The Yin and Yang of ACE/ACE2 Pathways: The Rationale for the Use of Renin-Angiotensin System Inhibitors in COVID-19 Patients


{kind=link}
{kind=link}
Abstract
1. Introduction
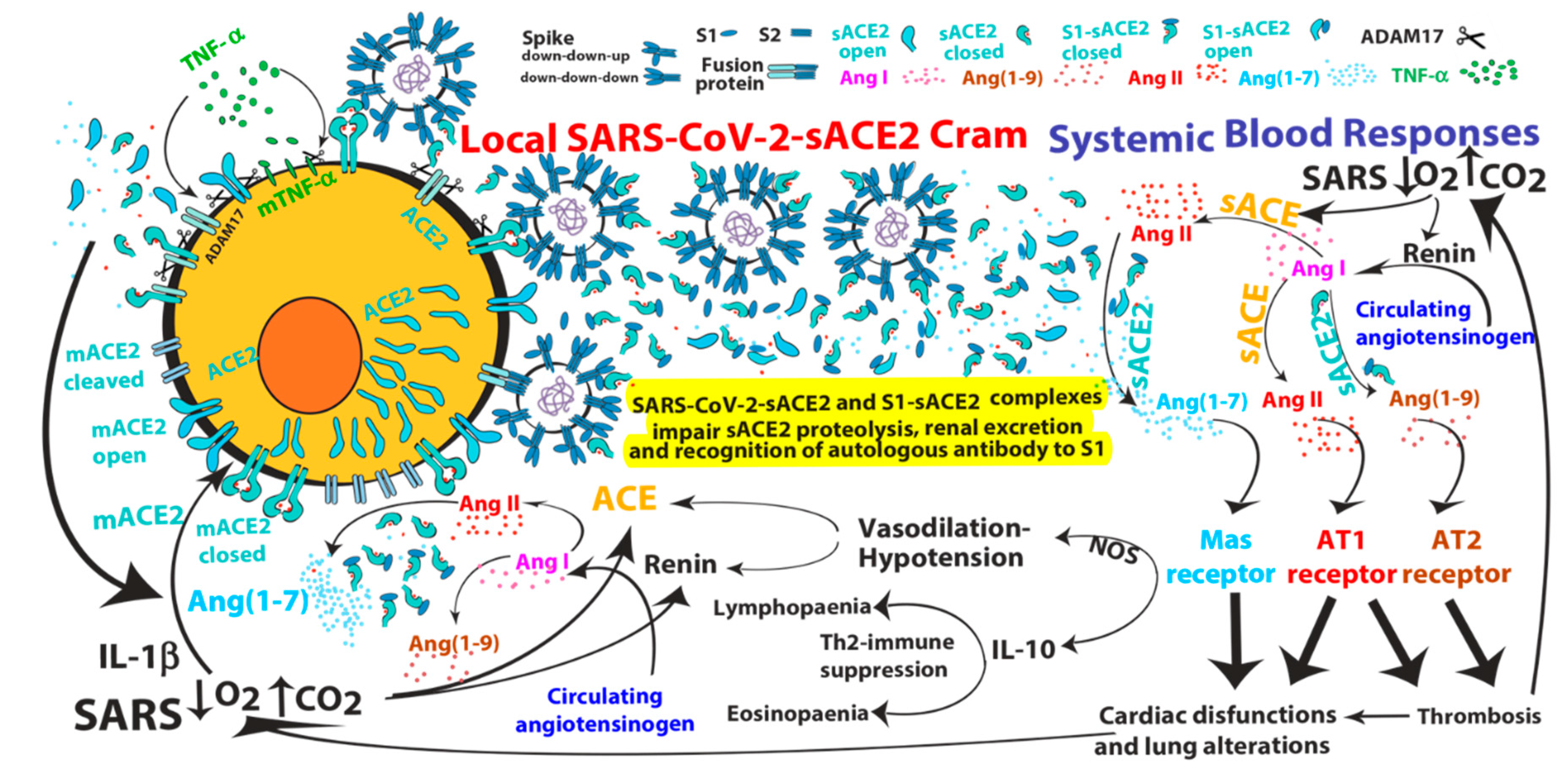
2. ACE2-mediated SARS-CoV and SARS-CoV-2 Infections
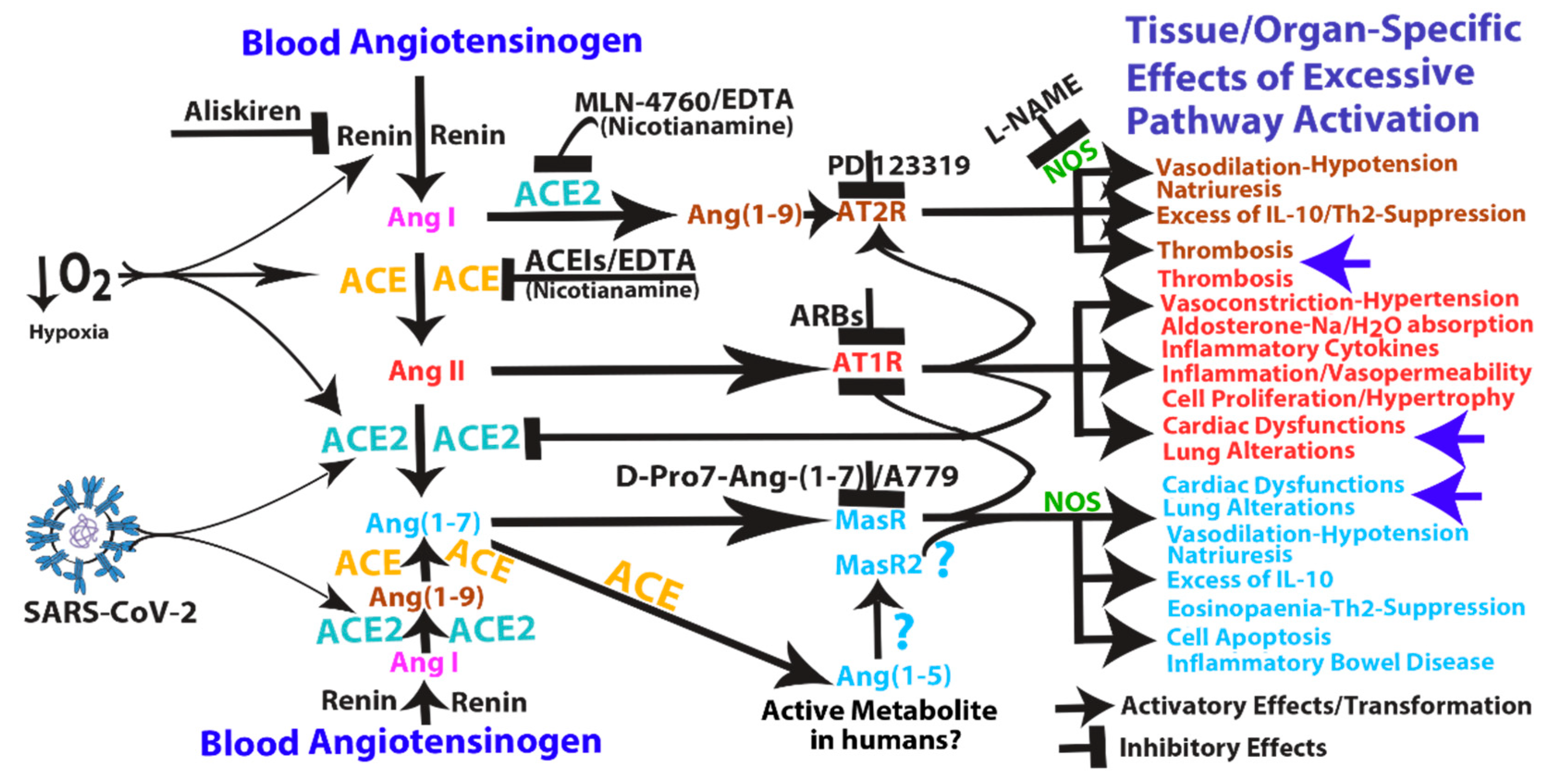
3. Pathological Effects of ACE2 Pathway Upregulation
4. COVID-19 Can Induce RAS-mediated Positive Feedback Loops at Different Levels
- (1)
- SARS-CoV can induce IL-1β and TNF-α systemic secretion that can mediate viral-independent surface membrane ACE2 upregulation and shedding. Of note, ACE2 shedding, on one hand, protects from viral infection but, on the other hand, increases circulating/systemic active sACE2, leading to its downstream pathway activation.
- (2)
- Hypoxia in combination or not with hypercapnia can upregulate the activity of both arms of the renin–angiotensin system by inducing renin, ACE and ACE2 synthesis, which can increase expression of Ang I, Ang II, Ang (1–7), Ang (1–9), Ang (1–5) and the inactive metabolite bradykinin (1–7), but also membrane bound ACE2, finally giving more chances to SARS-CoV-2 entry.
- (3)
- ACE2 can induce vasodilatative hypotensive effects by Ang II catabolism. Hypotension can induce again renin and ACE upregulation finally providing further Ang II, as a ACE2 substrate for further Ang (1–7) production.
- (4)
- Ang (1–7) antiproliferative and apoptotic effects, possibly in part through IL-10, may mediate eosinopaenia and lymphopaenia that, on one hand, reduce inflammatory responses but, on the other hand, impair immune system ability to counter virus infection, finally predisposing the organism to further infections. Ang (1–7) immunosuppressive activity, mediated or not by IL-10, may also support the reduced ability to generate an effective immunization to SARS-CoV-2 infection.
- (5)
- Ang (1–7)/MasR pathway can sustain ACE2 synthesis even in the presence of elevated concentrations of Ang II (such as in hypoxia), by inhibiting Ang II/AT1R-mediated down-modulation of ACE2 activity (see Figure 1).
- (6)
- Ang (1–7)/MasR pathway can produce cardiac dysfunction and lung alteration leading to systemic hypoxia, which, in turn, upregulates the activity of both arms of the RAS.
- (7)
- Although the ACE2 catalytic efficiency is 400-fold lower with Ang I than with Ang II [79], high concentration of circulating ACE2 may be able to produce significant increase of Ang (1–9) that, by binding AT2 receptors, can produce arteriolar microvascular thrombosis and local hypoxic conditions finally inducing local upregulation both arms of the RAS.
