Current Status of microRNA-Based Therapeutic Approaches in Neurodegenerative Disorders

 ,
,  , and
, and 
Abstract
1. Introduction
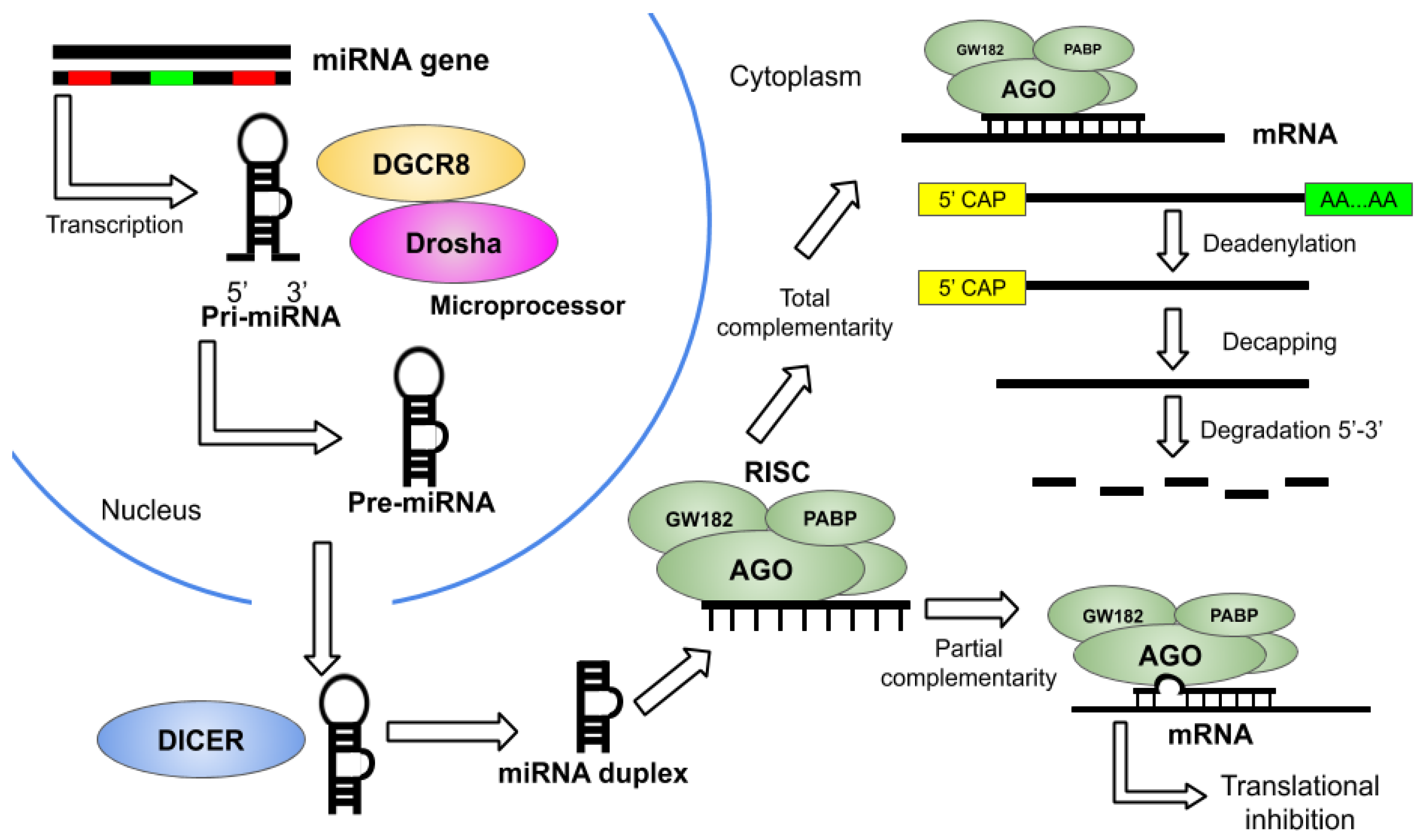
2. Overview of miRNA Therapeutics
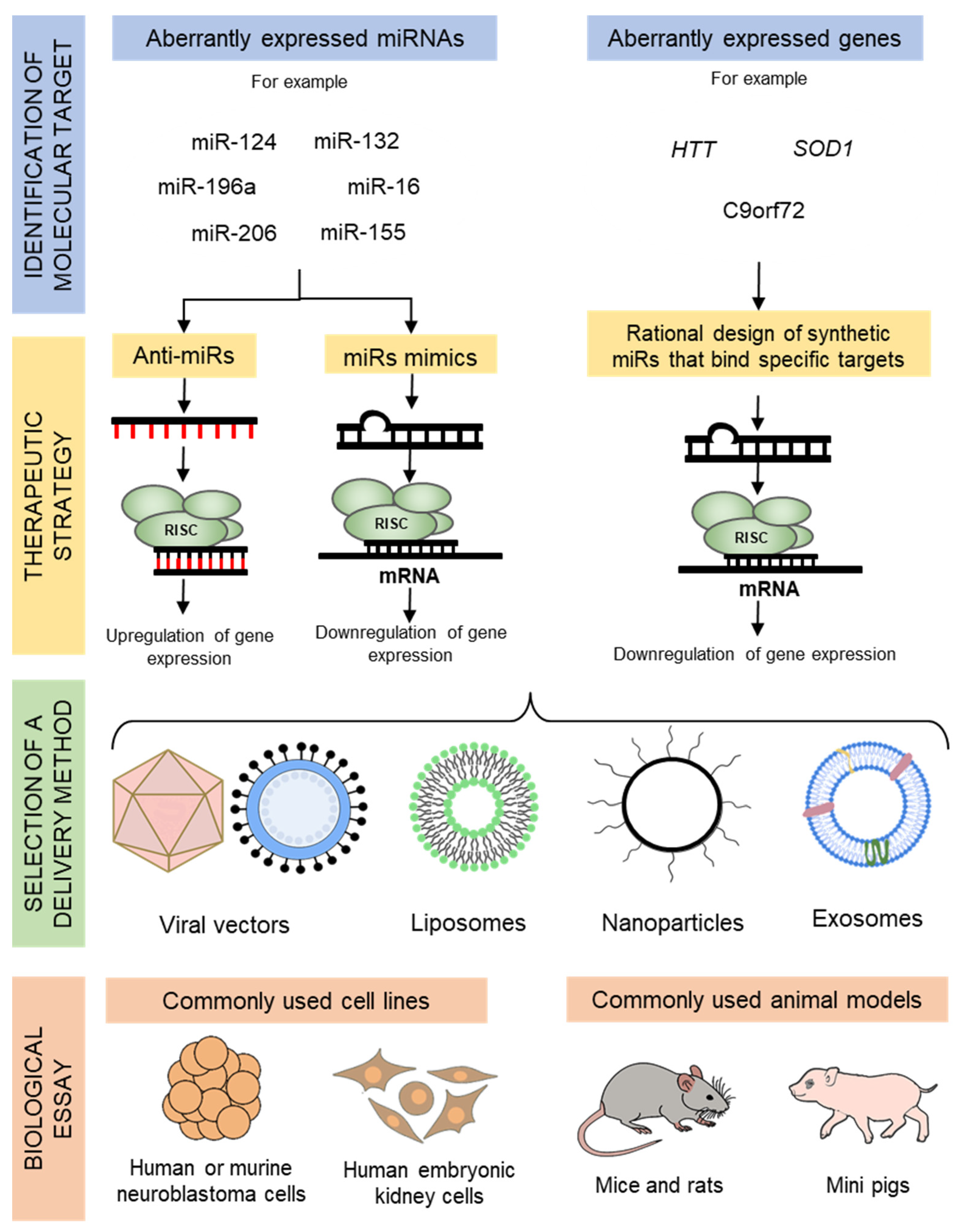
3. Delivery Techniques for miRNA Therapeutics
4. Alzheimer’s Disease (AD)
5. Parkinson’s Disease (PD)
6. Huntington’s Disease (HD)
7. Amyotrophic Lateral Sclerosis (ALS)
8. Friedreich’s Ataxia (FRDA)
9. Spinal Muscular Atrophy (SMA)
10. Frontotemporal Dementia
11. Conclusions
Author Contributions
Funding
Conflicts of Interest
References
- Michlewski, G.; Cáceres, J.F. Post-transcriptional control of miRNA biogenesis. RNA 2019, 25, 1–16. [Google Scholar] [CrossRef] [PubMed]
- Sun, P.; Liu, D.Z.; Jickling, G.C.; Sharp, F.R.; Yin, K.J. MicroRNA-based therapeutics in central nervous system injuries. J. Cereb. Blood Flow Metab. 2018, 38, 1125–1148. [Google Scholar] [CrossRef] [PubMed]
- Paul, S.; Reyes, P.R.; Garza, B.S.; Sharma, A. MicroRNAs and Child Neuropsychiatric Disorders: A Brief Review. Neurochem. Res. 2020, 45, 232–240. [Google Scholar] [CrossRef]
- Bandiera, S.; Cartault, F.; Jannot, A.S.; Hatem, E.; Girard, M.; Rifai, L.; Loiseau, C.; Munnich, A.; Lyonnet, S.; Henrion-Caude, A. Genetic Variations Creating MicroRNA Target Sites in the FXN 3′-UTR Affect Frataxin Expression in Friedreich Ataxia. PLoS ONE 2013, 8, e54791. [Google Scholar] [CrossRef] [PubMed]
- Sharma, S.; Lu, H.C. microRNAs in Neurodegeneration: Current Findings and Potential Impacts. J. Alzheimer’s Dis. Park. 2018, 8, 420. [Google Scholar] [CrossRef]
- Santhekadur, P.K.; Kumar, D.P. RISC assembly and post-transcriptional gene regulation in Hepatocellular Carcinoma. Genes Dis. 2020, 7, 199–204. [Google Scholar] [CrossRef] [PubMed]
- Gupta, S.; Kulhara, P. What is schizophrenia: A neurodevelopmental or neurodegenerative disorder or a combination of both A critical analysis. Indian J. Psychiatry 2010, 52, 21–27. [Google Scholar] [CrossRef]
- Kim, J.; Mook-Jung, I. Special issue on neurodegenerative diseases and their therapeutic approaches. Exp. Mol. Med. 2015, 47, e146. [Google Scholar] [CrossRef][Green Version]
- Kovacs, G.G. Molecular pathology of neurodegenerative diseases: Principles and practice. J. Clin. Pathol. 2019, 72, 725–735. [Google Scholar] [CrossRef]
- Qiu, L.; Tan, E.K.; Zeng, L. microRNAs and Neurodegenerative Diseases. Adv. Exp. Med. Biol. 2015, 888, 85–105. [Google Scholar] [CrossRef]
- Walayat, A.; Yang, M.; Xiao, D. Therapeutic Implication of miRNA in Human Disease. In Antisense Therapy; Sharad, S., Kapur, S., Eds.; IntechOpen: London, UK, 2019; ISBN 978-1-78984-532. [Google Scholar]
- Christopher, A.F.; Kaur, R.P.; Kaur, G.; Kaur, A.; Gupta, V.; Bansal, P. MicroRNA therapeutics: Discovering novel targets and developing specific therapy. Perspect. Clin. Res. 2016, 7, 68–74. [Google Scholar] [CrossRef] [PubMed]
- Liu, E.Y.; Cali, C.P.; Lee, E.B. RNA metabolism in neurodegenerative disease. DMM Dis. Model. Mech. 2017, 10, 509–518. [Google Scholar] [CrossRef] [PubMed]
- Bernardo, B.C.; Ooi, J.Y.Y.; Lin, R.C.Y.; McMullen, J.R. miRNA therapeutics: A new class of drugs with potential therapeutic applications in the heart. Future Med. Chem. 2015, 7, 1771–1792. [Google Scholar] [CrossRef]
- Leclercq, M.; Diallo, A.B.; Blanchette, M. Prediction of human miRNA target genes using computationally reconstructed ancestral mammalian sequences. Nucleic Acids Res. 2017, 45, 556–566. [Google Scholar] [CrossRef]
- Bajan, S.; Hutvagner, G. RNA-Based Therapeutics: From Antisense Oligonucleotides to miRNAs. Cells 2020, 9, 137. [Google Scholar] [CrossRef]
- Campbell, K.; Booth, S.A. MicroRNA in neurodegenerative drug discovery: The way forward? Expert Opin. Drug Discov. 2015, 10, 9–16. [Google Scholar] [CrossRef]
- Goldgraben, M.A.; Russell, R.; Rueda, O.M.; Caldas, C.; Git, A. Double-stranded microRNA mimics can induce length-and passenger strand-dependent effects in a cell type-specific manner. RNA 2016, 22, 193–203. [Google Scholar] [CrossRef]
- Lin, H.M.; Nikolic, I.; Yang, J.; Castillo, L.; Deng, N.; Chan, C.L.; Yeung, N.K.; Dodson, E.; Elsworth, B.; Spielman, C.; et al. MicroRNAs as potential therapeutics to enhance chemosensitivity in advanced prostate cancer. Sci. Rep. 2018, 8, 7820. [Google Scholar] [CrossRef]
- Tang, L.; Chen, H.Y.; Hao, N.B.; Tang, B.; Guo, H.; Yong, X.; Dong, H.; Yang, S.M. microRNA inhibitors: Natural and artificial sequestration of microRNA. Cancer Lett. 2017, 407, 139–147. [Google Scholar] [CrossRef]
- Lima, J.F.; Cerqueira, L.; Figueiredo, C.; Oliveira, C.; Azevedo, N.F. Anti-miRNA oligonucleotides: A comprehensive guide for design. RNA Biol. 2018, 15, 338–352. [Google Scholar] [CrossRef] [PubMed]
- Ebert, M.S.; Sharp, P.A. MicroRNA sponges: Progress and possibilities. RNA 2010, 16, 2043–2050. [Google Scholar] [CrossRef]
- Simonson, B.; Das, S. MicroRNA Therapeutics: The Next Magic Bullet? Mini Rev. Med. Chem. 2015, 15, 467–474. [Google Scholar] [CrossRef] [PubMed]
- Pereira, D.M.; Rodrigues, P.M.; Borralho, P.M.; Rodrigues, C.M.P. Delivering the promise of miRNA cancer therapeutics. Drug Discov. Today 2013, 18, 282–289. [Google Scholar] [CrossRef] [PubMed]
- Sun, X.; Guo, Q.; Wei, W.; Robertson, S.; Yuan, Y.; Luo, X. Current progress on microRNA-based gene delivery in the treatment of osteoporosis and osteoporotic fracture. Int. J. Endocrinol. 2019, 2019, 6782653. [Google Scholar] [CrossRef] [PubMed]
- Yang, N. An overview of viral and nonviral delivery systems for microRNA. Int. J. Pharm. Investig. 2015, 5, 179–181. [Google Scholar] [CrossRef]
- Myoung, S.; Kasinski, A.L. Strategies for Safe and Targeted Delivery of MicroRNA Therapeutics. In MicroRNAs in Diseases and Disorders: Emerging Therapeutic Targets; The Royal Society of Chemistry: London, UK, 2019; pp. 386–415. ISBN 9781782621454. [Google Scholar]
- Ojala, D.S.; Amara, D.P.; Schaffer, D.V. Adeno-associated virus vectors and neurological gene therapy. Neuroscientist 2015, 21, 84–98. [Google Scholar] [CrossRef]
