Senescence and Host–Pathogen Interactions



Abstract

{kind=link}
{kind=link}
{kind=link}
1. Introduction
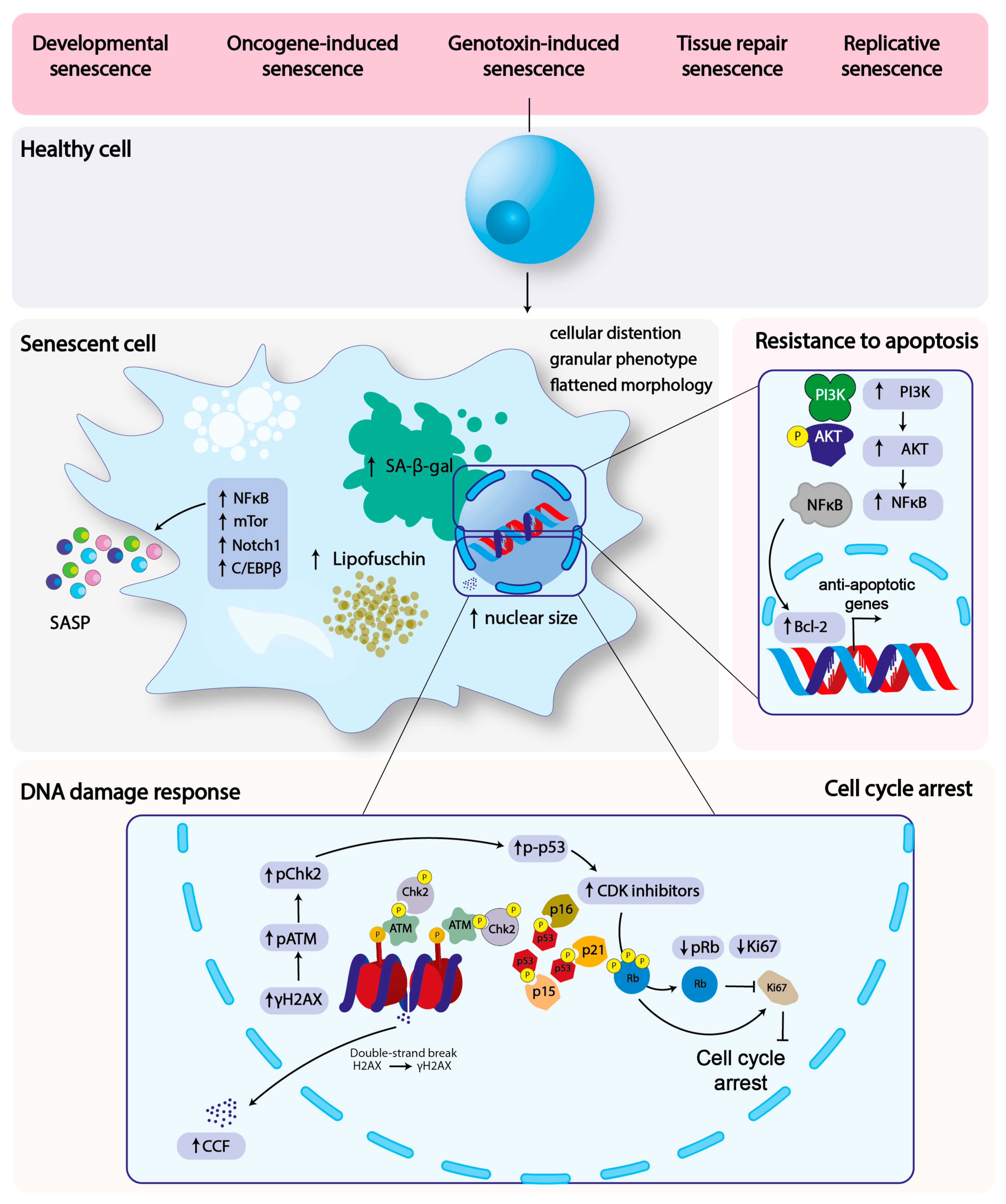
2. Hallmarks of Cellular Senescence
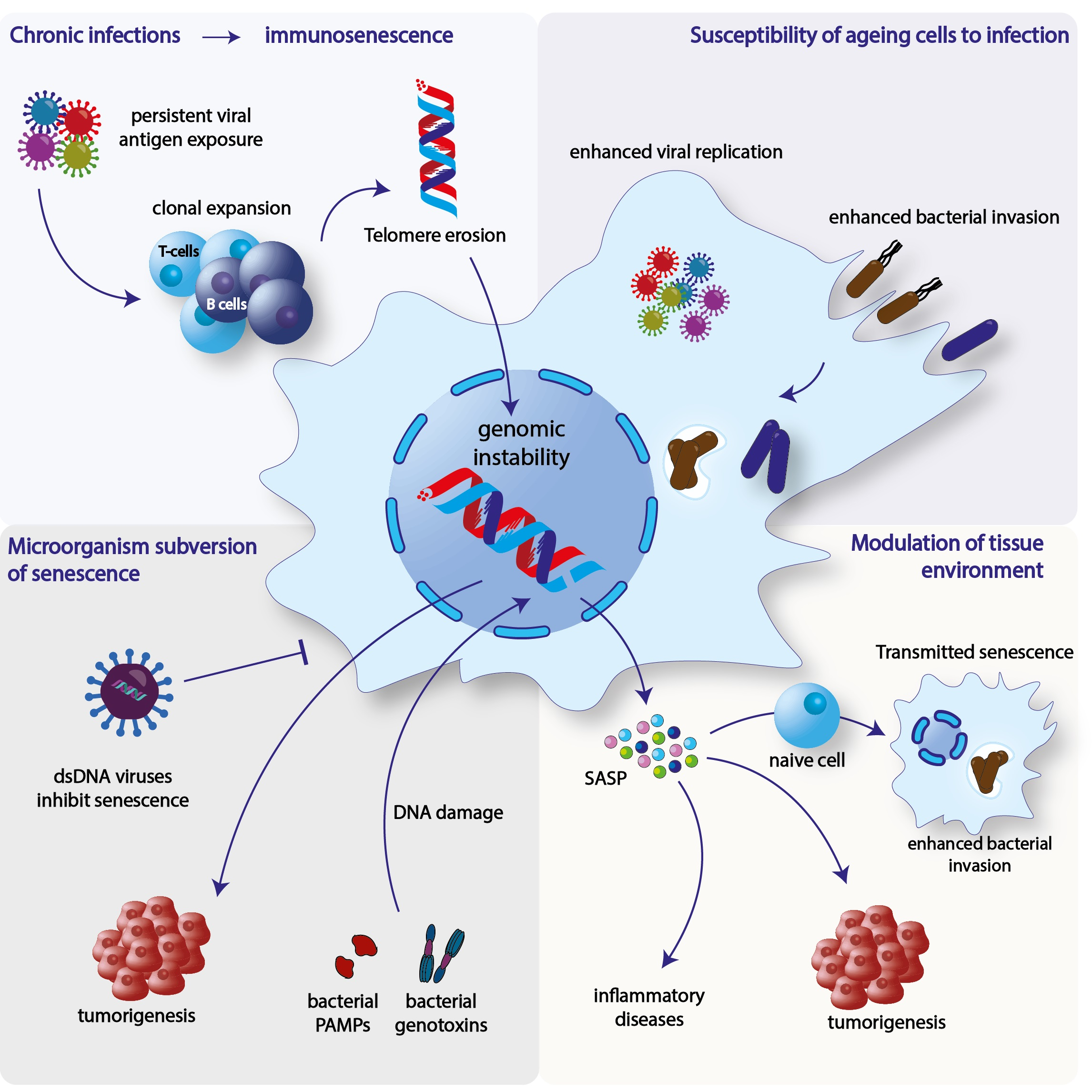
3. Infection and Senescence
3.1. Immunosenescence
Immunosenescence and Chronic Viral Infections
3.2. Effect of Senescence in Microbial Replication and Invasion
3.2.1. Bacterial Infections
3.2.2. Viral Infections
3.3. Microorganism-Induced Interference of Senescence
3.3.1. Induction of Senescence
3.3.2. Inhibition of Senescence
3.4. Bacterial-Induced Senescence and Modulation of the Tissue Microenvironment
4. Concluding Remarks
Author Contributions
Funding
Conflicts of Interest
References
- He, S.H.; Sharpless, N.E. Senescence in Health and Disease. Cell 2017, 169, 1000–1011. [Google Scholar] [CrossRef]
- Gorgoulis, V.; Adams, P.D.; Alimonti, A.; Bennett, D.C.; Bischof, O.; Bishop, C.; Campisi, J.; Collado, M.; Evangelou, K.; Ferbeyre, G.; et al. Cellular Senescence: Defining a Path Forward. Cell 2019, 179, 813–827. [Google Scholar] [CrossRef] [PubMed]
- Munoz-Espin, D.; Canamero, M.; Maraver, A.; Gomez-Lopez, G.; Contreras, J.; Murillo-Cuesta, S.; Rodriguez-Baeza, A.; Varela-Nieto, I.; Ruberte, J.; Collado, M.; et al. Programmed Cell Senescence during Mammalian Embryonic Development. Cell 2013, 155, 1104–1118. [Google Scholar] [CrossRef] [PubMed]
- Demaria, M.; Ohtani, N.; Youssef, S.A.; Rodier, F.; Toussaint, W.; Mitchell, J.R.; Laberge, R.M.; Vijg, J.; Van Steeg, H.; Dolle, M.E.T.; et al. An Essential Role for Senescent Cells in Optimal Wound Healing through Secretion of PDGF-AA. Dev. Cell 2014, 31, 722–733. [Google Scholar] [CrossRef]
- Coppe, J.P.; Desprez, P.Y.; Krtolica, A.; Campisi, J. The Senescence-Associated Secretory Phenotype: The Dark Side of Tumor Suppression. Annu. Rev. Pathol. Mech. Dis. 2010, 5, 99–118. [Google Scholar] [CrossRef]
- Coppe, J.P.; Patil, C.K.; Rodier, F.; Sun, Y.; Munoz, D.P.; Goldstein, J.; Nelson, P.S.; Desprez, P.Y.; Campisi, J. Senescence-Associated Secretory Phenotypes Reveal Cell-Nonautonomous Functions of Oncogenic RAS and the p53 Tumor Suppressor. PLoS Biol. 2008, 6, e301. [Google Scholar] [CrossRef]
- Acosta, J.C.; Banito, A.; Wuestefeld, T.; Georgilis, A.; Janich, P.; Morton, J.P.; Athineos, D.; Kang, T.W.; Lasitschka, F.; Andrulis, M.; et al. A complex secretory program orchestrated by the inflammasome controls paracrine senescence. Nat. Cell Biol. 2013, 15, 978–990. [Google Scholar] [CrossRef]
- Van Deursen, J.M. The role of senescent cells in ageing. Nature 2014, 509, 439–446. [Google Scholar] [CrossRef]
- Jun, J.I.; Lau, L.F. The matricellular protein CCN1 induces fibroblast senescence and restricts fibrosis in cutaneous wound healing. Nat. Cell Biol. 2010, 12, 676–685. [Google Scholar] [CrossRef]
- Serrano, M.; Lin, A.W.; McCurrach, M.E.; Beach, D.; Lowe, S.W. Oncogenic ras provokes premature cell senescence associated with accumulation of p53 and p16(INK4a). Cell 1997, 88, 593–602. [Google Scholar] [CrossRef]
- Le, O.N.L.; Rodier, F.; Fontaine, F.; Coppe, J.P.; Campisi, J.; DeGregori, J.; Laverdiere, C.; Kokta, V.; Haddad, E.; Beausejour, C.M. Ionizing radiation-induced long-term expression of senescence markers in mice is independent of p53 and immune status. Aging Cell 2010, 9, 398–409. [Google Scholar] [CrossRef] [PubMed]
- Baker, D.J.; Wijshake, T.; Tchkonia, T.; LeBrasseur, N.K.; Childs, B.G.; Van de Sluis, B.; Kirkland, J.L.; Van Deursen, J.M. Clearance of p16(Ink4a)-positive senescent cells delays ageing-associated disorders. Nature 2011, 479, 232–236. [Google Scholar] [CrossRef] [PubMed]
