Regorafenib Regulates AD Pathology, Neuroinflammation, and Dendritic Spinogenesis in Cells and a Mouse Model of AD


 , and
, and {kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
{kind=link}
Abstract
1. Introduction
2. Experimental Procedures
2.1. Ethics Statement
2.2. Cell Culture
2.3. Cell Viability Assay
2.4. mRNA Quantification by RT-PCR
2.5. Real-Time PCR (q-PCR)
2.6. Enzyme-Linked Immunosorbent Assay (ELISA)
2.7. Antibodies and Inhibitors
2.8. Western Blotting
2.9. Immunocytochemistry
2.10. Cytosolic and Nuclear Fractionation
2.11. Primary Microglial Cell and Astrocyte Culture
2.12. Animals
2.13. Immunohistochemistry
2.14. Golgi Staining
2.15. Statistical Analyses
3. Results
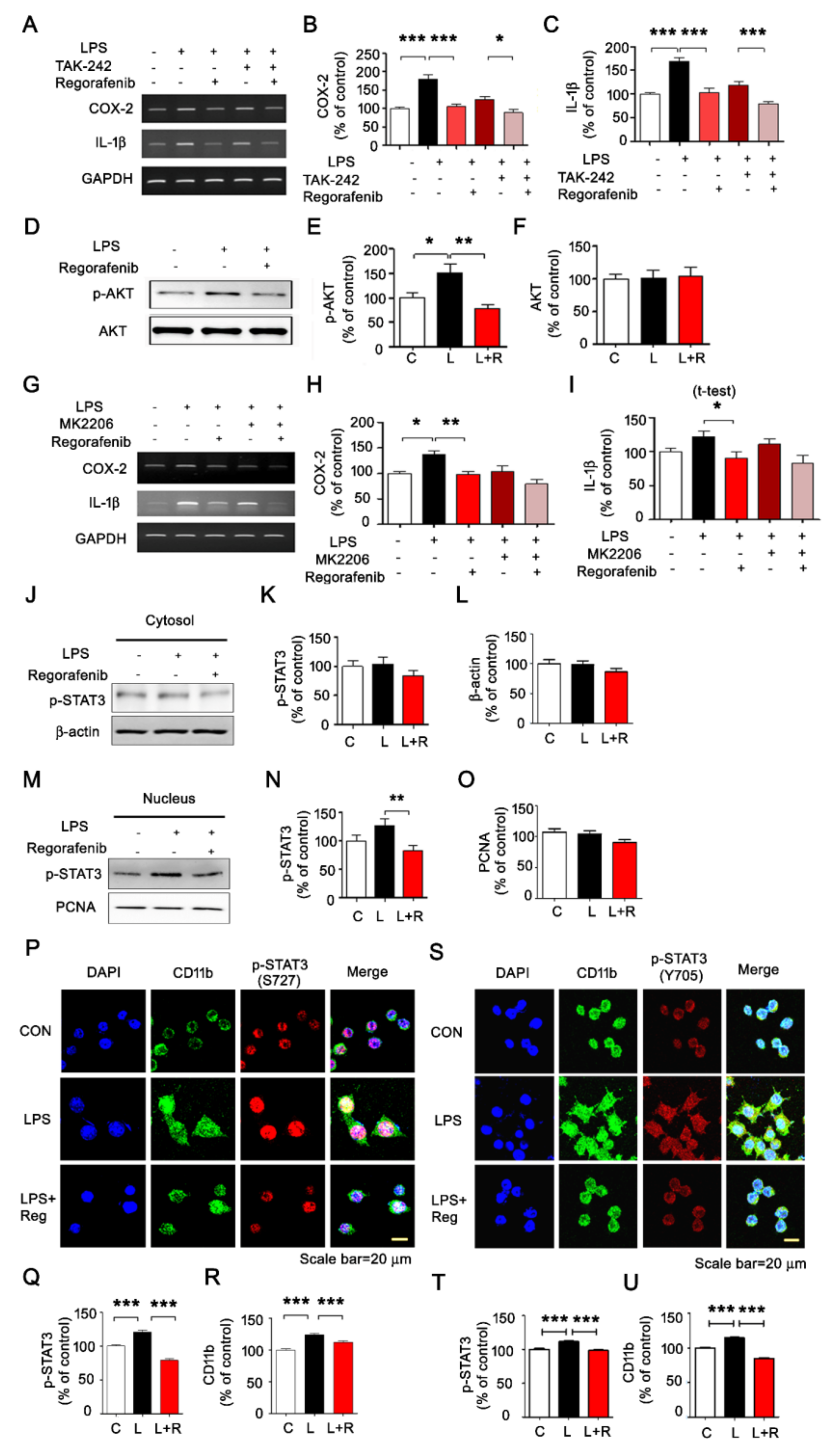
3.1. Regorafenib Regulates LPS-Induced Expression of Proinflammatory Cytokines
3.2. Regorafenib Modulates AKT/STAT3 Signaling to Alter LPS-Induced Neuroinflammatory Responses
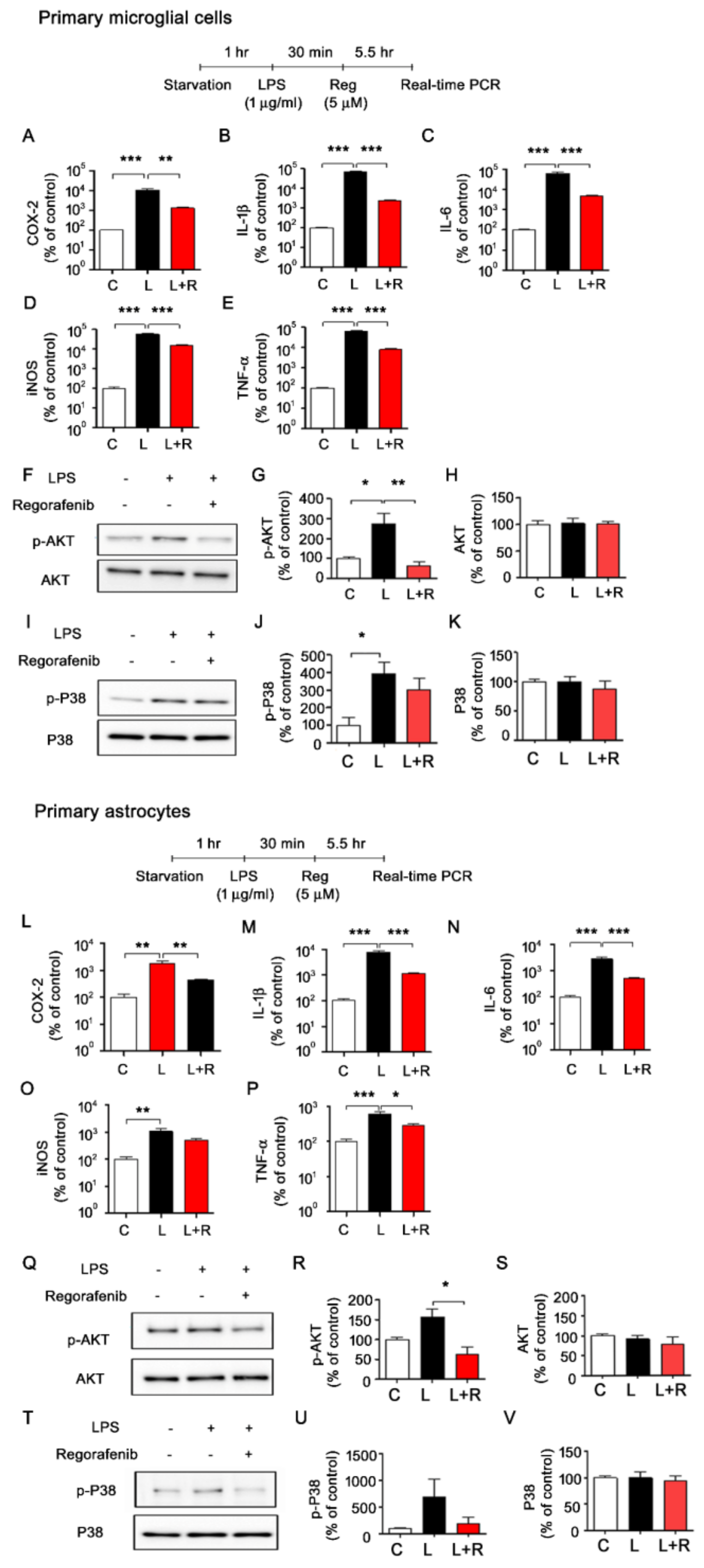
3.3. Regorafenib Alters LPS-Induced Expression of Proinflammatory Cytokines in Primary Microglia and Astrocytes
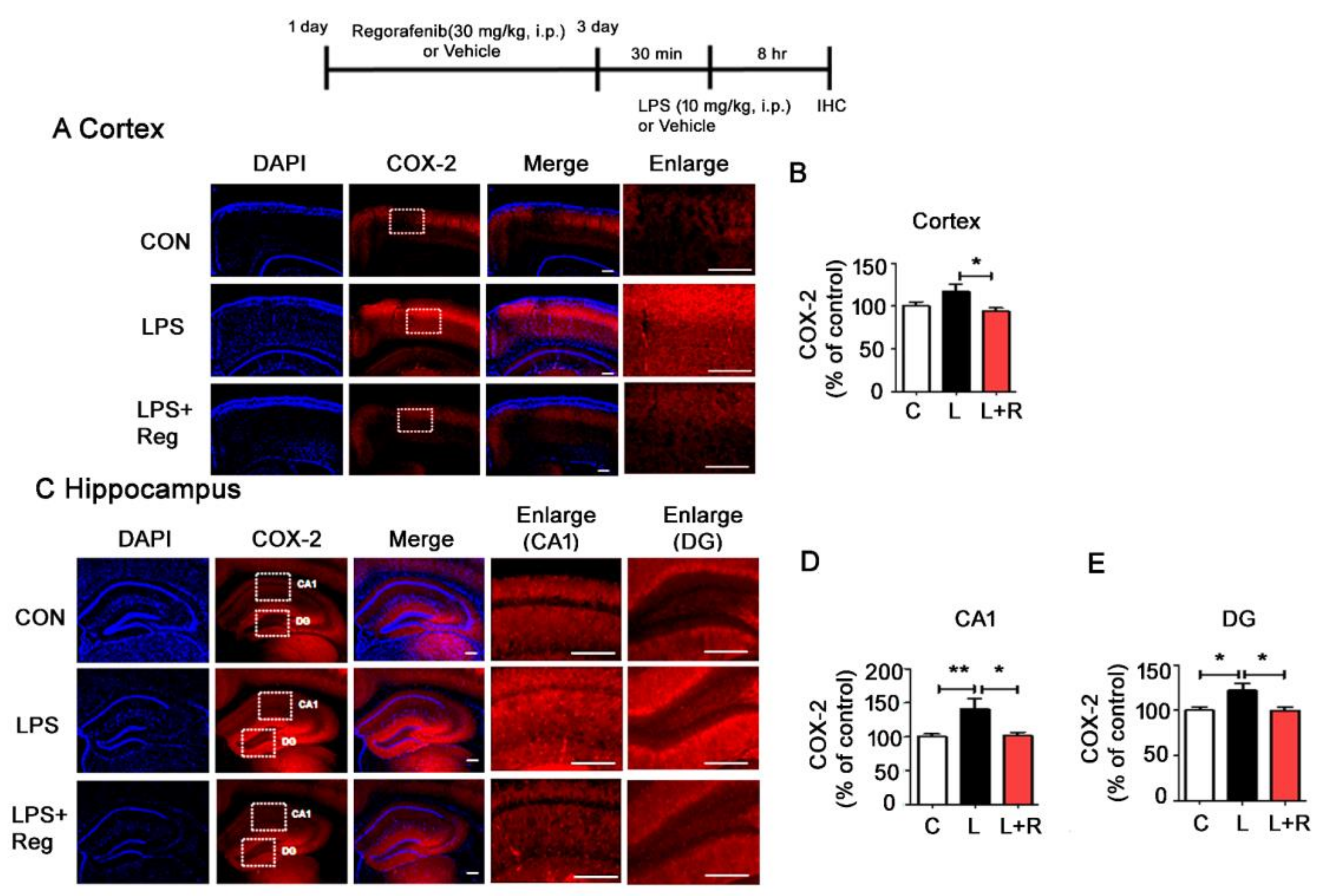
3.4. Regorafenib Attenuates LPS-Induced Glial Activation and the Expression of Proinflammatory Cytokines in Wild-Type Mice
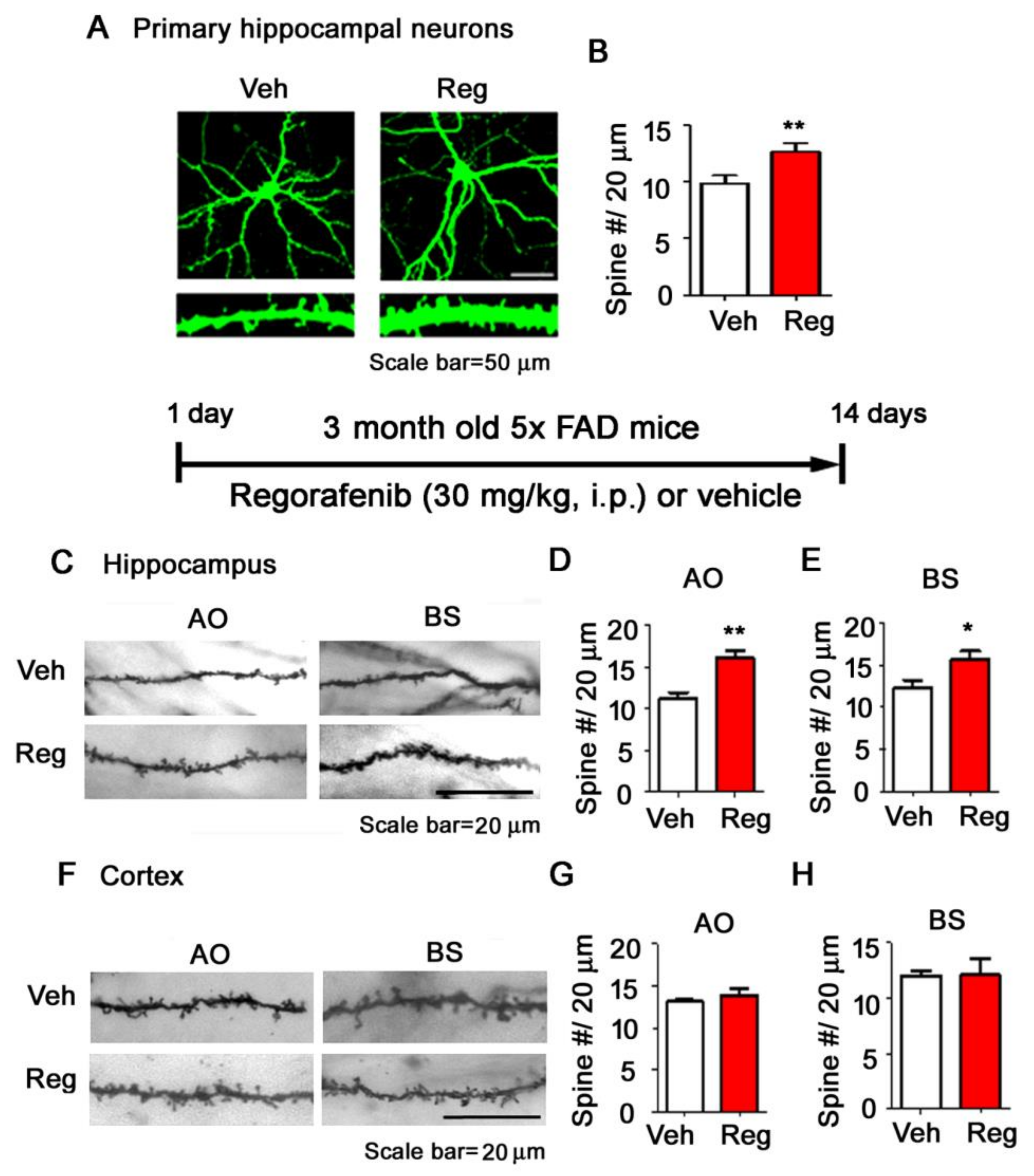
3.5. Regorafenib Significantly Increases Dendritic Spine Density in Primary Hippocampal Neurons and in the Brains of 5x FAD Mice
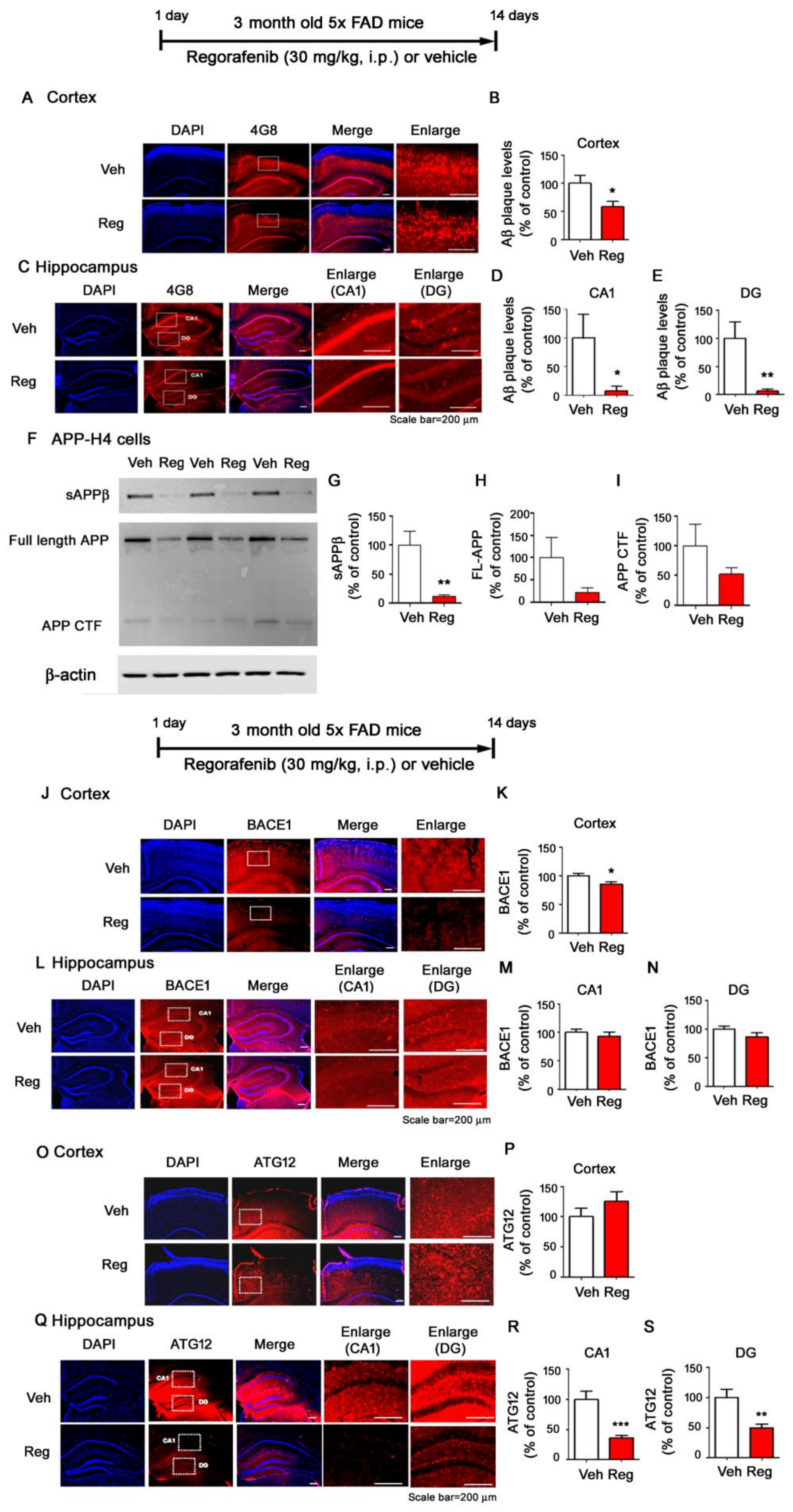
3.6. Regorafenib Significantly Downregulates Aβ Plaque Levels in 5x FAD Mice
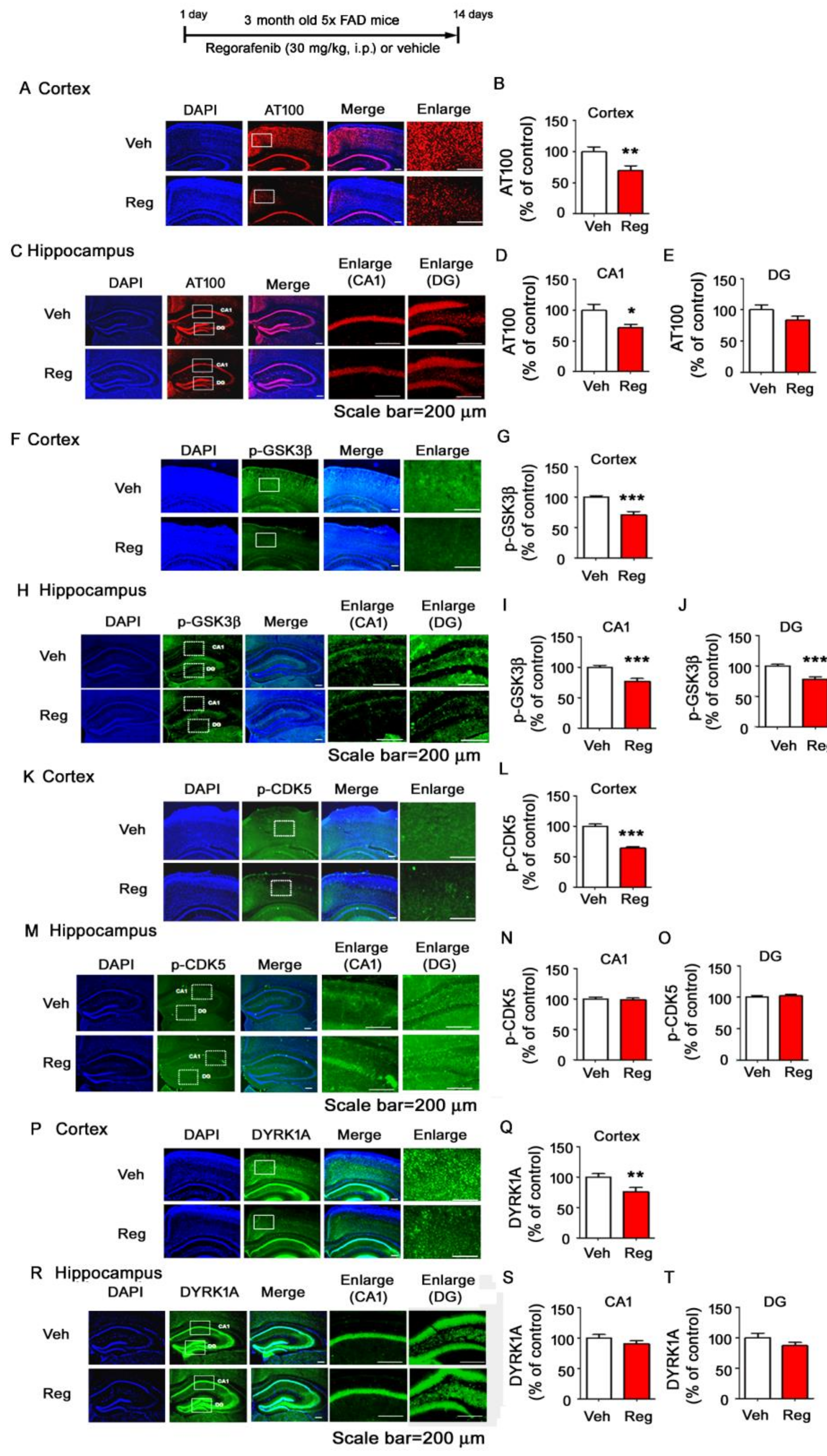
3.7. Regorafenib Significantly Reduces Tau Phosphorylation at T212 and S214 and Tau Kinase GSK3β Activity in the Brains of 5x FAD Mice
4. Discussion
Supplementary Materials
