CD38: T Cell Immuno-Metabolic Modulator




{kind=link}
{kind=link}
Abstract
1. Introduction
2. CD38-NAD+ Axis in Health and Diseases
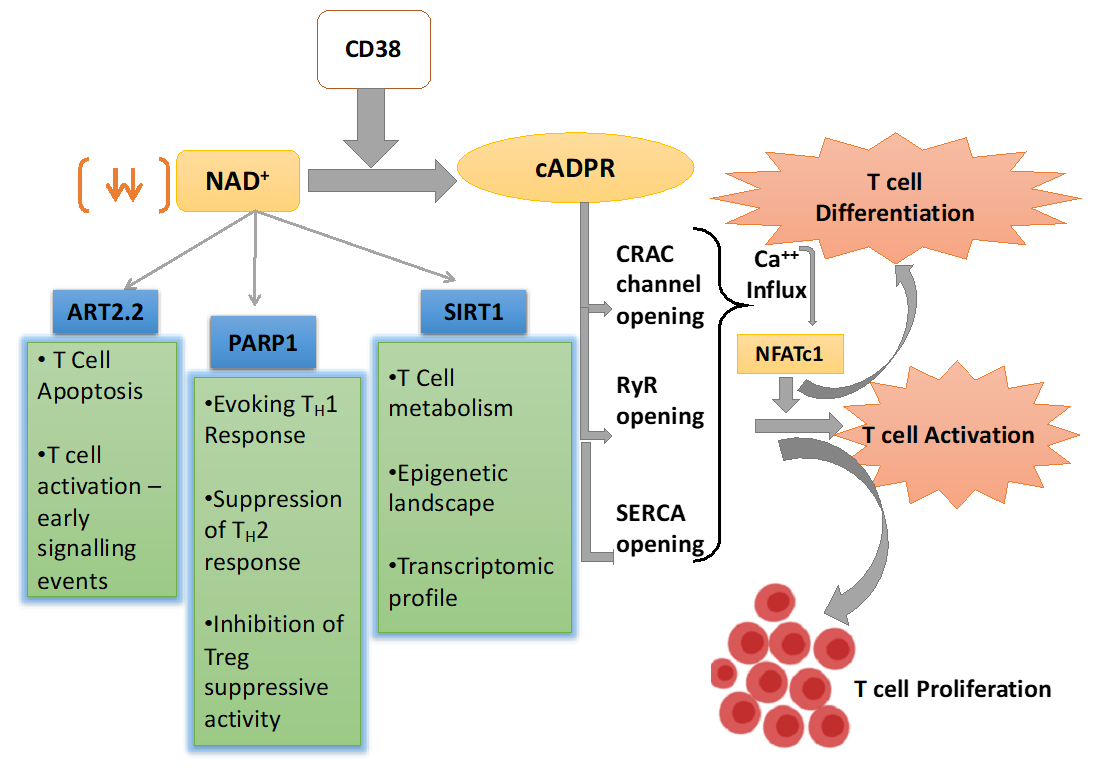
3. CD38 Mediated Signaling in Activated T Cells
4. CD38-NAD+ Axis in Regulating T cell Fate and Function
4.1. NAD+ Dependent Mono-ADP-Ribosyl Transferases (ARTs) in T Cells
4.2. NAD+ Dependent Poly-ADP-Ribose Polymerases in T Cells
4.3. NAD+-Sirt1 Dependent Regulation of T Cell Function
4.3.1. Regulation of T Cell Effector Function
4.3.2. Th2 Response
4.3.3. Th17 Response
4.3.4. Treg Differentiation
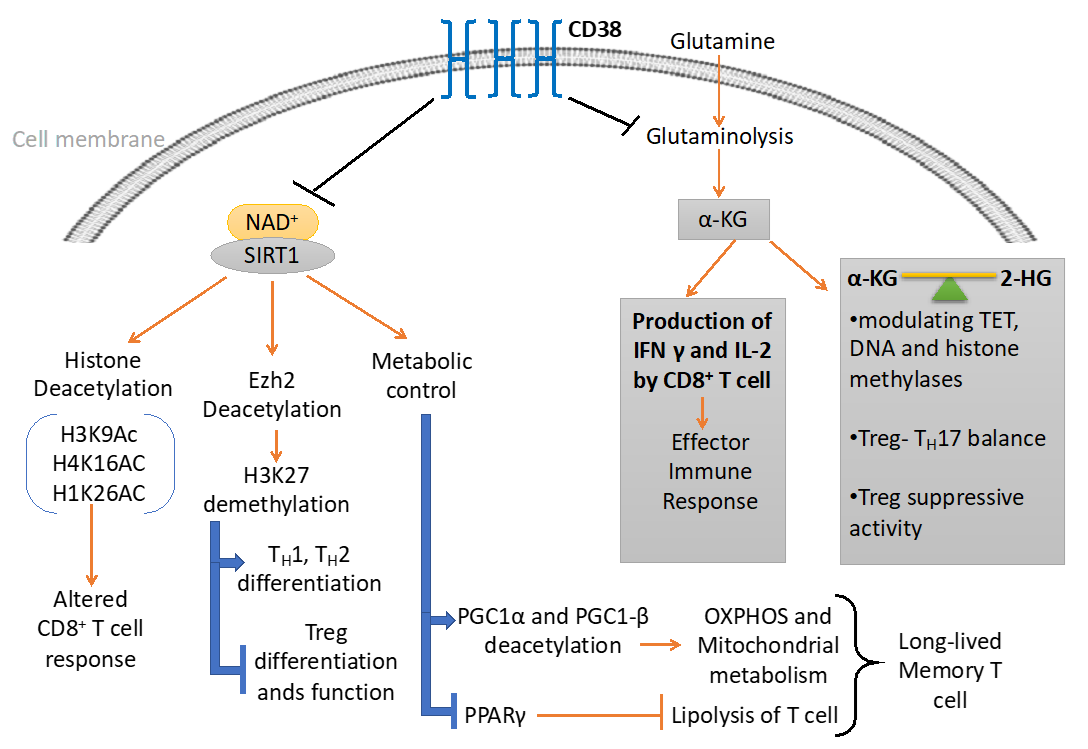
5. CD38-NAD+ Axis in T Cell Immuno-Metabolism
6. CD38-NAD+ Axis and T Cell Epigenetic Modifications
6.1. Metabolites Mediated Epigenetic Regulation
6.2. Sirt1 Dependent Epigenetic Regulation
7. Conclusions
Author Contributions
Funding
Conflicts of Interest
Abbreviations
| NAD | Nicotinamide Adenine Dinucleotide |
| NADP | Nicotinamide Adenine Dinucleotide Phosphate |
| NAADP | Nicotinic Acid Adenine Dinucleotide Phosphate |
| ADPR | Adenosine Di-phosphate Ribose |
| cADPR | cyclic Adenosine Di-phosphate Ribose |
| TCR | T Cell Receptor |
| PD1 | Programmed Death 1 |
| CTLA4 | Cytotoxic T-Lymphocyte-Associated protein 4 |
| Lag3 | Lymphocyte Activation Gene-3 |
| Tim3 | T-cell Immunoglobulin domain and Mucin domain 3 |
| ATP | Adenosine Tri-Phosphate |
| ADP | Adenosine Di-Phosphate |
| AMP | Adenosine Mono-Phosphate |
| MM | Multiple Myeloma |
| DC | Dendritic Cell |
| MDSC | Myeloid Derived Suppressor Cells |
| Treg | Regulatory T cells |
| CRC | Colo-Rectal Cancer |
| SOCE | Store-Operated Calcium Entry |
| CRAC | Calcium Release-Activated Channel |
| IP3 | Inositol Tri-Phosphate |
| IP3R | Inositol Tri-Phosphate Receptor |
| ER | Endoplasmic Reticulum |
| RyR | Ryanodine Receptor |
| SERCA | Sarcoendoplasmic Reticulum Ca2+ ATPase |
| M. avium | Mycobacterium avium |
| IL | Interleukin |
| IFN-γ | Interferon γ |
| GM-CSF | Granulocyte Macrophage Colony Stimulating Factor |
| NFAT | Nuclear factor of activated T-cells |
| NFATc1 | Nuclear factor of activated T-cells cytoplasmic 1 |
| NFATc2 | Nuclear factor of activated T-cells cytoplasmic 2 |
| ChIP | Chromatin Immunoprecipitation |
| MAPK | Mitogen Activated Protein Kinase |
| PTK | Protein Tyrosine Kinase |
| ZAP70 | Zeta-chain-associated protein kinase 70 |
| PLCg1 | Phospho-Lipase C Gamma 1 |
| PARP | Poly-ADP Ribose Polymerase |
| ART | ADP Ribosyl Transferase |
| SIRT | Sirtuin |
| LEF-1 | Lymphoid enhancer-binding factor 1 |
| NICD | NAD+-induced cell death |
| P2RX7 | P2X purinoceptor 7 |
| R35 | Arginine 35 |
| STAT | Signal Transducer and Activator of Transcription |
| TNFα | Tumor Necrosis Factor α |
| NF-kB | Nuclear Factor kappa-light-chain-enhancer of Activated B cells |
| FoxP3 | Forkhead box Protein3 |
| CNS2 | Conserved Non-coding DNA Sequence 2 |
| HDAC | Histone Deacetylase |
| EAE | Experimental Autoimmune Encephalomyelitis |
| Bclaf1 | BCL2 Associated Transcription Factor 1 |
| SLE | Systemic Lupus Erythematosus |
| RORγ | RAR-related Orphan Receptor Gamma |
| OXPHOS | Oxidative Phosphorylation |
| PGC1 | Peroxisome Proliferators γ Co-activator 1 |
| PPARγ | Peroxisome Proliferator-activated Receptor Gamma |
| α-KG | α Keto Gluterate |
| 2-HG | 2-Hydroxy Gluterate |
| IDH1/2 | Isocitrate Dehydrogenase1/2 |
| EZH2 | Enhancer of Zeste Homolog 2 |
| RA | Rheumatoid Arthritis |
References
- Schumacher, T.N.; Gerlach, C.; van Heijst, J.W. Mapping the life histories of T cells. Nat. Rev. Immunol. 2010, 10, 621–631. [Google Scholar] [CrossRef] [PubMed]
- Xing, Y.; Hogquist, K.A. T-cell tolerance: Central and peripheral. Cold Spring Harb. Perspect. Biol. 2012, 4. [Google Scholar] [CrossRef] [PubMed]
- Chen, L.; Flies, D.B. Molecular mechanisms of T cell co-stimulation and co-inhibition. Nat. Rev. Immunol. 2013, 13, 227–242. [Google Scholar] [CrossRef] [PubMed]
- Smith-Garvin, J.E.; Koretzky, G.A.; Jordan, M.S. T cell activation. Annu. Rev. Immunol. 2009, 27, 591–619. [Google Scholar] [CrossRef]
- Fulop, T.; Le Page, A.; Garneau, H.; Azimi, N.; Baehl, S.; Dupuis, G.; Pawelec, G.; Larbi, A. Aging, immunosenescence and membrane rafts: The lipid connection. Longev. Healthspan 2012, 1, 6. [Google Scholar] [CrossRef]
- Curtsinger, J.M.; Mescher, M.F. Inflammatory cytokines as a third signal for T cell activation. Curr. Opin. Immunol. 2010, 22, 333–340. [Google Scholar] [CrossRef]
- Schwartz, R.H. T cell anergy. Annu. Rev. Immunol. 2003, 21, 305–334. [Google Scholar] [CrossRef]
- Powell, J.D. The induction and maintenance of T cell anergy. Clin. Immunol. 2006, 120, 239–246. [Google Scholar] [CrossRef]
- Attanasio, J.; Wherry, E.J. Costimulatory and Coinhibitory Receptor Pathways in Infectious Disease. Immunity 2016, 44, 1052–1068. [Google Scholar] [CrossRef]
- Zhang, Q.; Vignali, D.A. Co-stimulatory and Co-inhibitory Pathways in Autoimmunity. Immunity 2016, 44, 1034–1051. [Google Scholar] [CrossRef]