- Lee, S.W.L.; Paoletti, C.; Campisi, M.; Osaki, T.; Adriani, G.; Kamm, R.D.; Mattu, C.; Chiono, V. MicroRNA delivery through nanoparticles. J. Control. Release 2019, 313, 80–95. [Google Scholar] [CrossRef]
- Zhang, Y.; Wang, Z.; Gemeinhart, R.A. Progress in microRNA delivery. J. Control. Release 2013, 172, 962–974. [Google Scholar] [CrossRef]
- Carter, M.; Shieh, J. Gene Delivery Strategies. In Guide to Research Techniques in Neuroscience; Academic Press: San Diego, CA, USA, 2015; pp. 239–252. ISBN 978-0-12-800511-8. [Google Scholar]
- Wen, M.M. Getting miRNA therapeutics into the target cells for neurodegenerative diseases: A mini-review. Front. Mol. Neurosci. 2016, 9, 129. [Google Scholar] [CrossRef]
- Wang, H.; Jiang, Y.; Peng, H.; Chen, Y.; Zhu, P.; Huang, Y. Recent progress in microRNA delivery for cancer therapy by non-viral synthetic vectors. Adv. Drug Deliv. Rev. 2015, 81, 142–160. [Google Scholar] [CrossRef]
- Magen, I.; Hornstein, E. Oligonucleotide-based therapy for neurodegenerative diseases. Brain Res. 2014, 1584, 116–128. [Google Scholar] [CrossRef]
- Campani, V.; De Rosa, G.; Misso, G.; RZarone, M.; Grimaldi, A. Lipid Nanoparticles to Deliver miRNA in Cancer. Curr. Pharm. Biotechnol. 2016, 17, 741–749. [Google Scholar] [CrossRef] [PubMed]
- Estanqueiro, M.; Vasconcelos, H.; Lobo, J.M.S.; Amaral, H. Delivering miRNA modulators for cancer treatment. In Drug Targeting and Stimuli Sensitive Drug Delivery Systems; Grumezescu, A.M., Ed.; William Andrew: Norwich, NY, USA, 2018; pp. 517–565. ISBN 9780128136898. [Google Scholar]
- Huynh, N.; VonMoss, L.; Smith, D.; Rahman, I.; Felemban, M.F.; Zuo, J.; Rody, W.J.; McHugh, K.P.; Holliday, L.S. Characterization of regulatory extracellular vesicles from osteoclasts. J. Dent. Res. 2016, 95, 673–679. [Google Scholar] [CrossRef] [PubMed]
- Khanahmadi, M.; Farhud, D.D.; Malmir, M. Genetic of Alzheimer’s disease: A narrative review article. Iran. J. Public Health 2015, 44, 892–901. [Google Scholar] [PubMed]
- Chen, J.J.; Zhao, B.; Zhao, J.; Li, S. Potential Roles of Exosomal MicroRNAs as Diagnostic Biomarkers and Therapeutic Application in Alzheimer’s Disease. Neural Plast. 2017, 2017, 7027380. [Google Scholar] [CrossRef]
- Bekris, L.M.; Leverenz, J.B. The biomarker and therapeutic potential of miRNA in Alzheimer’s disease. Neurodegener. Dis. Manag. 2015, 5, 61–74. [Google Scholar] [CrossRef]
- Angelucci, F.; Cechova, K.; Valis, M.; Kuca, K.; Zhang, B.; Hort, J. MicroRNAs in Alzheimer’s disease: Diagnostic markers or therapeutic agents? Front. Pharmacol. 2019, 10, 665. [Google Scholar] [CrossRef] [PubMed]
- Parsi, S.; Smith, P.Y.; Goupil, C.; Dorval, V.; Hébert, S.S. Preclinical evaluation of miR-15/107 family members as multifactorial drug targets for Alzheimer’s disease. Mol. Ther. Nucleic Acids 2015, 4, e256. [Google Scholar] [CrossRef] [PubMed]
- Smith, P.Y.; Hernandez-Rapp, J.; Jolivette, F.; Lecours, C.; Bisht, K.; Goupil, C.; Dorval, V.; Parsi, S.; Morin, F.; Planel, E.; et al. MiR-132/212 deficiency impairs tau metabolism and promotes pathological aggregation in vivo. Hum. Mol. Genet. 2015, 24, 6721–6735. [Google Scholar] [CrossRef] [PubMed]
- Salta, E.; Sierksma, A.; Vanden Eynden, E.; De Strooper, B. miR-132 loss de-represses ITPKB and aggravates amyloid and TAU pathology in Alzheimer’s brain. EMBO Mol. Med. 2016, 8, 1005–1018. [Google Scholar] [CrossRef]
- Fang, M.; Wang, J.; Zhang, X.; Geng, Y.; Hu, Z.; Rudd, J.A.; Ling, S.; Chen, W.; Han, S. The miR-124 regulates the expression of BACE1/β-secretase correlated with cell death in Alzheimer’s disease. Toxicol. Lett. 2012, 209, 94–105. [Google Scholar] [CrossRef] [PubMed]
- Cao, F.; Liu, Z.; Sun, G. Diagnostic value of miR-193a-3p in Alzheimer’s disease and miR-193a-3p attenuates amyloid-β induced neurotoxicity by targeting PTEN. Exp. Gerontol. 2020, 130, 110814. [Google Scholar] [CrossRef] [PubMed]
- Sun, L.; Zhang, T.; Xiu, W.; Cao, W.; He, M.; Sun, W.; Zhao, W. MiR-107 overexpression attenuates neurotoxicity induced by 6-hydroxydopamine both in vitro and in vivo. Chem. Biol. Interact. 2020, 315, 108908. [Google Scholar] [CrossRef] [PubMed]
- He, B.; Chen, W.; Zeng, J.; Tong, W.; Zheng, P. MicroRNA-326 decreases tau phosphorylation and neuron apoptosis through inhibition of the JNK signaling pathway by targeting VAV1 in Alzheimer’s disease. J. Cell. Physiol. 2020, 235, 480–493. [Google Scholar] [CrossRef] [PubMed]
- Zovoilis, A.; Agbemenyah, H.Y.; Agis-Balboa, R.C.; Stilling, R.M.; Edbauer, D.; Rao, P.; Farinelli, L.; Delalle, I.; Schmitt, A.; Falkai, P.; et al. MicroRNA-34c is a novel target to treat dementias. EMBO J. 2011, 30, 4299–4308. [Google Scholar] [CrossRef]
- Shi, Z.; Zhang, K.; Zhou, H.; Jiang, L.; Xie, B.; Wang, R.; Xia, W.; Yin, Y.; Gao, Z.; Cui, D.; et al. Increased miR-34c mediates synaptic deficits by targeting synaptotagmin 1 through ROS-JNK-p53 pathway in Alzheimer’s Disease. Aging Cell 2020, 19, e13125. [Google Scholar] [CrossRef]
- Lee, K.; Kim, H.; An, K.; Kwon, O.B.; Park, S.; Cha, J.H.; Kim, M.H.; Lee, Y.; Kim, J.H.; Cho, K.; et al. Replenishment of microRNA-188-5p restores the synaptic and cognitive deficits in 5XFAD Mouse Model of Alzheimer’s Disease. Sci. Rep. 2016, 6, 34433. [Google Scholar] [CrossRef]
- Zolochevska, O.; Taglialatela, G. Selected microRNAs Increase Synaptic Resilience to the Damaging Binding of the Alzheimer’s Disease Amyloid Beta Oligomers. Mol. Neurobiol. 2020, 57, 2232–2243. [Google Scholar] [CrossRef]
- Higaki, S.; Muramatsu, M.; Matsuda, A.; Matsumoto, K.; Satoh, J.I.; Michikawa, M.; Niida, S. Defensive effect of microRNA-200b/c against amyloid-beta peptide-induced toxicity in Alzheimer’s disease models. PLoS ONE 2018, 13, e0196929. [Google Scholar] [CrossRef]
- Barros-Viegas, A.T.; Carmona, V.; Ferreiro, E.; Guedes, J.; Cardoso, A.M.; Cunha, P.; Pereira de Almeida, L.; Resende de Oliveira, C.; Pedro de Magalhães, J.; Peça, J.; et al. miRNA-31 Improves Cognition and Abolishes Amyloid-β Pathology by Targeting APP and BACE1 in an Animal Model of Alzheimer’s Disease. Mol. Ther. Nucleic Acids 2020, 19, 1219–1236. [Google Scholar] [CrossRef] [PubMed]