Author Contributions
Funding
Acknowledgments
Conflicts of Interest
References
- Fischer, P.; Zehetmayer, S.; Jungwirth, S.; Weissgram, S.; Krampla, W.; Hinterberger, M.; Torma, S.; Rainer, M.; Huber, K.; Hoenigschnabl, S.; et al. Risk factors for Alzheimer dementia in a community-based birth cohort at the age of 75 years. Dement Geriatr. Cogn. Disord. 2008, 25, 501–507. [Google Scholar] [CrossRef] [PubMed]
- Hamley, I.W. The amyloid beta peptide: A chemist’s perspective. Role in Alzheimer’s and fibrillization. Chem. Rev. 2012, 112, 5147–5192. [Google Scholar] [CrossRef] [PubMed]
- Zempel, H.; Mandelkow, E. Lost after translation: Missorting of Tau protein and consequences for Alzheimer disease. Trends Neurosci. 2014, 37, 721–732. [Google Scholar] [CrossRef] [PubMed]
- Kumar, K.; Kumar, A.; Keegan, R.M.; Deshmukh, R. Recent advances in the neurobiology and neuropharmacology of Alzheimer’s disease. Biomed. Pharm. 2018, 98, 297–307. [Google Scholar] [CrossRef]
- Roberson, E.D.; Halabisky, B.; Yoo, J.W.; Yao, J.; Chin, J.; Yan, F.; Wu, T.; Hamto, P.; Devidze, N.; Yu, G.Q.; et al. Amyloid-beta/Fyn-induced synaptic, network, and cognitive impairments depend on tau levels in multiple mouse models of Alzheimer’s disease. J. Neurosci. 2011, 31, 700–711. [Google Scholar] [CrossRef]
- Neumann, H.; Kotter, M.R.; Franklin, R.J. Debris clearance by microglia: An essential link between degeneration and regeneration. Brain 2009, 132, 288–295. [Google Scholar] [CrossRef]
- Cai, Z.; Hussain, M.D.; Yan, L.J. Microglia, neuroinflammation, and beta-amyloid protein in Alzheimer’s disease. Int. J. Neurosci. 2014, 124, 307–321. [Google Scholar] [CrossRef]