- Bono, M.R.; Fernandez, D.; Flores-Santibanez, F.; Rosemblatt, M.; Sauma, D. CD73 and CD39 ectonucleotidases in T cell differentiation: Beyond immunosuppression. FEBS Lett. 2015, 589, 3454–3460. [Google Scholar] [CrossRef] [PubMed]
- Deaglio, S.; Robson, S.C. Ectonucleotidases as regulators of purinergic signaling in thrombosis, inflammation, and immunity. Adv. Pharm. 2011, 61, 301–332. [Google Scholar] [CrossRef]
- Chatterjee, S.; Daenthanasanmak, A.; Chakraborty, P.; Wyatt, M.W.; Dhar, P.; Selvam, S.P.; Fu, J.; Zhang, J.; Nguyen, H.; Kang, I.; et al. CD38-NAD(+)Axis Regulates Immunotherapeutic Anti-Tumor T Cell Response. Cell Metab. 2018, 27, 85–100.e8. [Google Scholar] [CrossRef] [PubMed]
- Hogan, K.A.; Chini, C.C.S.; Chini, E.N. The Multi-faceted Ecto-enzyme CD38: Roles in Immunomodulation, Cancer, Aging, and Metabolic Diseases. Front. Immunol. 2019, 10, 1187. [Google Scholar] [CrossRef] [PubMed]
- Aksoy, P.; White, T.A.; Thompson, M.; Chini, E.N. Regulation of intracellular levels of NAD: A novel role for CD38. Biochem. Biophys. Res. Commun. 2006, 345, 1386–1392. [Google Scholar] [CrossRef]
- Chini, E.N. CD38 as a regulator of cellular NAD: A novel potential pharmacological target for metabolic conditions. Curr. Pharm. Des. 2009, 15, 57–63. [Google Scholar] [CrossRef]
- Aarhus, R.; Graeff, R.M.; Dickey, D.M.; Walseth, T.F.; Lee, H.C. ADP-ribosyl cyclase and CD38 catalyze the synthesis of a calcium-mobilizing metabolite from NADP. J. Biol. Chem. 1995, 270, 30327–30333. [Google Scholar] [CrossRef]
- Schmid, F.; Bruhn, S.; Weber, K.; Mittrucker, H.W.; Guse, A.H. CD38: A NAADP degrading enzyme. FEBS Lett. 2011, 585, 3544–3548. [Google Scholar] [CrossRef]
- Lin, W.K.; Bolton, E.L.; Cortopassi, W.A.; Wang, Y.; O’Brien, F.; Maciejewska, M.; Jacobson, M.P.; Garnham, C.; Ruas, M.; Parrington, J.; et al. Synthesis of the Ca(2+)-mobilizing messengers NAADP and cADPR by intracellular CD38 enzyme in the mouse heart: Role in beta-adrenoceptor signaling. J. Biol. Chem. 2017, 292, 13243–13257. [Google Scholar] [CrossRef]
- Kwong, A.K.; Chen, Z.; Zhang, H.; Leung, F.P.; Lam, C.M.; Ting, K.Y.; Zhang, L.; Hao, Q.; Zhang, L.H.; Lee, H.C. Catalysis-based inhibitors of the calcium signaling function of CD38. Biochemistry 2012, 51, 555–564. [Google Scholar] [CrossRef]
- States, D.J.; Walseth, T.F.; Lee, H.C. Similarities in amino acid sequences of Aplysia ADP-ribosyl cyclase and human lymphocyte antigen CD38. Trends Biochem. Sci. 1992, 17, 495. [Google Scholar] [CrossRef]
- Munoz, P.; Mittelbrunn, M.; de la Fuente, H.; Perez-Martinez, M.; Garcia-Perez, A.; Ariza-Veguillas, A.; Malavasi, F.; Zubiaur, M.; Sanchez-Madrid, F.; Sancho, J. Antigen-induced clustering of surface CD38 and recruitment of intracellular CD38 to the immunologic synapse. Blood 2008, 111, 3653–3664. [Google Scholar] [CrossRef] [PubMed]
- Reinherz, E.L.; Kung, P.C.; Goldstein, G.; Levey, R.H.; Schlossman, S.F. Discrete stages of human intrathymic differentiation: Analysis of normal thymocytes and leukemic lymphoblasts of T-cell lineage. Proc. Natl. Acad. Sci. USA 1980, 77, 1588–1592. [Google Scholar] [CrossRef] [PubMed]
- Terhorst, C.; van Agthoven, A.; LeClair, K.; Snow, P.; Reinherz, E.; Schlossman, S. Biochemical studies of the human thymocyte cell-surface antigens T6, T9 and T10. Cell 1981, 23, 771–780. [Google Scholar] [CrossRef]
- Berthelier, V.; Tixier, J.M.; Muller-Steffner, H.; Schuber, F.; Deterre, P. Human CD38 is an authentic NAD(P)+ glycohydrolase. Biochem. J. 1998, 330, 1383–1390. [Google Scholar] [CrossRef]
- De Flora, A.; Guida, L.; Franco, L.; Zocchi, E. The CD38/cyclic ADP-ribose system: A topological paradox. Int. J. Biochem. Cell Biol. 1997, 29, 1149–1166. [Google Scholar] [CrossRef]
- Van de Donk, N.; Richardson, P.G.; Malavasi, F. CD38 antibodies in multiple myeloma: Back to the future. Blood 2018, 131, 13–29. [Google Scholar] [CrossRef]
- Morandi, F.; Horenstein, A.L.; Costa, F.; Giuliani, N.; Pistoia, V.; Malavasi, F. CD38: A Target for Immunotherapeutic Approaches in Multiple Myeloma. Front. Immunol. 2018, 9, 2722. [Google Scholar] [CrossRef]
- Stevenson, F.K.; Bell, A.J.; Cusack, R.; Hamblin, T.J.; Slade, C.J.; Spellerberg, M.B.; Stevenson, G.T. Preliminary studies for an immunotherapeutic approach to the treatment of human myeloma using chimeric anti-CD38 antibody. Blood 1991, 77, 1071–1079. [Google Scholar] [CrossRef]
- Costa, F.; Vescovini, R.; Bolzoni, M.; Marchica, V.; Storti, P.; Toscani, D.; Accardi, F.; Notarfranchi, L.; Dalla Palma, B.; Manferdini, C.; et al. Lenalidomide increases human dendritic cell maturation in multiple myeloma patients targeting monocyte differentiation and modulating mesenchymal stromal cell inhibitory properties. Oncotarget 2017, 8, 53053–53067. [Google Scholar] [CrossRef]
- Deaglio, S.; Mehta, K.; Malavasi, F. Human CD38: A (r)evolutionary story of enzymes and receptors. Leuk. Res. 2001, 25, 1–12. [Google Scholar] [CrossRef]