5. Mechanism of Action and Potential Risk of Using RAS Pathway Inhibitors Targeting ACE2
5.1. ACE2 (and ACE) Hyperactivity: Is It a Matter of (Free) Zinc?
5.2. Safety and Efficacy Concerns of MLN4760 and Dx600 ACE2 Inhibitors.
5.3. Safety and Efficacy Concerns of Chelating Agents
6. Correlation of Pre-existing Circulating ACE2 Activity and Increased Potential to Develop Severe Forms of COVID-19
7. Monitoring ACE/ACE2 Activity in COVID-19 in Order to Determine a Rationale Use of the Specific RAS Inhibitors
- (1)
- Organ-specific local microenvironment might not reflect the systemic one;
- (2)
- S1-sACE2 and SARS-CoV-2-sACE2 complexes, formed by viral-induced ACE2 shedding or by subsequent binding of sACE2 with viral particles or S1 fragments in the circulation, might not be detectable by some anti-ACE2 antibodies in ELISA. Therefore, since the complexes might prevent/mask sACE2 antibody recognition but not the enzymatic activity, sACE2 detection (and its real concentration) by ELISA in blood samples of COVID-19 patients might not be reliable;
- (3)
- (4)
- circulating concentrations ACE/ACE2 substrates/products likely depend on the:
- (a)
- level of ACE/ACE2 pathway activity and availability of substrates;
- (b)
- level of expression of the respective receptors (AT1R, AT2R and MasR) that bind and remove ligand products away from circulation.
8. Hypothesizing Pharmacological Treatments for COVID-19
8.1. Inhibition of ACE2: MLN-4760
- (1)
- It has been shown to bind/inhibit ACE2 enzymatic activity even at low/acidic pH (pH 6.5, [115]) typical of hypercapnia (as it might occur in lungs of COVID-19 patients) when human ACE2 activity is maximal [79]; nevertheless, it retains its inhibitory effects on soluble ACE2 bound to spike proteins [24], indicating that it is able to bind and inhibit ACE2 activity regardless ACE2 binding to SARS-CoV-2 particles or to S1 fragments.
- (2)
- No significant adverse effects were described upon its chronic administration neither alone nor in combination with ACE2 activators (while inhibiting their activating effects) nor after inducing functional impairment of ACE2 activity in rodent experiments in vivo [52,117,118,122,183,184] nor in a clinical Phase I trial in humans (http://oreholdings.com/wp-content/uploads/2013/06/09.10.09-425.pdf);
- (3)
- Its administration by different route is well described in rodents and humans. In particular:
- (a)
- Chronic administration (about 4 weeks) of C-16/DLM-4760 in combination with ACE2 activating treatments was performed by daily intraperitoneal injection at a dose of 25mg/kg in distilled water (as a solution of 42 mg/mL) or 0.9% sterile saline (as a solution of 84 mg/mL using a 0.5-mL insulin syringe) freshly prepared [52,183,184].
- (b)
- Alternatively, chronic administration (about 8 days) of GL1001/DLM-4760 disodium salt in combination with an ACE2 activating treatment was performed by subcutaneous injection (5mL/kg) containing up to a dose of 300 mg/kg, twice a day, formulated in a vehicle solution [15% 2-hydroxypropyl-β-cyclodextrin (HPBDC)/85% H2O] [122]. Subchronic doses of GL1001 indicate no adverse effects up to 1,000 mg/kg (see [122]).
- (c)
- In humans ORE1001/GL1001/MLN-4760 was already proposed and tested in clinical trials. Its pharmaceutical indication was for digestive tract inflammations (Inflammatory bowel disease, gastritis and colitis) that are correlated with overexpression of ACE2. In a Phase I clinical testing up 14 days dosing, ORE1001 was well tolerated. Subjects received drug (dosing up to 2100 mg) with no side adverse effects reported. In particular, 47 subjects received single-dose from 2.1 to 2100 mg and 24 subjects received 14 day multiple doses from 50 mg to 1800 mg. All doses were well tolerated, with no significant side effects including blood pressure. Pharmacokinetics of orally administered capsules was consistent with once-daily dosing. (http://oreholdings.com/wp-content/uploads/2013/06/09.10.09-425.pdf). 300 mg (active drug) oral capsules were used in a Phase Ib/IIa clinical trial that was, however, abandoned. (https://clinicaltrials.gov/ct2/show/NCT01039597).
- (d)
- Finally, MLN-4760 was also administered (2.5 mg/kg per day) by nasal inhalation for 2–3 days in lung-infected mice by Pseudomonas bacteria [185]. Interestingly, the report underscores the role played by local concentration of molecules (ACE2) in modulating lung inflammation and disease. For these reasons, in diseases involving respiratory tract, like SARS, inhalation treatment is preferable, even for the lower concentration (and hopefully lower toxicity) of MLN-4760 needed for this route of treatment administration.
8.2. Inhibition of Renin Activity: Aliskiren
8.3. Chelating Agents: CaNa2EDTA
8.4. Safety and Efficacy Concerns
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Zhang, J.-J.; Dong, X.; Cao, Y.-Y.; Yuan, Y.-D.; Yang, Y.-B.; Yan, Y.-Q.; Akdis, C.A.; Gao, Y.-D. Clinical characteristics of 140 patients infected with SARS-CoV-2 in Wuhan, China. Allergy 2020. [Google Scholar] [CrossRef] [PubMed]
- Qin, C.; Zhou, L.; Hu, Z.; Zhang, S.; Yang, S.; Tao, Y.; Xie, C.; Ma, K.; Shang, K.; Wang, W.; et al. Dysregulation of Immune Response in Patients With Coronavirus 2019 (COVID-19) in Wuhan, China. Clin. Infect. Dis. 2020, ciaa248. [Google Scholar] [CrossRef] [PubMed]
- Zhou, F.; Yu, T.; Du, R.; Fan, G.; Liu, Y.; Liu, Z.; Xiang, J.; Wang, Y.; Song, B.; Gu, X.; et al. Clinical course and risk factors for mortality of adult inpatients with COVID-19 in Wuhan, China: A retrospective cohort study. Lancet 2020, 395, 1054–1062. [Google Scholar] [CrossRef]
- Wang, D.; Hu, B.; Hu, C.; Zhu, F.; Liu, X.; Zhang, J.; Wang, B.; Xiang, H.; Cheng, Z.; Xiong, Y.; et al. Clinical Characteristics of 138 Hospitalized Patients With 2019 Novel Coronavirus–Infected Pneumonia in Wuhan, China. JAMA 2020, 323, 1061. [Google Scholar] [CrossRef]
- Guan, W.-J.; Ni, Z.-Y.; Hu, Y.; Liang, W.-H.; Ou, C.-Q.; He, J.-X.; Liu, L.; Shan, H.; Lei, C.-L.; Hui, D.S.; et al. Clinical Characteristics of Coronavirus Disease 2019 in China. New Engl. J. Med. 2020, 382, 1708–1720. [Google Scholar] [CrossRef]
- Li, X.; Xu, S.; Yu, M.; Wang, K.; Tao, Y.; Zhou, Y.; Shi, J.; Zhou, M.; Wu, B.; Yang, Z.; et al. Risk factors for severity and mortality in adult COVID-19 inpatients in Wuhan. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wu, Z.; McGoogan, J.M. Characteristics of and Important Lessons from the Coronavirus Disease 2019 (COVID-19) Outbreak in China: Summary of a Report of 72314 Cases from the Chinese Center for Disease Control and Prevention. JAMA 2020, 323, 1239–1242. [Google Scholar] [CrossRef] [PubMed]
- Huang, C.; Wang, Y.; Li, X.; Ren, L.; Zhao, J.; Hu, Y.; Zhang, L.; Fan, G.; Xu, J.; Gu, X.; et al. Clinical features of patients infected with 2019 novel coronavirus in Wuhan, China. Lancet 2020, 395, 497–506. [Google Scholar] [CrossRef]
- Wan, S.; Yi, Q.; Fan, S.; Lv, J.; Zhang, X.; Guo, L.; Lang, C.; Xiao, Q.; Xiao, K.; Yi, Z.; et al. Relationships among lymphocyte subsets, cytokines, and the pulmonary inflammation index in coronavirus (COVID-19) infected patients. Br. J. Haematol. 2020, 189, 428–437. [Google Scholar] [CrossRef] [PubMed]
- Huang, K.; Yang, T.; Xu, J.; Yang, L.; Zhao, J.; Zhang, X.; Bai, C.; Kang, J.; Ran, P.; Shen, H.; et al. Prevalence, risk factors, and management of asthma in China: A national cross-sectional study. Lancet 2019, 394, 407–418. [Google Scholar] [CrossRef]
- Wang, Z.-W.; Chen, Z.; Zhang, L.; Wang, X.; Hao, G.; Zhang, Z.; Shao, L.; Tian, Y.; Dong, Y.; Zheng, C.; et al. Status of Hypertension in China. Circulation 2018, 137, 2344–2356. [Google Scholar] [CrossRef] [PubMed]
- Zhang, L.; Wang, F.; Wang, L.; Wang, W.; Liu, B.; Liu, J.; Chen, M.; He, Q.; Liao, Y.; Yu, X.; et al. Prevalence of chronic kidney disease in China: A cross-sectional survey. Lancet 2012, 379, 815–822. [Google Scholar] [CrossRef]
- Hoffmann, M.; Kleine-Weber, H.; Schroeder, S.; Mü, M.A.; Drosten, C.; Pö, S.; Krü, N.; Herrler, T.; Erichsen, S.; Schiergens, T.S.; et al. SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor Article SARS-CoV-2 Cell Entry Depends on ACE2 and TMPRSS2 and Is Blocked by a Clinically Proven Protease Inhibitor. Cell 2020, 181, 271–280. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Rao, S.; Gao, H.; Guo, F.; Guan, B.; Huan, Y.; Yang, P.; Zhang, Y.; Deng, W.; et al. A crucial role of angiotensin converting enzyme 2 (ACE2) in SARS coronavirus–induced lung injury. Nat. Med. 2005, 11, 875–879. [Google Scholar] [CrossRef]