- Franceschi, C.; Campisi, J. Chronic Inflammation (Inflammaging) and Its Potential Contribution to Age-Associated Diseases. J. Gerontol. Ser. Biomed. Sci. Med. Sci. 2014, 69, S4–S9. [Google Scholar] [CrossRef] [PubMed]
- Jurk, D.; Wilson, C.; Passos, J.F.; Oakley, F.; Correia-Melo, C.; Greaves, L.; Saretzki, G.; Fox, C.; Lawless, C.; Anderson, R.; et al. Chronic inflammation induces telomere dysfunction and accelerates ageing in mice. Nat. Commun. 2014, 5. [Google Scholar] [CrossRef]
- McHugh, D.; Gil, J. Senescence and aging: Causes, consequences, and therapeutic avenues. J. Cell Biol. 2018, 217, 65–77. [Google Scholar] [CrossRef]
- Yang, X.H.; He, Z.M.; Xin, B.Z.; Cao, L. LMP1 of Epstein-Barr virus suppresses cellular senescence associated with the inhibition of p16(INK4a) expression. Oncogene 2000, 19, 2002–2013. [Google Scholar] [CrossRef]
- Blazkova, H.; Krejcikova, K.; Moudry, P.; Frisan, T.; Hodny, Z.; Bartek, J. Bacterial Intoxication Evokes Cellular Senescence with Persistent DNA Damage and Cytokine Signaling. J. Cell Mol. Med. 2010, 14, 357–367. [Google Scholar] [CrossRef]
- Cougnoux, A.; Dalmasso, G.; Martinez, R.; Buc, E.; Delmas, J.; Gibold, L.; Sauvanet, P.; Darcha, C.; Dechelotte, P.; Bonnet, M.; et al. Bacterial genotoxin colibactin promotes colon tumour growth by inducing a senescence-associated secretory phenotype. Gut 2014, 63, 1932–1942. [Google Scholar] [CrossRef]
- Ibler, A.E.M.; ElGhazaly, M.; Naylor, K.L.; Bulgakova, N.A.; Sherif, F.E.-K.; Humphreys, D. Typhoid toxin exhausts the RPA response to DNA replication stress driving senescence and Salmonella infection. Nat. Commun. 2019, 10, 4040. [Google Scholar] [CrossRef]
- DiMaio, T.A.; Vogt, D.T.; Lagunoff, M. KSHV requires vCyclin to overcome replicative senescence in primary human lymphatic endothelial cells. PLoS Pathog. 2020, 16, e1008634. [Google Scholar] [CrossRef]
- Leida, A.M.; Cyr, D.P.; Hill, R.J.; Lee, P.W.K.; McCormick, C. Subversion of Autophagy by Kaposi’s Sarcoma-Associated Herpesvirus Impairs Oncogene-Induced Senescence. Cell Host Microbe 2012, 11, 167–180. [Google Scholar] [CrossRef] [PubMed]
- Secher, T.; Samba-Louaka, A.; Oswald, E.; Nougayrede, J.P. Escherichia coli producing colibactin triggers premature and transmissible senescence in mammalian cells. PLoS ONE 2013, 8, e77157. [Google Scholar] [CrossRef] [PubMed]
- Weitzman, M.D.; Weitzman, J.B. What’s the Damage? The Impact of Pathogens on Pathways that Maintain Host Genome Integrity. Cell Host Microbe 2014, 15, 283–294. [Google Scholar] [CrossRef] [PubMed]
- Soto-Gamez, A.; Quax, W.J.; Demaria, M. Regulation of Survival Networks in Senescent Cells: From Mechanisms to Interventions. J. Mol. Biol. 2019, 431, 2629–2643. [Google Scholar] [CrossRef]
- Georgakilas, A.G.; Martin, O.A.; Bonner, W.M. P21: A Two-Faced Genome Guardian. Trends Mol. Med. 2017, 23, 310–319. [Google Scholar] [CrossRef]
- Rayess, H.; Wang, M.B.; Srivatsan, E.S. Cellular senescence and tumor suppressor gene p16. Int. J. Cancer 2012, 130, 1715–1725. [Google Scholar] [CrossRef]
- Hirai, H.; Roussel, M.F.; Kato, J.Y.; Ashmun, R.A.; Sherr, C.J. Novel Ink4 Proteins, P19 and P18, Are Specific Inhibitors of the Cyclin D-Dependent Kinases Cdk4 and Cdk6. Mol. Cell. Biol. 1995, 15, 2672–2681. [Google Scholar] [CrossRef]
- d’Adda di Fagagna, F. Living on a break: Cellular senescence as a DNA-damage response. Nat. Rev. Cancer 2008, 8, 512–522. [Google Scholar] [CrossRef]
- Bartkova, J.; Rezaei, N.; Liontos, M.; Karakaidos, P.; Kletsas, D.; Issaeva, N.; Vassiliou, L.V.; Kolettas, E.; Niforou, K.; Zoumpourlis, V.C.; et al. Oncogene-induced senescence is part of the tumorigenesis barrier imposed by DNA damage checkpoints. Nature 2006, 444, 633–637. [Google Scholar] [CrossRef]