- Van de Donk, N.W.; Moreau, P.; Plesner, T.; Palumbo, A.; Gay, F.; Laubach, J.P.; Malavasi, F.; Avet-Loiseau, H.; Mateos, M.V.; Sonneveld, P.; et al. Clinical efficacy and management of monoclonal antibodies targeting CD38 and SLAMF7 in multiple myeloma. Blood 2016, 127, 681–695. [Google Scholar] [CrossRef] [PubMed]
- de Weers, M.; Tai, Y.T.; van der Veer, M.S.; Bakker, J.M.; Vink, T.; Jacobs, D.C.; Oomen, L.A.; Peipp, M.; Valerius, T.; Slootstra, J.W.; et al. Daratumumab, a novel therapeutic human CD38 monoclonal antibody, induces killing of multiple myeloma and other hematological tumors. J. Immunol. 2011, 186, 1840–1848. [Google Scholar] [CrossRef] [PubMed]
- Konen, J.M.; Fradette, J.J.; Gibbons, D.L. The Good, the Bad and the Unknown of CD38 in the Metabolic Microenvironment and Immune Cell Functionality of Solid Tumors. Cells 2019, 9. [Google Scholar] [CrossRef] [PubMed]
- Karakasheva, T.A.; Waldron, T.J.; Eruslanov, E.; Kim, S.B.; Lee, J.S.; O’Brien, S.; Hicks, P.D.; Basu, D.; Singhal, S.; Malavasi, F.; et al. CD38-Expressing Myeloid-Derived Suppressor Cells Promote Tumor Growth in a Murine Model of Esophageal Cancer. Cancer. Res. 2015, 75, 4074–4085. [Google Scholar] [CrossRef] [PubMed]
- Lischke, T.; Heesch, K.; Schumacher, V.; Schneider, M.; Haag, F.; Koch-Nolte, F.; Mittrucker, H.W. CD38 controls the innate immune response against Listeria monocytogenes. Infect. Immun. 2013, 81, 4091–4099. [Google Scholar] [CrossRef]
- Viegas, M.S.; do Carmo, A.; Silva, T.; Seco, F.; Serra, V.; Lacerda, M.; Martins, T.C. CD38 plays a role in effective containment of mycobacteria within granulomata and polarization of Th1 immune responses against Mycobacterium avium. Microbes. Infect. 2007, 9, 847–854. [Google Scholar] [CrossRef]
- Matalonga, J.; Glaria, E.; Bresque, M.; Escande, C.; Carbo, J.M.; Kiefer, K.; Vicente, R.; Leon, T.E.; Beceiro, S.; Pascual-Garcia, M.; et al. The Nuclear Receptor LXR Limits Bacterial Infection of Host Macrophages through a Mechanism that Impacts Cellular NAD Metabolism. Cell Rep. 2017, 18, 1241–1255. [Google Scholar] [CrossRef]
- Amici, S.A.; Young, N.A.; Narvaez-Miranda, J.; Jablonski, K.A.; Arcos, J.; Rosas, L.; Papenfuss, T.L.; Torrelles, J.B.; Jarjour, W.N.; Guerau-de-Arellano, M. CD38 Is Robustly Induced in Human Macrophages and Monocytes in Inflammatory Conditions. Front. Immunol. 2018, 9, 1593. [Google Scholar] [CrossRef]
- Fedele, G.; Frasca, L.; Palazzo, R.; Ferrero, E.; Malavasi, F.; Ausiello, C.M. CD38 is expressed on human mature monocyte-derived dendritic cells and is functionally involved in CD83 expression and IL-12 induction. Eur. J. Immunol. 2004, 34, 1342–1350. [Google Scholar] [CrossRef]
- Lande, R.; Urbani, F.; Di Carlo, B.; Sconocchia, G.; Deaglio, S.; Funaro, A.; Malavasi, F.; Ausiello, C.M. CD38 ligation plays a direct role in the induction of IL-1beta, IL-6, and IL-10 secretion in resting human monocytes. Cell Immunol. 2002, 220, 30–38. [Google Scholar] [CrossRef]
- Feng, X.; Zhang, L.; Acharya, C.; An, G.; Wen, K.; Qiu, L.; Munshi, N.C.; Tai, Y.T.; Anderson, K.C. Targeting CD38 Suppresses Induction and Function of T Regulatory Cells to Mitigate Immunosuppression in Multiple Myeloma. Clin. Cancer Res. 2017, 23, 4290–4300. [Google Scholar] [CrossRef] [PubMed]
- Karakasheva, T.A.; Dominguez, G.A.; Hashimoto, A.; Lin, E.W.; Chiu, C.; Sasser, K.; Lee, J.W.; Beatty, G.L.; Gabrilovich, D.I.; Rustgi, A.K. CD38+ M-MDSC expansion characterizes a subset of advanced colorectal cancer patients. JCI Insight 2018, 3. [Google Scholar] [CrossRef] [PubMed]
- Tenca, C.; Merlo, A.; Zarcone, D.; Saverino, D.; Bruno, S.; De Santanna, A.; Ramarli, D.; Fabbi, M.; Pesce, C.; Deaglio, S.; et al. Death of T cell precursors in the human thymus: A role for CD38. Int. Immunol. 2003, 15, 1105–1116. [Google Scholar] [CrossRef]
- Malavasi, F.; Funaro, A.; Alessio, M.; DeMonte, L.B.; Ausiello, C.M.; Dianzani, U.; Lanza, F.; Magrini, E.; Momo, M.; Roggero, S. CD38: A multi-lineage cell activation molecule with a split personality. Int. J. Clin. Lab. Res. 1992, 22, 73–80. [Google Scholar] [CrossRef]
- Deterre, P.; Berthelier, V.; Bauvois, B.; Dalloul, A.; Schuber, F.; Lund, F. CD38 in T- and B-cell functions. Chem. Immunol. 2000, 75, 146–168. [Google Scholar] [CrossRef]
- Funaro, A.; Spagnoli, G.C.; Ausiello, C.M.; Alessio, M.; Roggero, S.; Delia, D.; Zaccolo, M.; Malavasi, F. Involvement of the multilineage CD38 molecule in a unique pathway of cell activation and proliferation. J. Immunol. 1990, 145, 2390–2396. [Google Scholar]
- Ausiello, C.M.; la Sala, A.; Ramoni, C.; Urbani, F.; Funaro, A.; Malavasi, F. Secretion of IFN-gamma, IL-6, granulocyte-macrophage colony-stimulating factor and IL-10 cytokines after activation of human purified T lymphocytes upon CD38 ligation. Cell Immunol. 1996, 173, 192–197. [Google Scholar] [CrossRef]
