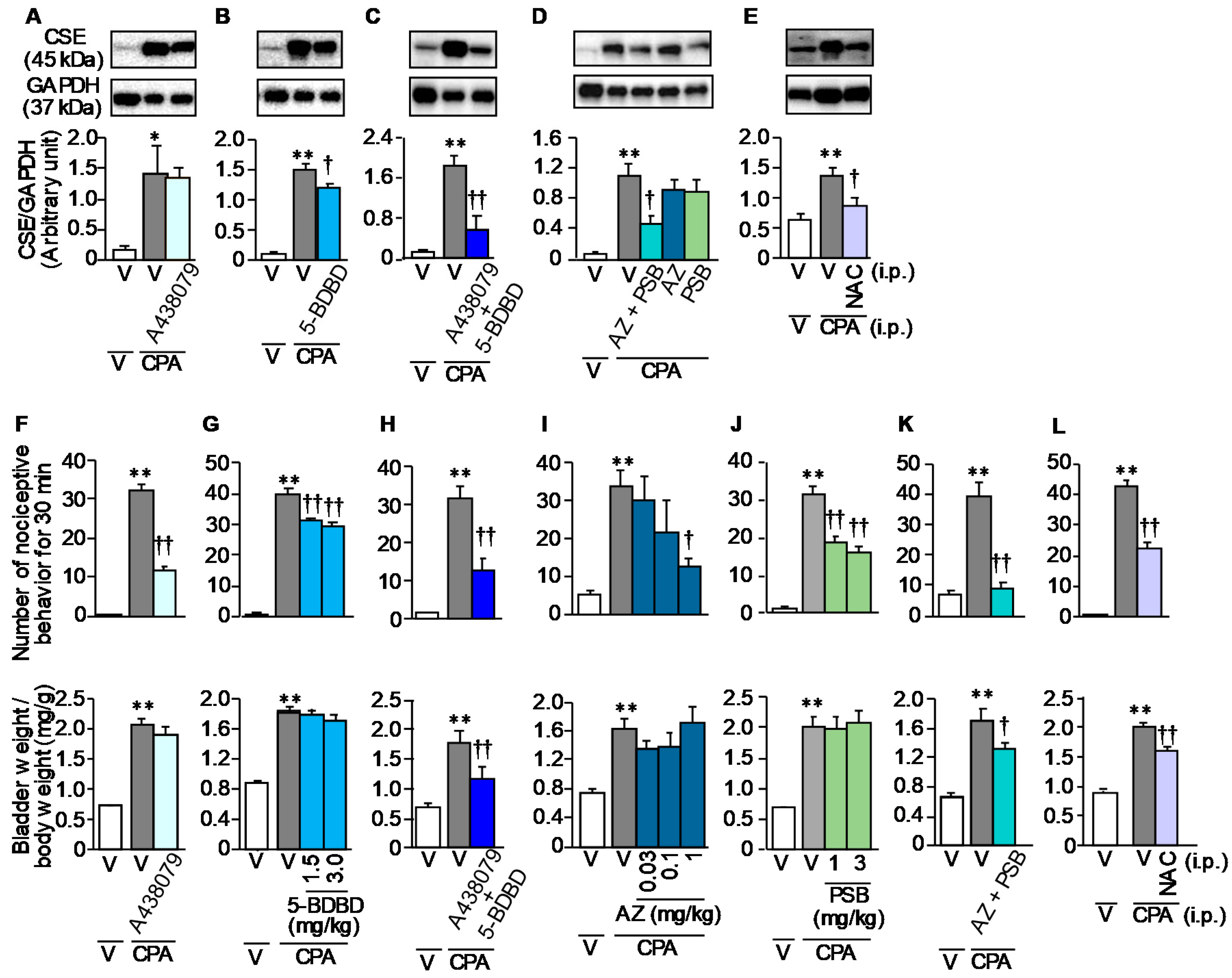
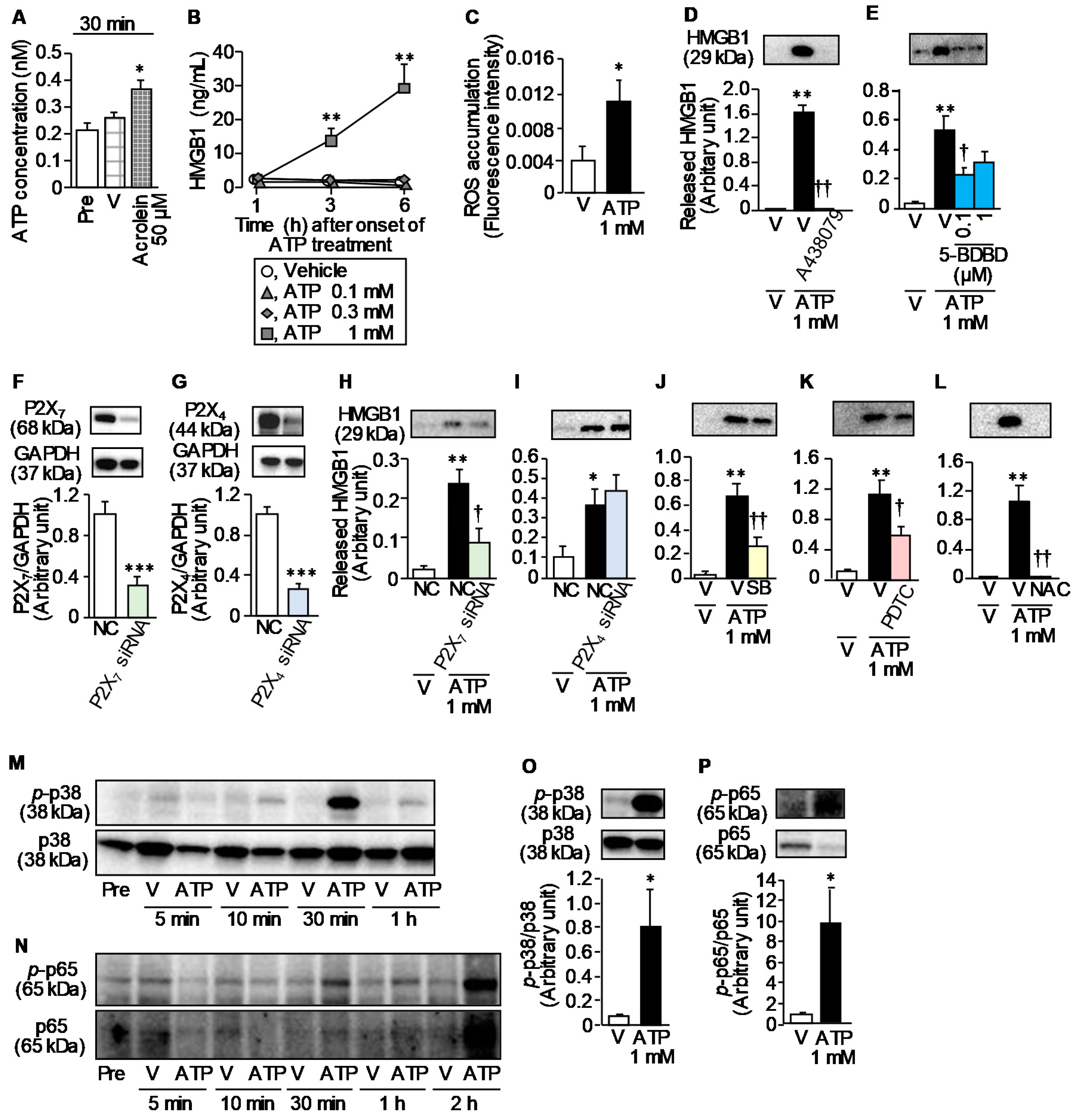
3.4. Involvement of ATP and ROS in the CPA-Induced CSE Over-Expression and Bladder Pain Symptoms in Mice
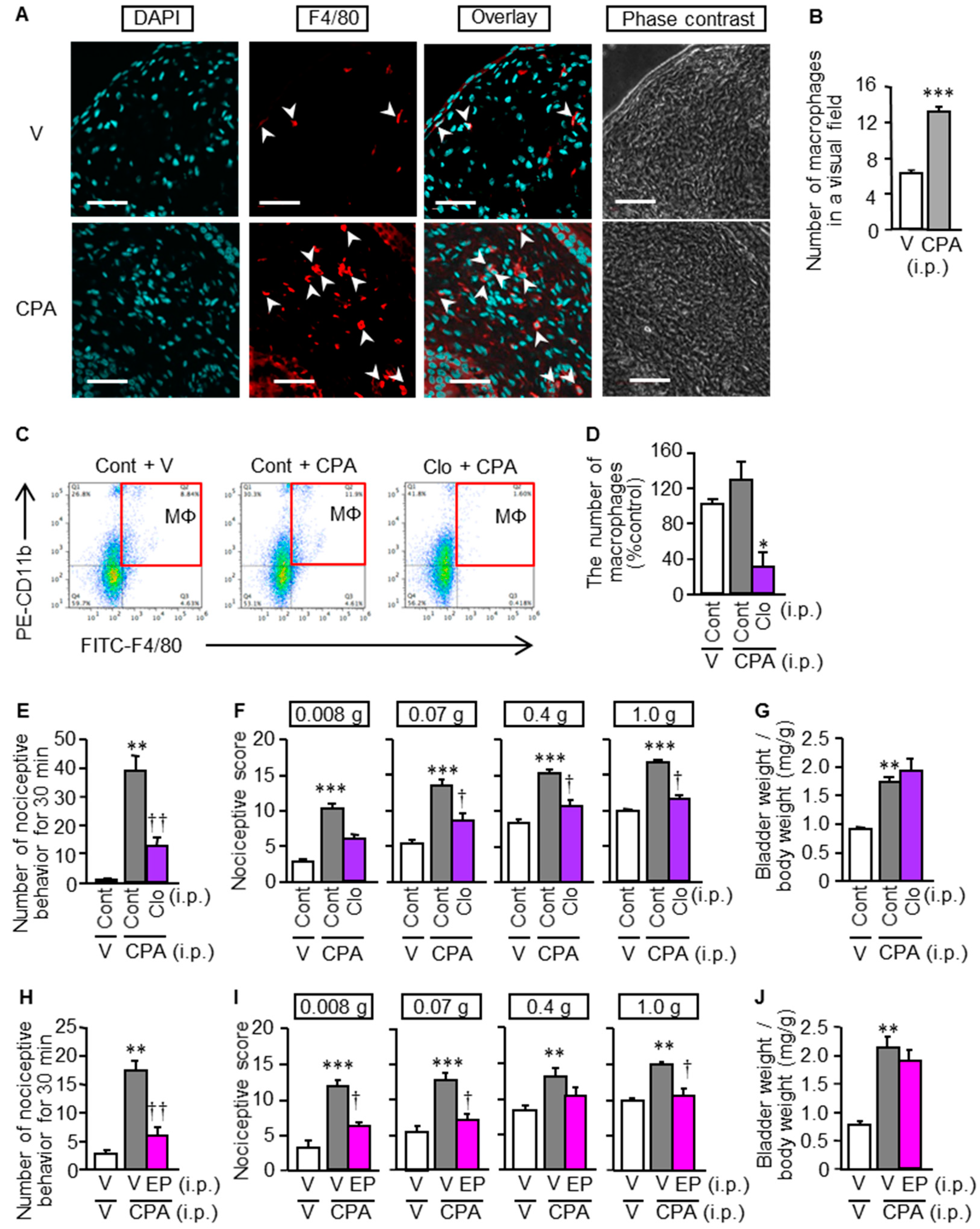
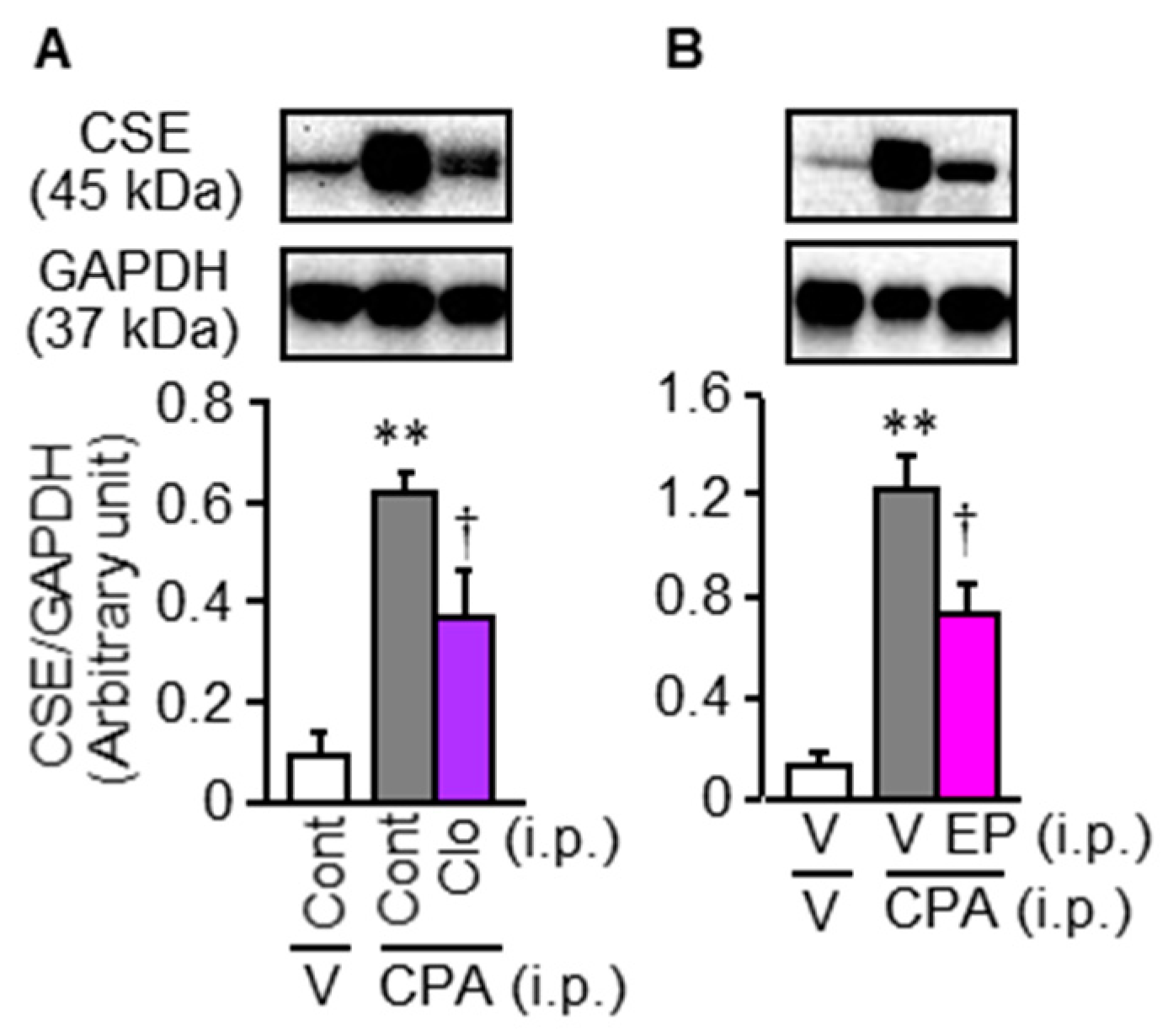
3.5. Involvement of Macrophages in the CPA-Induced Bladder Pain Symptoms and CSE Upregulation in Mice
3.6. ATP Release from Urothelial T24 Cells in Response to Acrolein, a Hepatic Metabolite of CPA, and ATP-Induced HMGB1 Release from Macrophage-Like RAW264.7 Cells
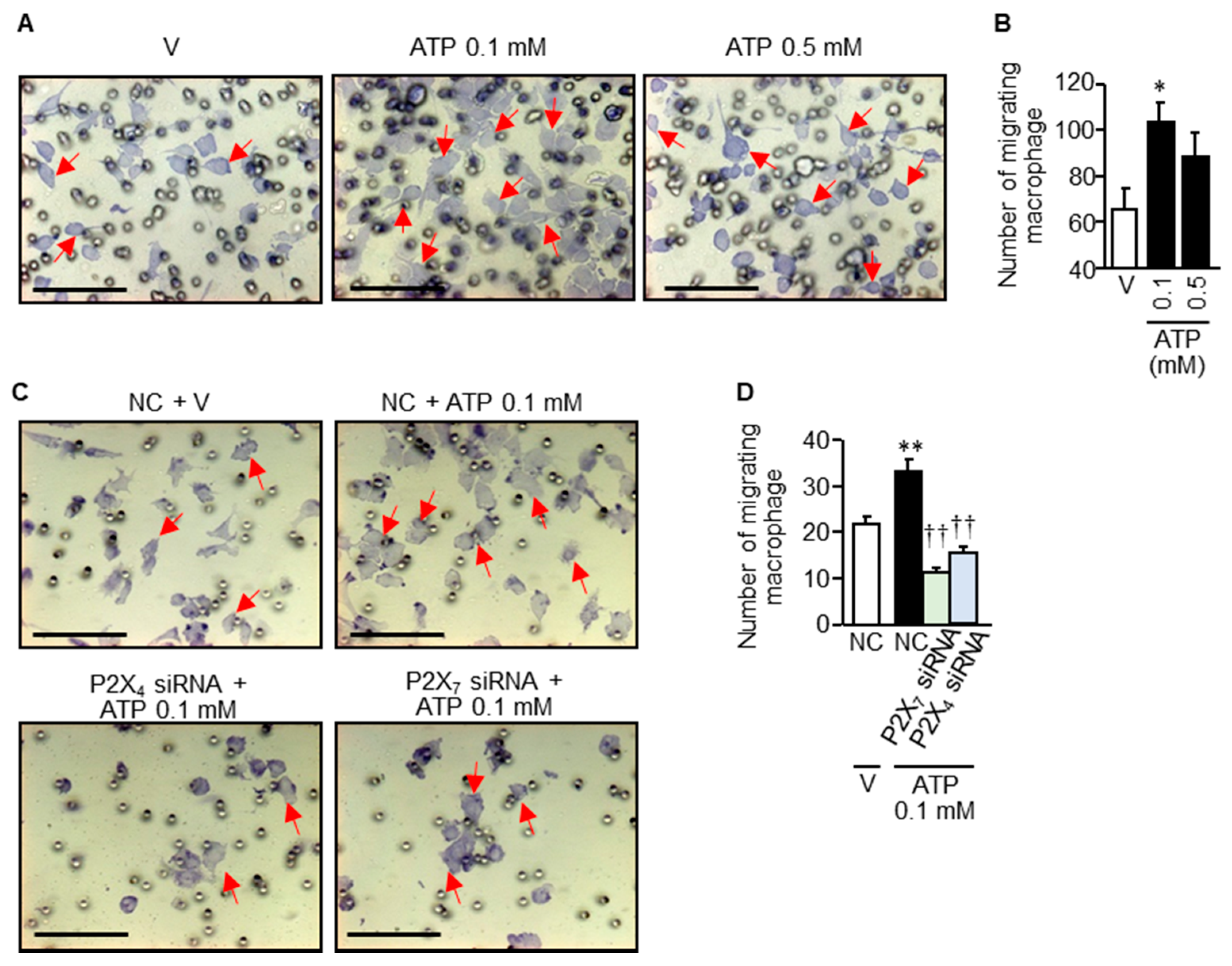
3.7. Rapid Infiltration/Migration of Macrophages in Response to Relatively Low Concentrations of ATP
4. Discussion
Supplementary Materials
Author Contributions
Funding
Conflicts of Interest
References
- Pinto, R.A.; Lopes, T.; Costa, D.; Barros, S.; Silva, J.; Silva, C.M.; Cruz, C.D.; Dinis, P.; Cruz, F.J. Ulcerative and Nonulcerative Forms of Bladder Pain Syndrome/Interstitial Cystitis Do Not Differ in Symptom Intensity or Response to Onabotulinum Toxin A. Urology 2014, 83, 1030–1034. [Google Scholar] [CrossRef]
- Tanaka, J.; Yamaguchi, K.; Ishikura, H.; Tsubota, M.; Sekiguchi, F.; Seki, Y.; Tsujiuchi, T.; Murai, A.; Umemura, T.; Kawabata, A. Bladder pain relief by HMGB1 neutralization and soluble thrombomodulin in mice with cyclophosphamide-induced cystitis. Neuropharmacology 2014, 79, 112–118. [Google Scholar] [CrossRef]