- Wang, B.; Kohli, J.; Demaria, M. Senescent Cells in Cancer Therapy: Friends or Foes? Trends Cancer 2020. [Google Scholar] [CrossRef]
- Rodier, F.; Munoz, D.P.; Teachenor, R.; Chu, V.; Le, O.; Bhaumik, D.; Coppe, J.P.; Campeau, E.; Beausejour, C.M.; Kim, S.H.; et al. DNA-SCARS: Distinct nuclear structures that sustain damage-induced senescence growth arrest and inflammatory cytokine secretion. J. Cell Sci. 2011, 124, 68–81. [Google Scholar] [CrossRef] [PubMed]
- Ahn, J.; Gutman, D.; Saijo, S.; Barber, G.N. STING manifests self DNA-dependent inflammatory disease. Proc. Natl. Acad. Sci. USA 2012, 109, 19386–19391. [Google Scholar] [CrossRef] [PubMed]
- Lan, Y.Y.; Londono, D.; Bouley, R.; Rooney, M.S.; Hacohen, N. Dnase2a deficiency uncovers lysosomal clearance of damaged nuclear DNA via autophagy. Cell Rep. 2014, 9, 180–192. [Google Scholar] [CrossRef] [PubMed]
- Hartlova, A.; Erttmann, S.F.; Raffi, F.A.; Schmalz, A.M.; Resch, U.; Anugula, S.; Lienenklaus, S.; Nilsson, L.M.; Kroger, A.; Nilsson, J.A.; et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015, 42, 332–343. [Google Scholar] [CrossRef]
- Harding, S.M.; Benci, J.L.; Irianto, J.; Discher, D.E.; Minn, A.J.; Greenberg, R.A. Mitotic progression following DNA damage enables pattern recognition within micronuclei. Nature 2017, 548, 466–470. [Google Scholar] [CrossRef]
- Krtolica, A.; Parrinello, S.; Lockett, S.; Desprez, P.Y.; Campisi, J. Senescent fibroblasts promote epithelial cell growth and tumorigenesis: A link between cancer and aging. Proc. Natl. Acad. Sci. USA 2001, 98, 12072–12077. [Google Scholar] [CrossRef]
- Campisi, J.; d’Adda di Fagagna, F. Cellular senescence: When bad things happen to good cells. Nat. Rev. Mol. Cell Biol. 2007, 8, 729–740. [Google Scholar] [CrossRef]
- Hubackova, S.; Krejcikova, K.; Bartek, J.; Hodny, Z. IL1-and TGF beta-Nox4 signaling, oxidative stress and DNA damage response are shared features of replicative, oncogene-induced, and drug-induced paracrine ‘Bystander senescence’. Aging 2012, 4, 932–951. [Google Scholar] [CrossRef]
- Hoare, M.; Ito, Y.; Kang, T.W.; Weekes, M.P.; Matheson, N.J.; Patten, D.A.; Shetty, S.; Parry, A.J.; Menon, S.; Salama, R.; et al. NOTCH1 mediates a switch between two distinct secretomes during senescence. Nat. Cell Biol. 2016, 18, 979–992. [Google Scholar] [CrossRef]
- Ohanna, M.; Giuliano, S.; Bonet, C.; Imbert, V.; Hofman, V.; Zangari, J.; Bille, K.; Robert, C.; Bressac-de Paillerets, B.; Hofman, P.; et al. Senescent cells develop a PARP-1 and nuclear factor-{kappa}B-associated secretome (PNAS). Genes Dev. 2011, 25, 1245–1261. [Google Scholar] [CrossRef]
- Anerillas, C.; Abdelmohsen, K.; Gorospe, M. Regulation of senescence traits by MAPKs. Geroscience 2020, 42, 397–408. [Google Scholar] [CrossRef] [PubMed]
- Toso, A.; Revandkar, A.; Di Mitri, D.; Guccini, I.; Proietti, M.; Sarti, M.; Pinton, S.; Zhang, J.; Kalathur, M.; Civenni, G.; et al. Enhancing chemotherapy efficacy in Pten-deficient prostate tumors by activating the senescence-associated antitumor immunity. Cell Rep. 2014, 9, 75–89. [Google Scholar] [CrossRef] [PubMed]
- McCool, K.W.; Miyamoto, S. DNA damage-dependent NF-kappaB activation: NEMO turns nuclear signaling inside out. Immunol. Rev. 2012, 246, 311–326. [Google Scholar] [CrossRef] [PubMed]
- Datta, S.R.; Dudek, H.; Tao, X.; Masters, S.; Fu, H.A.; Gotoh, Y.; Greenberg, M.E. Akt phosphorylation of BAD couples survival signals to the cell-intrinsic death machinery. Cell 1997, 91, 231–241. [Google Scholar] [CrossRef]
- Zhang, X.B.; Tang, N.M.; Hadden, T.J.; Rishi, A.K. Akt, FoxO and regulation of apoptosis. Mol. Cell Res. 2011, 1813, 1978–1986. [Google Scholar] [CrossRef]