- Ausiello, C.M.; Urbani, F.; la Sala, A.; Funaro, A.; Malavasi, F. CD38 ligation induces discrete cytokine mRNA expression in human cultured lymphocytes. Eur. J. Immunol. 1995, 25, 1477–1480. [Google Scholar] [CrossRef]
- Thevarajan, I.; Nguyen, T.H.O.; Koutsakos, M.; Druce, J.; Caly, L.; van de Sandt, C.E.; Jia, X.; Nicholson, S.; Catton, M.; Cowie, B.; et al. Breadth of concomitant immune responses prior to patient recovery: A case report of non-severe COVID-19. Nat. Med. 2020, 26, 453–455. [Google Scholar] [CrossRef]
- Chen, L.; Diao, L.; Yang, Y.; Yi, X.; Rodriguez, B.L.; Li, Y.; Villalobos, P.A.; Cascone, T.; Liu, X.; Tan, L.; et al. CD38-Mediated Immunosuppression as a Mechanism of Tumor Cell Escape from PD-1/PD-L1 Blockade. Cancer. Discov. 2018, 8, 1156–1175. [Google Scholar] [CrossRef] [PubMed]
- Philip, M.; Fairchild, L.; Sun, L.; Horste, E.L.; Camara, S.; Shakiba, M.; Scott, A.C.; Viale, A.; Lauer, P.; Merghoub, T.; et al. Chromatin states define tumour-specific T cell dysfunction and reprogramming. Nature 2017, 545, 452–456. [Google Scholar] [CrossRef] [PubMed]
- Malavasi, F.; Funaro, A.; Roggero, S.; Horenstein, A.; Calosso, L.; Mehta, K. Human CD38: A glycoprotein in search of a function. Immunol. Today 1994, 15, 95–97. [Google Scholar] [CrossRef]
- Jackson, D.G.; Bell, J.I. Isolation of a cDNA encoding the human CD38 (T10) molecule, a cell surface glycoprotein with an unusual discontinuous pattern of expression during lymphocyte differentiation. J. Immunol. 1990, 144, 2811–2815. [Google Scholar]
- Zhao, Y.J.; Lam, C.M.; Lee, H.C. The membrane-bound enzyme CD38 exists in two opposing orientations. Sci. Signal. 2012, 5, ra67. [Google Scholar] [CrossRef]
- Feske, S. Calcium signalling in lymphocyte activation and disease. Nat. Rev. Immunol. 2007, 7, 690–702. [Google Scholar] [CrossRef]
- Iino, M.; Endo, M. Calcium-dependent immediate feedback control of inositol 1,4,5-triphosphate-induced Ca2+ release. Nature 1992, 360, 76–78. [Google Scholar] [CrossRef]
- Bezprozvanny, I.; Watras, J.; Ehrlich, B.E. Bell-shaped calcium-response curves of Ins(1,4,5)P3- and calcium-gated channels from endoplasmic reticulum of cerebellum. Nature 1991, 351, 751–754. [Google Scholar] [CrossRef]
- Harnick, D.J.; Jayaraman, T.; Ma, Y.; Mulieri, P.; Go, L.O.; Marks, A.R. The human type 1 inositol 1,4,5-trisphosphate receptor from T lymphocytes. Structure, localization, and tyrosine phosphorylation. J. Biol. Chem. 1995, 270, 2833–2840. [Google Scholar] [CrossRef]
- Guse, A.H.; Roth, E.; Emmrich, F. Intracellular Ca2+ pools in Jurkat T-lymphocytes. Biochem. J. 1993, 291, 447–451. [Google Scholar] [CrossRef]
- Donnadieu, E.; Cefai, D.; Tan, Y.P.; Paresys, G.; Bismuth, G.; Trautmann, A. Imaging early steps of human T cell activation by antigen-presenting cells. J. Immunol. 1992, 148, 2643–2653. [Google Scholar] [PubMed]
- Guse, A.H. Ca2+ signaling in T-lymphocytes. Crit. Rev. Immunol. 1998, 18, 419–448. [Google Scholar] [CrossRef] [PubMed]
- Guse, A.H.; da Silva, C.P.; Berg, I.; Skapenko, A.L.; Weber, K.; Heyer, P.; Hohenegger, M.; Ashamu, G.A.; Schulze-Koops, H.; Potter, B.V.; et al. Regulation of calcium signalling in T lymphocytes by the second messenger cyclic ADP-ribose. Nature 1999, 398, 70–73. [Google Scholar] [CrossRef] [PubMed]
- Langhorst, M.F.; Schwarzmann, N.; Guse, A.H. Ca2+ release via ryanodine receptors and Ca2+ entry: Major mechanisms in NAADP-mediated Ca2+ signaling in T-lymphocytes. Cell Signal. 2004, 16, 1283–1289. [Google Scholar] [CrossRef] [PubMed]
- Park, S.A.; Hong, B.Z.; Ha, K.C.; Kim, U.H.; Han, M.K.; Kwak, Y.G. Protein tyrosine phosphatase 1B is a mediator of cyclic ADP ribose-induced Ca(2+) signaling in ventricular myocytes. Exp. Mol. Med. 2017, 49, e341. [Google Scholar] [CrossRef]
- Gwack, Y.; Feske, S.; Srikanth, S.; Hogan, P.G.; Rao, A. Signalling to transcription: Store-operated Ca2+ entry and NFAT activation in lymphocytes. Cell Calcium 2007, 42, 145–156. [Google Scholar] [CrossRef]
- Klein-Hessling, S.; Muhammad, K.; Klein, M.; Pusch, T.; Rudolf, R.; Floter, J.; Qureischi, M.; Beilhack, A.; Vaeth, M.; Kummerow, C.; et al. NFATc1 controls the cytotoxicity of CD8(+) T cells. Nat. Commun. 2017, 8, 511. [Google Scholar] [CrossRef]
- Zubiaur, M.; Guirado, M.; Terhorst, C.; Malavasi, F.; Sancho, J. The CD3-gamma delta epsilon transducing module mediates CD38-induced protein-tyrosine kinase and mitogen-activated protein kinase activation in Jurkat T cells. J. Biol. Chem. 1999, 274, 20633–20642. [Google Scholar] [CrossRef]
- Hurtado-Bages, S.; Knobloch, G.; Ladurner, A.G.; Buschbeck, M. The taming of PARP1 and its impact on NAD(+) metabolism. Mol. Metab. 2020, 100950. [Google Scholar] [CrossRef]
- Yaku, K.; Okabe, K.; Nakagawa, T. NAD metabolism: Implications in aging and longevity. Age. Res. Rev. 2018, 47, 1–17. [Google Scholar] [CrossRef]
- Kupis, W.; Palyga, J.; Tomal, E.; Niewiadomska, E. The role of sirtuins in cellular homeostasis. J. Physiol. Biochem. 2016, 72, 371–380. [Google Scholar] [CrossRef]