- Liu, W.; Liu, C.; Zhu, J.; Shu, P.; Yin, B.; Gong, Y.; Qiang, B.; Yuan, J.; Peng, X. MicroRNA-16 targets amyloid precursor protein to potentially modulate Alzheimer’s-associated pathogenesis in SAMP8 mice. Neurobiol. Aging 2012, 33, 522–534. [Google Scholar] [CrossRef]
- Lee, S.T.; Chu, K.; Jung, K.H.; Kim, J.H.; Huh, J.Y.; Yoon, H.; Park, D.K.; Lim, J.Y.; Kim, J.M.; Jeon, D.; et al. MiR-206 regulates brain-derived neurotrophic factor in Alzheimer disease model. Ann. Neurol. 2012, 72, 269–277. [Google Scholar] [CrossRef]
- Wu, G.D.; Li, Z.H.; Li, X.; Zheng, T.; Zhang, D.K. microRNA-592 blockade inhibits oxidative stress injury in Alzheimer’s disease astrocytes via the KIAA0319-mediated Keap1/Nrf2/ARE signaling pathway. Exp. Neurol. 2020, 324, 113128. [Google Scholar] [CrossRef] [PubMed]
- Long, J.M.; Lahiri, D.K. MicroRNA-101 downregulates Alzheimer’s amyloid-β precursor protein levels in human cell cultures and is differentially expressed. Biochem. Biophys. Res. Commun. 2011, 404, 889–895. [Google Scholar] [CrossRef] [PubMed]
- Aarsland, D.; Creese, B.; Politis, M.; Chaudhuri, K.R.; Ffytche, D.H.; Weintraub, D.; Ballard, C. Cognitive decline in Parkinson disease. Nat. Rev. Neurol. 2017, 13, 217–231. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Reed, X.; Krohn, L.; Heilbron, K.; Bandres-Ciga, S.; Tan, M.; Gibbs, J.R.; Hernandez, D.G.; Kumaran, R.; Langston, R.; et al. Genetic modifiers of risk and age at onset in GBA associated Parkinson’s disease and Lewy body dementia. Brain 2020, 143, 234–248. [Google Scholar] [CrossRef] [PubMed]
- Blauwendraat, C.; Reed, X.; Kia, D.A.; Gan-Or, Z.; Lesage, S.; Pihlstrøm, L.; Guerreiro, R.; Gibbs, J.R.; Sabir, M.; Ahmed, S.; et al. Frequency of loss of function variants in LRRK2 in Parkinson disease. JAMA Neurol. 2018, 75, 1416–1422. [Google Scholar] [CrossRef]
- Cho, H.J.; Liu, G.; Jin, S.M.; Parisiadou, L.; Xie, C.; Yu, J.; Sun, L.; Ma, B.; Ding, J.; Vancraenenbroeck, R.; et al. Microrna-205 regulates the expression of parkinson’s disease-related leucine-rich repeat kinase 2 protein. Hum. Mol. Genet. 2013, 22, 608–620. [Google Scholar] [CrossRef]
- Khodr, C.E.; Becerra, A.; Han, Y.; Bohn, M.C. Targeting alpha-synuclein with a microRNA-embedded silencing vector in the rat substantia nigra: Positive and negative effects. Brain Res. 2014, 1550, 47–60. [Google Scholar] [CrossRef]
- Li, D.; Yang, H.; Ma, J.; Luo, S.; Chen, S.; Gu, Q. MicroRNA-30e regulates neuroinflammation in MPTP model of Parkinson’s disease by targeting Nlrp3. Hum. Cell 2018, 31, 106–115. [Google Scholar] [CrossRef]
- Wang, H.; Ye, Y.; Zhu, Z.; Mo, L.; Lin, C.; Wang, Q.; Wang, H.; Gong, X.; He, X.; Lu, G.; et al. MiR-124 regulates apoptosis and autophagy process in MPTP model of Parkinson’s disease by targeting to bim. Brain Pathol. 2016, 26, 167–176. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Ferreira, L.; Bernardino, L. Traceable microRNA-124 loaded nanoparticles as a new promising therapeutic tool for Parkinson’s disease. Neurogenesis 2016, 3, e1256855. [Google Scholar] [CrossRef] [PubMed]
- Saraiva, C.; Paiva, J.; Santos, T.; Ferreira, L.; Bernardino, L. MicroRNA-124 loaded nanoparticles enhance brain repair in Parkinson’s disease. J. Control. Release 2016, 235, 291–305. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.; Song, Y.; Zhu, X. MicroRNA-181a regulates apoptosis and autophagy process in Parkinson’s disease by inhibiting p38 mitogen-activated protein kinase (MAPK)/c-Jun N-terminal kinases (JNK) signaling pathways. Med. Sci. Monit. 2017, 23, 1597–1606. [Google Scholar] [CrossRef] [PubMed]
- Wen, Z.; Zhang, J.; Tang, P.; Tu, N.; Wang, K.; Wu, G. Overexpression of miR-185 inhibits autophagy and apoptosis of dopaminergic neurons by regulating the AMPK/mTOR signaling pathway in Parkinson’s disease. Mol. Med. Rep. 2018, 17, 131–137. [Google Scholar] [CrossRef] [PubMed]
- Gao, J.X.; Li, Y.; Wang, S.N.; Chen, X.C.; Lin, L.L.; Zhang, H. Overexpression of microRNA-183 promotes apoptosis of substantia nigra neurons via the inhibition of OSMR in a mouse model of Parkinson’s disease. Int. J. Mol. Med. 2019, 43, 209–220. [Google Scholar] [CrossRef]
- Thome, A.D.; Harms, A.S.; Volpicelli-Daley, L.A.; Standaert, D.G. MicroRNA-155 regulates alpha-synuclein-induced inflammatory responses in models of Parkinson disease. J. Neurosci. 2016, 36, 2383–2390. [Google Scholar] [CrossRef]
- Zhou, Y.; Lu, M.; Du, R.-H.; Qiao, C.; Jiang, C.-Y.; Zhang, K.-Z.; Ding, J.-H.; Hu, G. MicroRNA-7 targets Nod-like receptor protein 3 inflammasome to modulate neuroinflammation in the pathogenesis of Parkinson’s disease. Mol. Neurodegener. 2016, 11, 28. [Google Scholar] [CrossRef]
- Geng, L.; Zhang, T.; Liu, W.; Chen, Y. miR-494-3p modulates the progression of in vitro and in vivo Parkinson’s disease models by targeting SIRT3. Neurosci. Lett. 2018, 675, 23–30. [Google Scholar] [CrossRef]
- Ansari, A.; Rahman, M.S.; Saha, S.K.; Saikot, F.K.; Deep, A.; Kim, K.H. Function of the SIRT3 mitochondrial deacetylase in cellular physiology, cancer, and neurodegenerative disease. Aging Cell 2017, 16, 4–16. [Google Scholar] [CrossRef]
- Wang, Z.; Sun, L.; Jia, K.; Wang, H.; Wang, X. miR-9-5p modulates the progression of Parkinson’s disease by targeting SIRT1. Neurosci. Lett. 2019, 701, 226–233. [Google Scholar] [CrossRef] [PubMed]
- Dong, Y.; Han, L.L.; Xu, Z.X. Suppressed microRNA-96 inhibits iNOS expression and dopaminergic neuron apoptosis through inactivating the MAPK signaling pathway by targeting CACNG5 in mice with Parkinson’s disease. Mol. Med. 2018, 24, 61. [Google Scholar] [CrossRef] [PubMed]
- Yang, X.; Zhang, M.; Wei, M.; Wang, A.; Deng, Y.; Cao, H. MicroRNA-216a inhibits neuronal apoptosis in a cellular Parkinson’s disease model by targeting Bax. Metab. Brain Dis. 2020, 35, 627–635. [Google Scholar] [CrossRef] [PubMed]
- Roos, R.A.C. Huntington’s disease: A clinical review. Orphanet J. Rare Dis. 2010, 5, 40. [Google Scholar] [CrossRef] [PubMed]