- Pluquet, O.; Pourtier, A.; Abbadie, C. The unfolded protein response and cellular senescence. A Review in the Theme: Cellular Mechanisms of Endoplasmic Reticulum Stress Signaling in Health and Disease. Am. J. Physiol. Cell Physiol. 2015, 308, C415–C425. [Google Scholar] [CrossRef]
- Aw, D.; Silva, A.B.; Palmer, D.B. Immunosenescence: Emerging challenges for an ageing population. Immunology 2007, 120, 435–446. [Google Scholar] [CrossRef]
- Aiello, A.; Farzaneh, F.; Candore, G.; Caruso, C.; Davinelli, S.; Gambino, C.M.; Ligotti, M.E.; Zareian, N.; Accardi, G. Immunosenescence and Its Hallmarks: How to Oppose Aging Strategically? A Review of Potential Options for Therapeutic Intervention. Front. Immunol. 2019, 10, 2247. [Google Scholar] [CrossRef]
- Fagiolo, U.; Cossarizza, A.; Scala, E.; Fanalesbelasio, E.; Ortolani, C.; Cozzi, E.; Monti, D.; Franceschi, C.; Paganelli, R. Increased Cytokine Production in Mononuclear-Cells of Healthy Elderly People. Eur. J. Immunol. 1993, 23, 2375–2378. [Google Scholar] [CrossRef]
- Licastro, F.; Candore, G.; Lio, D.; Porcellini, E.; Colonna-Romano, G.; Franceschi, C.; Caruso, C. Innate immunity and inflammation in ageing: A key for understanding age-related diseases. Immun. Ageing 2005, 2, 8. [Google Scholar] [CrossRef]
- Franceschi, C.; Bonafe, M.; Valensin, S.; Olivieri, F.; De Luca, M.; Ottaviani, E.; De Benedictis, G. Inflamm-aging—An evolutionary perspective on immunosenescence. Ann. N. Y. Acad. Sci. 2000, 908, 244–254. [Google Scholar] [CrossRef] [PubMed]
- Shaw, A.C.; Goldstein, D.R.; Montgomery, R.R. Age-dependent dysregulation of innate immunity. Nat. Rev. Immunol. 2013, 13, 875–887. [Google Scholar] [CrossRef] [PubMed]
- Crough, T.; Khanna, R. Immunobiology of Human Cytomegalovirus: From Bench to Bedside. Clin. Microbiol. Rev. 2009, 22, 76. [Google Scholar] [CrossRef] [PubMed]
- Czesnikiewicz-Guzik, M.; Lee, W.W.; Cui, D.; Hiruma, Y.; Lamar, D.L.; Yang, Z.Z.; Ouslander, J.G.; Weyand, C.M.; Goronzy, J.J. T cell subset-specific susceptibility to aging. Clin. Immunol. 2008, 127, 107–118. [Google Scholar] [CrossRef]
- Bellon, M.; Nicot, C. Telomere Dynamics in Immune Senescence and Exhaustion Triggered by Chronic Viral Infection. Viruses 2017, 9. [Google Scholar] [CrossRef]
- Heath, J.J.; Grant, M.D. The Immune Response Against Human Cytomegalovirus Links Cellular to Systemic Senescence. Cell 2020, 9. [Google Scholar] [CrossRef]
- Fletcher, J.M.; Vukmanovic-Stejic, M.; Dunne, P.J.; Birch, K.E.; Cook, J.E.; Jackson, S.E.; Salmon, M.; Rustin, M.H.; Akbar, A.N. Cytomegalovirus-specific CD4(+) T cells in healthy carriers are continuously driven to replicative exhaustion. J. Immunol. 2005, 175, 8218–8225. [Google Scholar] [CrossRef]
- Van de Berg, P.J.E.J.; Griffiths, S.J.; Yong, S.L.; Macaulay, R.; Bemelman, F.J.; Jackson, S.; Henson, S.M.; Ten Berge, I.J.M.; Akbar, A.N.; Van Lier, R.A.W. Cytomegalovirus Infection Reduces Telomere Length of the Circulating T Cell Pool. J. Immunol. 2010, 184, 3417–3423. [Google Scholar] [CrossRef]
- Riddell, N.E.; Griffiths, S.J.; Rivino, L.; King, D.C.B.; Teo, G.H.; Henson, S.M.; Cantisan, S.; Solana, R.; Kemeny, D.M.; MacAry, P.A.; et al. Multifunctional cytomegalovirus (CMV)-specific CD8(+) T cells are not restricted by telomere-related senescence in young or old adults. Immunology 2015, 144, 549–560. [Google Scholar] [CrossRef]
- Bukh, J. The history of hepatitis C virus (HCV): Basic research reveals unique features in phylogeny, evolution and the viral life cycle with new perspectives for epidemic control. J. Hepatol. 2016, 65 (Suppl. 1), S2–S21. [Google Scholar] [CrossRef]
- Hoare, M.; Gelson, W.T.H.; Das, A.; Fletcher, J.M.; Davies, S.E.; Curran, M.D.; Vowler, S.L.; Maini, M.K.; Akbar, A.N.; Alexander, G.J.M. CD4+ T-lymphocyte telomere length is related to fibrosis stage, clinical outcome and treatment response in chronic hepatitis C virus infection. J. Hepatol. 2010, 53, 252–260. [Google Scholar] [CrossRef] [PubMed]