- Morales, J.; Li, L.; Fattah, F.J.; Dong, Y.; Bey, E.A.; Patel, M.; Gao, J.; Boothman, D.A. Review of poly (ADP-ribose) polymerase (PARP) mechanisms of action and rationale for targeting in cancer and other diseases. Crit. Rev. Eukaryot. Gene. Expr. 2014, 24, 15–28. [Google Scholar] [CrossRef]
- Bheda, P.; Jing, H.; Wolberger, C.; Lin, H. The Substrate Specificity of Sirtuins. Annu. Rev. Biochem. 2016, 85, 405–429. [Google Scholar] [CrossRef]
- Okamoto, S.; Azhipa, O.; Yu, Y.; Russo, E.; Dennert, G. Expression of ADP-ribosyltransferase on normal T lymphocytes and effects of nicotinamide adenine dinucleotide on their function. J. Immunol. 1998, 160, 4190–4198. [Google Scholar] [PubMed]
- Liu, Z.X.; Yu, Y.; Dennert, G. A cell surface ADP-ribosyltransferase modulates T cell receptor association and signaling. J. Biol. Chem. 1999, 274, 17399–17401. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, E.; Yu, Y.; Dennert, G. Cell surface ADP-ribosyltransferase regulates lymphocyte function-associated molecule-1 (LFA-1) function in T cells. J. Immunol. 1996, 157, 3341–3349. [Google Scholar]
- Wang, J.; Nemoto, E.; Kots, A.Y.; Kaslow, H.R.; Dennert, G. Regulation of cytotoxic T cells by ecto-nicotinamide adenine dinucleotide (NAD) correlates with cell surface GPI-anchored/arginine ADP-ribosyltransferase. J. Immunol. 1994, 153, 4048–4058. [Google Scholar]
- Rissiek, B.; Haag, F.; Boyer, O.; Koch-Nolte, F.; Adriouch, S. ADP-ribosylation of P2X7: A matter of life and death for regulatory T cells and natural killer T cells. Curr. Top. Microbiol. Immunol. 2015, 384, 107–126. [Google Scholar] [CrossRef]
- Seman, M.; Adriouch, S.; Scheuplein, F.; Krebs, C.; Freese, D.; Glowacki, G.; Deterre, P.; Haag, F.; Koch-Nolte, F. NAD-induced T cell death: ADP-ribosylation of cell surface proteins by ART2 activates the cytolytic P2X7 purinoceptor. Immunity 2003, 19, 571–582. [Google Scholar] [CrossRef]
- Gu, B.; Bendall, L.J.; Wiley, J.S. Adenosine triphosphate-induced shedding of CD23 and L-selectin (CD62L) from lymphocytes is mediated by the same receptor but different metalloproteases. Blood 1998, 92, 946–951. [Google Scholar] [CrossRef]
- Taylor, S.R.; Gonzalez-Begne, M.; Dewhurst, S.; Chimini, G.; Higgins, C.F.; Melvin, J.E.; Elliott, J.I. Sequential shrinkage and swelling underlie P2X7-stimulated lymphocyte phosphatidylserine exposure and death. J. Immunol. 2008, 180, 300–308. [Google Scholar] [CrossRef] [PubMed]
- Ohlrogge, W.; Haag, F.; Lohler, J.; Seman, M.; Littman, D.R.; Killeen, N.; Koch-Nolte, F. Generation and characterization of ecto-ADP-ribosyltransferase ART2.1/ART2.2-deficient mice. Mol. Cell Biol. 2002, 22, 7535–7542. [Google Scholar] [CrossRef] [PubMed]
- Adriouch, S.; Ohlrogge, W.; Haag, F.; Koch-Nolte, F.; Seman, M. Rapid induction of naive T cell apoptosis by ecto-nicotinamide adenine dinucleotide: Requirement for mono(ADP-ribosyl)transferase 2 and a downstream effector. J. Immunol. 2001, 167, 196–203. [Google Scholar] [CrossRef] [PubMed]
- Nemoto, E.; Stohlman, S.; Dennert, G. Release of a glycosylphosphatidylinositol-anchored ADP-ribosyltransferase from cytotoxic T cells upon activation. J. Immunol. 1996, 156, 85–92. [Google Scholar]
- Krebs, C.; Adriouch, S.; Braasch, F.; Koestner, W.; Leiter, E.H.; Seman, M.; Lund, F.E.; Oppenheimer, N.; Haag, F.; Koch-Nolte, F. CD38 controls ADP-ribosyltransferase-2-catalyzed ADP-ribosylation of T cell surface proteins. J. Immunol. 2005, 174, 3298–3305. [Google Scholar] [CrossRef]
- Teege, S.; Hann, A.; Miksiewicz, M.; MacMillan, C.; Rissiek, B.; Buck, F.; Menzel, S.; Nissen, M.; Bannas, P.; Haag, F.; et al. Tuning IL-2 signaling by ADP-ribosylation of CD25. Sci. Rep. 2015, 5, 8959. [Google Scholar] [CrossRef]
- Ray Chaudhuri, A.; Nussenzweig, A. The multifaceted roles of PARP1 in DNA repair and chromatin remodelling. Nat. Rev. Mol. Cell Biol. 2017, 18, 610–621. [Google Scholar] [CrossRef]
- Rosado, M.M.; Bennici, E.; Novelli, F.; Pioli, C. Beyond DNA repair, the immunological role of PARP-1 and its siblings. Immunology 2013, 139, 428–437. [Google Scholar] [CrossRef]
- Valdor, R.; Schreiber, V.; Saenz, L.; Martinez, T.; Munoz-Suano, A.; Dominguez-Villar, M.; Ramirez, P.; Parrilla, P.; Aguado, E.; Garcia-Cozar, F.; et al. Regulation of NFAT by poly(ADP-ribose) polymerase activity in T cells. Mol. Immunol. 2008, 45, 1863–1871. [Google Scholar] [CrossRef]
- Olabisi, O.A.; Soto-Nieves, N.; Nieves, E.; Yang, T.T.; Yang, X.; Yu, R.Y.; Suk, H.Y.; Macian, F.; Chow, C.W. Regulation of transcription factor NFAT by ADP-ribosylation. Mol. Cell Biol. 2008, 28, 2860–2871. [Google Scholar] [CrossRef]
- Bai, P.; Virag, L. Role of poly(ADP-ribose) polymerases in the regulation of inflammatory processes. FEBS Lett. 2012, 586, 3771–3777. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Hottiger, M.O. The functional role of poly(ADP-ribose)polymerase 1 as novel coactivator of NF-kappaB in inflammatory disorders. Cell Mol. Life. Sci. 2002, 59, 1534–1553. [Google Scholar] [CrossRef] [PubMed]