- Nopoulos, P.C. Huntington disease: A single-gene degenerative disorder of the striatum. Dialogues Clin. Neurosci. 2016, 18, 91–98. [Google Scholar]
- McColgan, P.; Tabrizi, S.J. Huntington’s disease: A clinical review. Eur. J. Neurol. 2018, 25, 24–34. [Google Scholar] [CrossRef]
- Zhang, Y.; Friedlander, R.M. Using non-coding small RNAs to develop therapies for Huntington’s disease. Gene Ther. 2011, 18, 1139–1149. [Google Scholar] [CrossRef] [PubMed]
- McBride, J.L.; Pitzer, M.R.; Boudreau, R.L.; Dufour, B.; Hobbs, T.; Ojeda, S.R.; Davidson, B.L. Preclinical safety of RNAi-mediated HTT suppression in the rhesus macaque as a potential therapy for Huntington’s disease. Mol. Ther. 2011, 19, 2152–2162. [Google Scholar] [CrossRef]
- Boudreau, R.L.; McBride, J.L.; Martins, I.; Shen, S.; Xing, Y.; Carter, B.J.; Davidson, B.L. Nonallele-specific silencing of mutant and wild-type huntingtin demonstrates therapeutic efficacy in Huntington’s disease mice. Mol. Ther. 2009, 17, 1053–1063. [Google Scholar] [CrossRef]
- Monteys, A.M.; Wilson, M.J.; Boudreau, R.L.; Spengler, R.M.; Davidson, B.L. Artificial miRNAs targeting mutant huntingtin show preferential silencing in vitro and in vivo. Mol. Ther. Nucleic Acids 2015, 4, e234. [Google Scholar] [CrossRef]
- Ban, J.J.; Chung, J.Y.; Lee, M.; Im, W.; Kim, M. MicroRNA-27a reduces mutant hutingtin aggregation in an in vitro model of Huntington’s disease. Biochem. Biophys. Res. Commun. 2017, 488, 316–321. [Google Scholar] [CrossRef]
- Cheng, P.H.; Li, C.L.; Chang, Y.F.; Tsai, S.J.; Lai, Y.Y.; Chan, A.W.S.; Chen, C.M.; Yang, S.H. MiR-196a ameliorates phenotypes of huntington disease in cell, transgenic mouse, and induced pluripotent stem cell models. Am. J. Hum. Genet. 2013, 93, 306–312. [Google Scholar] [CrossRef] [PubMed]
- Her, L.S.; Mao, S.H.; Chang, C.Y.; Cheng, P.H.; Chang, Y.F.; Yang, H.I.; Chen, C.M.; Yang, S.H. miR-196a enhances neuronal morphology through suppressing RANBP10 to provide neuroprotection in Huntington’s disease. Theranostics 2017, 7, 2452–2462. [Google Scholar] [CrossRef] [PubMed]
- Fukuoka, M.; Takahashi, M.; Fujita, H.; Chiyo, T.; Popiel, H.A.; Watanabe, S.; Furuya, H.; Murata, M.; Wada, K.; Okada, T.; et al. Supplemental Treatment for Huntington’s Disease with miR-132 that Is Deficient in Huntington’s Disease Brain. Mol. Ther. Nucleic Acids 2018, 11, 79–90. [Google Scholar] [CrossRef]
- Lee, S.-T.; Im, W.; Ban, J.-J.; Lee, M.; Jung, K.-H.; Lee, S.K.; Chu, K.; Kim, M. Exosome-Based Delivery of miR-124 in a Huntington’s Disease Model. J. Mov. Disord. 2017, 10, 45–52. [Google Scholar] [CrossRef] [PubMed]
- Liu, T.; Im, W.; Mook-Jung, I.; Kim, M. MicroRNA-124 slows down the progression of huntington’s disease by promoting neurogenesis in the striatum. Neural Regen. Res. 2015, 10, 786–791. [Google Scholar] [CrossRef]
- Jovicic, A.; Zaldivar Jolissaint, J.F.; Moser, R.; Silva Santos, M.D.; Luthi-Carter, R. MicroRNA-22 (miR-22) Overexpression Is Neuroprotective via General Anti-Apoptotic Effects and May also Target Specific Huntington’s Disease-Related Mechanisms. PLoS ONE 2013, 8, e54222. [Google Scholar] [CrossRef] [PubMed]
- Pfister, E.L.; Dinardo, N.; Mondo, E.; Borel, F.; Conroy, F.; Fraser, C.; Gernoux, G.; Han, X.; Hu, D.; Johnson, E.; et al. Artificial miRNAs Reduce Human Mutant Huntingtin Throughout the Striatum in a Transgenic Sheep Model of Huntington’s Disease. Hum. Gene Ther. 2018, 29, 663–673. [Google Scholar] [CrossRef]
- Miniarikova, J.; Zimmer, V.; Martier, R.; Brouwers, C.C.; Pythoud, C.; Richetin, K.; Rey, M.; Lubelski, J.; Evers, M.M.; Van Deventer, S.J.; et al. AAV5-miHTT gene therapy demonstrates suppression of mutant huntingtin aggregation and neuronal dysfunction in a rat model of Huntington’s disease. Gene Ther. 2017, 24, 630–639. [Google Scholar] [CrossRef]
- Evers, M.M.; Miniarikova, J.; Juhas, S.; Vallès, A.; Bohuslavova, B.; Juhasova, J.; Skalnikova, H.K.; Vodicka, P.; Valekova, I.; Brouwers, C.; et al. AAV5-miHTT Gene Therapy Demonstrates Broad Distribution and Strong Human Mutant Huntingtin Lowering in a Huntington’s Disease Minipig Model. Mol. Ther. 2018, 26, 2163–2177. [Google Scholar] [CrossRef]
- Spronck, E.A.; Brouwers, C.C.; Vallès, A.; de Haan, M.; Petry, H.; van Deventer, S.J.; Konstantinova, P.; Evers, M.M. AAV5-miHTT Gene Therapy Demonstrates Sustained Huntingtin Lowering and Functional Improvement in Huntington Disease Mouse Models. Mol. Ther. Methods Clin. Dev. 2019, 13, 334–343. [Google Scholar] [CrossRef] [PubMed]
- Caron, N.S.; Southwell, A.L.; Brouwers, C.C.; Cengio, L.D.; Xie, Y.; Black, H.F.; Anderson, L.M.; Ko, S.; Zhu, X.; Van Deventer, S.J.; et al. Potent and sustained huntingtin lowering via AAV5 encoding miRNA preserves striatal volume and cognitive function in a humanized mouse model of Huntington disease. Nucleic Acids Res. 2020, 48, 36–54. [Google Scholar] [CrossRef] [PubMed]
- Brown, R.H.; Al-Chalabi, A. Amyotrophic Lateral Sclerosis. N. Engl. J. Med. 2017, 377, 162–172. [Google Scholar] [CrossRef]
- Van Es, M.A.; Hardiman, O.; Chio, A.; Al-Chalabi, A.; Pasterkamp, R.J.; Veldink, J.H.; van den Berg, L.H. Amyotrophic lateral sclerosis. Lancet 2017, 390, 2084–2098. [Google Scholar] [CrossRef]
- Martin, S.; Al Khleifat, A.; Al-Chalabi, A. What causes amyotrophic lateral sclerosis? F1000Research 2017, 6, 371. [Google Scholar] [CrossRef] [PubMed]
- De Felice, B.; Guida, M.; Guida, M.; Coppola, C.; De Mieri, G.; Cotrufo, R. A miRNA signature in leukocytes from sporadic amyotrophic lateral sclerosis. Gene 2012, 508, 35–40. [Google Scholar] [CrossRef]
- Ricci, C.; Marzocchi, C.; Battistini, S. MicroRNAs as Biomarkers in Amyotrophic Lateral Sclerosis. Cells 2018, 7, 219. [Google Scholar] [CrossRef] [PubMed]
- Kovanda, A.; Leonardis, L.; Zidar, J.; Koritnik, B.; Dolenc-Groselj, L.; Ristic Kovacic, S.; Curk, T.; Rogelj, B. Differential expression of microRNAs and other small RNAs in muscle tissue of patients with ALS and healthy age-matched controls. Sci. Rep. 2018, 8, 5609. [Google Scholar] [CrossRef]