- Hoare, M.; Shankar, A.; Shah, M.; Rushbrook, S.; Gelson, W.; Davies, S.; Akbar, A.; Alexander, G.J.M. gamma-H2AX+CD8+T lymphocytes cannot respond to IFN-alpha, IL-2 or IL-6 in chronic hepatitis C virus infection. J. Hepatol. 2013, 58, 868–874. [Google Scholar] [CrossRef]
- Deeks, S.G.; Overbaugh, J.; Phillips, A.; Buchbinder, S. HIV infection. Nat. Rev. Dis. Primers 2015, 1. [Google Scholar] [CrossRef] [PubMed]
- Deeks, S.G. HIV Infection, Inflammation, Immunosenescence, and Aging. Annu. Rev. Med. 2011, 62, 141–155. [Google Scholar] [CrossRef] [PubMed]
- Nelson, J.A.E.; Krishnamurthy, J.; Menezes, P.; Liu, Y.; Hudgens, M.G.; Sharpless, N.E.; Eron, J.J. Expression of p16INK4a as a biomarker of T-cell aging in HIV-infected patients prior to and during antiretroviral therapy. Aging Cell 2012, 11, 916–918. [Google Scholar] [CrossRef] [PubMed]
- Ribeiro, S.P.; Milush, J.M.; Cunha-Neto, E.; Kailas, E.G.; Kalil, J.; Passero, L.F.D.; Hunt, P.W.; Deeks, S.G.; Nixon, D.F.; SenGupta, D. p16(INK4a) Expression and Immunologic Aging in Chronic HIV Infection. PLoS ONE 2016, 11, e0166759. [Google Scholar] [CrossRef]
- Lichterfeld, M.; Mou, D.; Cung, T.D.H.; Williams, K.L.; Waring, M.T.; Huang, J.; Pereyra, F.; Trocha, A.; Freeman, G.J.; Rosenberg, E.S.; et al. Telomerase activity of HIV-1-specific CD8(+) T cells: Constitutive up-regulation in controllers and selective increase by blockade of PD ligand 1 in progressors. Blood 2008, 112, 3679–3687. [Google Scholar] [CrossRef]
- Bangham, C.R.M. Human T Cell Leukemia Virus Type 1: Persistence and Pathogenesis. Annu. Rev. Immunol. 2018, 36, 43–71. [Google Scholar] [CrossRef]
- Bellon, M.; Datta, A.; Brown, M.; Pouliquen, J.F.; Couppie, P.; Kazanji, M.; Nicot, C. Increased expression of telomere length regulating factors TRF1, TRF2 and TIN2 in patients with adult T-cell leukemia. Int. J. Cancer 2006, 119, 2090–2097. [Google Scholar] [CrossRef]
- Datta, A.; Nicot, C. Telomere attrition induces a DNA double-strand break damage signal that reactivates p53 transcription in HTLV-I leukemic cells. Oncogene 2008, 27, 1135–1141. [Google Scholar] [CrossRef]
- Kanasi, E.; Ayilavarapu, S.; Jones, J. The aging population: Demographics and the biology of aging. Periodontology 2000 2016, 72, 13–18. [Google Scholar] [CrossRef] [PubMed]
- Bradley, S.F.; Kauffman, C.A. Aging and the Response to Salmonella Infection. Exp. Gerontol. 1990, 25, 75–80. [Google Scholar] [CrossRef]
- Gal-Mor, O.; Boyle, E.C.; Grassl, G.A. Same species, different diseases: How and why typhoidal and non-typhoidal Salmonella enterica serovars differ. Front. Microbiol. 2014, 5, 391. [Google Scholar] [CrossRef] [PubMed]
- Lim, J.S.; Choy, H.E.; Park, S.C.; Han, J.M.; Jang, I.S.; Cho, K.A. Caveolae-mediated entry of Salmonella typhimurium into senescent nonphagocytotic host cells. Aging Cell 2010, 9, 243–251. [Google Scholar] [CrossRef]
- Ahn, S.H.; Cho, S.H.; Song, J.E.; Kim, S.; Oh, S.S.; Jung, S.; Cho, K.A.; Lee, T.H. Caveolin-1 serves as a negative effector in senescent human gingival fibroblasts during Fusobacterium nucleatum infection. Mol. Oral Microbiol. 2017, 32, 236–249. [Google Scholar] [CrossRef]
- Troeger, C.; Blacker, B.F.; Khalil, I.A.; Rao, P.C.; Cao, S.J.; Zimsen, S.R.M.; Albertson, S.; Stanaway, J.D.; Deshpande, A.; Farag, T.; et al. Estimates of the global, regional, and national morbidity, mortality, and aetiologies of lower respiratory infections in 195 countries, 1990–2016: A systematic analysis for the Global Burden of Disease Study 2016. Lancet Infect. Dis. 2018, 18, 1191–1210. [Google Scholar] [CrossRef]