- Hassa, P.O.; Covic, M.; Hasan, S.; Imhof, R.; Hottiger, M.O. The enzymatic and DNA binding activity of PARP-1 are not required for NF-kappa B coactivator function. J. Biol. Chem. 2001, 276, 45588–45597. [Google Scholar] [CrossRef] [PubMed]
- Zerfaoui, M.; Errami, Y.; Naura, A.S.; Suzuki, Y.; Kim, H.; Ju, J.; Liu, T.; Hans, C.P.; Kim, J.G.; Abd Elmageed, Z.Y.; et al. Poly(ADP-ribose) polymerase-1 is a determining factor in Crm1-mediated nuclear export and retention of p65 NF-kappa B upon TLR4 stimulation. J. Immunol. 2010, 185, 1894–1902. [Google Scholar] [CrossRef]
- Saenz, L.; Lozano, J.J.; Valdor, R.; Baroja-Mazo, A.; Ramirez, P.; Parrilla, P.; Aparicio, P.; Sumoy, L.; Yelamos, J. Transcriptional regulation by poly(ADP-ribose) polymerase-1 during T cell activation. BMC Genom. 2008, 9, 171. [Google Scholar] [CrossRef]
- Datta, R.; Naura, A.S.; Zerfaoui, M.; Errami, Y.; Oumouna, M.; Kim, H.; Ju, J.; Ronchi, V.P.; Haas, A.L.; Boulares, A.H. PARP-1 deficiency blocks IL-5 expression through calpain-dependent degradation of STAT-6 in a murine asthma model. Allergy 2011, 66, 853–861. [Google Scholar] [CrossRef]
- Zhang, P.; Maruyama, T.; Konkel, J.E.; Abbatiello, B.; Zamarron, B.; Wang, Z.Q.; Chen, W. PARP-1 controls immunosuppressive function of regulatory T cells by destabilizing Foxp3. PLoS ONE 2013, 8, e71590. [Google Scholar] [CrossRef]
- Nasta, F.; Laudisi, F.; Sambucci, M.; Rosado, M.M.; Pioli, C. Increased Foxp3+ regulatory T cells in poly(ADP-Ribose) polymerase-1 deficiency. J. Immunol. 2010, 184, 3470–3477. [Google Scholar] [CrossRef]
- Thoren, F.B.; Romero, A.I.; Hellstrand, K. Oxygen radicals induce poly(ADP-ribose) polymerase-dependent cell death in cytotoxic lymphocytes. J. Immunol. 2006, 176, 7301–7307. [Google Scholar] [CrossRef]
- Carafa, V.; Nebbioso, A.; Altucci, L. Sirtuins and disease: The road ahead. Front. Pharm. 2012, 3, 4. [Google Scholar] [CrossRef]
- Zhang, J.; Lee, S.M.; Shannon, S.; Gao, B.; Chen, W.; Chen, A.; Divekar, R.; McBurney, M.W.; Braley-Mullen, H.; Zaghouani, H.; et al. The type III histone deacetylase Sirt1 is essential for maintenance of T cell tolerance in mice. J. Clin. Investig. 2009, 119, 3048–3058. [Google Scholar] [CrossRef] [PubMed]
- Nimmagadda, V.K.; Bever, C.T.; Vattikunta, N.R.; Talat, S.; Ahmad, V.; Nagalla, N.K.; Trisler, D.; Judge, S.I.; Royal, W., 3rd; Chandrasekaran, K.; et al. Overexpression of SIRT1 protein in neurons protects against experimental autoimmune encephalomyelitis through activation of multiple SIRT1 targets. J. Immunol. 2013, 190, 4595–4607. [Google Scholar] [CrossRef] [PubMed]
- Gao, B.; Kong, Q.; Kemp, K.; Zhao, Y.S.; Fang, D. Analysis of sirtuin 1 expression reveals a molecular explanation of IL-2-mediated reversal of T-cell tolerance. Proc. Natl. Acad. Sci. USA 2012, 109, 899–904. [Google Scholar] [CrossRef]
- Gao, Z.; Ye, J. Inhibition of transcriptional activity of c-JUN by SIRT1. Biochem. Biophys. Res. Commun. 2008, 376, 793–796. [Google Scholar] [CrossRef]
- Attema, J.L.; Reeves, R.; Murray, V.; Levichkin, I.; Temple, M.D.; Tremethick, D.J.; Shannon, M.F. The human IL-2 gene promoter can assemble a positioned nucleosome that becomes remodeled upon T cell activation. J. Immunol. 2002, 169, 2466–2476. [Google Scholar] [CrossRef]
- Kong, S.; Kim, S.J.; Sandal, B.; Lee, S.M.; Gao, B.; Zhang, D.D.; Fang, D. The type III histone deacetylase Sirt1 protein suppresses p300-mediated histone H3 lysine 56 acetylation at Bclaf1 promoter to inhibit T cell activation. J. Biol. Chem. 2011, 286, 16967–16975. [Google Scholar] [CrossRef]
- Sequeira, J.; Boily, G.; Bazinet, S.; Saliba, S.; He, X.; Jardine, K.; Kennedy, C.; Staines, W.; Rousseaux, C.; Mueller, R.; et al. sirt1-null mice develop an autoimmune-like condition. Exp. Cell Res. 2008, 314, 3069–3074. [Google Scholar] [CrossRef]
- Legutko, A.; Marichal, T.; Fievez, L.; Bedoret, D.; Mayer, A.; de Vries, H.; Klotz, L.; Drion, P.V.; Heirman, C.; Cataldo, D.; et al. Sirtuin 1 promotes Th2 responses and airway allergy by repressing peroxisome proliferator-activated receptor-gamma activity in dendritic cells. J. Immunol. 2011, 187, 4517–4529. [Google Scholar] [CrossRef]
- Lim, H.W.; Kang, S.G.; Ryu, J.K.; Schilling, B.; Fei, M.; Lee, I.S.; Kehasse, A.; Shirakawa, K.; Yokoyama, M.; Schnolzer, M.; et al. SIRT1 deacetylates RORgammat and enhances Th17 cell generation. J. Exp. Med. 2015, 212, 607–617. [Google Scholar] [CrossRef]
- Limagne, E.; Thibaudin, M.; Euvrard, R.; Berger, H.; Chalons, P.; Vegan, F.; Humblin, E.; Boidot, R.; Rebe, C.; Derangere, V.; et al. Sirtuin-1 Activation Controls Tumor Growth by Impeding Th17 Differentiation via STAT3 Deacetylation. Cell Rep. 2017, 19, 746–759. [Google Scholar] [CrossRef]