- Philips, T.; Rothstein, J.D. Rodent Models of Amyotrophic Lateral Sclerosis. Curr. Protoc. Pharmacol. 2015, 69, 5.67.1–5.67.21. [Google Scholar] [CrossRef] [PubMed]
- Martier, R.; Liefhebber, J.M.; Miniarikova, J.; van der Zon, T.; Snapper, J.; Kolder, I.; Petry, H.; van Deventer, S.J.; Evers, M.M.; Konstantinova, P. Artificial MicroRNAs Targeting C9orf72 Can Reduce Accumulation of Intra-nuclear Transcripts in ALS and FTD Patients. Mol. Ther. Nucleic Acids 2019, 14, 593–608. [Google Scholar] [CrossRef] [PubMed]
- Koval, E.D.; Shaner, C.; Zhang, P.; du Maine, X.; Fischer, K.; Tay, J.; Chau, B.N.; Wu, G.F.; Miller, T.M. Method for widespread microRNA-155 inhibition prolongs survival in ALS-model mice. Hum. Mol. Genet. 2013, 22, 4127–4135. [Google Scholar] [CrossRef] [PubMed]
- Nolan, K.; Mitchem, M.R.; Jimenez-Mateos, E.M.; Henshall, D.C.; Concannon, C.G.; Prehn, J.H.M. Increased Expression of MicroRNA-29a in ALS Mice: Functional Analysis of Its Inhibition. J. Mol. Neurosci. 2014, 53, 231–241. [Google Scholar] [CrossRef] [PubMed]
- Shioya, M.; Obayashi, S.; Tabunoki, H.; Arima, K.; Saito, Y.; Ishida, T.; Satoh, J. Aberrant microRNA expression in the brains of neurodegenerative diseases: miR-29a decreased in Alzheimer disease brains targets neurone navigator 3. Neuropathol. Appl. Neurobiol. 2010, 36, 320–330. [Google Scholar] [CrossRef] [PubMed]
- Stoica, L.; Todeasa, S.H.; Cabrera, G.T.; Salameh, J.S.; ElMallah, M.K.; Mueller, C.; Brown Jr, R.H.; Sena-Esteves, M. Adeno-associated virus–delivered artificial microRNA extends survival and delays paralysis in an amyotrophic lateral sclerosis mouse model. Ann. Neurol. 2016, 79, 687–700. [Google Scholar] [CrossRef]
- Borel, F.; Gernoux, G.; Cardozo, B.; Metterville, J.P.; Cabrera, G.T.; Song, L.; Su, Q.; Gao, G.P.; Elmallah, M.K.; Brown, R.H.; et al. Therapeutic rAAVrh10 Mediated SOD1 Silencing in Adult SOD1(G93A) Mice and Nonhuman Primates. Hum. Gene Ther. 2016, 27, 19–31. [Google Scholar] [CrossRef]
- Keeler, A.M.; Zieger, M.; Semple, C.; Pucci, L.; Veinbachs, A.; Brown, R.H.; Mueller, C.; ElMallah, M.K. Intralingual and Intrapleural AAV Gene Therapy Prolongs Survival in a SOD1 ALS Mouse Model. Mol. Ther. Methods Clin. Dev. 2020, 17, 246–257. [Google Scholar] [CrossRef]
- Li, D.; Liu, C.; Yang, C.; Wang, D.; Wu, D.; Qi, Y.; Su, Q.; Gao, G.; Xu, Z.; Guo, Y. Slow Intrathecal Injection of rAAVrh10 Enhances its Transduction of Spinal Cord and Therapeutic Efficacy in a Mutant SOD1 Model of ALS. Neuroscience 2017, 365, 192–205. [Google Scholar] [CrossRef] [PubMed]
- La Rosa, P.; Bertini, E.S.; Piemonte, F. The NRF2 signaling network defines clinical biomarkers and therapeutic opportunity in Friedreich’s Ataxia. Int. J. Mol. Sci. 2020, 21, 916. [Google Scholar] [CrossRef]
- Dantham, S.; Srivastava, A.K.; Gulati, S.; Rajeswari, M.R. Differentially Regulated Cell-Free MicroRNAs in the Plasma of Friedreich’s Ataxia Patients and Their Association with Disease Pathology. Neuropediatrics 2018, 49, 35–43. [Google Scholar] [CrossRef]
- Pandolfo, M. Friedreich ataxia: The clinical picture. J. Neurol. 2009, 256, 3–8. [Google Scholar] [CrossRef]
- Koeppen, A.H. Friedreich’s ataxia: Pathology, pathogenesis, and molecular genetics. J. Neurol. Sci. 2011, 303, 1–12. [Google Scholar] [CrossRef] [PubMed]
- Harding, A.E. Friedreich’s ataxia: A clinical and genetic study of 90 families with an analysis of early diagnostic criteria and intrafamilial clustering of clinical features. Brain 1981, 104, 589–620. [Google Scholar] [CrossRef] [PubMed]
- Mahishi, L.H.; Hart, R.P.; Lynch, D.R.; Ratan, R.R. miR-886-3p levels are elevated in Friedreich ataxia. J. Neurosci. 2012, 32, 9369–9373. [Google Scholar] [CrossRef]
- Shen, J.; Zhou, W.; Bi, N.; Song, Y.-M.; Zhang, F.-Q.; Zhan, Q.-M.; Wang, L.-H. MicroRNA-886-3P functions as a tumor suppressor in small cell lung cancer. Cancer Biol. Ther. 2018, 19, 1185–1192. [Google Scholar] [CrossRef] [PubMed]
- Groen, E.J.N.; Talbot, K.; Gillingwater, T.H. Advances in therapy for spinal muscular atrophy: Promises and challenges. Nat. Rev. Neurol. 2018, 14, 214–224. [Google Scholar] [CrossRef] [PubMed]
- Schellino, R.; Boido, M.; Vercelli, A. JNK Signaling Pathway Involvement in Spinal Cord Neuron Development and Death. Cells 2019, 8, 1576. [Google Scholar] [CrossRef]
- Haramati, S.; Chapnik, E.; Sztainberg, Y.; Eilam, R.; Zwang, R.; Gershoni, N.; McGlinn, E.; Heiser, P.W.; Wills, A.-M.; Wirguin, I.; et al. miRNA malfunction causes spinal motor neuron disease. Proc. Natl. Acad. Sci. USA 2010, 107, 13111–13116. [Google Scholar] [CrossRef]
- Magri, F.; Vanoli, F.; Corti, S. miRNA in spinal muscular atrophy pathogenesis and therapy. J. Cell. Mol. Med. 2018, 22, 755–767. [Google Scholar] [CrossRef]
- Luchetti, A.; Ciafrè, S.; Murdocca, M.; Malgieri, A.; Masotti, A.; Sanchez, M.; Farace, M.; Novelli, G.; Sangiuolo, F. A Perturbed MicroRNA Expression Pattern Characterizes Embryonic Neural Stem Cells Derived from a Severe Mouse Model of Spinal Muscular Atrophy (SMA). Int. J. Mol. Sci. 2015, 16, 18312–18327. [Google Scholar] [CrossRef]
- Amin, N.D.; Bai, G.; Klug, J.R.; Bonanomi, D.; Pankratz, M.T.; Gifford, W.D.; Hinckley, C.A.; Sternfeld, M.J.; Driscoll, S.P.; Dominguez, B.; et al. Loss of motoneuron-specific microRNA-218 causes systemic neuromuscular failure. Science 2015, 350, 1525–1529. [Google Scholar] [CrossRef]
- Sison, S.L.; Patitucci, T.N.; Seminary, E.R.; Villalon, E.; Lorson, C.L.; Ebert, A.D. Astrocyte-produced miR-146a as a mediator of motor neuron loss in spinal muscular atrophy. Hum. Mol. Genet. 2017, 26, 3409–3420. [Google Scholar] [CrossRef]
- Wertz, M.H.; Winden, K.; Neveu, P.; Ng, S.-Y.; Ercan, E.; Sahin, M. Cell-type-specific miR-431 dysregulation in a motor neuron model of spinal muscular atrophy. Hum. Mol. Genet. 2016, 25, 2168–2181. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, V.; Boido, M.; De Amicis, E.; Piras, A.; Vercelli, A. Expression of Muscle-Specific MiRNA 206 in the Progression of Disease in a Murine SMA Model. PLoS ONE 2015, 10, e0128560. [Google Scholar] [CrossRef] [PubMed]