- Matsunami, M.; Miki, T.; Nishiura, K.; Hayashi, Y.; Okawa, Y.; Nishikawa, H.; Sekiguchi, F.; Kubo, L.; Ozaki, T.; Tsujiuchi, T.; et al. Involvement of the endogenous hydrogen sulfide/Cav3.2 T-type Ca2+ channel pathway in cystitis-related bladder pain in mice. Br. J. Pharmacol. 2012, 167, 917–928. [Google Scholar] [CrossRef]
- Paudel, Y.N.; Angelopoulou, E.; Piperi, C.; Balasubramaniam, V.; Othman, I.; Shaikh, M.F. Enlightening the role of high mobility group box 1 (HMGB1) in inflammation: Updates on receptor signalling. Eur. J. Pharmacol. 2019, 858, 172487. [Google Scholar] [CrossRef]
- Palanissami, G.; Paul, S.F.D. RAGE and Its Ligands: Molecular Interplay between Glycation, Inflammation, and Hallmarks of Cancer—A Review. Horm. Cancer 2018, 9, 295–325. [Google Scholar] [CrossRef]
- Agalave, N.M.; Svensson, C.I. Extracellular High-Mobility Group Box 1 Protein (HMGB1) as a Mediator of Persistent Pain. Mol. Med. 2014, 20, 569–578. [Google Scholar] [CrossRef]
- Yamasoba, D.; Tsubota, M.; Domoto, R.; Sekiguchi, F.; Nishikawa, H.; Liu, K.; Nishibori, M.; Ishikura, H.; Yamamoto, T.; Taga, A.; et al. Peripheral HMGB1-induced hyperalgesia in mice: Redox state-dependent distinct roles of RAGE and TLR4. J. Pharmacol. Sci. 2016, 130, 139–142. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, F.; Domoto, R.; Nakashima, K.; Yamasoba, D.; Yamanishi, H.; Tsubota, M.; Wake, H.; Nishibori, M.; Kawabata, A. Paclitaxel-induced HMGB1 release from macrophages and its implication for peripheral neuropathy in mice: Evidence for a neuroimmune crosstalk. Neuropharmacology 2018, 141, 201–213. [Google Scholar] [CrossRef]