- Shivshankar, P.; Boyd, A.R.; Le Saux, C.J.; Yeh, I.T.; Orihuela, C.J. Cellular senescence increases expression of bacterial ligands in the lungs and is positively correlated with increased susceptibility to pneumococcal pneumonia. Aging Cell 2011, 10, 798–806. [Google Scholar] [CrossRef]
- Hinojosa, C.A.; Mgbemena, V.; Van Roekel, S.; Austad, S.N.; Miller, R.A.; Bose, S.; Orihuela, C.J. Enteric-delivered rapamycin enhances resistance of aged mice to pneumococcal pneumonia through reduced cellular senescence. Exp. Gerontol. 2012, 47, 958–965. [Google Scholar] [CrossRef]
- Kline, K.A.; Bowdish, D.M.E. Infection in an aging population. Curr. Opin. Microbiol. 2016, 29, 63–67. [Google Scholar] [CrossRef]
- Wilhelm, M. Influenza in Older Patients: A Call to Action and Recent Updates for Vaccinations. Am. J. Manag. Care 2018, 24 (Suppl. 2), S15–S24. [Google Scholar]
- Kim, J.A.; Seong, R.K.; Shin, O.S. Enhanced Viral Replication by Cellular Replicative Senescence. Immune Netw. 2016, 16, 286–295. [Google Scholar] [CrossRef]
- Weiland, T.; Lampe, J.; Essmann, F.; Venturelli, S.; Berger, A.; Bossow, S.; Berchtold, S.; Schulze-Osthoff, K.; Lauer, U.M.; Bitzer, M. Enhanced killing of therapy-induced senescent tumor cells by oncolytic measles vaccine viruses. Int. J. Cancer 2014, 134, 235–243. [Google Scholar] [CrossRef] [PubMed]
- Zhou, P.; Yang, X.L.; Wang, X.G.; Hu, B.; Zhang, L.; Zhang, W.; Si, H.R.; Zhu, Y.; Li, B.; Huang, C.L.; et al. A pneumonia outbreak associated with a new coronavirus of probable bat origin. Nature 2020, 579, 270. [Google Scholar] [CrossRef] [PubMed]
- Khemais-Benkhiat, S.; Idris-Khodja, N.; Ribeiro, T.P.; Silva, G.C.; Abbas, M.; Kheloufi, M.; Lee, J.O.; Toti, F.; Auger, C.; Schini-Kerth, V.B. The Redox-sensitive Induction of the Local Angiotensin System Promotes Both Premature and Replicative Endothelial Senescence: Preventive Effect of a Standardized Crataegus Extract. J. Gerontol. Ser. A Biomed. Sci. Med Sci. 2016, 71, 1581–1590. [Google Scholar] [CrossRef]
- Baz-Martinez, M.; Da Silva-Alvarez, S.; Rodriguez, E.; Guerra, J.; El Motiam, A.; Vidal, A.; Garcia-Caballero, T.; Gonzalez-Barcia, M.; Sanchez, L.; Munoz-Fontela, C.; et al. Cell senescence is an antiviral defense mechanism. Sci. Rep. 2016, 6, 37007. [Google Scholar] [CrossRef] [PubMed]
- Siebels, S.; Czech-Sioli, M.; Spohn, M.; Schmidt, C.; Theiss, J.; Indenbirken, D.; Gunther, T.; Grundhoff, A.; Fischer, N. Merkel Cell Polyomavirus DNA Replication Induces Senescence in Human Dermal Fibroblasts in a Kap1/Trim28-Dependent Manner. Mbio 2020, 11. [Google Scholar] [CrossRef] [PubMed]
- Feng, G.J.; Zheng, K.; Cao, T.; Zhang, J.L.; Lian, M.; Huang, D.; Wei, C.B.; Gu, Z.F.; Feng, X.M. Repeated stimulation by LPS promotes the senescence of DPSCs via TLR4/MyD88-NF-kappa B-p53/p21 signaling. Cytotechnology 2018, 70, 1023–1035. [Google Scholar] [CrossRef]
- Bezine, E.; Vignard, J.; Mirey, G. The Cytolethal Distending Toxin Effects on Mammalian Cells: A DNA Damage Perspective. Cells 2014, 3, 592–615. [Google Scholar] [CrossRef]
- Martin, O.C.B.; Frisan, T. Bacterial Genotoxin-Induced DNA Damage and Modulation of the Host Immune Microenvironment. Toxins 2020, 12. [Google Scholar] [CrossRef] [PubMed]
- Nougayrede, J.P.; Homburg, S.; Taieb, F.; Boury, M.; Brzuszkiewicz, E.; Gottschalk, G.; Buchrieser, C.; Hacker, J.; Dobrindt, U.; Oswald, E. Escherichia coli induces DNA double-strand breaks in eukaryotic cells. Science 2006, 313, 848–851. [Google Scholar] [CrossRef]