- Kirkley, K.S.; Popichak, K.A.; Afzali, M.F.; Legare, M.E.; Tjalkens, R.B. Microglia amplify inflammatory activation of astrocytes in manganese neurotoxicity. J. Neuroinflamm. 2017, 14, 99. [Google Scholar] [CrossRef]
- Hauss-Wegrzyniak, B.; Dobrzanski, P.; Stoehr, J.D.; Wenk, G.L. Chronic neuroinflammation in rats reproduces components of the neurobiology of Alzheimer’s disease. Brain Res. 1998, 780, 294–303. [Google Scholar] [CrossRef]
- Ettrich, T.J.; Seufferlein, T. Regorafenib. Recent Results Cancer Res. 2018, 211, 45–56. [Google Scholar] [CrossRef]
- Ravi, S.; Singal, A.K. Regorafenib: An evidence-based review of its potential in patients with advanced liver cancer. Core Evid. 2014, 9, 81–87. [Google Scholar] [CrossRef]
- Strumberg, D.; Scheulen, M.E.; Schultheis, B.; Richly, H.; Frost, A.; Buchert, M.; Christensen, O.; Jeffers, M.; Heinig, R.; Boix, O.; et al. Regorafenib (BAY 73-4506) in advanced colorectal cancer: A phase I study. Br. J. Cancer 2012, 106, 1722–1727. [Google Scholar] [CrossRef]
- Nam, J.H.; Cho, H.J.; Kang, H.; Lee, J.Y.; Jung, M.; Chang, Y.C.; Kim, K.; Hoe, H.S. A Mercaptoacetamide-Based Class II Histone Deacetylase Inhibitor Suppresses Cell Migration and Invasion in Monomorphic malignant human Glioma cells by inhibiting FAK/STAT3 signaling. J. Cell Biochem. 2017, 118, 4672–4685. [Google Scholar] [CrossRef] [PubMed]
- Lee, J.Y.; Nam, J.H.; Nam, Y.; Nam, H.Y.; Yoon, G.; Ko, E.; Kim, S.B.; Bautista, M.R.; Capule, C.C.; Koyanagi, T.; et al. The small molecule CA140 inhibits the neuroinflammatory response in wild-type mice and a mouse model of AD. J. Neuroinflamm. 2018, 15, 286. [Google Scholar] [CrossRef] [PubMed]