- Tsubota, M.; Fukuda, R.; Hayashi, Y.; Miyazaki, T.; Ueda, S.; Yamashita, R.; Koike, N.; Sekiguchi, F.; Wake, H.; Wakatsuki, S.; et al. Role of non-macrophage cell-derived HMGB1 in oxaliplatin-induced peripheral neuropathy and its prevention by the thrombin/thrombomodulin system in rodents: Negative impact of anticoagulants. J. Neuroinflamm. 2019, 16, 199. [Google Scholar] [CrossRef]
- Irie, Y.; Tsubota, M.; Ishikura, H.; Sekiguchi, F.; Terada, Y.; Tsujiuchi, T.; Liu, K.; Nishibori, M.; Kawabata, A. Macrophage-derived HMGB1 as a Pain Mediator in the Early Stage of Acute Pancreatitis in Mice: Targeting RAGE and CXCL12/CXCR4 Axis. J. Neuroimmune Pharmacol. 2017, 12, 693–707. [Google Scholar] [CrossRef]
- Ozaki, T.; Matsuoka, J.; Tsubota, M.; Tomita, S.; Sekiguchi, F.; Minami, T.; Kawabata, A. Zinc deficiency promotes cystitis-related bladder pain by enhancing function and expression of Cav 3.2 in mice. Toxicology 2018, 393, 102–112. [Google Scholar] [CrossRef]
- Ozaki, T.; Tsubota, M.; Sekiguchi, F.; Kawabata, A. Involvement of NF-κB in the upregulation of cystathionine-γ-lyase, a hydrogen sulfide-forming enzyme, and bladder pain accompanying cystitis in mice. Clin. Exp. Pharmacol. Physiol. 2017, 45, 355–361. [Google Scholar] [CrossRef]
- Martins, J.; Silva, R.; Coutinho-Silva, R.; Takiya, C.; Battastini, A.; Morrone, F.B.; Campos, M.M. The role of P2X7 purinergic receptors in inflammatory and nociceptive changes accompanying cyclophosphamide-induced haemorrhagic cystitis in mice. Br. J. Pharmacol. 2011, 165, 183–196. [Google Scholar] [CrossRef] [PubMed]