- Imai, Y.; Kuba, K.; Rao, S.; Huan, Y.; Guo, F.; Guan, B.; Yang, P.; Sarao, R.; Wada, T.; Leong-Poi, H.; et al. Angiotensin-converting enzyme 2 protects from severe acute lung failure. Nature 2005, 436, 112–116. [Google Scholar] [CrossRef] [PubMed]
- Jia, H.P.; Look, D.C.; Tan, P.; Shi, L.; Hickey, M.; Gakhar, L.; Chappell, M.C.; Wohlford-Lenane, C.; McCray, P.B. Ectodomain shedding of angiotensin converting enzyme 2 in human airway epithelia. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L84–L96. [Google Scholar] [CrossRef]
- Haga, S.; Yamamoto, N.; Nakai-Murakami, C.; Osawa, Y.; Tokunaga, K.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. Modulation of TNF-α-converting enzyme by the spike protein of SARS-CoV and ACE2 induces TNF-α production and facilitates viral entry. Proc. Natl. Acad. Sci. USA 2008, 105, 7809–7814. [Google Scholar] [CrossRef]
- Glowacka, I.; Bertram, S.; Herzog, P.; Pfefferle, S.; Steffen, I.; Muench, M.O.; Simmons, G.; Hofmann, H.; Kuri, T.; Weber, F.; et al. Differential Downregulation of ACE2 by the Spike Proteins of Severe Acute Respiratory Syndrome Coronavirus and Human Coronavirus NL63. J. Virol. 2009, 84, 1198–1205. [Google Scholar] [CrossRef]
- Moore, M.J.; Dorfman, T.; Li, W.; Wong, S.K.; Li, Y.; Kuhn, J.H.; Coderre, J.; Vasilieva, N.; Han, Z.C.; Greenough, T.C.; et al. Retroviruses Pseudotyped with the Severe Acute Respiratory Syndrome Coronavirus Spike Protein Efficiently Infect Cells Expressing Angiotensin-Converting Enzyme 2. J. Virol. 2004, 78, 10628–10635. [Google Scholar] [CrossRef]
- Haga, S.; Nagata, N.; Okamura, T.; Yamamoto, N.; Sata, T.; Yamamoto, N.; Sasazuki, T.; Ishizaka, Y. TACE antagonists blocking ACE2 shedding caused by the spike protein of SARS-CoV are candidate antiviral compounds. Antivir. Res. 2010, 85, 551–555. [Google Scholar] [CrossRef]
- Han, D.P.; Penn-Nicholson, A.; Cho, M.W. Identification of critical determinants on ACE2 for SARS-CoV entry and development of a potent entry inhibitor. Virology 2006, 350, 15–25. [Google Scholar] [CrossRef] [PubMed]
- Epelman, S.; Shrestha, K.; Troughton, R.W.; Francis, G.S.; Sen, S.; Klein, A.L.; Tang, W.W. Soluble Angiotensin-Converting Enzyme 2 in Human Heart Failure: Relation With Myocardial Function and Clinical Outcomes. J. Card. Fail. 2009, 15, 565–571. [Google Scholar] [CrossRef] [PubMed]
- Yu, C.-M.; Wong, R.S.; Wu, E.B.; Kong, S.-L.; Wong, J.; Yip, G.; Soo, Y.O.Y.; Chiu, M.L.S.; Chan, Y.-S.; Hui, D.S.; et al. Cardiovascular complications of severe acute respiratory syndrome. Postgrad. Med J. 2006, 82, 140–144. [Google Scholar] [CrossRef]
- Li, W.; Zhang, C.; Sui, J.; Kuhn, J.H.; Moore, M.J.; Luo, S.; Wong, S.-K.; Huang, I.-C.; Xu, K.; Vasilieva, N.; et al. Receptor and viral determinants of SARS-coronavirus adaptation to human ACE2. EMBO J. 2005, 24, 1634–1643. [Google Scholar] [CrossRef] [PubMed]
- He, X.; Zhang, L.; Ran, Q.; Xiong, A.; Wang, J.; Wu, D.; Chen, F.; Li, G. Integrative Bioinformatics Analysis Provides Insight into the Molecular Mechanisms of 2019-nCoV. MedRxiv 2020. [Google Scholar]
- Song, W.; Gui, M.; Wang, X.; Xiang, Y. Cryo-EM structure of the SARS coronavirus spike glycoprotein in complex with its host cell receptor ACE2. PLOS Pathog. 2018, 14, e1007236. [Google Scholar] [CrossRef]
- Wrapp, D.; Wang, N.; Corbett, K.; Goldsmith, J.A.; Hsieh, C.-L.; Abiona, O.; Graham, B.S.; McLellan, J.S. Cryo-EM structure of the 2019-nCoV spike in the prefusion conformation. Science 2020, 367, 1260–1263. [Google Scholar] [CrossRef]
- Yan, R.; Zhang, Y.; Li, Y.; Xia, L.; Guo, Y.; Zhou, Q. Structural basis for the recognition of SARS-CoV-2 by full-length human ACE2. Science 2020, 367, 1444–1448. [Google Scholar] [CrossRef]
- Inoue, Y.; Tanaka, N.; Tanaka, Y.; Inoue, S.; Morita, K.; Zhuang, M.; Hattori, T.; Sugamura, K. Clathrin-Dependent Entry of Severe Acute Respiratory Syndrome Coronavirus into Target Cells Expressing ACE2 with the Cytoplasmic Tail Deleted. J. Virol. 2007, 81, 8722–8729. [Google Scholar] [CrossRef]
- Chang, L.; Yan, Y.; Wang, L. Coronavirus Disease 2019: Coronaviruses and Blood Safety. Transfus. Med. Rev. 2020, 34, 75–80. [Google Scholar] [CrossRef]
- Cereda, D.; Tirani, M.; Rovida, F.; Demicheli, V.; Ajelli, M.; Poletti, P.; Trentini, F.; Guzzetta, G.; Marziano, V.; Barone, A.; et al. The Early Phase of the COVID-19 Outbreak in Lombardy, Italy. Available online: https://arxiv.org/ftp/arxiv/papers/2003/2003.09320.pdf (accessed on 20 March 2020).
- Tian, X.; Li, C.; Huang, A.; Xia, S.; Lu, S.; Shi, Z.; Lu, L.; Jiang, S.; Yang, Z.; Wu, Y.; et al. Potent binding of 2019 novel coronavirus spike protein by a SARS coronavirus-specific human monoclonal antibody. Emerg. Microbes Infect. 2020, 9, 382–385. [Google Scholar] [CrossRef] [PubMed]
- Wang, C.; Li, W.; Drabek, D.; Okba, N.M.; Van Haperen, R.; Osterhaus, A.D.M.E.; Van Kuppeveld, F.J.M.; Haagmans, B.L.; Grosveld, F.; Bosch, B.-J. A human monoclonal antibody blocking SARS-CoV-2 infection. Nat. Commun. 2020, 11, 2251–2256. [Google Scholar] [CrossRef] [PubMed]
- Wu, Y.; Wang, F.; Shen, C.; Peng, W.; Li, D.; Zhao, C.; Li, Z.; Li, S.; Bi, Y.; Yang, Y.; et al. A noncompeting pair of human neutralizing antibodies block COVID-19 virus binding to its receptor ACE2. Science 2020, 368, 1274–1278. [Google Scholar] [CrossRef] [PubMed]
- Shen, C.; Wang, Z.; Zhao, F.; Yang, Y.; Li, J.; Yuan, J.; Wang, F.; Li, D.; Yang, M.; Xing, L.; et al. Treatment of 5 Critically Ill Patients With COVID-19 With Convalescent Plasma. JAMA 2020, 323, 1582. [Google Scholar] [CrossRef]
- Duan, K.; Liu, B.; Li, C.; Zhang, H.; Yu, T.; Qu, J.; Zhou, M.; Chen, L.; Meng, S.; Hu, Y.; et al. Effectiveness of convalescent plasma therapy in severe COVID-19 patients. Proc. Natl. Acad. Sci. USA 2020, 117, 9490–9496. [Google Scholar] [CrossRef]
- Zhang, B.; Liu, S.; Tan, T.; Huang, W.; Dong, Y.; Chen, L.; Chen, Q.; Zhang, L.; Zhong, Q.; Zhang, X.; et al. Treatment With Convalescent Plasma for Critically Ill Patients With Severe Acute Respiratory Syndrome Coronavirus 2 Infection. Chest 2020, 158, e9–e13. [Google Scholar] [CrossRef]
- Jia, H. Pulmonary Angiotensin-Converting Enzyme 2 (ACE2) and Inflammatory Lung Disease. Shock 2016, 46, 239–248. [Google Scholar] [CrossRef]
- Xu, P.; Sriramula, S.; Lazartigues, E. ACE2/ANG-(1-7)/Mas pathway in the brain: The axis of good. Am. J. Physiol. 2011, 300, R804–R817. [Google Scholar] [CrossRef]
- Magalhaes, G.S.; Barroso, L.C.; Reis, A.C.; Rodrigues-Machado, M.G.; Gregório, J.; Motta-Santos, D.; Oliveira, A.C.; Perez, D.A.; Barcelos, L.D.S.; Teixeira, M.M.; et al. Angiotensin-(1–7) Promotes Resolution of Eosinophilic Inflammation in an Experimental Model of Asthma. Front. Immunol. 2018, 9, 58. [Google Scholar] [CrossRef]
- Sharma, N.; Anders, H.-J.; Gaikwad, A.B. Fiend and friend in the renin angiotensin system: An insight on acute kidney injury. Biomed. Pharmacother. 2019, 110, 764–774. [Google Scholar] [CrossRef]
- Ocaranza, M.P.; Riquelme, J.A.; García, L.; Jalil, J.E.; Chiong, M.; Santos, R.A.S.; Lavandero, S. Counter-regulatory renin–angiotensin system in cardiovascular disease. Nat. Rev. Cardiol. 2019, 17, 116–129. [Google Scholar] [CrossRef] [PubMed]
- Ocaranza, M.P.; Jalil, J.E. Protective role of the ACE2/Ang-(19) axis in cardiovascular remodeling. Int. J. Hypertens. 2012, 594361. [Google Scholar] [CrossRef]
- Kuba, K.; Imai, Y.; Ohto-Nakanishi, T.; Penninger, J.M. Trilogy of ACE2: A peptidase in the renin–angiotensin system, a SARS receptor, and a partner for amino acid transporters. Pharmacol. Ther. 2010, 128, 119–128. [Google Scholar] [CrossRef] [PubMed]
- Passos-Silva, D.G.; Verano-Braga, T.; Santos, R.A.S. Angiotensin-(1–7): Beyond the cardio-renal actions. Clin. Sci. 2012, 124, 443–456. [Google Scholar] [CrossRef] [PubMed]
- Santos, R.A.S.; Ferreira, A.J.; Verano-Braga, T.; Bader, M. Angiotensin-converting enzyme 2, angiotensin-(1–7) and Mas: New players of the renin–angiotensin system. J. Endocrinol. 2012, 216, R1–R17. [Google Scholar] [CrossRef]
- Esteban, V.; Heringer-Walther, S.; Sterner-Kock, A.; De Bruin, R.; Engel, S.V.D.; Wang, Y.; Mezzano, S.; Egido, J.; Schultheiss, H.-P.; Ruiz-Ortega, M.; et al. Angiotensin-(1–7) and the G Protein-Coupled Receptor Mas Are Key Players in Renal Inflammation. PLoS ONE 2009, 4, e5406. [Google Scholar] [CrossRef]
- Velkoska, E.; Dean, R.G.; Griggs, K.; Burchill, L.; Burrell, L. Angiotensin-(1–7) infusion is associated with increased blood pressure and adverse cardiac remodelling in rats with subtotal nephrectomy. Clin. Sci. 2010, 120, 335–345. [Google Scholar] [CrossRef][Green Version]