- Wang, J.; Zhao, C.; Kong, P.; Sun, H.; Sun, Z.; Bian, G.; Sun, Y.; Guo, L. Treatment with NAD(+) inhibited experimental autoimmune encephalomyelitis by activating AMPK/SIRT1 signaling pathway and modulating Th1/Th17 immune responses in mice. Int. Immunopharmacol. 2016, 39, 287–294. [Google Scholar] [CrossRef] [PubMed]
- Van Loosdregt, J.; Vercoulen, Y.; Guichelaar, T.; Gent, Y.Y.; Beekman, J.M.; van Beekum, O.; Brenkman, A.B.; Hijnen, D.J.; Mutis, T.; Kalkhoven, E.; et al. Regulation of Treg functionality by acetylation-mediated Foxp3 protein stabilization. Blood 2010, 115, 965–974. [Google Scholar] [CrossRef] [PubMed]
- Beier, U.H.; Wang, L.; Bhatti, T.R.; Liu, Y.; Han, R.; Ge, G.; Hancock, W.W. Sirtuin-1 targeting promotes Foxp3+ T-regulatory cell function and prolongs allograft survival. Mol. Cell Biol. 2011, 31, 1022–1029. [Google Scholar] [CrossRef] [PubMed]
- Deng, G.; Song, X.; Fujimoto, S.; Piccirillo, C.A.; Nagai, Y.; Greene, M.I. Foxp3 Post-translational Modifications and Treg Suppressive Activity. Front. Immunol. 2019, 10, 2486. [Google Scholar] [CrossRef]
- Van Loosdregt, J.; Brunen, D.; Fleskens, V.; Pals, C.E.; Lam, E.W.; Coffer, P.J. Rapid temporal control of Foxp3 protein degradation by sirtuin-1. PLoS ONE 2011, 6, e19047. [Google Scholar] [CrossRef]
- Kwon, H.S.; Lim, H.W.; Wu, J.; Schnolzer, M.; Verdin, E.; Ott, M. Three novel acetylation sites in the Foxp3 transcription factor regulate the suppressive activity of regulatory T cells. J. Immunol. 2012, 188, 2712–2721. [Google Scholar] [CrossRef]
- Ruan, Q.; Kameswaran, V.; Tone, Y.; Li, L.; Liou, H.C.; Greene, M.I.; Tone, M.; Chen, Y.H. Development of Foxp3(+) regulatory t cells is driven by the c-Rel enhanceosome. Immunity 2009, 31, 932–940. [Google Scholar] [CrossRef]
- Elibol, B.; Kilic, U. High Levels of SIRT1 Expression as a Protective Mechanism Against Disease-Related Conditions. Front. Endocrinol. 2018, 9, 614. [Google Scholar] [CrossRef]
- Chang, H.C.; Guarente, L. SIRT1 and other sirtuins in metabolism. Trends Endocrinol. Metab. 2014, 25, 138–145. [Google Scholar] [CrossRef]
- Geltink, R.I.K.; Kyle, R.L.; Pearce, E.L. Unraveling the Complex Interplay Between T Cell Metabolism and Function. Annu. Rev. Immunol. 2018, 36, 461–488. [Google Scholar] [CrossRef]
- Camacho-Pereira, J.; Tarrago, M.G.; Chini, C.C.S.; Nin, V.; Escande, C.; Warner, G.M.; Puranik, A.S.; Schoon, R.A.; Reid, J.M.; Galina, A.; et al. CD38 Dictates Age-Related NAD Decline and Mitochondrial Dysfunction through an SIRT3-Dependent Mechanism. Cell Metab. 2016, 23, 1127–1139. [Google Scholar] [CrossRef] [PubMed]
- Hu, Y.; Wang, H.; Wang, Q.; Deng, H. Overexpression of CD38 decreases cellular NAD levels and alters the expression of proteins involved in energy metabolism and antioxidant defense. J. Proteome Res. 2014, 13, 786–795. [Google Scholar] [CrossRef] [PubMed]
- Mak, T.W.; Grusdat, M.; Duncan, G.S.; Dostert, C.; Nonnenmacher, Y.; Cox, M.; Binsfeld, C.; Hao, Z.; Brustle, A.; Itsumi, M.; et al. Glutathione Primes T Cell Metabolism for Inflammation. Immunity 2017, 46, 675–689. [Google Scholar] [CrossRef] [PubMed]
- Vellinga, T.T.; Borovski, T.; de Boer, V.C.; Fatrai, S.; van Schelven, S.; Trumpi, K.; Verheem, A.; Snoeren, N.; Emmink, B.L.; Koster, J.; et al. SIRT1/PGC1alpha-Dependent Increase in Oxidative Phosphorylation Supports Chemotherapy Resistance of Colon Cancer. Clin. Cancer Res. 2015, 21, 2870–2879. [Google Scholar] [CrossRef]
- Abraham, A.; Qiu, S.; Chacko, B.K.; Li, H.; Paterson, A.; He, J.; Agarwal, P.; Shah, M.; Welner, R.; Darley-Usmar, V.M.; et al. SIRT1 regulates metabolism and leukemogenic potential in CML stem cells. J. Clin. Investig. 2019, 129, 2685–2701. [Google Scholar] [CrossRef]
- Brenmoehl, J.; Hoeflich, A. Dual control of mitochondrial biogenesis by sirtuin 1 and sirtuin 3. Mitochondrion 2013, 13, 755–761. [Google Scholar] [CrossRef] [PubMed]
- Tang, B.L. Sirt1 and the Mitochondria. Mol. Cells 2016, 39, 87–95. [Google Scholar] [CrossRef]
- Van der Windt, G.J.; Everts, B.; Chang, C.H.; Curtis, J.D.; Freitas, T.C.; Amiel, E.; Pearce, E.J.; Pearce, E.L. Mitochondrial respiratory capacity is a critical regulator of CD8+ T cell memory development. Immunity 2012, 36, 68–78. [Google Scholar] [CrossRef] [PubMed]
- Buck, M.D.; O’Sullivan, D.; Klein Geltink, R.I.; Curtis, J.D.; Chang, C.H.; Sanin, D.E.; Qiu, J.; Kretz, O.; Braas, D.; van der Windt, G.J.; et al. Mitochondrial Dynamics Controls T Cell Fate through Metabolic Programming. Cell 2016, 166, 63–76. [Google Scholar] [CrossRef]
- Kelly, T.J.; Lerin, C.; Haas, W.; Gygi, S.P.; Puigserver, P. GCN5-mediated transcriptional control of the metabolic coactivator PGC-1beta through lysine acetylation. J. Biol. Chem. 2009, 284, 19945–19952. [Google Scholar] [CrossRef]
- Picard, F.; Kurtev, M.; Chung, N.; Topark-Ngarm, A.; Senawong, T.; Machado De Oliveira, R.; Leid, M.; McBurney, M.W.; Guarente, L. Sirt1 promotes fat mobilization in white adipocytes by repressing PPAR-gamma. Nature 2004, 429, 771–776. [Google Scholar] [CrossRef]