- De Oliveira, M.G.; Calmasini, F.B.; Alexandre, E.C.; Denucci, G.; Mónica, F.Z.; Antunes, E. Activation of soluble guanylyl cyclase by BAY 58-2667 improves bladder function in cyclophosphamide-induced cystitis in mice. Am. J. Physiol. Physiol. 2016, 311, F85–F93. [Google Scholar] [CrossRef]
- Du Nguyen, H.; Okada, T.; Kitamura, S.; Yamaoka, S.; Horaguchi, Y.; Kasanami, Y.; Sekiguchi, F.; Tsubota, M.; Yoshida, S.; Nishikawa, H.; et al. Design and synthesis of novel anti-hyperalgesic agents based on 6-prenylnaringenin as the T-type calcium channel blockers. Bioorg. Med. Chem. 2018, 26, 4410–4427. [Google Scholar] [CrossRef]
- Liu, K.; Mori, S.; Takahashi, H.; Tomono, Y.; Wake, H.; Kanke, T.; Sato, Y.; Hiraga, N.; Adachi, N.; Yoshino, T.; et al. Anti-high mobility group box 1 monoclonal antibody ameliorates brain infarction induced by transient ischemia in rats. FASEB J. 2007, 21, 3904–3916. [Google Scholar] [CrossRef] [PubMed]
- Miki, T.; Matsunami, M.; Nakamura, S.; Okada, H.; Matsuya, H.; Kawabata, A. ONO-8130, a selective prostanoid EP1 receptor antagonist, relieves bladder pain in mice with cyclophosphamide-induced cystitis. Pain 2011, 152, 1373–1381. [Google Scholar] [CrossRef]
- Olivar, T.; Laird, J.M.A. Cyclophosphamide cystitis in mice: Behavioural characterisation and correlation with bladder inflammation. Eur. J. Pain 1999, 3, 141–149. [Google Scholar] [CrossRef]
- Fischer, W.; Franke, H.; Krügel, U.; Muller, H.; Dinkel, K.; Lord, B.; Letavic, M.A.; Henshall, D.C.; Engel, T. Critical Evaluation of P2X7 Receptor Antagonists in Selected Seizure Models. PLoS ONE 2016, 11, e0156468. [Google Scholar] [CrossRef]