- Ortiz-Pérez, J.T.; Riera, M.; Bosch, X.; De Caralt, T.M.; Perea, R.J.; Pascual, J.; Soler, M.J. Role of Circulating Angiotensin Converting Enzyme 2 in Left Ventricular Remodeling following Myocardial Infarction: A Prospective Controlled Study. PLoS ONE 2013, 8, e61695. [Google Scholar] [CrossRef]
- Donoghue, M.; Wakimoto, H.; Maguire, C.T.; Acton, S.; Hales, P.; Stagliano, N.; Fairchild-Huntress, V.; Xu, J.; Lorenz, J.N.; Kadambi, V.; et al. Heart block, ventricular tachycardia, and sudden death in ACE2 transgenic mice with downregulated connexins. J. Mol. Cell. Cardiol. 2003, 35, 1043–1053. [Google Scholar] [CrossRef]
- Masson, R.; A Nicklin, S.; Craig, M.A.; McBride, M.; Gilday, K.; Gregorevic, P.; Allen, J.M.; Chamberlain, J.S.; Smith, G.; Graham, D.; et al. Onset of Experimental Severe Cardiac Fibrosis Is Mediated by Overexpression of Angiotensin-Converting Enzyme 2. Hypertension 2009, 53, 694–700. [Google Scholar] [CrossRef]
- Kim, M.-A.; Yang, D.; Kida, K.; Molotkova, N.; Yeo, S.J.; Varki, N.; Iwata, M.; Dalton, N.D.; Peterson, K.L.; Siems, W.-E.; et al. Effects of ACE2 Inhibition in the Post-Myocardial Infarction Heart. J. Card. Fail. 2010, 16, 777–785. [Google Scholar] [CrossRef] [PubMed]
- Mogielnicki, A.; Kramkowski, K.; Hermanowicz, J.; Leszczynska, A.; Przyborowski, K.; Buczko, W. Angiotensin-(1–9) enhances stasis-induced venous thrombosis in the rat because of the impairment of fibrinolysis. J. Renin-Angiotensin-Aldosterone Syst. 2013, 15, 13–21. [Google Scholar] [CrossRef]
- Senchenkova, E.Y.; Russell, J.; Almeida-Paula, L.D.; Harding, J.W.; Granger, D.N. Angiotensin II-mediated microvascular thrombosis. Hypertension 2010, 56, 1089–1095. [Google Scholar] [CrossRef]
- Paizis, G.; Tikellis, C.; E Cooper, M.; Schembri, J.M.; A Lew, R.; I Smith, A.; Shaw, T.; Warner, F.J.; Zuilli, A.; Burrell, L.M.; et al. Chronic liver injury in rats and humans upregulates the novel enzyme angiotensin converting enzyme 2. Gut 2005, 54, 1790–1796. [Google Scholar] [CrossRef] [PubMed]
- Clarke, N.E.; Belyaev, N.D.; Lambert, D.W.; Turner, A.J. Epigenetic regulation of angiotensin-converting enzyme 2 (ACE2) by SIRT1 under conditions of cell energy stress. Clin. Sci. 2013, 126, 507–516. [Google Scholar] [CrossRef] [PubMed]
- Joshi, S.; Wollenzien, H.; Leclerc, E.; Jarajapu, Y.P. Hypoxic regulation of angiotensin-converting enzyme 2 and Mas receptor in human CD34+ cells. J. Cell. Physiol. 2019, 234, 20420–20431. [Google Scholar] [CrossRef] [PubMed]
- Zhang, R.; Wu, Y.; Zhao, M.; Liu, C.; Zhou, L.; Shen, S.; Liao, S.; Yang, K.; Li, Q.; Wan, H. Role of HIF-1α in the regulation ACE and ACE2 expression in hypoxic human pulmonary artery smooth muscle cells. Am. J. Physiol. Cell. Mol. Physiol. 2009, 297, L631–L640. [Google Scholar] [CrossRef]
- Burrell, L.M.; Risvanis, J.; Kubota, E.; Dean, R.G.; Macdonald, P.S.; Lu, S.; Tikellis, C.; Grant, S.L.; Lew, R.A.; Smith, A.I.; et al. Myocardial infarction increases ACE2 expression in rat and humans. Eur. Hear. J. 2005, 26, 369–375. [Google Scholar] [CrossRef]
- Hampl, V.; Herget, J.; Bíbová, J.; Banasová, A.; Husková, Z.; Vańourková, Z.; Jíchová, S.; Kujal, P.; Vernerová, Z.; Sadowski, J.; et al. Intrapulmonary activation of the angiotensin-converting enzyme type 2/angiotensin 1-7/g-protein-coupled mas receptor axis attenuates pulmonary hypertension in ren-2 transgenic rats exposed to chronic hypoxia. Physiol. Res. 2015, 64, 25–38. [Google Scholar] [CrossRef]
- Krämer, B.K.; Ritthaler, T.; Schweda, F.; Kees, F.; Schricker, K.; Holmer, S.R.; Kurtz, A.; Kr, T.R.B.K. Effects of hypoxia on renin secretion and renal renin gene expression. Kidney Int. 1998, 54, S155–S158. [Google Scholar] [CrossRef]
- Wood, C.E.; Kane, C.; Raff, H. Peripheral chemoreceptor control of fetal renin responses to hypoxia and hypercapnia. Circ. Res. 1990, 67, 722–732. [Google Scholar] [CrossRef] [PubMed]
- Yamaguchi, K.; Suzuki, K.; Naoki, K.; Nishio, K.; Sato, N.; Takeshita, K.; Kudo, H.; Aoki, T.; Suzuki, Y.; Miyata, A.; et al. Response of intra-acinar pulmonary microvessels to hypoxia, hypercapnic acidosis, and isocapnic acidosis. Circ. Res. 1998, 82, 722–728. [Google Scholar] [CrossRef] [PubMed]
- Liao, X.; Wang, L.; Yang, C.; He, J.; Wang, X.; Guo, R.; Lan, A.; Dong, X.; Yang, Z.; Wang, H.; et al. Cyclooxygenase mediates cardioprotection of angiotensin-(1-7) against ischemia/reperfusion-induced injury through the inhibition of oxidative stress. Mol. Med. Rep. 2011, 4, 1145–1150. [Google Scholar] [CrossRef] [PubMed]
- Yu, L.; Yuan, K.; Phuong, H.T.A.; Park, B.M.; Kim, S.H. Angiotensin-(1-5), an active mediator of renin-angiotensin system, stimulates ANP secretion via Mas receptor. Peptides 2016, 86, 33–41. [Google Scholar] [CrossRef] [PubMed]
- Garg, M.; Burrell, L.M.; Velkoska, E.; Griggs, K.; Angus, P.W.; Gibson, P.R.; Lubel, J.S. Upregulation of circulating components of the alternative renin-angiotensin system in inflammatory bowel disease: A pilot study. J. Renin Angiotensin Aldosterone Syst. 2014, 16, 559–569. [Google Scholar] [CrossRef] [PubMed]
- Meng, Y.; Yu, C.H.; Li, W.; Li, T.; Luo, W.; Huang, S.; Wu, P.S.; Cai, S.X.; Li, X. Angiotensin-converting enzyme 2/angiotensin-(1-7)/mas axis protects against lung fibrosis by inhibiting the MAPK/NF-κB pathway. Am. J. Respir. Cell Mol. Biol. 2014, 50, 723–736. [Google Scholar] [CrossRef]
- El-Hashim, A.Z.; Renno, W.M.; Raghupathy, R.; Abduo, H.T.; Akhtar, S.; Benter, I.F. Angiotensin-(1-7) inhibits allergic inflammation, via the MAS1 receptor, through suppression of ERK1/2- and NF-kB-dependent pathways. Br. J. Pharmacol. 2012, 166, 1964–1976. [Google Scholar] [CrossRef]
- Rodrigues-Machado, M.G.; Magalhães, G.S.; A Cardoso, J.; Kangussu, L.M.; Murari, A.; Caliari, M.V.; Oliveira, M.L.; Cara, D.C.; Noviello, M.L.M.; Marques, F.D.; et al. AVE 0991, a non-peptide mimic of angiotensin-(1–7) effects, attenuates pulmonary remodelling in a model of chronic asthma. Br. J. Pharmacol. 2013, 170, 835–846. [Google Scholar] [CrossRef]
- Simões-E-Silva, A.C.; Silveira, K.; Ferreira, A.; Teixeira, M. ACE2, angiotensin-(1-7) and Mas receptor axis in inflammation and fibrosis. Br. J. Pharmacol. 2013, 169, 477–492. [Google Scholar] [CrossRef] [PubMed]
- Dhande, I.; Ali, Q.; Hussain, T. Proximal tubule angiotensin AT2 receptors mediate an anti-inflammatory response via interleukin-10: Role in renoprotection in obese rats. Hypertension 2013, 61, 1218–1226. [Google Scholar] [CrossRef]
- Schülke, S. Induction of Interleukin-10 Producing Dendritic Cells As a Tool to Suppress Allergen-Specific T Helper 2 Responses. Front. Immunol. 2018, 9, 455. [Google Scholar] [CrossRef] [PubMed]
- Van Scott, M.R.; Justice, J.P.; Bradfield, J.F.; Enright, E.; Sigounas, A.; Sur, S. IL-10 reduces Th2 cytokine production and eosinophilia but augments airway reactivity in allergic mice. Am. J. Physiol. Cell. Mol. Physiol. 2000, 278, L667–L674. [Google Scholar] [CrossRef] [PubMed]
- Wakahara, S.; Konoshita, T.; Mizuno, S.; Motomura, M.; Aoyama, C.; Makino, Y.; Kato, N.; Koni, I.; Miyamori, I. Synergistic Expression of Angiotensin-Converting Enzyme (ACE) and ACE2 in Human Renal Tissue and Confounding Effects of Hypertension on the ACE to ACE2 Ratio. Endocrinology 2007, 148, 2453–2457. [Google Scholar] [CrossRef] [PubMed]
- Ferrario, C.M.; Jessup, J.; Chappell, M.C.; Averill, D.B.; Brosnihan, K.B.; Tallant, E.A.; Diz, D.I.; Gallagher, P.E. Effect of Angiotensin-Converting Enzyme Inhibition and Angiotensin II Receptor Blockers on Cardiac Angiotensin-Converting Enzyme 2. Circulation 2005, 111, 2605–2610. [Google Scholar] [CrossRef] [PubMed]
- Gallagher, P.E.; Ferrario, C.M.; Tallant, E.A. Regulation of ACE2 in cardiac myocytes and fibroblasts. Am. J. Physiol. Heart Circ. Physiol. 2008, 295, H2373–H2379. [Google Scholar] [CrossRef]
- E Gallagher, P.; Ferrario, C.M.; Tallant, E.A. MAP kinase/phosphatase pathway mediates the regulation of ACE2 by angiotensin peptides. Am. J. Physiol. Cell Physiol. 2008, 295, C1169–C1174. [Google Scholar] [CrossRef]
- Wu, K.; Li, W.; Peng, G.; Li, F. Crystal structure of NL63 respiratory coronavirus receptor-binding domain complexed with its human receptor. Proc. Natl. Acad. Sci. USA 2009, 106, 19970–19974. [Google Scholar] [CrossRef]
- Vickers, C. Hydrolysis of Biological Peptides by Human Angiotensin-converting Enzyme-related Carboxypeptidase. J. Boil. Chem. 2002, 277, 14838–14843. [Google Scholar] [CrossRef]
- Zamai, L. Unveiling Human Non-random Genome Editing Mechanisms Activated in Response to Chronic Environmental Changes. Available online: https://www.researchgate.net/publication/331791652 (accessed on 20 February 2020).