- Ryu, K.Y.; Lee, H.J.; Woo, H.; Kang, R.J.; Han, K.M.; Park, H.; Lee, S.M.; Lee, J.Y.; Jeong, Y.J.; Nam, H.W.; et al. Dasatinib regulates LPS-induced microglial and astrocytic neuroinflammatory responses by inhibiting AKT/STAT3 signaling. J. Neuroinflamm. 2019, 16, 190. [Google Scholar] [CrossRef] [PubMed]
- Nam, Y.; Joo, B.; Lee, J.Y.; Han, K.M.; Ryu, K.Y.; Koh, Y.H.; Kim, J.; Koo, J.W.; We, Y.M.; Hoe, H.S. ALWPs improve cognitive function and regulate Abeta plaque and tau Hyperphosphorylation in a mouse model of Alzheimer’s disease. Front. Mol. Neurosci. 2019, 12, 192. [Google Scholar] [CrossRef] [PubMed]
- Uschner, F.E.; Schueller, F.; Nikolova, I.; Klein, S.; Schierwagen, R.; Magdaleno, F.; Groschl, S.; Loosen, S.; Ritz, T.; Roderburg, C.; et al. The multikinase inhibitor regorafenib decreases angiogenesis and improves portal hypertension. Oncotarget 2018, 9, 36220–36237. [Google Scholar] [CrossRef]
- Yeh, C.N.; Chen, M.H.; Chen, Y.Y.; Yang, C.Y.; Yen, C.C.; Tzen, C.Y.; Chen, L.T.; Chen, J.S. A phase II trial of regorafenib in patients with metastatic and/or a unresectable gastrointestinal stromal tumor harboring secondary mutations of exon 17. Oncotarget 2017, 8, 44121–44130. [Google Scholar] [CrossRef]
- Cyran, C.C.; Kazmierczak, P.M.; Hirner, H.; Moser, M.; Ingrisch, M.; Havla, L.; Michels, A.; Eschbach, R.; Schwarz, B.; Reiser, M.F.; et al. Regorafenib effects on human colon carcinoma xenografts monitored by dynamic contrast-enhanced computed tomography with immunohistochemical validation. PLoS ONE 2013, 8, e76009. [Google Scholar] [CrossRef]
- Dhawan, G.; Combs, C.K. Inhibition of Src kinase activity attenuates amyloid associated microgliosis in a murine model of Alzheimer’s disease. J. Neuroinflamm. 2012, 9, 117. [Google Scholar] [CrossRef]