- Yu, W.; Hill, W.; Robson, S.C.; Zeidel, M.L. Role of P2X4 Receptor in Mouse Voiding Function. Sci. Rep. 2018, 8, 1838. [Google Scholar] [CrossRef]
- Hernandez-Olmos, V.; Abdelrahman, A.; El-Tayeb, A.; Freudendahl, D.; Weinhausen, S.; Müller, C.E. N-Substituted Phenoxazine and Acridone Derivatives: Structure–Activity Relationships of Potent P2X4 Receptor Antagonists. J. Med. Chem. 2012, 55, 9576–9588. [Google Scholar] [CrossRef] [PubMed]
- Boeira, V.T.; Leite, C.E.; Santos, A.A.; Edelweiss, M.I.; Calixto, J.B.; Campos, M.M.; Morrone, F.B. Effects of the hydroalcoholic extract of Phyllanthus niruri and its isolated compounds on cyclophosphamide-induced hemorrhagic cystitis in mouse. Naunyn-Schmiedeberg’s Arch. Pharmacol. 2011, 384, 265–275. [Google Scholar] [CrossRef] [PubMed]
- Kawabata, A.; Ishiki, T.; Nagasawa, K.; Yoshida, S.; Maeda, Y.; Takahashi, T.; Sekiguchi, F.; Wada, T.; Ichida, S.; Nishikawa, H. Hydrogen sulfide as a novel nociceptive messenger. Pain 2007, 132, 74–81. [Google Scholar] [CrossRef] [PubMed]
- Sekiguchi, F.; Miyamoto, Y.; Kanaoka, D.; Ide, H.; Yoshida, S.; Ohkubo, T.; Kawabata, A. Endogenous and exogenous hydrogen sulfide facilitates T-type calcium channel currents in Cav3.2-expressing HEK293 cells. Biochem. Biophys. Res. Commun. 2014, 445, 225–229. [Google Scholar] [CrossRef]
- Matsui, K.; Tsubota, M.; Fukushi, S.; Koike, N.; Masuda, H.; Kasanami, Y.; Miyazaki, T.; Sekiguchi, F.; Ohkubo, T.; Yoshida, S.; et al. Genetic deletion of Cav3.2 T-type calcium channels abolishes H2S-dependent somatic and visceral pain signaling in C57BL/6 mice. J. Pharmacol. Sci. 2019, 140, 310–312. [Google Scholar] [CrossRef]