- Villalobos, L.A.; Hipólito-Luengo, Á.S.; Ramos-González, M.; Cercas, E.; Vallejo, S.; Romero, A.; Romacho, T.; Carraro, R.; Sánchez-Ferrer, C.F.; Peiró, C. The angiotensin-(1-7)/mas axis counteracts angiotensin II-dependent and -independent pro-inflammatory signaling in human vascular smooth muscle cells. Front. Pharmacol. 2016, 7, 482. [Google Scholar] [CrossRef]
- Wysocki, J.; Ye, M.; Rodriguez, E.; González-Pacheco, F.R.; Barrios, C.; Evora, K.; Schuster, M.; Loibner, H.; Brosnihan, K.B.; Ferrario, C.M.; et al. Targeting the degradation of angiotensin II with recombinant angiotensin-converting enzyme 2: Prevention of angiotensin II-dependent hypertension. Hypertension 2009, 55, 90–98. [Google Scholar] [CrossRef]
- Sullivan, J.C.; Bhatia, K.; Yamamoto, T.; Elmarakby, A.A. Angiotensin (1-7) receptor antagonism equalizes angiotensin II-induced hypertension in male and female spontaneously hypertensive rats. Hypertension 2010, 56, 658–666. [Google Scholar] [CrossRef] [PubMed]
- Liao, W.; Fan, H.; Davidge, S.T.; Wu, J. Egg White–Derived Antihypertensive Peptide IRW (Ile-Arg-Trp) Reduces Blood Pressure in Spontaneously Hypertensive Rats via the ACE2/Ang (1-7)/Mas Receptor Axis. Mol. Nutr. Food Res. 2019, 63, 1900063. [Google Scholar] [CrossRef] [PubMed]
- Joviano-Santos, J.V.; Santos-Miranda, A.; Joca, H.C.; Cruz, J.S.; Ferreira, A.J. New insights into the elucidation of angiotensin-(1–7) in vivo antiarrhythmic effects and its related cellular mechanisms. Exp. Physiol. 2016, 101, 1506–1516. [Google Scholar] [CrossRef] [PubMed]
- Donoghue, M.; Hsieh, F.; Baronas, E.; Godbout, K.; Gosselin, M.; Stagliano, N.; Donovan, M.; Woolf, B.; Robison, K.; Jeyaseelan, R.; et al. A Novel Angiotensin-Converting Enzyme—Related to Angiotensin 1–9. Circ. Res. 2000, 87, e1–e9. [Google Scholar] [CrossRef]
- Tipnis, S.R.; Hooper, N.M.; Hyde, R.; Karran, E.; Christie, G.; Turner, A.J. A Human Homolog of Angiotensin-converting Enzyme. J. Boil. Chem. 2000, 275, 33238–33243. [Google Scholar] [CrossRef] [PubMed]
- Mossel, E.C.; Wang, J.; Jeffers, S.; Edeen, K.E.; Wang, S.; Cosgrove, G.P.; Funk, C.; Manzer, R.; Miura, T.A.; Pearson, L.D.; et al. SARS-CoV replicates in primary human alveolar type II cell cultures but not in type I-like cells. Virology 2008, 372, 127–135. [Google Scholar] [CrossRef] [PubMed]
- Sims, A.C.; Baric, R.S.; Yount, B.; Burkett, S.E.; Collins, P.L.; Pickles, R.J. Severe Acute Respiratory Syndrome Coronavirus Infection of Human Ciliated Airway Epithelia: Role of Ciliated Cells in Viral Spread in the Conducting Airways of the Lungs. J. Virol. 2005, 79, 15511–15524. [Google Scholar] [CrossRef]
- Towler, P.; Staker, B.; Prasad, S.G.; Menon, S.; Tang, J.; Parsons, T.; Ryan, D.; Fisher, M.; Williams, D.; Dales, N.A.; et al. ACE2 X-Ray Structures Reveal a Large Hinge-bending Motion Important for Inhibitor Binding and Catalysis. J. Boil. Chem. 2004, 279, 17996–18007. [Google Scholar] [CrossRef]
- Coverdale, J.P.C.; Khazaipoul, S.; Arya, S.; Stewart, A.J.; Blindauer, C.A. Crosstalk between zinc and free fatty acids in plasma. Biochim. Biophys. Acta Mol. Cell Biol. Lipids. 2019, 1864, 532–542. [Google Scholar] [CrossRef]
- Reddy, R.; Asante, I.; Liu, S.; Parikh, P.; Liebler, J.; Borok, Z.; Rodgers, K.; Baydur, A.; Louie, S.G. Circulating angiotensin peptides levels in Acute Respiratory Distress Syndrome correlate with clinical outcomes: A pilot study. PLoS ONE 2019, 14, e0213096. [Google Scholar] [CrossRef]
- Linko, R.; Karlsson, S.; Pettilä, V.; Varpula, T.; Okkonen, M.; Lund, V.; Ala-Kokko, T.; Ruokonen, E.; Ala-Kokko, T. Serum zinc in critically ill adult patients with acute respiratory failure. Acta Anaesthesiol. Scand. 2011, 55, 615–621. [Google Scholar] [CrossRef] [PubMed]
- Boudreault, F.; Pinilla-Vera, M.; Englert, J.A.; Kho, A.T.; Isabelle, C.; Arciniegas, A.J.; Barragan-Bradford, D.; Quintana, C.; Amador, D.P.; Guan, J.; et al. Zinc deficiency primes the lung for ventilator-induced injury. JCI Insight 2017, 2, 1–14. [Google Scholar] [CrossRef] [PubMed]
- Alker, W.; Schwerdtle, T.; Schomburg, L.; Haase, H. A Zinpyr-1-based Fluorimetric Microassay for Free Zinc in Human Serum. Int. J. Mol. Sci. 2019, 20, 4006. [Google Scholar] [CrossRef] [PubMed]
- Anguiano, L.; Riera, M.; Pascual, J.; Valdivielso, J.; Barrios, C.; Betriu, A.; Mojal, S.; Fernández, E.; Soler, M.J.; Castro, E.; et al. Circulating angiotensin-converting enzyme 2 activity in patients with chronic kidney disease without previous history of cardiovascular disease. Nephrol. Dial. Transplant. 2015, 30, 1176–1185. [Google Scholar] [CrossRef] [PubMed]
- Damianaki, K.; Lourenco, J.M.; Braconnier, P.; Ghobril, J.-P.; Devuyst, O.; Burnier, M.; Lenglet, S.; Augsburger, M.; Thomas, A.; Pruijm, M. Renal handling of zinc in chronic kidney disease patients and the role of circulating zinc levels in renal function decline. Nephrol. Dial. Transplant. 2019, gfz065. [Google Scholar] [CrossRef]
- Yousef, A.M.; Elmorsy, E. Serum zinc level in bronchial asthma. Egypt. J. Chest Dis. Tuberc. 2017, 66, 1–4. [Google Scholar] [CrossRef]
- Jackson, D.J.; Busse, W.W.; Bacharier, L.B.; Kattan, M.; O’Connor, G.T.; Wood, R.A.; Visness, C.M.; Durham, S.R.; Larson, D.; Esnault, S.; et al. Association of respiratory allergy, asthma, and expression of the SARS-CoV-2 receptor ACE2. J. Allergy Clin. Immunol. 2020. [Google Scholar] [CrossRef]
- Xie, F.; Zhang, X.; Xie, L. Prognostic value of serum zinc levels in patients with acute HC/zinc chloride smoke inhalation. Medicine 2017, 96, e8156. [Google Scholar] [CrossRef]
- Hjortsø, E.; Ovist, J.; Bud, M.I.; Thomsen, J.L.; Andersen, J.B.; Wiberg-Jørgensen, F.; Jensen, N.K.; Jones, R.; Reid, L.M.; Zapol, W.M. ARDS after accidental inhalation of zinc chloride smoke. Intensiv. Care Med. 1988, 14, 17–24. [Google Scholar] [CrossRef]
- Plum, L.M.; Rink, L.; Haase, H. The Essential Toxin: Impact of Zinc on Human Health. Int. J. Environ. Res. Public Health 2010, 7, 1342–1365. [Google Scholar] [CrossRef]
- Riccelli, M.G.; Goldoni, M.; Poli, D.; Mozzoni, P.; Cavallo, D.; Corradi, A.M.A.M. Welding Fumes, a Risk Factor for Lung Diseases. Int. J. Environ. Res. Public Health 2020, 17, 2552. [Google Scholar] [CrossRef] [PubMed]
- Bleidorn, J.; Alamzad-Krabbe, H.; Gerhards, B.; Kraus, T.; Brand, P.; Krabbe, J.; Martin, C. The pro-inflammatory stimulus of zinc- and copper-containing welding fumes in whole blood assay via protein tyrosine phosphatase 1B inhibition. Sci. Rep. 2019, 9, 1–10. [Google Scholar] [CrossRef] [PubMed]
- Skalny, A.V.; Rink, L.; Ajsuvakova, O.P.; Aschner, M.; Gritsenko, V.A.; Alekseenko, S.I.; Svistunov, A.A.; Petrakis, D.; Spandidos, D.A.; Aaseth, J.; et al. Zinc and respiratory tract infections: Perspectives for COVID-19 (Review). Int. J. Mol. Med. 2020, 19, 17–26. [Google Scholar] [CrossRef]
- Moseley, H.N.B. Current Evidence Supporting the Use of Orally Administered Zinc in the Treatment of COVID-19. OSF Preprints 2020. Available online: https://osf.io/z8wvq (accessed on 20 March 2020).