- Bossuet-Greif, N.; Vignard, J.; Taieb, F.; Mirey, G.; Dubois, D.; Petit, C.; Oswald, E.; Nougayrede, J.P. The Colibactin Genotoxin Generates DNA Interstrand Cross-Links in Infected Cells. Mbio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Nesic, D.; Hsu, Y.; Stebbins, C.E. Assembly and function of a bacterial genotoxin. Nature 2004, 429, 429–433. [Google Scholar] [CrossRef] [PubMed]
- Nesic, D.; Stebbins, C.E. Mechanisms of Assembly and Cellular Interactions for the Bacterial Genotoxin CDT. PLoS Pathog. 2005, 1, e28. [Google Scholar] [CrossRef]
- Frisan, T. Bacterial genotoxins: The long journey to the nucleus of mammalian cells. Biochim. Biophys. Acta 2015, 1858, 567–575. [Google Scholar] [CrossRef]
- Johnson, W.M.; Lior, H. Response of Chinese hamster ovary cells to a cytolethal distending toxin (CDT) of Escherichia coli and possible misinterpretation as heat-labile (LT) enterotoxin. FEMS Microbiol. Lett. 1987, 43, 19–23. [Google Scholar] [CrossRef]
- Elwell, C.A.; Dreyfus, L.A. DNAase I homologous residues in CdtB are critical for cytolethal distending toxin-mediated cell cycle arrest. Mol. Microbiol. 2000, 37, 952–963. [Google Scholar] [CrossRef] [PubMed]
- Lara-Tejero, M.; Galan, J.E. CdtA, CdtB and CdtC form a tripartite complex that is required for cytolethal distending toxin activity. Infect. Immun. 2001, 69, 4358–4365. [Google Scholar] [CrossRef]
- Cortes-Bratti, X.; Frisan, T.; Thelestam, M. The cytolethal distending toxins induce DNA damage and cell cycle arrest. Toxicon 2001, 39, 1729–1736. [Google Scholar] [CrossRef]
- Fedor, Y.; Vignard, J.; Nicolau-Travers, M.L.; Boutet-Robinet, E.; Watrin, C.; Salles, B.; Mirey, G. From single-strand breaks to double-strand breaks during S-phase: A new mode of action of the Escherichia coli Cytolethal Distending Toxin. Cell Microbiol. 2013, 15, 1–15. [Google Scholar] [CrossRef]
- Pere-Vedrenne, C.; Prochazkova-Carlotti, M.; Rousseau, B.; He, W.; Chambonnier, L.; Sifre, E.; Buissonniere, A.; Dubus, P.; Megraud, F.; Varon, C.; et al. The Cytolethal Distending Toxin Subunit CdtB of Helicobacter hepaticus Promotes Senescence and Endoreplication in Xenograft Mouse Models of Hepatic and Intestinal Cell Lines. Front. Cell Infect. Microbiol. 2017, 7, 268. [Google Scholar] [CrossRef]
- Den Bakker, H.C.; Switt, A.I.M.; Govoni, G.; Cummings, C.A.; Ranieri, M.L.; Degoricija, L.; Hoelzer, K.; Rodriguez-Rivera, L.D.; Brown, S.; Bolchacova, E.; et al. Genome sequencing reveals diversification of virulence factor content and possible host adaptation in distinct subpopulations of Salmonella enterica. BMC Genom. 2011, 12, 425. [Google Scholar] [CrossRef] [PubMed]
- Spano, S.; Ugalde, J.E.; Galan, J.E. Delivery of a Salmonella Typhi exotoxin from a host intracellular compartment. Cell Host Microbe 2008, 3, 30–38. [Google Scholar] [CrossRef] [PubMed]
- Song, J.; Gao, X.; Galan, J.E. Structure and function of the Salmonella Typhi chimaeric A(2)B(5) typhoid toxin. Nature 2013, 499, 350–354. [Google Scholar] [CrossRef]
- Del Bel Belluz, L.; Guidi, R.; Pateras, I.S.; Levi, L.; Mihaljevic, B.; Rouf, S.F.; Wrande, M.; Candela, M.; Turroni, S.; Nastasi, C.; et al. The Typhoid Toxin Promotes Host Survival and the Establishment of a Persistent Asymptomatic Infection. PLoS Pathog. 2016, 12, e1005528. [Google Scholar] [CrossRef]
- Miller, R.A.; Betteken, M.I.; Guo, X.; Altier, C.; Duhamel, G.E.; Wiedmann, M. The Typhoid Toxin Produced by the Nontyphoidal Salmonella enterica Serotype Javiana Is Required for Induction of a DNA Damage Response In Vitro and Systemic Spread In Vivo. Mbio 2018, 9. [Google Scholar] [CrossRef] [PubMed]
- Gibani, M.M.; Jones, E.; Barton, A.; Jin, C.; Meek, J.; Camara, S.; Galal, U.; Heinz, E.; Rosenberg-Hasson, Y.; Obermoser, G.; et al. Investigation of the role of typhoid toxin in acute typhoid fever in a human challenge model. Nat. Med. 2019, 25, 1082. [Google Scholar] [CrossRef] [PubMed]