- Valsecchi, V.; Anzilotti, S.; Serani, A.; Laudati, G.; Brancaccio, P.; Guida, N.; Cuomo, O.; Pignataro, G.; Annunziato, L. miR-206 Reduces the Severity of Motor Neuron Degeneration in the Facial Nuclei of the Brainstem in a Mouse Model of SMA. Mol. Ther. 2020, 28, 1154–1166. [Google Scholar] [CrossRef] [PubMed]
- Greaves, C.V.; Rohrer, J.D. An update on genetic frontotemporal dementia. J. Neurol. 2019, 266, 2075–2086. [Google Scholar] [CrossRef] [PubMed]
- Olney, N.T.; Spina, S.; Miller, B.L. Frontotemporal Dementia. Neurol. Clin. 2017, 35, 339–374. [Google Scholar] [CrossRef]
- Goldman, J.S.; Van Deerlin, V.M. Alzheimer’s Disease and Frontotemporal Dementia: The Current State of Genetics and Genetic Testing Since the Advent of Next-Generation Sequencing. Mol. Diagnosis Ther. 2018, 22, 505–513. [Google Scholar] [CrossRef]
- Bennion Callister, J.; Pickering-Brown, S.M. Pathogenesis/genetics of frontotemporal dementia and how it relates to ALS. Exp. Neurol. 2014, 262, 84–90. [Google Scholar] [CrossRef]
- Bott, N.T.; Radke, A.; Stephens, M.L.; Kramer, J.H. Frontotemporal dementia: Diagnosis, deficits and management. Neurodegener. Dis. Manag. 2014, 4, 439–454. [Google Scholar] [CrossRef]
- Olszewska, D.A.; Lonergan, R.; Fallon, E.M.; Lynch, T. Genetics of Frontotemporal Dementia. Curr. Neurol. Neurosci. Rep. 2016, 16, 107. [Google Scholar] [CrossRef]
- Piscopo, P.; Albani, D.; Castellano, A.E.; Forloni, G.; Confaloni, A. Frontotemporal lobar degeneration and microRNAs. Front. Aging Neurosci. 2016, 8, 7. [Google Scholar] [CrossRef] [PubMed]
- Rabinovici, G.D.; Miller, B.L. Frontotemporal lobar degeneration: Epidemiology, pathophysiology, diagnosis and management. CNS Drugs 2010, 24, 375–398. [Google Scholar] [CrossRef] [PubMed]
- Kocerha, J.; Kouri, N.; Baker, M.; Finch, N.C.; DeJesus-Hernandez, M.; Gonzalez, J.; Chidamparam, K.; Josephs, K.A.; Boeve, B.F.; Graff-Radford, N.R.; et al. Altered microRNA expression in frontotemporal lobar degeneration with TDP-43 pathology caused by progranulin mutations. BMC Genom. 2011, 12, 527. [Google Scholar] [CrossRef] [PubMed]
- Jiao, J.; Herl, L.D.; Farese, R.V.; Gao, F.-B. MicroRNA-29b Regulates the Expression Level of Human Progranulin, a Secreted Glycoprotein Implicated in Frontotemporal Dementia. PLoS ONE 2010, 5, e10551. [Google Scholar] [CrossRef] [PubMed]
- Gascon, E.; Lynch, K.; Ruan, H.; Almeida, S.; Verheyden, J.M.; Seeley, W.W.; Dickson, D.W.; Petrucelli, L.; Sun, D.; Jiao, J.; et al. Alterations in microRNA-124 and AMPA receptors contribute to social behavioral deficits in frontotemporal dementia. Nat. Med. 2014, 20, 1444–1451. [Google Scholar] [CrossRef] [PubMed]
- Chen-Plotkin, A.S.; Unger, T.L.; Gallagher, M.D.; Bill, E.; Kwong, L.K.; Volpicelli-Daley, L.; Busch, J.I.; Akle, S.; Grossman, M.; Van Deerlin, V.; et al. TMEM106B, the risk gene for frontotemporal dementia, is regulated by the microRNA-132/212 cluster and affects progranulin pathways. J. Neurosci. 2012, 32, 11213–11227. [Google Scholar] [CrossRef]
- Wang, W.X.; Wilfred, B.R.; Madathil, S.K.; Tang, G.; Hu, Y.; Dimayuga, J.; Stromberg, A.J.; Huang, Q.; Saatman, K.E.; Nelson, P.T. miR-107 regulates granulin/progranulin with implications for traumatic brain injury and neurodegenerative disease. Am. J. Pathol. 2010, 177, 334–345. [Google Scholar] [CrossRef]
- Rademakers, R.; Eriksen, J.L.; Baker, M.; Robinson, T.; Ahmed, Z.; Lincoln, S.J.; Finch, N.; Rutherford, N.J.; Crook, R.J.; Josephs, K.A.; et al. Common variation in the miR-659 binding-site of GRN is a major risk factor for TDP43-positive frontotemporal dementia. Hum. Mol. Genet. 2008, 17, 3631–3642. [Google Scholar] [CrossRef]
- Hashimoto, Y.; Akiyama, Y.; Yuasa, Y. Multiple-to-multiple relationships between microRNAs and target genes in gastric cancer. PLoS ONE 2013, 8, e62589. [Google Scholar] [CrossRef] [PubMed]
- Titze-de-Almeida, S.S.; Soto-Sánchez, C.; Fernandez, E.; Koprich, J.B.; Brotchie, J.M.; Titze-de-Almeida, R. The Promise and Challenges of Developing miRNA-Based Therapeutics for Parkinson’s Disease. Cells 2020, 9, 841. [Google Scholar] [CrossRef]
- Guo, L.; Zhang, Q.; Ma, X.; Wang, J.; Liang, T. miRNA and mRNA expression analysis reveals potential sex-biased miRNA expression. Sci. Rep. 2017, 7, 39812. [Google Scholar] [CrossRef] [PubMed]
- Keller, A.; Rounge, T.; Backes, C.; Ludwig, N.; Gislefoss, R.; Leidinger, P.; Langseth, H.; Meese, E. Sources to variability in circulating human miRNA signatures. RNA Biol. 2017, 14, 1791–1798. [Google Scholar] [CrossRef] [PubMed]



{kind=link}
{kind=link}
| Disorder | Targeted MiRNA | Antagonist or Mimics | Target Gene | Delivery Method | Biological Model or Tissue | Functional Implication | Reference |
|---|---|---|---|---|---|---|---|
| Alzheimer’s Disease (AD) | miR-16 | Mimics | APP, BACE1 | Liposomes | HEK293 cells | Reduced expression of BACE1 and APP | [42] |
| miR-16,15,195 | HEK293 cells overexpressing APP Swedish mutation | Suppression of Aβ production | |||||
| miR-16 | Neuro2a cells | Reduction of BACE1 and APP protein expression, decrease in Tau phosphorylation | |||||
| HT22 cells | Reduction of BACE1 and APP protein expression | ||||||
| APP, BACE1, Tau | Oligonucleotide | Mice | APP downregulated in cortex, brainstem, and striatum. BACE1 reduced in the hippocampus, brainstem, and striatum. Tau downregulation in the hippocampus, brainstem, and striatum. ERK1 downregulated in the hippocampus and cortex. Nicastrin, Gfap, and Aif1 downregulation | ||||
| miR-132 | Mimics | Tau | Liposomes | Neuro2a cells cotransfected with a reporter construct containing mouse 3’UTR | Downregulation of tau expression | [43] | |
| Neuro2a cells | |||||||
| Cell transfection | 3xTg-AD mice | Improve long term memory and reduction of phosphorylated tau | |||||
| miR-132 | Mimics | ITPKB | Oligonucleotide | Mice | Amelioration of amyloidosis and Tau hyperphosphorylation | [44] | |
| miR-124 | Mimics | BACE1 | Oligonucleotide | Hippocampal neurons, PC12 cells | Reduction of BACE1 expression and Aβ production | [45] | |