- Derwand, R.; Scholz, M. Does zinc supplementation enhance the clinical efficacy of chloroquine/hydroxychloroquine to win todays battle against COVID-19? Med. Hypotheses 2020, 142, 109815. [Google Scholar] [CrossRef]
- Hemnes, A.; Rathinasabapathy, A.; Austin, E.A.; Brittain, E.L.; Carrier, E.J.; Chen, X.; Fessel, J.P.; Fike, C.D.; Fong, P.; Fortune, N.; et al. A potential therapeutic role for angiotensin-converting enzyme 2 in human pulmonary arterial hypertension. Eur. Respir. J. 2018, 51, 1702638. [Google Scholar] [CrossRef] [PubMed]
- Soon, E.; Holmes, A.; Treacy, C.M.; Doughty, N.J.; Southgate, L.; Machado, R.D.; Trembath, R.C.; Jennings, S.; Barker, L.; Nicklin, P.; et al. Elevated Levels of Inflammatory Cytokines Predict Survival in Idiopathic and Familial Pulmonary Arterial Hypertension. Circulation 2010, 122, 920–927. [Google Scholar] [CrossRef] [PubMed]
- Li, C.K.-F.; Wu, H.; Yan, H.; Ma, S.; Wang, L.; Zhang, M.; Tang, X.; Temperton, N.J.; Weiss, R.A.; Brenchley, J.M.; et al. T Cell Responses to Whole SARS Coronavirus in Humans1. J. Immunol. 2008, 181, 5490–5500. [Google Scholar] [CrossRef] [PubMed]
- Sauer, A.K.; Pfaender, S.; Hagmeyer, S.; Tarana, L.; Mattes, A.-K.; Briel, F.; Küry, S.; Boeckers, T.M.; Grabrucker, A. Characterization of zinc amino acid complexes for zinc delivery in vitro using Caco-2 cells and enterocytes from hiPSC. BioMetals 2017, 30, 643–661. [Google Scholar] [CrossRef] [PubMed]
- Eid, R.; Arab, N.T.; Greenwood, M.T. Iron mediated toxicity and programmed cell death: A review and a re-examination of existing paradigms. Biochim. Biophys. Acta Bioenerg. 2017, 1864, 399–430. [Google Scholar] [CrossRef]
- Dales, N.A.; Gould, A.E.; Brown, J.A.; Calderwood, E.F.; Guan, B.; Minor, C.A.; Gavin, J.M.; Hales, P.; Kaushik, V.K.; Stewart, M.; et al. Substrate-Based Design of the First Class of Angiotensin-Converting Enzyme-Related Carboxypeptidase (ACE2) Inhibitors. J. Am. Chem. Soc. 2002, 124, 11852–11853. [Google Scholar] [CrossRef]
- Joshi, S.; Balasubramanian, N.; Vasam, G.; Jarajapu, Y.P. Angiotensin converting enzyme versus angiotensin converting enzyme-2 selectivity of MLN-4760 and DX600 in human and murine bone marrow-derived cells. Eur. J. Pharmacol. 2016, 774, 25–33. [Google Scholar] [CrossRef] [PubMed]
- Ye, M.; Wysocki, J.; Gonzalez-Pacheco, F.R.; Salem, M.; Evora, K.; Garcia-Halpin, L.; Poglitsch, M.; Schuster, M.; Batlle, D. Murine Recombinant ACE2: Effect on Angiotensin II Dependent Hypertension and Distinctive ACE2 Inhibitor Characteristics on rodent and human ACE2. Hypertension 2012, 60, 730–740. [Google Scholar] [CrossRef] [PubMed]
- Lew, R.A.; Warner, F.J.; Hanchapola, I.; Yarski, M.A.; Manohar, J.; Burrell, L.M.; Smith, A.I.; Ramchand, J. Angiotensin-converting enzyme 2 catalytic activity in human plasma is masked by an endogenous inhibitor. Exp. Physiol. 2008, 93, 685–693. [Google Scholar] [CrossRef] [PubMed]
- Tikellis, C.; Bialkowski, K.; Pete, J.; Sheehy, K.; Su, Q.; Johnston, C.; E Cooper, M.; Thomas, M. ACE2 Deficiency Modifies Renoprotection Afforded by ACE Inhibition in Experimental Diabetes. Diabetes 2008, 57, 1018–1025. [Google Scholar] [CrossRef]
- Trask, A.J.; Groban, L.; Westwood, B.M.; Varagic, J.; Ganten, D.; Gallagher, P.E.; Chappell, M.C.; Ferrario, C.M. Inhibition of angiotensin-converting enzyme 2 exacerbates cardiac hypertrophy and fibrosis in Ren-2 hypertensive rats. Am. J. Hypertens. 2010, 23, 687–693. [Google Scholar] [CrossRef]
- Soler, M.; Wysocki, J.; Ye, M.; Lloveras, J.; Kanwar, Y.; Batlle, D. ACE2 inhibition worsens glomerular injury in association with increased ACE expression in streptozotocin-induced diabetic mice. Kidney Int. 2007, 72, 614–623. [Google Scholar] [CrossRef]
- Huang, L.; VanLeeuwen, D.; Steffey, M.E.; Donahue, C.; Ho, G.; MacKenzie, R.G.; Sexton, D.J.; Skogerson, K.; Devlin, M.; Smith, R.; et al. Novel Peptide Inhibitors of Angiotensin-converting Enzyme 2. J. Boil. Chem. 2003, 278, 15532–15540. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Wohlford-Lenane, C.; Yamaguchi, Y.; Prindle, T.; Fulton, W.B.; Wang, S.; McCray, P.B.; Chappell, M.C.; Hackam, D.J.; Jia, H. Attenuation of pulmonary ACE2 activity impairs inactivation of des-Arg9 bradykinin/BKB1R axis and facilitates LPS-induced neutrophil infiltration. Am. J. Physiol. Cell. Mol. Physiol. 2017, 314, L17–L31. [Google Scholar] [CrossRef]
- Byrnes, J.J.; Gross, S.; Ellard, C.; Connolly, K.; Donahue, S.; Picarella, D. Effects of the ACE2 inhibitor GL1001 on acute dextran sodium sulfate-induced colitis in mice. Inflamm. Res. 2009, 58, 819–827. [Google Scholar] [CrossRef]
- Flora, S.; Pachauri, V. Chelation in Metal Intoxication. Int. J. Environ. Res. Public Health 2010, 7, 2745–2788. [Google Scholar] [CrossRef]
- Xue, J.; Moyer, A.; Peng, B.; Wu, J.; Hannafon, B.N.; Ding, W.-Q. Chloroquine Is a Zinc Ionophore. PLoS ONE 2014, 9, e109180. [Google Scholar] [CrossRef] [PubMed]
- Belizna, C.; Pregnolato, F.; Abad, S.; Alijotas-Reig, J.; Amital, H.; Amoura, Z.; Andreoli, L.; Andrès, O.K.A.E.; Aouba, A.; Bilgen, S.A.; et al. HIBISCUS: Hydroxychloroquine for the secondary prevention of thrombotic and obstetrical events in primary antiphospholipid syndrome. Autoimmun. Rev. 2018, 17, 1153–1168. [Google Scholar] [CrossRef]
- Gotru, S.K.; Van Geffen, J.P.; Nagy, M.; Mammadova-Bach, E.; Eilenberger, J.; Volz, J.; Manukjan, G.; Schulze, H.; Wagner, L.; Eber, S.; et al. Defective Zn2+ homeostasis in mouse and human platelets with α- and δ-storage pool diseases. Sci. Rep. 2019, 9, 8333. [Google Scholar] [CrossRef] [PubMed]
- Huentelman, M.J.; Zubcevic, J.; Hernández Prada, J.A.; Xiao, X.; Dimitrov, D.S.; Raizada, M.K.; Ostrov, D.A. Sructure-based discovery of a novel angiotensin-converting enzyme 2 inhibitor. Hypertension 2004, 44, 903–906. [Google Scholar] [CrossRef] [PubMed]
- Piletz, J.E.; Aricioglu, F.; Cheng, J.-T.; Fairbanks, C.A.; Gilad, V.H.; Haenisch, B.; Halaris, A.; Hong, S.; Lee, W.T.; Li, J.; et al. Agmatine: Clinical applications after 100 years in translation. Drug Discov. Today 2013, 18, 880–893. [Google Scholar] [CrossRef] [PubMed]
- Rosenberg, M.L.; Tohidi, V.; Sherwood, K.; Gayen, S.; Medel, R.; Gilad, G.M. Evidence for Dietary Agmatine Sulfate Effectiveness in Neuropathies Associated with Painful Small Fiber Neuropathy. A Pilot Open-Label Consecutive Case Series Study. Nutrients. 2020, 12, 576. [Google Scholar] [CrossRef]
- Takahashi, S.; Yoshiya, T.; Yoshizawa-Kumagaye, K.; Sugiyama, T. Nicotianamine is a novel angiotensin-converting enzyme 2 inhibitor in soybean. Biomed. Res. 2015, 36, 219–224. [Google Scholar] [CrossRef]
- Pavelić, S.K.; Medica, J.S.; Gumbarević, D.; Filošević, A.; Pržulj, N.; Pavelić, K. Critical Review on Zeolite Clinoptilolite Safety and Medical Applications in vivo. Front. Pharmacol. 2018, 9, 1–15. [Google Scholar] [CrossRef]
- Mastinu, A.; Kumar, A.; Maccarinelli, G.; Bonini, S.A.; Premoli, M.; Aria, F.; Gianoncelli, A.; Memo, M. Zeolite Clinoptilolite: Therapeutic Virtues of an Ancient Mineral. Molecules 2019, 24, 1517. [Google Scholar] [CrossRef]
- Monteil, V.; Kwon, H.; Prado, P.; Hagelkrüys, A.; Wimmer, R.A.; Stahl, M.; Leopoldi, A.; Garreta, E.; Del Pozo, C.H.; Prosper, F.; et al. Inhibition of SARS-CoV-2 Infections in Engineered Human Tissues Using Clinical-Grade Soluble Human ACE2. Cell 2020, 181, 905–913.e7. [Google Scholar] [CrossRef]
- Khan, A.; Benthin, C.; Zeno, B.; Albertson, T.E.; Boyd, J.; Christie, J.D.; Hall, R.; Poirier, G.; Ronco, J.J.; Tidswell, M.; et al. A pilot clinical trial of recombinant human angiotensin-converting enzyme 2 in acute respiratory distress syndrome. Crit. Care 2017, 21, 234. [Google Scholar] [CrossRef] [PubMed]
- Fang, L.; Karakiulakis, G.; Roth, M. Are patients with hypertension and diabetes mellitus at increased risk for COVID-19 infection? Lancet Respir. Med. 2020, 8, e21. [Google Scholar] [CrossRef]
- Brown, J.D. Antihypertensive drugs and risk of COVID-19? Lancet Respir. Med. 2020, 8, e28. [Google Scholar] [CrossRef]
- Fang, L.; Karakiulakis, G.; Roth, M. Antihypertensive drugs and risk of COVID-19?—Authors’ reply. Lancet Respir. Med. 2020, 8, e32–e33. [Google Scholar] [CrossRef]
- Bosso, M.; Thanaraj, T.A.; Abu-Farha, M.; Alanbaei, M.; Abubaker, J.; Al-Mulla, F. The two faces of ACE2: The Role of ACE2 receptor and its polymorphisms in hypertension and COVID-19. Mol. Ther.-Methods Clin. Dev. 2020, 18, 321–327. [Google Scholar] [CrossRef]
- Ghosal, S.; Mukherjee, J.; Sinha, B.; Gangopadhyay, K. The Effect of Angiotensin Converting Enzyme Inhibitors and Angiotensin Receptor Blockers on Death and Severity of Disease in Patients with Coronavirus Disease 2019 (COVID-19): A Meta-analysis. medRxiv 2020. Available online: https://www.medrxiv.org/content/10.1101/2020.04.23.20076661v2 (accessed on 3 March 2020).