- Davé, S.H.; Tilstra, J.S.; Matsuoka, K.; Li, F.; Demarco, R.A.; Beer-Stolz, D.; Sepulveda, A.R.; Fink, M.P.; Lotze, M.T.; Plevy, S.E. Ethyl pyruvate decreases HMGB1 release and ameliorates murine colitis. J. Leukoc. Biol. 2009, 86, 633–643. [Google Scholar] [CrossRef]
- Korkmaz, A.; Topal, T.; Oter, S. Pathophysiological aspects of cyclophosphamide and ifosfamide induced hemorrhagic cystitis; implication of reactive oxygen and nitrogen species as well as PARP activation. Cell Biol. Toxicol. 2007, 23, 303–312. [Google Scholar] [CrossRef]
- Wu, C.-X.; Sun, H.; Liu, Q.; Guo, H.; Gong, J.-P. LPS Induces HMGB1 Relocation and Release by Activating the NF-κB-CBP Signal Transduction Pathway in the Murine Macrophage-Like Cell Line RAW264.7. J. Surg. Res. 2012, 175, 88–100. [Google Scholar] [CrossRef]
- Yang, Z.; Li, L.; Chen, L.; Yuan, W.; Dong, L.; Zhang, Y.; Wu, H.; Wang, C. PARP-1 Mediates LPS-Induced HMGB1 Release by Macrophages through Regulation of HMGB1 Acetylation. J. Immunol. 2014, 193, 6114–6123. [Google Scholar] [CrossRef]
- Nazif, O.; Teichman, J.M.; Gebhart, G. Neural Upregulation in Interstitial Cystitis. Urology 2007, 69, S24–S33. [Google Scholar] [CrossRef]
- Layhadi, J.A.; Turner, J.; Crossman, D.; Fountain, S. ATP Evokes Ca2+ Responses and CXCL5 Secretion via P2X4 Receptor Activation in Human Monocyte-Derived Macrophages. J. Immunol. 2017, 200, 1159–1168. [Google Scholar] [CrossRef] [PubMed]