- Nam, H.Y.; Nam, J.H.; Yoon, G.; Lee, J.Y.; Nam, Y.; Kang, H.J.; Cho, H.J.; Kim, J.; Hoe, H.S. Ibrutinib suppresses LPS-induced neuroinflammatory responses in BV2 microglial cells and wild-type mice. J. Neuroinflamm. 2018, 15, 271. [Google Scholar] [CrossRef] [PubMed]
- Le, Q.; Daniel, R.; Chung, S.W.; Kang, A.D.; Eisenstein, T.K.; Sultzer, B.M.; Simpkins, H.; Wong, P.M. Involvement of C-Abl tyrosine kinase in lipopolysaccharide-induced macrophage activation. J. Immunol. 1998, 160, 3330–3336. [Google Scholar] [PubMed]
- Roseweir, A.K.; Powell, A.; Horstman, S.L.; Inthagard, J.; Park, J.H.; McMillan, D.C.; Horgan, P.G.; Edwards, J. Src family kinases, HCK and FGR, associate with local inflammation and tumour progression in colorectal cancer. Cell. Signal. 2019, 56, 15–22. [Google Scholar] [CrossRef] [PubMed]
- Li, Y.; Weng, Y.; Zhong, L.; Chong, H.; Chen, S.; Sun, Y.; Li, W.; Shi, Q. VEGFR3 inhibition chemosensitizes lung adenocarcinoma A549 cells in the tumor-associated macrophage microenvironment through upregulation of p53 and PTEN. Oncol. Rep. 2017, 38, 2761–2773. [Google Scholar] [CrossRef] [PubMed]
- Liu, Y.C.; Wu, R.H.; Wang, W.S. Regorafenib diminishes the expression and secretion of angiogenesis and metastasis associated proteins and inhibits cell invasion via NF-kappaB inactivation in SK-Hep1 cells. Oncol. Lett. 2017, 14, 461–467. [Google Scholar] [CrossRef] [PubMed]
- Edwards, J.P.; Emens, L.A. The multikinase inhibitor sorafenib reverses the suppression of IL-12 and enhancement of IL-10 by PGE (2) in murine macrophages. Int. Immunopharmacol. 2010, 10, 1220–1228. [Google Scholar] [CrossRef]
- Wang, H.; Huang, W.; Liang, M.; Shi, Y.; Zhang, C.; Li, Q.; Liu, M.; Shou, Y.; Yin, H.; Zhu, X.; et al. (+)-JQ1 attenuated LPS-induced microglial inflammation via MAPK/NFkappaB signaling. Cell Biosci. 2018, 8, 60. [Google Scholar] [CrossRef]