- Chakraborty, P.; Vaena, S.G.; Thyagarajan, K.; Chatterjee, S.; Al-Khami, A.; Selvam, S.P.; Nguyen, H.; Kang, I.; Wyatt, M.W.; Baliga, U.; et al. Pro-Survival Lipid Sphingosine-1-Phosphate Metabolically Programs T Cells to Limit Anti-tumor Activity. Cell Rep. 2019, 28, 1879–1893. [Google Scholar] [CrossRef] [PubMed]
- O’Sullivan, D.; van der Windt, G.J.W.; Huang, S.C.; Curtis, J.D.; Chang, C.H.; Buck, M.D.; Qiu, J.; Smith, A.M.; Lam, W.Y.; DiPlato, L.M.; et al. Memory CD8(+) T Cells Use Cell-Intrinsic Lipolysis to Support the Metabolic Programming Necessary for Development. Immunity 2018, 49, 375–376. [Google Scholar] [CrossRef] [PubMed]
- Chen, Y.; Zander, R.; Khatun, A.; Schauder, D.M.; Cui, W. Transcriptional and Epigenetic Regulation of Effector and Memory CD8 T Cell Differentiation. Front. Immunol. 2018, 9, 2826. [Google Scholar] [CrossRef] [PubMed]
- Lu, C.; Thompson, C.B. Metabolic regulation of epigenetics. Cell Metab. 2012, 16, 9–17. [Google Scholar] [CrossRef] [PubMed]
- Kietzmann, T.; Petry, A.; Shvetsova, A.; Gerhold, J.M.; Gorlach, A. The epigenetic landscape related to reactive oxygen species formation in the cardiovascular system. Br. J. Pharm. 2017, 174, 1533–1554. [Google Scholar] [CrossRef]
- Suzuki, J.; Yamada, T.; Inoue, K.; Nabe, S.; Kuwahara, M.; Takemori, N.; Takemori, A.; Matsuda, S.; Kanoh, M.; Imai, Y.; et al. The tumor suppressor menin prevents effector CD8 T-cell dysfunction by targeting mTORC1-dependent metabolic activation. Nat. Commun. 2018, 9, 3296. [Google Scholar] [CrossRef]
- Xu, T.; Stewart, K.M.; Wang, X.; Liu, K.; Xie, M.; Ryu, J.K.; Li, K.; Ma, T.; Wang, H.; Ni, L.; et al. Metabolic control of TH17 and induced Treg cell balance by an epigenetic mechanism. Nature 2017, 548, 228–233. [Google Scholar] [CrossRef]
- Klysz, D.; Tai, X.; Robert, P.A.; Craveiro, M.; Cretenet, G.; Oburoglu, L.; Mongellaz, C.; Floess, S.; Fritz, V.; Matias, M.I.; et al. Glutamine-dependent alpha-ketoglutarate production regulates the balance between T helper 1 cell and regulatory T cell generation. Sci. Signal. 2015, 8, ra97. [Google Scholar] [CrossRef] [PubMed]
- Jing, H.; Lin, H. Sirtuins in epigenetic regulation. Chem. Rev. 2015, 115, 2350–2375. [Google Scholar] [CrossRef]
- Rifai, K.; Judes, G.; Idrissou, M.; Daures, M.; Bignon, Y.J.; Penault-Llorca, F.; Bernard-Gallon, D. SIRT1-dependent epigenetic regulation of H3 and H4 histone acetylation in human breast cancer. Oncotarget 2018, 9, 30661–30678. [Google Scholar] [CrossRef] [PubMed]
- Araki, Y.; Fann, M.; Wersto, R.; Weng, N.P. Histone acetylation facilitates rapid and robust memory CD8 T cell response through differential expression of effector molecules (eomesodermin and its targets: Perforin and granzyme B). J. Immunol. 2008, 180, 8102–8108. [Google Scholar] [CrossRef] [PubMed]
- Katsuyama, E.; Suarez-Fueyo, A.; Bradley, S.J.; Mizui, M.; Marin, A.V.; Mulki, L.; Krishfield, S.; Malavasi, F.; Yoon, J.; Sui, S.J.H.; et al. The CD38/NAD/SIRTUIN1/EZH2 Axis Mitigates Cytotoxic CD8 T Cell Function and Identifies Patients with SLE Prone to Infections. Cell Rep. 2020, 30, 112–123.e4. [Google Scholar] [CrossRef] [PubMed]
- Tumes, D.J.; Onodera, A.; Suzuki, A.; Shinoda, K.; Endo, Y.; Iwamura, C.; Hosokawa, H.; Koseki, H.; Tokoyoda, K.; Suzuki, Y.; et al. The polycomb protein Ezh2 regulates differentiation and plasticity of CD4(+) T helper type 1 and type 2 cells. Immunity 2013, 39, 819–832. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.P.; Jiang, K.; Hirahara, K.; Vahedi, G.; Afzali, B.; Sciume, G.; Bonelli, M.; Sun, H.W.; Jankovic, D.; Kanno, Y.; et al. EZH2 is crucial for both differentiation of regulatory T cells and T effector cell expansion. Sci. Rep. 2015, 5, 10643. [Google Scholar] [CrossRef] [PubMed]
- Xiao, X.Y.; Li, Y.T.; Jiang, X.; Ji, X.; Lu, X.; Yang, B.; Wu, L.J.; Wang, X.H.; Guo, J.B.; Zhao, L.D.; et al. EZH2 deficiency attenuates Treg differentiation in rheumatoid arthritis. J. Autoimmun. 2020, 108, 102404. [Google Scholar] [CrossRef]
- Van de Donk, N.; Usmani, S.Z. CD38 Antibodies in Multiple Myeloma: Mechanisms of Action and Modes of Resistance. Front. Immunol. 2018, 9, 2134. [Google Scholar] [CrossRef]


© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Kar, A.; Mehrotra, S.; Chatterjee, S. CD38: T Cell Immuno-Metabolic Modulator. Cells 2020, 9, 1716. https://doi.org/10.3390/cells9071716
Kar A, Mehrotra S, Chatterjee S. CD38: T Cell Immuno-Metabolic Modulator. Cells. 2020; 9(7):1716. https://doi.org/10.3390/cells9071716
Chicago/Turabian StyleKar, Anwesha, Shikhar Mehrotra, and Shilpak Chatterjee. 2020. "CD38: T Cell Immuno-Metabolic Modulator" Cells 9, no. 7: 1716. https://doi.org/10.3390/cells9071716
APA StyleKar, A., Mehrotra, S., & Chatterjee, S. (2020). CD38: T Cell Immuno-Metabolic Modulator. Cells, 9(7), 1716. https://doi.org/10.3390/cells9071716