- Bean, D.; Kraljevic, Z.; Searle, T.; Bendayan, R.; O′, G.K.; Pickles, A.; Folarin, A.; Roguski, L.; Noor, K.; Shek, A.; et al. ACE-inhibitors and Angiotensin-2 Receptor Blockers are not associated with severe SARS-COVID19 infection in a multi-site UK acute Hospital Trust. Eur. J. Hear. Fail. 2020. [Google Scholar] [CrossRef]
- Mackey, K.; King, V.J.; Gurley, S.; Kiefer, M.; Liederbauer, E.; Vela, M.K.; Sonnen, B.P.; Kansagara, D. Risks and Impact of Angiotensin-Converting Enzyme Inhibitors or Angiotensin-Receptor Blockers on SARS-CoV-2 Infection in Adults. Ann. Intern. Med. 2020, M20-1515. [Google Scholar] [CrossRef]
- Li, J.; Wang, X.; Chen, J.; Zhang, H.; Deng, A. Association of Renin-Angiotensin System Inhibitors with Severity or Risk of Death in Patients with Hypertension Hospitalized for Coronavirus Disease 2019 (COVID-19) Infection in Wuhan, China. JAMA Cardiol. 2020. [Google Scholar] [CrossRef]
- Reynolds, H.R.; Adhikari, S.; Pulgarin, C.; Troxel, A.B.; Iturrate, E.; Johnson, S.B.; Hausvater, A.; Newman, J.D.; Berger, J.S.; Bangalore, S.; et al. Renin–angiotensin–aldosterone system inhibitors and risk of covid-19. N. Engl. J. Med. 2020, 382, 2441–2448. [Google Scholar] [CrossRef]
- Liu, Y.; Huang, F.; Xu, J.; Yang, P.; Qin, Y.; Cao, M.; Wang, Z.; Li, X.; Zhang, S.; Ye, L.; et al. Anti-hypertensive Angiotensin II Receptor Blockers Associated to Mitigation of Disease Severity in Elderly COVID-19 Patients. Available online: https://www.medrxiv.org/content/10.1101/2020.03.20.20039586v1 (accessed on 27 March 2020).
- Meng, J.; Xiao, G.; Zhang, J.; He, X.; Ou, M.; Bi, J.; Yang, R.; Di, W.; Wang, Z.; Li, Z.; et al. Renin-angiotensin system inhibitors improve the clinical outcomes of COVID-19 patients with hypertension. Emerg. Microbes Infect. 2020, 9, 757–760. [Google Scholar] [CrossRef]
- Pirola, C.J.; Sookoian, S. Estimation of Renin-Angiotensin-Aldosterone-System (RAAS)-Inhibitor effect on COVID-19 outcome: A Meta-analysis. J. Infect. 2020. [Google Scholar] [CrossRef] [PubMed]
- Zhang, X.; Yu, J.; Pan, L.Y.; Jiang, H.Y. ACEI/ARB use and risk of infection or severity or mortality of COVID-19: A systematic review and meta-analysis. Pharmacol. Res. 2020, 158, 104927. [Google Scholar] [CrossRef] [PubMed]
- Zhang, P.; Zhu, L.; Cai, J.; Lei, F.; Qin, J.J.; Xie, J.; Liu, Y.M.; Zhao, Y.C.; Huang, X.; Lin, L.; et al. Association of Inpatient Use of Angiotensin-Converting Enzyme Inhibitors and Angiotensin II Receptor Blockers with Mortality among Patients with Hypertension Hospitalized with COVID-19. Circ. Res. 2020, 126, 1671–1681. [Google Scholar] [CrossRef] [PubMed]
- Magalhães, D.M.; Nunes-Silva, A.; Rocha, G.C.; Vaz, L.N.; De Faria, M.H.S.; Vieira, E.L.M.; Rocha, N.P.; E Silva, A.C.S. Two protocols of aerobic exercise modulate the counter-regulatory axis of the renin-angiotensin system. Heliyon 2020, 6, 03208. [Google Scholar] [CrossRef] [PubMed]
- Agarwal, D.; Elks, C.M.; Reed, S.D.; Mariappan, N.; Majid, D.S.; Francis, J. Chronic Exercise Preserves Renal Structure and Hemodynamics in Spontaneously Hypertensive Rats. Antioxid. Redox Signal. 2012, 16, 139–152. [Google Scholar] [CrossRef] [PubMed]
- Ramchand, J.; Patel, S.K.; Srivastava, P.M.; Farouque, O.; Burrell, L.M. Elevated plasma angiotensin converting enzyme 2 activity is an independent predictor of major adverse cardiac events in patients with obstructive coronary artery disease. PLoS ONE 2018, 13, e0198144. [Google Scholar] [CrossRef]
- Walters, T.; Kalman, J.M.; Patel, S.; Mearns, M.; Velkoska, E.; Burrell, L.M. Angiotensin converting enzyme 2 activity and human atrial fibrillation: Increased plasma angiotensin converting enzyme 2 activity is associated with atrial fibrillation and more advanced left atrial structural remodelling. Europace 2017, 19, 1280–1287. [Google Scholar] [CrossRef]
- Filha, R.D.S.; Pinheiro, S.V.B.; Macedo e Cordeiro, T.; Feracin, V.; Vieira, E.L.M.; Miranda, A.S.; Simões e Silva, A.C. Evidence for a role of angiotensin converting enzyme 2 in proteinuria of idiopathic nephrotic syndrome. Biosci. Rep. 2019, 39, 1–13. [Google Scholar] [CrossRef]
- Furuhashi, M.; Moniwa, N.; Mita, T.; Fuseya, T.; Ishimura, S.; Ohno, K.; Shibata, S.; Tanaka, M.; Watanabe, Y.; Akasaka, H.; et al. Urinary Angiotensin-Converting Enzyme 2 in Hypertensive Patients May Be Increased by Olmesartan, an Angiotensin II Receptor Blocker. Am. J. Hypertens. 2014, 28, 15–21. [Google Scholar] [CrossRef]
- Liu, C.; Li, Y.; Guan, T.; Lai, Y.; Shen, Y.; Zeyaweiding, A.; Zhao, H.; Li, F.; Maimaiti, T. ACE2 polymorphisms associated with cardiovascular risk in Uygurs with type 2 diabetes mellitus 11 Medical and Health Sciences 1103 Clinical Sciences. Cardiovasc. Diabetol. 2018, 17, 127. [Google Scholar] [CrossRef]
- Luo, Y.; Liu, C.; Guan, T.; Li, Y.; Lai, Y.; Li, F.; Zhao, H.; Maimaiti, T.; Zeyaweiding, A. Correction: Association of ACE2 genetic polymorphisms with hypertension-related target organ damages in south Xinjiang. Hypertens. Res. 2019, 42, 744. [Google Scholar] [CrossRef] [PubMed]
- Pan, Y.; Wang, T.; Li, Y.; Guan, T.; Lai, Y.; Shen, Y.; Zeyaweiding, A.; Maimaiti, T.; Li, F.; Zhao, H.; et al. Association of ACE2 polymorphisms with susceptibility to essential hypertension and dyslipidemia in Xinjiang, China. Lipids Heal. Dis. 2018, 17, 241. [Google Scholar] [CrossRef]
- Zhang, Q.; Cong, M.; Wang, N.; Li, X.; Zhang, H.; Zhang, K.; Jin, M.; Wu, N.; Qiu, C.; Li, J. Association of angiotensin-converting enzyme 2 gene polymorphism and enzymatic activity with essential hypertension in different gender A case–control study. Medicine 2018, 97, e12917. [Google Scholar] [CrossRef] [PubMed]
- Lieb, W.; Graf, J.; Götz, A.; König, I.R.; Mayer, B.; Fischer, M.; Stritzke, J.; Hengstenberg, C.; Holmer, S.R.; Döring, A.; et al. Association of angiotensin-converting enzyme 2 (ACE2) gene polymorphisms with parameters of left ventricular hypertrophy in men: Results of the MONICA Augsburg echocardiographic substudy. J. Mol. Med. 2006, 84, 88–96. [Google Scholar] [CrossRef] [PubMed]
- Liu, D.; Chen, Y.; Zhang, P.; Zhong, J.; Jin, L.; Zhang, C.; Lin, S.; Wu, S.; Yu, H. Association between circulating levels of ACE2-Ang-(1–7)-MAS axis and ACE2 gene polymorphisms in hypertensive patients. Medicine 2016, 95, e3876. [Google Scholar] [CrossRef] [PubMed]
- Fan, Z.; Wu, G.; Yue, M.; Ye, J.; Chen, Y.; Xu, B.; Shu, Z.; Zhu, J.; Lu, N.; Tan, X. Hypertension and hypertensive left ventricular hypertrophy are associated with ACE2 genetic polymorphism. Life Sci. 2019, 225, 39–45. [Google Scholar] [CrossRef]
- Cao, Y.; Li, L.; Feng, Z.; Wan, S.; Huang, P.; Sun, X.; Wen, F.; Huang, X.; Ning, G.; Wang, W. Comparative genetic analysis of the novel coronavirus (2019-nCoV/SARS-CoV-2) receptor ACE2 in different populations. Cell Discov. 2020, 6, 1–4. [Google Scholar] [CrossRef]
- Zou, X.; Chen, K.; Zou, J.; Han, P.; Hao, J.; Han, Z.-G. Single-cell RNA-seq data analysis on the receptor ACE2 expression reveals the potential risk of different human organs vulnerable to 2019-nCoV infection. Front. Med. 2020, 14, 185–192. [Google Scholar] [CrossRef]
- Pazarlı, A.C.; Ekiz, T.; Ilik, F. Coronavirus disease 2019 and obstructive sleep apnea syndrome. Sleep Breath. 2020, 1. [Google Scholar] [CrossRef] [PubMed]
- Feuth, T.; Saaresranta, T.; Karlsson, A.; Valtonen, M.; Peltola, V.; Rintala, E.; Oksi, J. Is Sleep Apnoea a Risk Factor for Covid-19? Findings from a Retrospective Cohort Study. Available online: https://www.medrxiv.org/content/10.1101/2020.05.14.20098319v1.abstract (accessed on 18 May 2020).