- Chai, H.; Luo, A.Z.; Weerasinghe, P.; Brown, R.E. Sorafenib downregulates ERK/Akt and STAT3 survival pathways and induces apoptosis in a human neuroblastoma cell line. Int. J. Clin. Exp. Pathol. 2010, 3, 408–415. [Google Scholar]
- Huang, S.P.; Guan, X.; Kai, G.Y.; Xu, Y.Z.; Xu, Y.; Wang, H.J.; Pang, T.; Zhang, L.Y.; Liu, Y. Broussonin E suppresses LPS-induced inflammatory response in macrophages via inhibiting MAPK pathway and enhancing JAK2-STAT3 pathway. Chin. J. Nat. Med. 2019, 17, 372–380. [Google Scholar] [CrossRef]
- Coppo, P.; Flamant, S.; De Mas, V.; Jarrier, P.; Guillier, M.; Bonnet, M.L.; Lacout, C.; Guilhot, F.; Vainchenker, W.; Turhan, A.G. BCR-ABL activates STAT3 via JAK and MEK pathways in human cells. Br. J. Haematol. 2006, 134, 171–179. [Google Scholar] [CrossRef]
- Bisht, K.; Sharma, K.; Tremblay, M.E. Chronic stress as a risk factor for Alzheimer’s disease: Roles of microglia-mediated synaptic remodeling, inflammation, and oxidative stress. Neurobiol. Stress 2018, 9, 9–21. [Google Scholar] [CrossRef] [PubMed]
- Echeverria, V.; Burgess, S.; Gamble-George, J.; Zeitlin, R.; Lin, X.; Cao, C.; Arendash, G.W. Sorafenib inhibits nuclear factor kappa B, decreases inducible nitric oxide synthase and cyclooxygenase-2 expression, and restores working memory in APPswe mice. Neuroscience 2009, 162, 1220–1231. [Google Scholar] [CrossRef] [PubMed]
- Mulder, S.F.; Bertens, D.; Desar, I.M.; Vissers, K.C.; Mulders, P.F.; Punt, C.J.; van Spronsen, D.J.; Langenhuijsen, J.F.; Kessels, R.P.; van Herpen, C.M. Impairment of cognitive functioning during Sunitinib or Sorafenib treatment in cancer patients: A cross sectional study. BMC Cancer 2014, 14, 219. [Google Scholar] [CrossRef] [PubMed]