| miR-193a-3p | Mimics | PTEN | Liposomes | PC12 cells, SH-SY5Y cells | Attenuation of β-amyloid induced neurotoxicity | [46] | |
| miR-107 | Mimics | PDCD10 | Liposomes | PC12 cells, SH-SY5Y cells | Increase of cell viability, Reduction of Caspase-3 activity, LDH release, and ROS levels, increase SOD levels. Reduction of induced cytotoxicity | [47] | |
| Lentivirus | 6-OHDA induced mouse model | Suppression of 6-OHDA induced motor disorder | |||||
| miR-326 | Mimics | VAV1 | Lentivirus | Mice | Improvement in cognitive function, inhibition of JNK signaling pathway, decrease in Aβ deposition, inhibit protein expression of VAV1 | [48] | |
| miR-34c | Inhibitor | SIRT1 | Hippocampal injection | C57Bl/6J mice | Enhance memory, reinstate learning behavior and SIRT1 levels | [49] | |
| miR-34c | Inhibitor | SYT1 | Intranasal administration | SAMP8 mice | Increase levels of SYT1, amelioration of cognitive function, enhance memory function, recovery of dendritic spine density | [50] | |
| miR-188-5p | Mimics | NRP2 | Oligonucleotide | Primary hippocampal neurons from 5XFAD mice | Reverse synaptic damage induced by Aβ 1-42 and reduction in dendritic spine density, enhance cognitive function impairments | [51] | |
| miR-149 | Mimics | APP, BACE1, Tau | Cell transfection | SH-SY5Y cells | Resilience to Aβ oligomer | [52] | |
| mir-485 | BACE1, Tau | ||||||
| miR-485 | APP, Syn1, Ppp3ca, Mapt, Snap25, Snca, DNM1 | ICV | C57B6 mice | Upregulation of APP, Syn1, Ppp3ca, Mapt, Snap25, Snca and protection against Aβ oligomers in males, downregulation of DNM1, Mapt and Snca in females | |||
| miR-4723 | Snap25 | Downregulation of Snap25 in males | |||||
| miR-149 | Creb1 | Increase of Creb1 in males | |||||
| miR-200b,miR-200c | Mimics | S6K1 | Liposomes | Primary neuronal cells and SH-SY5Y cells | Suppression of cytotoxic damage caused by Aβ | [53] | |
| miR-200b/c | Lipid nanoparticle | C57BL/6J and Tg2576 mice | Defensive effect against Aβ-induced toxicity | ||||
| miR-31 | Mimics | BACE1 | Cell transfection | HT-22 cells | Decrease in BACE1 mRNA levels | [54] | |
| APP, BACE1 | Lentivirus | 3xTg-AD mice | Amelioration of deficits in memory, reduced anxiety and could prevent the progression of cognitive decline | ||||
| miR-16 | Mimics | APP | Liposomes | Neuroblastoma2a, NIH3T3 murine cells | Reduction of APP protein level | [55] | |
| Infusion | SAMP and SAMR1 mice | ||||||
| miR-592 | Inhibitor | KIAA0319 | Liposomes | Astrocytes | Promotion of astrocytes viability, decrease oxidative stress injury in ASTs, reduction in C-keap1 expression, activation of Keap1/Nrf2/ARE signaling pathway | [57] | |
| miR-101 | Mimics | APP | Liposomes | HeLa cells | Reduction of APP levels | [58] | |
| Liposomes | Hela, U373, PC12 cells | ||||||
| miR-206 | Inhibitor | BDNF | Cell transfection | Neuro2a cells | Increase of BDNF protein levels | [56] | |
| Primary hippocampal neurons | Increase of dendritic spine density, enhanced hippocampal neurogenesis | ||||||
| Intranasal administration | Mice | Increase DFNF protein levels, enhanced synaptogenesis, and neurogenesis, improvement of memory function | |||||
| Parkinson’s Disease (PD) | miR-205 | Mimics | LRRK2 | Liposomes | Cell lines, primary neuron cultures, and mouse midbrain dopaminergic neurons | Reducted expression of LRRK2 and prevention of neurite outgrowth defects | [62] |
| mir30-hSNCA | Mimics | SNCA | AAV2/8 (unilateral SN injection) | Rats | Protection against the deficit in the forelimb and partial protection against TH-IR cell loss in the SN | [63] | |
| miR-30e | Mimics | NLRP3 | miR-30e agomir injected using a catheter | MPTP-treated PD mice | Reduction of neuroinflammation and lowered expression of α-synuclein | [64] | |
| miR-124 | Mimics | BIM | Exogenous delivery | MPTP-treated PD mice | Reduction of autophagy, apoptosis and lysosomal exhausting | [65] | |
| miR-124 | Mimics | BIM | Cell transfection | MPP+-intoxicated SH-SY5Y cells | Reduction of autophagy, apoptosis and lysosomal exhausting | [65] | |
| miR-124 | Mimics | Sox9 Jagged1 | Cell transfection with NPs | Neural stem cells and neuroblasts | Neuronal maturation and proliferation | [66,67] | |
| miR-124 | Mimics | Sox9 Jagged1 | Injection of NPs in the lateral ventricle | (6-OHDA) lesioned mice | Neuronal migration to the olfactory bulb and damaged striatum | [66,67] | |
| miR-181a | Mimics | Regulation of the p38 MAPK and JNK signaling pathways | Liposomes | Human MPP+-induced SK-N-SH neuroblastoma cells | Diminution of the autophagy process and apoptosis | [68] | |
| miR-185 | Mimics | Regulation of the AMPK/mTOR signaling pathway | Liposomes | MPTP-treated SH-SY5Y cells | Diminution of the autophagy process and apoptosis | [69] | |
| miR-183 | Mimics | OSMR | Liposomes | SN neurons from MPTP-treated PD mice | Apoptosis of SN neurons | [70] | |
| miR-155 | - | MHCII | - | Mice with an entire deletion of the miR-155 | Regulation of the inflammation and neurodegeneration | [71] | |
| miR-155 | Mimics | MHCII | Oligonucleotide treatment | Microglial cells | Inflammatory response associated with α-synuclein | [71] | |
| miR-7 | Mimics | NLPR3 | Stereotactical injection into the striatum | MPTP-treated PD mice | Regulation of the NLPR3 inflammasome activation and reduction in the degeneration of dopaminergic neurons | [72] | |
| miR-494-3p | Inhibitor | SIRT3 | Liposomes | MPP+-induced SH-SY5Y cells | Decrease of neurotoxicity and motor deterioration | [73] | |
| miR-9-5p | Inhibitor | SIRT1 | Liposomes | MPP+-induced SH-SY5Y cells | Decrease of neurotoxicity, apoptosis, oxidative stress and inflammation | [75] | |
| miR-96 | Inhibitor | CACNG5 | Injection of inhibitor | MPTP-treated PD mice | Inhibition of both apoptosis and iNOS by blocking the MAPK signaling pathway | [76] | |
| miR-96 | Inhibitor | CACNG5 | Liposomes | SH-SY5Y cells | Increment of nigral cells | [76] | |
| miR-216a | Mimics | Bax | Liposomes | MPP+-treated SK-SY5Y neuroblastoma cells | Decrease of neurotoxicity and apoptosis | [77] | |