- Arentz, M.; Yim, E.; Klaff, L.; Lokhandwala, S.; Riedo, F.X.; Chong, M.; Lee, M. Characteristics and Outcomes of 21 Critically Ill Patients With COVID-19 in Washington State. JAMA 2020, 323, 1612. [Google Scholar] [CrossRef]
- Bonsignore, M.R.; Saaresranta, T.; Riha, R.L. Sex differences in obstructive sleep apnoea. Eur. Respir. Rev. 2019, 28, 190030. [Google Scholar] [CrossRef] [PubMed]
- Corman, V.M.; Muth, D.; Niemeyer, D.; Drosten, C. Hosts and Sources of Endemic Human Coronaviruses. Adv. Viruses Res. 2018, 100, 163–188. [Google Scholar] [CrossRef]
- Korber, B.; Fischer, W.M.; Gnanakaran, S.; Yoon, H.; Theiler, J.; Abfalterer, W.; Foley, B.; Giorgi, E.E.; Bhattacharya, T.; Parker, M.D.; et al. Spike Mutation pipeline Reveals the Emergence of a More Transmissible form of SARS-CoV-2. Available online: https://www.biorxiv.org/content/10.1101/2020.04.29.069054v2.abstract (accessed on 5 May 2020).
- Long, Q.-X.; Tang, X.-J.; Shi, Q.-L.; Li, Q.; Deng, H.-J.; Yuan, J.; Hu, J.-L.; Xu, W.; Zhang, Y.; Lv, F.-J.; et al. Clinical and immunological assessment of asymptomatic SARS-CoV-2 infections. Nat. Med. 2020. [Google Scholar] [CrossRef] [PubMed]
- Wang, X.; Guo, X.; Xin, Q.; Pan, Y.; Li, J.; Chu, Y.; Feng, Y.; Wang, Q. Neutralizing Antibodies Responses to SARS-CoV-2 in COVID-19 Inpatients and Convalescent Patients. Available online: https://www.medrxiv.org/content/10.1101/2020.04.15.20065623v1.full.pdf (accessed on 18 April 2020).
- Vabret, N.; Britton, G.J.; Gruber, C.; Hegde, S.; Kim, J.; Kuksin, M.; Levantovsky, R.; Malle, L.; Moreira, A.; Park, M.D.; et al. Immunology of COVID-19: Current State of the Science. Immunity 2020, 52, 910–941. [Google Scholar] [CrossRef]
- Andreano, E.; Nicastri, E.; Paciello, I.; Pileri, P.; Manganaro, N.; Piccini, G.; Manenti, A.; Pantano, E.; Kabanova, A.; Troisi, M.; et al. Identification of Neutralizing Human Monoclonal Antibodies from Italian Covid-19 Convalescent Patients. Available online: https://www.biorxiv.org/content/10.1101/2020.05.05.078154v1.abstract (accessed on 8 May 2020).
- Yu, J.; Tostanoski, L.H.; Peter, L.; Mercado, N.B.; Mcmahan, K.; Mahrokhian, S.H.; Nkolola, J.P.; Liu, J.; Li, Z.; Chandrashekar, A.; et al. DNA vaccine protection against SARS-CoV-2 in rhesus macaques. Science 2020, 6284, eabc6284. [Google Scholar] [CrossRef]
- Why We Might not Get a Coronavirus Vaccine. Available online: https://www.theguardian.com/world/2020/may/22/why-we-might-not-get-a-coronavirus-vaccine (accessed on 31 May 2020).
- Bolles, M.; E Deming, M.; Long, K.; Agnihothram, S.; Whitmore, A.; Ferris, M.; Funkhouser, W.; Gralinski, L.; Totura, A.; Heise, M.; et al. A Double-Inactivated Severe Acute Respiratory Syndrome Coronavirus Vaccine Provides Incomplete Protection in Mice and Induces Increased Eosinophilic Proinflammatory Pulmonary Response upon Challenge. J. Virol. 2011, 85, 12201–12215. [Google Scholar] [CrossRef]
- Liu, Y.; Yang, Y.; Zhang, C.; Huang, F.; Wang, F.; Yuan, J.; Wang, Z.; Li, J.; Li, J.; Feng, C.; et al. Clinical and biochemical indexes from 2019-nCoV infected patients linked to viral loads and lung injury. Sci. China Life Sci. 2020, 63, 364–374. [Google Scholar] [CrossRef]
- Haschke, M.; Schuster, M.; Poglitsch, M.; Loibner, H.; Salzberg, M.; Bruggisser, M.; Penninger, J.M.; Krähenbühl, S. Pharmacokinetics and Pharmacodynamics of Recombinant Human Angiotensin-Converting Enzyme 2 in Healthy Human Subjects. Clin. Pharmacokinet. 2013, 52, 783–792. [Google Scholar] [CrossRef]
- Kovarik, J.J.; Antlanger, M.; Domenig, O.; Kaltenecker, C.C.; Hecking, M.; Haidinger, M.; Werzowa, J.M.; Kopecky, C.; Säemann, M.D. Molecular regulation of the renin-angiotensin system in haemodialysis patients. Nephrol. Dial. Transplant. 2014, 30, 115–123. [Google Scholar] [CrossRef]
- Lawrence, A.C.; Evin, G.; Kladis, A.; Campbell, D.J. An alternative strategy for the radioimmunoassay of angiotensin peptides using amino-terminal-directed antisera: Measurement of eight angiotensin peptides in human plasma. J. Hypertens. 1990, 8, 715–724. [Google Scholar] [CrossRef]
- Poglitsch, M.; Domenig, O.; Schwager, C.; Stranner, S.; Peball, B.; Janzek, E.; Wagner, B.; Jungwirth, H.; Loibner, H.; Schuster, M. Recombinant Expression and Characterization of Human and Murine ACE2: Species-Specific Activation of the Alternative Renin-Angiotensin-System. Int. J. Hypertens. 2012, 2012, 1–8. [Google Scholar] [CrossRef] [PubMed]
- Li, W.; Li, J.; Hao, P.; Chen, W.; Meng, X.; Li, H.; Zhang, Y.; Zhang, C.; Yang, J. Imbalance between angiotensin II and angiotensin-(1–7) in human coronary atherosclerosis. J. Renin Angiotensin Aldosterone Syst. 2016, 17, 1–6. [Google Scholar] [CrossRef] [PubMed]
- Shenoy, V.; Gjymishka, A.; Jarajapu, Y.P.; Qi, Y.; Afzal, A.; Rigatto, K.; Ferreira, A.J.; Fraga-Silva, R.A.; Kearns, P.; Douglas, J.Y.; et al. Diminazene Attenuates Pulmonary Hypertension and Improves Angiogenic Progenitor Cell Functions in Experimental Models. Am. J. Respir. Crit. Care Med. 2013, 187, 648–657. [Google Scholar] [CrossRef] [PubMed]
- Evans, C.E.; Miners, J.S.; Piva, G.; Willis, C.L.; Heard, D.M.; Kidd, E.J.; Good, M.A.; Kehoe, P.G. ACE2 activation protects against cognitive decline and reduces amyloid pathology in the Tg2576 mouse model of Alzheimer’s disease. Acta Neuropathol. 2020, 139, 485–502. [Google Scholar] [CrossRef]
- Sodhi, C.P.; Nguyen, J.; Yamaguchi, Y.; Werts, A.D.; Lu, P.; Ladd, M.R.; Fulton, W.B.; Kovler, M.L.; Wang, S.; Prindle, T.; et al. A Dynamic Variation of Pulmonary ACE2 Is Required to Modulate Neutrophilic Inflammation in Response to Pseudomonas aeruginosa Lung Infection in Mice. J. Immunol. 2019, 203, 3000–3012. [Google Scholar] [CrossRef]
- Ramya, K.; Suresh, R.; Kumar, H.Y.; Kumar, B.P.; Murthy, N.S. Decades-old renin inhibitors are still struggling to find a niche in antihypertensive therapy. A fleeting look at the old and the promising new molecules. Bioorganic Med. Chem. 2020, 28, 115466. [Google Scholar] [CrossRef]
- Ding, W.; Li, X.; Wu, W.; He, H.; Li, Y.; Gao, L.; Gan, L.; Wang, M.; Ou, S.; Liu, J. Aliskiren inhibits angiotensin II/angiotensin 1-7(Ang II/Ang1-7) signal pathway in rats with diabetic nephropathy. Xi Bao Yu Fen Zi Mian Yi Xue Za Zhi = Chin. J. Cell. Mol. Immunol. 2018, 34, 891–895. (In Chinese) [Google Scholar]
- Williams, J.D.; Matthews, G.A.; Judd, A.W. Oral calcium disodium versenate in treatment of lead poisoning. Br. J. Ind. Med. 1962, 19, 211–215. [Google Scholar] [CrossRef] [PubMed]
- Ellithorpe, R.; Mazur, P.; Gum, G.; Button, G.; Le, J.; Pfadenhauer, E.H.; Settineri, R.A.; Nicolson, G. Comparison of the Absorption, Brain and Prostate Distribution, and Elimination of CaNa2EDTA of Rectal Chelation Suppositories to Intravenous Administration. JANA 2007, 10, 38–44. [Google Scholar]
- U.S. Department of Health and Human Services, F. and D.A. Prescribing Information Calcium Disodium Versenate. 2009; NDA 8-922/S-016, 3-10. Available online: https://www.accessdata.fda.gov/drugsatfda_docs/label/2009/008922s016lbl.pdf (accessed on 20 March 2020).
- Asmus, M.J.; Barros, M.D.; Liang, J.; Chesrown, S.E.; Hendeles, L. Pulmonary function response to EDTA, an additive in nebulized bronchodilators. J. Allergy Clin. Immunol. 2001, 107, 68–72. [Google Scholar] [CrossRef]
- Felton, D.J.; Kales, S.N.; Goldman, R.H. An update and review of unconventional metals testing and treatment. Toxics 2014, 2, 403–416. [Google Scholar] [CrossRef]


© 2020 by the author. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Zamai, L. The Yin and Yang of ACE/ACE2 Pathways: The Rationale for the Use of Renin-Angiotensin System Inhibitors in COVID-19 Patients. Cells 2020, 9, 1704. https://doi.org/10.3390/cells9071704
Zamai L. The Yin and Yang of ACE/ACE2 Pathways: The Rationale for the Use of Renin-Angiotensin System Inhibitors in COVID-19 Patients. Cells. 2020; 9(7):1704. https://doi.org/10.3390/cells9071704
Chicago/Turabian StyleZamai, Loris. 2020. "The Yin and Yang of ACE/ACE2 Pathways: The Rationale for the Use of Renin-Angiotensin System Inhibitors in COVID-19 Patients" Cells 9, no. 7: 1704. https://doi.org/10.3390/cells9071704
APA StyleZamai, L. (2020). The Yin and Yang of ACE/ACE2 Pathways: The Rationale for the Use of Renin-Angiotensin System Inhibitors in COVID-19 Patients. Cells, 9(7), 1704. https://doi.org/10.3390/cells9071704