- Du, C.; Shao, X.; Zhu, R.; Li, Y.; Zhao, Q.; Fu, D.; Gu, H.; Kong, J.; Luo, L.; Long, H.; et al. NMR-Based Metabolic Profiling Reveals Neurochemical Alterations in the Brain of Rats Treated with Sorafenib. Neurotox. Res. 2015, 28, 290–301. [Google Scholar] [CrossRef]
- Petry, F.D.S.; Coelho, B.P.; Gaelzer, M.M.; Kreutz, F.; Guma, F.; Salbego, C.G.; Trindade, V.M.T. Genistein protects against amyloid-beta-induced toxicity in SH-SY5Y cells by regulation of Akt and Tau phosphorylation. Phytother. Res. 2020, 34, 796–807. [Google Scholar] [CrossRef]
- Williamson, R.; Scales, T.; Clark, B.R.; Gibb, G.; Reynolds, C.H.; Kellie, S.; Bird, I.N.; Varndell, I.M.; Sheppard, P.W.; Everall, I.; et al. Rapid tyrosine phosphorylation of neuronal proteins including tau and focal adhesion kinase in response to amyloid-beta peptide exposure: Involvement of Src family protein kinases. J. Neurosci. 2002, 22, 10–20. [Google Scholar] [CrossRef]
- Bagheri, M.; Roghani, M.; Joghataei, M.T.; Mohseni, S. Genistein inhibits aggregation of exogenous amyloid-beta 1–40 and alleviates astrogliosis in the hippocampus of rats. Brain Res. 2012, 1429, 145–154. [Google Scholar] [CrossRef]
- Nilsson, P.; Saido, T.C. Dual roles for autophagy: Degradation and secretion of Alzheimer’s disease Abeta peptide. Bioessays 2014, 36, 570–578. [Google Scholar] [CrossRef]
- Cho, S.J.; Yun, S.M.; Jo, C.; Lee, D.H.; Choi, K.J.; Song, J.C.; Park, S.I.; Kim, Y.J.; Koh, Y.H. SUMO1 promotes Abeta production via the modulation of autophagy. Autophagy 2015, 11, 100–112. [Google Scholar] [CrossRef]