- Guidi, R.; Levi, L.; Rouf, S.F.; Puiac, S.; Rhen, M.; Frisan, T. Salmonella enterica delivers its genotoxin through outer membrane vesicles secreted from infected cells. Cell Microbiol. 2013, 15, 2034–2050. [Google Scholar] [CrossRef] [PubMed]
- Toledo, L.I.; Altmeyer, M.; Rask, M.B.; Lukas, C.; Larsen, D.H.; Povlsen, L.K.; Bekker-Jensen, S.; Mailand, N.; Bartek, J.; Lukas, J. ATR Prohibits Replication Catastrophe by Preventing Global Exhaustion of RPA. Cell 2013, 155, 1088–1103. [Google Scholar] [CrossRef]
- Pal, A.; Kundu, R. Human Papillomavirus E6 and E7: The Cervical Cancer Hallmarks and Targets for Therapy. Front. Microbiol. 2020, 10, 3116. [Google Scholar] [CrossRef]
- Kamranvar, S.A.; Masucci, M.G. Regulation of Telomere Homeostasis during Epstein-Barr virus Infection and Immortalization. Viruses 2017, 9, 217. [Google Scholar] [CrossRef]
- Cesarman, E.; Damania, B.; Krown, S.E.; Martin, J.; Bower, M.; Whitby, D. Kaposi sarcoma. Nat. Rev. Dis. Primers 2019, 5, 1–21. [Google Scholar] [CrossRef] [PubMed]
- Kamranvar, S.A.; Chen, X.; Masucci, M.G. Telomere dysfunction and activation of alternative lengthening of telomeres in B-lymphocytes infected by Epstein-Barr virus. Oncogene 2013, 32, 5522–5530. [Google Scholar] [CrossRef]
- Hafez, A.Y.; Luftig, M.A. Characterization of the EBV-Induced Persistent DNA Damage Response. Viruses 2017, 9, 366. [Google Scholar] [CrossRef] [PubMed]
- Padberg, I.; Janssen, S.; Meyer, T.F. Chlamydia trachomatis inhibits telomeric DNA damage signaling via transient hTERT upregulation. Int. J. Med. Microbiol. 2013, 303, 463–474. [Google Scholar] [CrossRef] [PubMed]
- Anttila, T.; Saikku, P.; Koskela, P.; Bloigu, A.; Dillner, J.; Ikaheimo, I.; Jellum, E.; Lehtinen, M.; Lenner, P.; Hakulinen, T.; et al. Serotypes of Chlamydia trachomatis and risk for development of cervical squamous cell carcinoma. JAMA 2001, 285, 47–51. [Google Scholar] [CrossRef]
- Aktories, K.; Schwan, C.; Jank, T. Clostridium difficile Toxin Biology. Annu. Rev. Microbiol. 2017, 71, 281–307. [Google Scholar] [CrossRef]
- Fettucciari, K.; Macchioni, L.; Davidescu, M.; Scarpelli, P.; Palumbo, C.; Corazzi, L.; Marchegiani, A.; Cerquetella, M.; Spaterna, A.; Marconi, P.; et al. Clostridium difficile toxin B induces senescence in enteric glial cells: A potential new mechanism of Clostridium difficile pathogenesis. Mol. Cell Res. 2018, 1865, 1945–1958. [Google Scholar] [CrossRef]
- Sargiacomo, C.; Sotgia, F.; Lisanti, M.P. COVID-19 and chronological aging: Senolytics and other anti-aging drugs for the treatment or prevention of corona virus infection? Aging 2020, 12, 6511–6517. [Google Scholar] [CrossRef]
- Malavolta, M.; Giacconi, R.; Brunetti, D.; Provinciali, M.; Maggi, F. Exploring the Relevance of Senotherapeutics for the Current SARS-CoV-2 Emergency and Similar Future Global Health Threats. Cells 2020, 9, 909. [Google Scholar] [CrossRef]
- Szaniawski, M.A.; Spivak, A.M. Senotherapeutics and HIV-1 Persistence. Curr. HIV/AIDS Rep. 2020, 17, 219–225. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Humphreys, D.; ElGhazaly, M.; Frisan, T. Senescence and Host–Pathogen Interactions. Cells 2020, 9, 1747. https://doi.org/10.3390/cells9071747
Humphreys D, ElGhazaly M, Frisan T. Senescence and Host–Pathogen Interactions. Cells. 2020; 9(7):1747. https://doi.org/10.3390/cells9071747
Chicago/Turabian StyleHumphreys, Daniel, Mohamed ElGhazaly, and Teresa Frisan. 2020. "Senescence and Host–Pathogen Interactions" Cells 9, no. 7: 1747. https://doi.org/10.3390/cells9071747
APA StyleHumphreys, D., ElGhazaly, M., & Frisan, T. (2020). Senescence and Host–Pathogen Interactions. Cells, 9(7), 1747. https://doi.org/10.3390/cells9071747