- Ulmann, L.; Hirbec, H.; Rassendren, F. P2X4 receptors mediate PGE2 release by tissue-resident macrophages and initiate inflammatory pain. EMBO J. 2010, 29, 2290–2300. [Google Scholar] [CrossRef] [PubMed]
- Dunn, P.M.; Zhong, Y.; Burnstock, G. P2X receptors in peripheral neurons. Prog. Neurobiol. 2001, 65, 107–134. [Google Scholar] [CrossRef]
- Sperlágh, B.; Vizi, E.S.; Wirkner, K.; Illes, P. P2X7 receptors in the nervous system. Prog. Neurobiol. 2006, 78, 327–346. [Google Scholar] [CrossRef]
- Lee, H.Y.; Bardini, M.; Burnstock, G. Distribution of P2X receptors in the urinary bladder and the ureter of the rat. J. Urol. 2000, 163, 2002–2007. [Google Scholar] [CrossRef]
- Rahman, I.; Marwick, J.; Kirkham, P. Redox modulation of chromatin remodeling: Impact on histone acetylation and deacetylation, NF-κB and pro-inflammatory gene expression. Biochem. Pharmacol. 2004, 68, 1255–1267. [Google Scholar] [CrossRef]
- Bonaldi, T.; Talamo, F.; Scaffidi, P.; Ferrera, D.; Porto, A.; Bachi, A.; Rubartelli, A.; Agresti, A.; Bianchi, M.E. Monocytic cells hyperacetylate chromatin protein HMGB1 to redirect it towards secretion. EMBO J. 2003, 22, 5551–5560. [Google Scholar] [CrossRef]
- Ong, S.P.; Lee, L.M.; Leong, Y.F.I.; Ng, M.L.; Chu, J.J.H. Dengue Virus Infection Mediates HMGB1 Release from Monocytes Involving PCAF Acetylase Complex and Induces Vascular Leakage in Endothelial Cells. PLoS ONE 2012, 7, e41932. [Google Scholar] [CrossRef]
- Feldman, P.; Due, M.; Ripsch, M.S.; Khanna, R.; White, F.A. The persistent release of HMGB1 contributes to tactile hyperalgesia in a rodent model of neuropathic pain. J. Neuroinflamm. 2012, 9, 180. [Google Scholar] [CrossRef]









© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Hiramoto, S.; Tsubota, M.; Yamaguchi, K.; Okazaki, K.; Sakaegi, A.; Toriyama, Y.; Tanaka, J.; Sekiguchi, F.; Ishikura, H.; Wake, H.; et al. Cystitis-Related Bladder Pain Involves ATP-Dependent HMGB1 Release from Macrophages and Its Downstream H2S/Cav3.2 Signaling in Mice. Cells 2020, 9, 1748. https://doi.org/10.3390/cells9081748
Hiramoto S, Tsubota M, Yamaguchi K, Okazaki K, Sakaegi A, Toriyama Y, Tanaka J, Sekiguchi F, Ishikura H, Wake H, et al. Cystitis-Related Bladder Pain Involves ATP-Dependent HMGB1 Release from Macrophages and Its Downstream H2S/Cav3.2 Signaling in Mice. Cells. 2020; 9(8):1748. https://doi.org/10.3390/cells9081748
Chicago/Turabian StyleHiramoto, Shiori, Maho Tsubota, Kaoru Yamaguchi, Kyoko Okazaki, Aya Sakaegi, Yuki Toriyama, Junichi Tanaka, Fumiko Sekiguchi, Hiroyasu Ishikura, Hidenori Wake, and et al. 2020. "Cystitis-Related Bladder Pain Involves ATP-Dependent HMGB1 Release from Macrophages and Its Downstream H2S/Cav3.2 Signaling in Mice" Cells 9, no. 8: 1748. https://doi.org/10.3390/cells9081748
APA StyleHiramoto, S., Tsubota, M., Yamaguchi, K., Okazaki, K., Sakaegi, A., Toriyama, Y., Tanaka, J., Sekiguchi, F., Ishikura, H., Wake, H., Nishibori, M., Nguyen, H. D., Okada, T., Toyooka, N., & Kawabata, A. (2020). Cystitis-Related Bladder Pain Involves ATP-Dependent HMGB1 Release from Macrophages and Its Downstream H2S/Cav3.2 Signaling in Mice. Cells, 9(8), 1748. https://doi.org/10.3390/cells9081748