- Wrasidlo, W.; Crews, L.A.; Tsigelny, I.F.; Stocking, E.; Kouznetsova, V.L.; Price, D.; Paulino, A.; Gonzales, T.; Overk, C.R.; Patrick, C.; et al. Neuroprotective effects of the anti-cancer drug sunitinib in models of HIV neurotoxicity suggests potential for the treatment of neurodegenerative disorders. Br. J. Pharm. 2014, 171, 5757–5773. [Google Scholar] [CrossRef]








© 2020 by the authors. Licensee MDPI, Basel, Switzerland. This article is an open access article distributed under the terms and conditions of the Creative Commons Attribution (CC BY) license (http://creativecommons.org/licenses/by/4.0/).
Share and Cite
Han, K.-M.; Kang, R.J.; Jeon, H.; Lee, H.-j.; Lee, J.-S.; Park, H.; Gak Jeon, S.; Suk, K.; Seo, J.; Hoe, H.-S. Regorafenib Regulates AD Pathology, Neuroinflammation, and Dendritic Spinogenesis in Cells and a Mouse Model of AD. Cells 2020, 9, 1655. https://doi.org/10.3390/cells9071655
Han K-M, Kang RJ, Jeon H, Lee H-j, Lee J-S, Park H, Gak Jeon S, Suk K, Seo J, Hoe H-S. Regorafenib Regulates AD Pathology, Neuroinflammation, and Dendritic Spinogenesis in Cells and a Mouse Model of AD. Cells. 2020; 9(7):1655. https://doi.org/10.3390/cells9071655
Chicago/Turabian StyleHan, Kyung-Min, Ri Jin Kang, Hyongjun Jeon, Hyun-ju Lee, Ji-Soo Lee, HyunHee Park, Seong Gak Jeon, Kyoungho Suk, Jinsoo Seo, and Hyang-Sook Hoe. 2020. "Regorafenib Regulates AD Pathology, Neuroinflammation, and Dendritic Spinogenesis in Cells and a Mouse Model of AD" Cells 9, no. 7: 1655. https://doi.org/10.3390/cells9071655
APA StyleHan, K.-M., Kang, R. J., Jeon, H., Lee, H.-j., Lee, J.-S., Park, H., Gak Jeon, S., Suk, K., Seo, J., & Hoe, H.-S. (2020). Regorafenib Regulates AD Pathology, Neuroinflammation, and Dendritic Spinogenesis in Cells and a Mouse Model of AD. Cells, 9(7), 1655. https://doi.org/10.3390/cells9071655