| Huntington’s Disease (HD) | mi306.12v16G | Artificial miRNA | HTT | AAV2/1 (bilateral injection in the striata) | Transgenic HD mice | Absence of preferential silencing | [84] |
| mi268.5 | Artificial miRNA | HTT | AAV2/1 (bilateral injection in the striata) | Transgenic HD mice | Targeting of the mutant allele | [84] | |
| miR-27a | Mimics | MDR-1 | Cell transfection | Derived neuronal stem cells | Reduction in the accumulation of mHTT | [85] | |
| miR-196a | Mimics | HTT | Cell transfection | Human embryonic renal cells and neuroblastoma cells of mice | Decrease in the expression of mHTT | [86] | |
| miR-196a | Mimics | HTT | Lentiviral transgenesis | Transgenic HD mice | Decrease in the expression of mHTT | [86] | |
| miR-196a | Mimics | HTT | Lentiviral infection | HD-iPSCs | Decrease in the expression of mHTT | [86] | |
| miR-196a | Mimics | RANBP10 | Liposomes | Primary cortical neurons | Differentiation and development of neuronal cells | [87] | |
| miR-196a | Mimics | RANBP10 | The hsa-miR-196a-2 precursor was in the transgenic mice | Transgenic HD mice | Differentiation and development of neuronal cells | [87] | |
| miR-132 | Mimics | MeCP2 | AAV9 (intracranial injection) | Transgenic HD mice | Symptom improvement and life extension | [88] | |
| miR-124 | Mimics | RE1 silencing transcription factor | Exosomal delivery | Transgenic HD mice | Reduced expression of the RE1 silencing transcription factor | [89] | |
| miR-124 | Mimics | SOX9 | Striatal injection | Transgenic HD mice | Increase of neurogenesis in the striatum and cortex and neuronal survival | [90] | |
| miR-22 | Mimics | MAPK14 Trp53inp1 Rcor1 HDAC4 Rgs2 | Lentiviral infection | Cultures of primary cortical and striatal neurons | Apoptosis inhibition, decrease in the cumulation of HTT and targeting of different genes | [91] | |
| miHTT | Artificial miRNA | HTT | AAV9 (striatal and cortical injection) | Transgenic HD sheep | Decrease in the cumulation of mHTT in the neostriatum | [92] | |
| miHTT | Artificial miRNA | HTT | AAV5 (intracerebral administration) | Lentiviral HD rats | Reduction in the accumulation of mHTT | [93] | |
| miHTT | Artificial miRNA | HTT | AAV5 (intracranial administration) | Transgenic HD minipig | Decrease in the cumulation of mHTT | [94] | |
| miHTT | Artificial miRNA | HTT | AAV5 (intracranial injection) | Transgenic HD mice | Decrease in the cumulation of HTT, weight gain, longer life expectancy, and functional improvement | [95] | |
| miHTT | Artificial miRNA | HTT | AAV5 (intrastriatal administration) | Hu128/21 mouse model of HD | Suppression of HTT | [96] | |
| Amyotrophic Lateral Sclerosis (ALS) | miR-155 | Inhibitor (antagomir) | Src homology-2 domain-containing SHIP1 and suppressor of cytokine signaling-1 | ICV | SOD1G93A mice | Prolonged survival rate, slower disease progress | [105] |
| miR-29a | Inhibitor (antagomir) | NAV3, BACE1 | ICV | SOD1G93A mice | Increased lifespan | [106] | |
| amiRSOD1 | Artificial miRNA | SOD1 | AAV9 | SOD1G93A mice | Extended survival, delayed hindlimb paralysis, reduction of mutant human SOD1 | [108] | |
| miC | Artificial miRNA | C9orf72 | AAV5 | HEK293T cells and iPSC-derived neurons | Silencing of C9orf72 | [104] | |
| miR-SOD1 | Artificial miRNA | SOD1 | rAAVrh10 | Marmoset | Silencing of SOD1 | [109] | |
| miR-SOD1 | Artificial miRNA | SOD1 | AAVrh10 (Intralingual and Intrapleural injection) | SOD1G93A mice | Delayed disease onset, prolonged survival, improved breathing | [110] | |
| miR-SOD1 | Artificial miRNA | SOD1 | rAAVrh10 | SOD1G93A mice | Delayed disease onset, extended survival | [111] | |
| Friedreich’s Ataxia (FRDA) | miR-886-3p | Inhibitor | FXN | Cell transfection | Fibroblast | Increase frataxin levels | [117] |
| miR-124 | Mimics | FXN | Cell transfection | HEK-293 | Down-regulation of FRDA-3-UTR haplotype | [4] | |
| Spinal Muscular Atrophy (SMA) | miR-146a | Inhibitor | Chodl | Endo-Porter peptide | iPSC-derived motor neurons | Restoration of neurite length | [125] |
| miR-431 | Inhibitor | Chodl | Cell transfection | Motor neurons | Restoration of neurite length | [126] | |
| miR-206 | Mimics | HDAC4 | Liposomes | Myoblast cell line C2C12 | FGFBP1 transcriptional up-regulation | [127] | |
| miR-206 | Mimics | HDAC4 | ICV | Pups and mice | Down-regulation of NCX2, reduction in the severity and progress of SMA pathology | [128] | |
| Frontotemporal Dementia (FTD) | miR-29b | Mimics | GRN | Liposomes | NIH3T3 and Hek293 cells | Regulation of the expression of hPGRN | [138] |
| miR-124 | Mimics | Gria2 and Gria4 | AAV-GFP injection | tTA: CHMP2BIntron5 mice | Reduction of the expression of Gria2, Gria4 and improvement of social behavior | [139] | |
| miR-132/212 | Mimics | TMEM106B | Liposomes | HEK293 and SHSY5Y cells | Reduction of TMEM106B mRNA levels | [140] | |
| miR-107 | Mimics | GRN | Liposomes | H4 and HeLa cells | Reduction of GRN expression | [141] | |
| miR-659 | Mimics | GRN | Liposomes | M17 and N2A neuroblastoma cells | Reduction of GRN expression | [142] |
© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Paul, S.; Bravo Vázquez, L.A.; Pérez Uribe, S.; Roxana Reyes-Pérez, P.; Sharma, A. Current Status of microRNA-Based Therapeutic Approaches in Neurodegenerative Disorders. Cells 2020, 9, 1698. https://doi.org/10.3390/cells9071698
Paul S, Bravo Vázquez LA, Pérez Uribe S, Roxana Reyes-Pérez P, Sharma A. Current Status of microRNA-Based Therapeutic Approaches in Neurodegenerative Disorders. Cells. 2020; 9(7):1698. https://doi.org/10.3390/cells9071698
Chicago/Turabian StylePaul, Sujay, Luis Alberto Bravo Vázquez, Samantha Pérez Uribe, Paula Roxana Reyes-Pérez, and Ashutosh Sharma. 2020. "Current Status of microRNA-Based Therapeutic Approaches in Neurodegenerative Disorders" Cells 9, no. 7: 1698. https://doi.org/10.3390/cells9071698
APA StylePaul, S., Bravo Vázquez, L. A., Pérez Uribe, S., Roxana Reyes-Pérez, P., & Sharma, A. (2020). Current Status of microRNA-Based Therapeutic Approaches in Neurodegenerative Disorders. Cells, 9(7), 1698. https://doi.org/10.3390/cells9071698